

Abstract
Genetic diseases involving overactivation of the mechanistic target of rapamycin (mTOR) pathway, so-called “mTORopathies,” often manifest with malformations of cortical development (MCDs), epilepsy, and cognitive impairment. How mTOR pathway hyperactivation results in abnormal human cortical development is poorly understood. To study the effect of mTOR hyperactivity on early stages of cortical development, we focused on Pretzel Syndrome (polyhydramnios, megalencephaly, symptomatic epilepsy; PMSE syndrome), a rare mTORopathy caused by homozygous germline mutations in the STRADA gene. We developed a human cortical organoid (hCO) model of PMSE and examined morphology and size for the first 2 weeks of organoid growth, and cell type composition at weeks 2, 8, and 12 of differentiation. In the second week, PMSE hCOs enlarged more rapidly than controls and displayed an abnormal Wnt pathway-dependent increase in neural rosette structures. PMSE hCOs also exhibited delayed neurogenesis, decreased subventricular zone progenitors, increased proliferation and cell death, and an abnormal architecture of primary cilia. At week 8, PMSE hCOs had fewer deep layer neurons. By week 12, neurogenesis recovered in PMSE organoids, but they displayed increased outer radial glia, a cell type thought to contribute to the expansion of the human cerebral cortex. Together, these findings suggest that megalencephaly in PMSE arises from the expansion of neural stem cells in early corticogenesis and potentially also from increased outer radial glial at later gestational stages. The delayed neuronal differentiation in PMSE organoids demonstrates the important role the mTOR pathway plays in the maintenance and expansion of the stem cell pool.
Keywords: cerebral organoids, corticogenesis, induced pluripotent stem cells, mechanistic target of rapamycin, megalencephaly, mTOR, neurogenesis, polyhydramnios, megalencephaly, symptomatic epilepsy, Pretzel Syndrome, stem cell model, STRADA
1 |. INTRODUCTION
Genetic mutations that result in hyperactivation of the mechanistic target of rapamycin (mTOR) pathway cause “mTORopathies,” a group of disorders that manifest with epilepsy, variable cognitive impairment, and malformations of cortical development (MCD) such as focal cortical dysplasia, hemimegalencephaly, and megalencephaly (Crino, 2016). Determining how mTOR pathway hyperactivation results in MCD is important to uncover novel therapeutic targets and to reveal basic mechanisms of cortical development. mTORopathies often manifest with focal MCD as a result of somatic mutations or heterozygous germline mutations, for example, in TSC1, TSC2, DEPDC5 genes, combined with a “second hit” somatic mutation (Ye et al., 2019). These somatic mutations can occur at various time points during corticogenesis, affecting the extent of the FCD. In order to learn how mTOR pathway hyperactivity affects the earliest stages of corticogenesis, we studied “Pretzel Syndrome” or polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE, OMIM#611087), a mTORopathy that is caused by homozygous, truncating, germline mutations in the STE20-related kinase adaptor alpha (STRADA) gene. PMSE was first described in the Old Order Mennonite community, where a series of individuals are homozygous for an inherited founder mutation that results in deletion of exons 9–13 of STRADA (Puffenberger et al., 2007). The cardinal features of PMSE include severe, global impairment of neurodevelopment, hypotonia, megalencephaly, cerebral ventriculomegaly, craniofacial dysmorphism, and early-onset, drug-resistant epilepsy. Five individuals with PMSE were treated with rapamycin, an mTOR complex 1 (mTORC1) inhibitor, with improvement in seizures and possible improvement of neurocognitive deficits (Parker et al., 2013). PMSE has also been identified outside the Mennonite population in two individuals with homozygous frameshift mutations in STRADA (Bi et al., 2016; Evers et al., 2017).
The STRADA protein is a pseudokinase and an upstream regulator of mTORC1 (Baas et al., 2003). STRADA complexes with LKB1 and MO25 and activates the kinase activity of LKB1 (Boudeau et al., 2003; Hawley et al., 2003). LKB1 phosphorylates and activates adenosine monophosphate-activated kinase (AMPK), which activates the tuberous sclerosis 1/2 complex (TSC1/TSC2), an inhibitor of mTORC1. Therefore, loss of STRADA results in disinhibition of mTORC1. An autopsy of one individual with PMSE demonstrated an enlarged brain size, cytomegalic cortical neurons, and heterotopic neurons in the subcortical white matter exhibiting markers of mTORC1 hyperactivation including increased phosphorylated S6 kinase1, phosphorylated S6 (pS6), and c-Myc proteins (Orlova et al., 2010).
Previous studies have used mouse models to determine the effect of Strada loss-of-function on cortical development. In utero electroporation of mouse fetal forebrain at embryonic day 14 (E14) with short hairpin RNA (shRNA) targeting Strada caused abnormal cortical lamination, with the accumulation of Strada knockdown cells in the ventricular zone (VZ) and subventricular zone (SVZ) at E17 and E19 (Orlova et al., 2010). These findings suggested an important role for mTOR signaling in cell fate determination or cell migration in neural stem cells. Germline homozygous knockout (KO) of Strada resulted in perinatal lethality, with the recapitulation of various features of PMSE including cerebral ventriculomegaly and enhanced numbers of white matter heterotopic neurons showing exuberant S6 phosphorylation (Dang et al., 2020).
Human neural development has important differences from that of the rodent including increased brain volume, a greater diversity of cell types, and a greatly expanded pool of outer radial glia (oRG), an SVZ progenitor cell that is thought to contribute to the significantly expanded superficial layers of the human cortex (Lui et al., 2011). To better understand the role of STRADA in human neural development and disease, a human-based model is needed to complement the animal models. Previously, we generated induced pluripotent stem cells (iPSCs) from two individuals with PMSE and differentiated them into cortical-like excitatory neurons in monolayer cultures (Dang et al., 2020). We found loss of STRADA expression, increased pS6 expression, and neuronal cytomegaly, validating that the iPSC neuron model of PMSE demonstrates mTORC1 hyperactivity.
In order to determine how the loss of STRADA and resultant mTOR pathway hyperactivity affects early corticogenesis in a three-dimensional model that better recapitulates aspects of the developing human cortex, we differentiated iPSCs from individuals with PMSE and controls into human cortical organoids (hCOs) (reviewed in Di Lullo & Kriegstein, 2017). hCOs allow for the in vitro analysis of early neurodevelopmental processes, an area of study that is otherwise limited by the difficulties in obtaining and studying human fetal tissue. hCOs display radially oriented neuroepithelium in neural rosette structures that correspond to the VZ of the developing cortex, followed by the development of an SVZ and cortical plate with deep and then superficial layer neurons at later stages. We found that PMSE hCOs displayed evidence of mTOR hyperactivation, increased size, increased and abnormal early neural rosette formation, delayed neurogenesis, disrupted primary cilia architecture, and an increase in oRG. Interestingly, the increase in neural rosette formation was dependent on the presence of a Wnt pathway agonist. These results demonstrate the importance of the mTOR pathway in neural stem cell regulation and suggest several potential mechanisms whereby loss of STRADA results in aberrant cortical development and megalencephaly in PMSE.
2 |. MATERIALS AND METHODS
2.1 |. Human subjects
Fibroblasts were isolated from skin biopsies of two female individuals with PMSE and reprogrammed into iPSCs as previously described (Dang et al., 2020). One iPSC line from each of the two individuals with PMSE was used in this study. Commercially available foreskin fibroblasts (GlobalStem, Rockville, Maryland, USA) were episomally reprogrammed using previously described methods (Tidball et al., 2017) and used as a control iPSC line. Additional control iPSC lines used in this study were derived from a 17-year-old female previously described as CC-1 (Tidball et al., 2016) and a healthy 1-year-old male subject described in a prior study (Liu et al., 2013). All skin biopsies were obtained with approval from Institutional Review Boards and with informed consent from the subjects or their parents.
2.2 |. Induced pluripotent stem cell culture
Stem cell colonies were maintained at 37°C, 5% CO2, in mTeSR1 media (Stemcell Technologies, Vancouver, British Columbia, Canada), on tissue culture plates pre-coated with Matrigel (1:100 dilution in DMEM/F12, BD Biosciences, San Jose, California, USA). iPSCs were passaged every 4–7 days using Dispase (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Cultures were confirmed myco-plasma negative via periodic testing with a PCR-based assay. Each line used for experiments had expression of pluripotency markers (NANOG, OCT3/4, SSEA, SOX2) (Dang et al., 2020; Liu et al., 2013; Tidball et al., 2016). Episomally reprogrammed lines tested negative for episomal reprogramming vector integration. Each line was assessed for chromosomal abnormalities by G-band karyotyping (Cell Line Genetics, Madison, Wisconsin, USA), single nucleotide polymorphism (SNP) chip microarray analysis (Infinium Core Exome-24 BeadChip, Illumina, San Diego, California, USA), or both. Both iPSC lines from the PMSE subjects had a normal karyotype. The SNP chip microarray revealed that the iPSC line from PMSE subject 1 contained a 3.9 Mb gain of genomic material at 3q25.32-q26.1, and the iPSC line from PMSE subject 2 contained a 12 Mb gain of genomic material at 12p13.33-p13.2.
2.3 |. Generation of cortical organoids
Cortical organoids were generated using a previously described protocol (Qian et al., 2016, 2018) with minor modifications. On day 0, iPSCs were dissociated to single cells using Accutase (Innovative Cell Technologies, San Diego, California, USA), and plated in mTeSR1 with 50 μM Y-27632 (Tocris Bioscience, Minneapolis, Minnesota, USA) into Aggrewell 800 microwell plates (Stemcell Technologies) at a density of 1–6 × 106 cells/well (each differentiation had a consistent cell density for all iPSC lines). On day 1, embryoid bodies were transferred to ultra-low attachment T-25 flasks (Corning, Corning, New York, USA), or 60 mm tissue culture suspension dishes (Corning), and the media was changed to A83/DM media comprised of DMEM/F-12, 20% knockout serum replacement, 1X GlutaMAX, 1X MEM non-essential amino acids, 55 μM 2-mercaptoethanol (Thermo Fisher Scientific), 2 μM Dorsomorphin (Tocris), and 2 μM A83–01 (Tocris). A83/DM media was changed daily on days 2–4. On days 5 and 6, half-volume of the media was replaced with CS media, containing DMEM/F-12 supplemented with 1X N2, 1X GlutaMAX, 1X MEM non-essential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml heparin, 1 μM CHIR-99021 (Cayman Chemical, Ann Arbor, Michigan, USA), and 1 μM SB-431542 (Cayman). On day 7, the hCOs were embedded in a 1:2 mixture of CS media and Matrigel in a 6-well plate, and after the diluted Matrigel solidified, CS media was added to each well. The media was changed on days 9, 11, and 13. On day 14, hCOs were mechanically dissociated from the Matrigel, and placed in a miniature bioreactor (Spin-Ω) with neural differentiation media (DMEM/F12, 1X N2, 1X B27 supplement without vitamin A, 1X GlutaMAX, 1X MEM non-essential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, 2.5 μg/ml insulin, and 55 μM 2-mercaptoethanol). From days 15–28, neural differentiation media without vitamin A was changed every other day. After day 28, the hCOs were cultured either in miniature bioreactors or on an orbital shaker. From day 29–70, the neural differentiation media included B27 supplement with vitamin A, and media was changed every other day. After day 70, the media was changed every other day with maturation media, consisting of Neurobasal, 1X B27 supplement with vitamin A, 1X GlutaMAX, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 ng/ml BDNF (Peprotech, Cranbury, New Jersey, USA), 20 ng/ml GDNF (Peprotech), 1 ng/ml TGF-β (Peprotech), 0.2 mM ascorbic acid (Sigma–Aldrich, St. Louis, Missouri, USA), and 0.5 mM dbcAMP (Sigma).
2.4 |. Immunohistochemistry
Cortical organoids were fixed in 4% paraformaldehyde for 20–30 min at 4°C, cryoprotected overnight with 30% sucrose in phosphate-buffered saline, and then embedded in Tissue-Tek OCT embedding medium (Sakura Finetek, Torrance, California, USA) and stored at −80°C. A Leica CM1850 cryostat was used to generate 20 μm thick sections that were collected on Superfrost Plus glass slides (Thermo Fisher) and stored at −20°C. For immunostaining, the sections were outlined with a lipid pen, and then washed 3 times with phosphate-buffered saline (PBS) for 5 min each. The sections were permeabilized with PBS with 0.2% Triton-X100 for 20 min and then incubated with blocking buffer (PBS with 0.05% Tween-20, 5% normal goat serum, and 1% bovine serum albumin) for 1 hr at room temperature (RT). The slides were incubated with primary antibodies (Table 1) diluted in blocking buffer overnight at 4°C in a humidified chamber. The slides were washed three times with PBS with 0.05% Tween-20 (PBS-T) for 10 min at RT, and incubated with AlexaFluor-conjugated secondary antibodies (Table 1) (Invitrogen, Carlsbad, California, USA) for 1–2 hr at RT. The slides were then washed once with PBS-T for 10 min at RT, and then incubated in 2 μg/ml bis-benzimide diluted in PBS for 5 min to label nuclei. The slides were washed three times with PBS-T for 10 min at RT, and mounted with Glycergel mounting medium (Agilent Dako, Santa Clara, California, USA). Images were obtained on a Leica SP5 confocal microscope using the Leica Application Suite Advanced Fluorescence software (Leica Microsystems Inc., Buffalo Grove, Illinois, USA).
TABLE 1.
Antibodies and dilutions
| Antigen | Species | Dilution | Vendor | Catalog# |
|---|---|---|---|---|
| Active Caspase-3 | Rabbit | 1:2,000 | BD Biosciences | 559565 |
| Arl13b | Mouse | 1:400 | NeuroMab | N295B/66 |
| Beta-3-tubulin | Rabbit | 1:1,000 | Abcam | Ab18207 |
| Beta-3-tubulin | Mouse | 1:2,000 | Biolegend | 801202 |
| CTIP2 | Rat | 1:600 | Abcam | Ab18465 |
| EOMES (TBR2) | Mouse | 1:400 | R&D Systems | MAB6166 |
| GFAP | Rabbit | 1:1,000 | Agilent Dako | Z0334 |
| HOPX | Rabbit | 1:1,000 | Sigma | HPA030180 |
| Ki67 | Rabbit | 1:2,000 | Thermo Fisher | MA5–14520 |
| p4E-BP1 | Rabbit | 1:200 | Cell Signaling | 2855 |
| PAX6 | Rabbit | 1:2,000 | MBL International | PD022 |
| pS6 | Rabbit | 1:100 | Cell Signaling | 2211 |
| SATB2 | Mouse | 1:200 | Abcam | Ab51502 |
| Anti-Mouse AlexaFluor 488 | Goat | 1:800 | Invitrogen | A-11001 |
| Anti-Mouse AlexaFluor 568 | Goat | 1:800 | Invitrogen | A-11031 |
| Anti-Mouse AlexaFluor 647 | Goat | 1:800 | Invitrogen | A-21236 |
| Anti-Rabbit AlexaFluor 488 | Goat | 1:800 | Invitrogen | A-11008 |
| Anti-Rabbit AlexaFluor 594 | Goat | 1:800 | Invitrogen | A-11037 |
| Anti-Rat AlexaFluor 488 | Goat | 1:800 | Invitrogen | A-11006 |
2.5 |. Image quantitation
Images were analyzed using ImageJ (NIH, Bethesda, MD, USA). With phase-contrast images, the area of hCOs was measured by manually outlining each hCO in ImageJ, and the number of neuroepithelial buds around the perimeter of each hCO was counted manually. The individual performing these analyses was blinded to the genotype and experimental conditions of the hCOs. Quantitation of Ki67 and active Capase-3 was performed by manually outlining each nucleus (by bisbenzimide staining) in three areas per image, and then using those regions of interest (ROIs) to measure the mean fluorescence of Ki67 or active Caspase-3. To adjust for batch-to-batch variability in background staining and overall fluorescence intensity, the mean fluorescence of each cell was converted to a Z-score by calculating the mean and standard deviation of all fluorescence values for a given batch of hCOs. Cells were determined to be positive for Ki67 or active Caspase-3 with a fluorescence Z-score of 1 or greater. The individual performing these analyses was blinded to the genotype of the hCOs.
For markers of mTORC1 activity, pS6 and phosphorylated 4E-BP1 (p4E-BP1), each hCO in an image was manually outlined based on the bisbenzimide staining and added as a ROI. The mean fluorescence of a given marker was then measured for each ROI. These values were also normalized to the bisbenzimide signal, in order to adjust for areas in the outlined ROI that did not contain cells. For the nuclear neurodevelopmental markers TBR2, PAX6, CTIP2, and SATB2, the percent of positive pixels in each image was measured for all pixels that also had bisbenzimide staining. A binary mask was created from the bisbenzimide image and added as a ROI. For each marker, the image was converted to binary, and then the percent area was measured for the ROI (which was limited to bisbenzimidepositive pixels). For the non-nuclear neural markers TUBB3, HOPX, and GFAP, each hCO in an image was manually outlined based on the bisbenzimide staining and added as a ROI. The mean fluorescence of a given marker was then measured for each ROI. These values were also normalized to the bisbenzimide signal, in order to adjust for areas in the outlined ROI that did not contain cells. These values were then converted to a Z-score to adjust for batch-to-batch variability. For Arl13b quantitation, the lumens of rosettes were manually outlined based on bisbenzimide and Arl13b staining, and this was used as the “intraluminal” ROI. The entirety of the hCOs was outlined, subtracting out the intraluminal regions, and this was used as the “extraluminal” ROI. The mean fluorescence of Arl13b was measured in the ROIs for each image and plotted.
Quantitation of each marker was performed on at least three independent biological replicates, with analysis of at least four organoids per cell line per replicate. Typically, one image was taken of each organoid. With the exception of the experiments to test the effect of CHIR-99021 on hCO size and budding, each experiment included data from both PMSE iPSC lines and multiple control lines.
2.6 |. Statistical methods
Results are expressed as mean ± SEM except for the Ki67 and active Caspase-3 data, which are mean ± standard deviation. Data were tabulated and analyzed using Excel (Microsoft, Seattle, Washington, USA). Prism (GraphPad, La Jolla, California, USA) was used for generating graphs and statistical analyses. Analysis of hCO size and number of buds over time was performed with two-way ANOVA with Sidak’s multiple comparisons test. Relative expression levels of pS6, p4E-BP1, TUBB3, TBR2, Ki67, active Caspase-3, Arl13b, PAX6, CTIP2, SATB2, HOPX, and GFAP were analyzed with a non-parametric Mann–Whitney test. For Ki67 and active Caspase-3, the proportion of positive cells was analyzed with Fisher’s exact test. The hCO size and number of buds with and without CHIR-99021 were analyzed with a non-parametric Kruskal–Wallis test with Dunn’s multiple comparisons test.
3 |. RESULTS
3.1 |. PMSE organoids exhibited increased mTORC1 activity
In order to validate that our PMSE organoid model demonstrated increased markers of mTORC1 activity, we assessed the levels of the downstream effectors pS6 and p4E-BP1. Consistent with our previous results from 2D neuronal differentiations of PMSE patient iPSCs (Dang et al., 2020), we found that PMSE hCOs at 14 days in vitro displayed robustly increased levels of pS6 and p4E-BP1 (Figure 1). Many of the positive cells were located around rosette lumens consistent with the location of neural stem cells, but the majority of cells in the organoids also showed increased expression.
FIGURE 1.
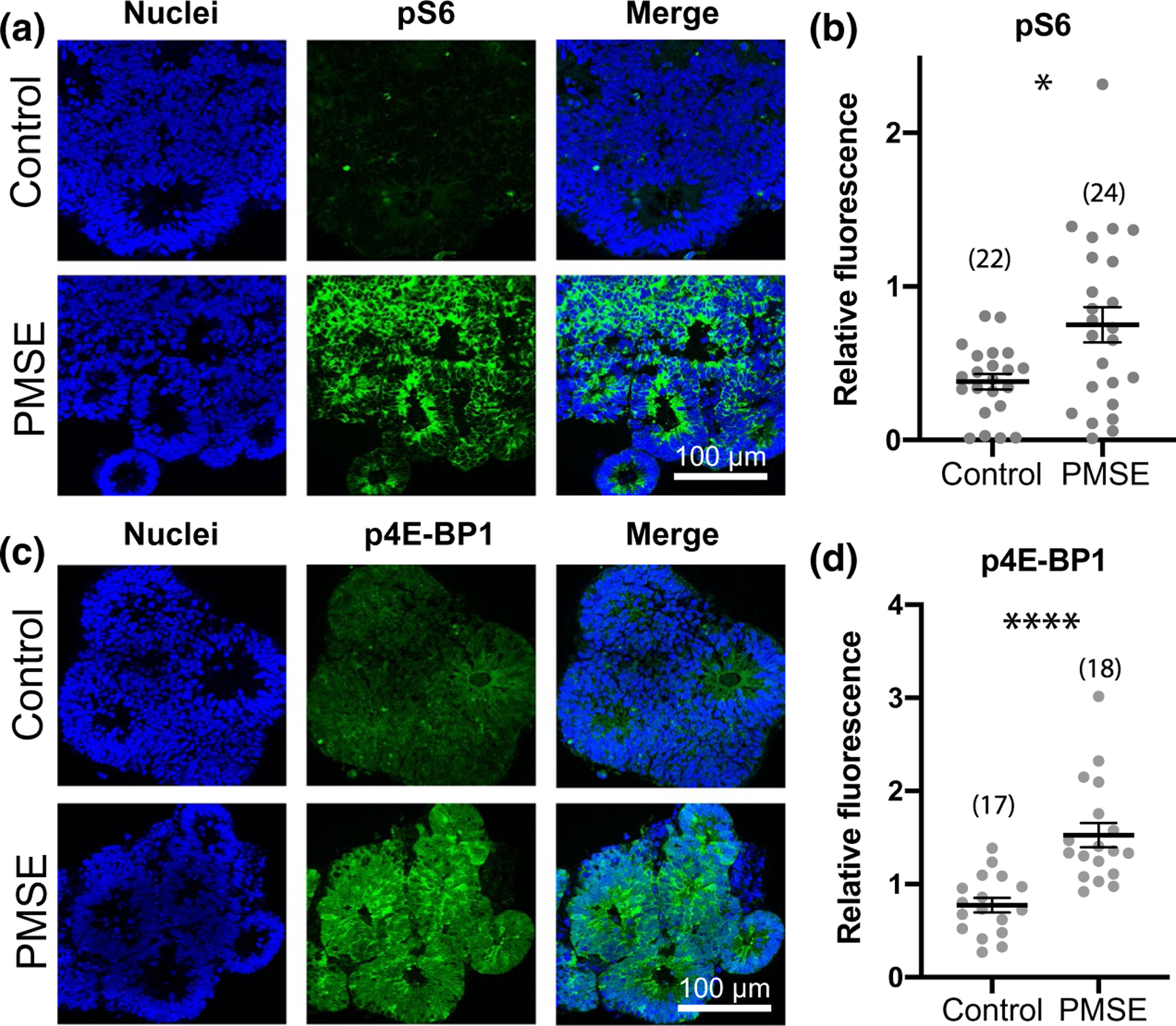
PMSE organoids at day 14 demonstrated increased mTOR complex 1 (mTORC1) activity. (a, c) PMSE hCOs exhibited increased phosphorylated S6 (pS6) and phosphorylated 4E-binding protein 1 (p4E-BP1) levels, most marked in the neural progenitors adjacent to the rosette lumens but also in most of the other cells present at this time point. (b, d) Quantification of pS6 and p4E-PB1 fluorescence, relative to the nuclear marker bisbenzimide revealed a significant increase in expression of mTORC1 activity markers in PMSE hCOs. Each dot represents one image of one organoid, and data are compiled from 3 separate differentiations with hCO numbers noted in parentheses. Mean ± SEM is displayed. *p < .05, ****p < .0001. Scale bars = 100 μm
3.2 |. PMSE organoids had increased size and neuroepithelial budding
To understand how the loss of STRADA and resultant mTOR pathway hyperactivity affect early corticogenesis, we analyzed hCOs generated from PMSE and control iPSCs. During the first week of differentiation, no differences were apparent in morphology of the embryoid bodies and hCOs. Starting on day 8, after Matrigel embedding of the hCOs, the PMSE group displayed altered morphology and increased size (approximately 35% larger by day 12) throughout the second week of differentiation (Figure 2a,b). The neural rosettes consisting of PAX6+ neural stem cells were 2.8-fold more numerous and appeared as small “buds” around the periphery of PMSE hCOs compared to the larger, fewer, and more contiguous radially organized PAX6+ neural stem cells in controls (Figure 2c,d). These differences persisted through the Matrigel-embedding period but abated in the third week of differentiation, after the hCOs were taken out of Matrigel and placed in the miniature spinning bioreactors at day 14 (data not shown).
FIGURE 2.
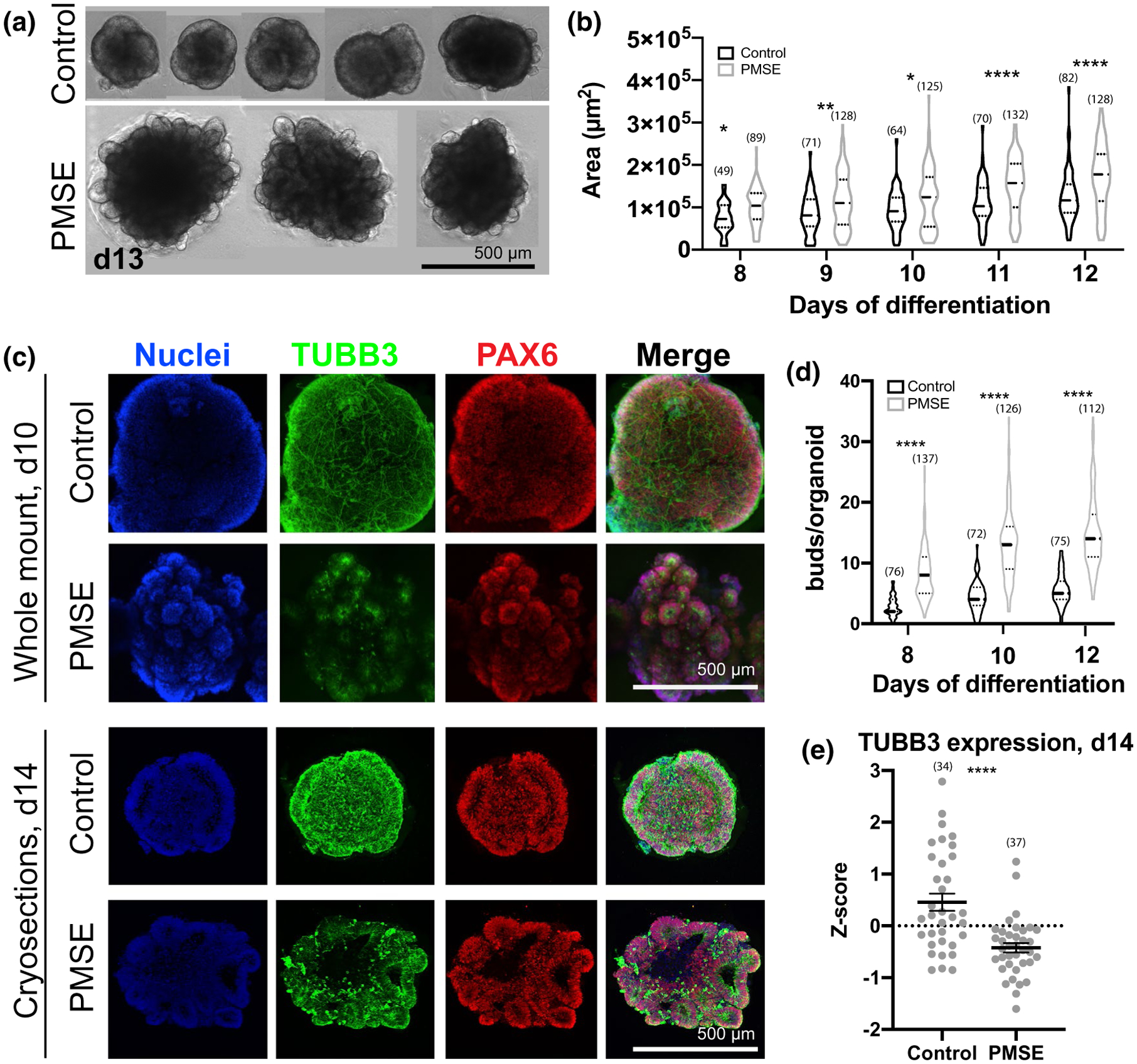
PMSE organoids at days 8–14 demonstrated increased size, increased neuroepithelial budding, and a decrease in neuronal differentiation. (a) Phase-contrast images of day (d) 13 hCOs demonstrated increased size, and prominent neuroepithelial budding in representative PMSE hCOs. (b) Violin plots showing quantitation of hCO size from phase-contrast images taken at 8–12 days in vitro. Each data point corresponded to a single hCO, and data were compiled from three separate differentiations. At each time point, PMSE hCOs were significantly larger than the controls. Solid lines represent the median, and the dotted lines represent the 25th and 75th percentiles. (c) Whole-mount day 10 (top two rows) and cryosectioned day 14 (bottom two rows) control and PMSE hCOs were immunostained with TUBB3 to label neurons and PAX6 to label neural stem cells. Control hCOs had increased TUBB3 expression and had neurons coursing through the hCO, particularly evident with whole-mount stains. The PAX6+ neuroepithelial buds were larger but fewer and more contiguous in control hCOs, and more numerous but smaller in PMSE. (d) Violin plots of the number of neuroepithelial buds around the perimeter of control and PMSE hCOs at days 8, 10, and 12, with each data point representing a single hCO. There were significantly greater neuroepithelial buds in PMSE at all time points. Solid lines represent the median, and the dotted lines represent the 25th and 75th percentiles. (e) Quantification of TUBB3 in day 14 cryosections demonstrated a lower level of expression in PMSE. Each dot represents one image, and data were compiled from four separate differentiations. Mean ± SEM is displayed. *p < .05, **p < .01, ***p < .001, ****p < .0001. Numbers of hCOs measured are noted in parentheses in all graphs. Scale bars = 500 μm
3.3 |. PMSE hCOs exhibited delayed neurogenesis, increased cell proliferation, and greater early apoptosis
At day 10, hCOs were harvested, fixed, and immunostained for whole-mount confocal imaging with maximum Z-stack projections (Figure 2c, top 2 rows). Control hCOs showed substantial numbers of Tubulin Beta 3 (TUBB3)-immunoreactive neurons and processes coursing through the hCOs, while PMSE hCOs had a paucity of TUBB3 expression, although immunoreactivity was more conspicuous in PMSE rosettes compared to controls (Figure 2c). While whole neurons were not as apparent with cryosectioned and immunostained day 14 hCOs (Figure 2c, bottom), a significant decrease in TUBB3 expression in the PMSE group was again apparent (Figure 2e). At day 14, we also examined TBR2+ SVZ progenitors, one of the earliest cell types to be generated from radial glial stem cells. Control hCOs had a few scattered TBR2+ SVZ progenitors outside of the neural rosettes, but TBR2+ cells were rarely found in PMSE hCOs (Figure 3).
FIGURE 3.
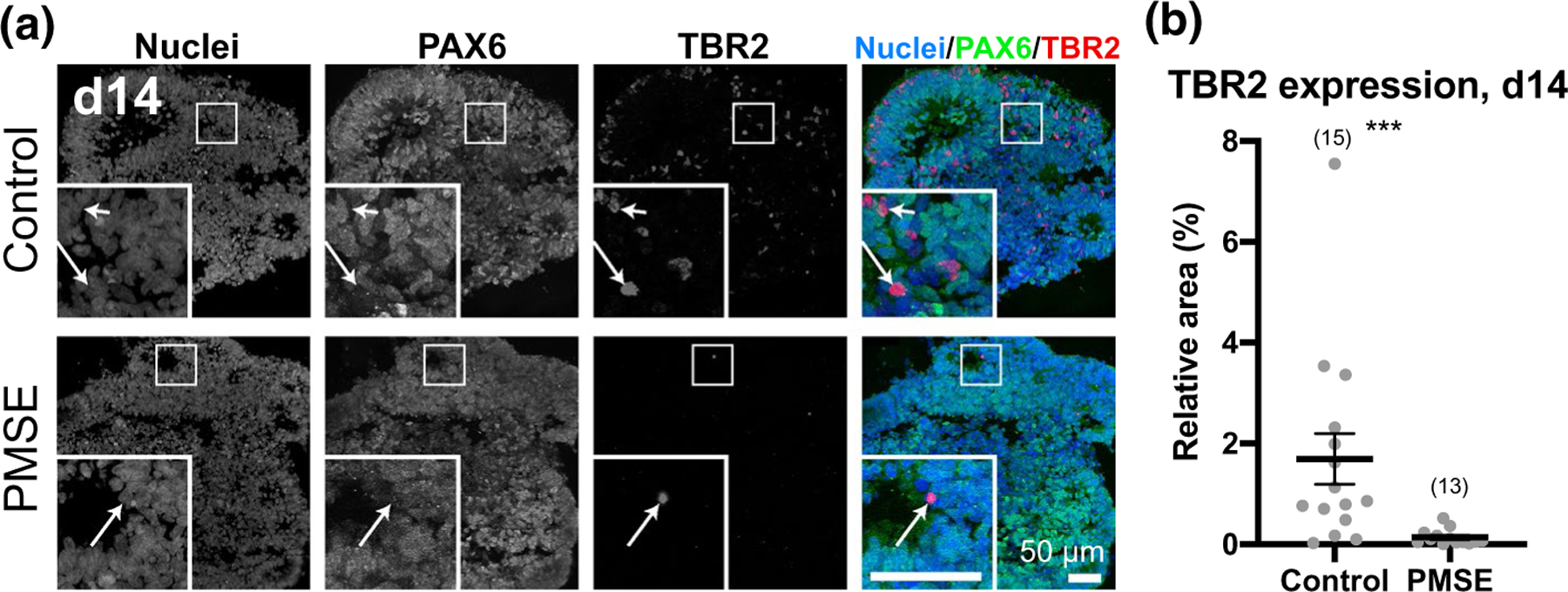
PMSE organoids exhibited decreased TBR2+ progenitors. (a) At day 14, control hCOs had scattered TBR2+ cells mostly at the outer edge of neural rosettes, and these were negative for PAX6 (insets, arrows). PMSE hCOs had very few TBR2+ cells at day 14. (b) Quantitation of the proportion of nuclear staining that was also TBR2+ revealed a significant decrease in TBR2 expression in PMSE hCOs. Each dot represents one image of one organoid, and data are compiled from three separate differentiations with hCO numbers noted in parentheses. Mean ± SEM is displayed. ***p < .001. Scale bars = 50 μm
In order to ascertain whether the increased size and neuroepithelial budding seen in PMSE hCOs results from increased cell proliferation, we immunostained day 14 hCOs for Ki67, a marker for proliferating cells. Both control and PMSE hCOs had Ki67+ cells located predominantly around the lumens of the neural rosettes (Figure 4a). We quantified the Ki67 signal on an individual cell level and found a continuum of expression levels rather than a bimodal distribution (Figure 4b). Therefore, for each of the four batches of immunostained slides, we used one standard deviation above the mean as a threshold for Ki67 positivity. In controls, 8.9% of cells were Ki67+ and in PMSE, 13.7% of cells were Ki67+, a significantly higher proportion (Figure 4c, p < .0001, Fisher’s exact test).
FIGURE 4.
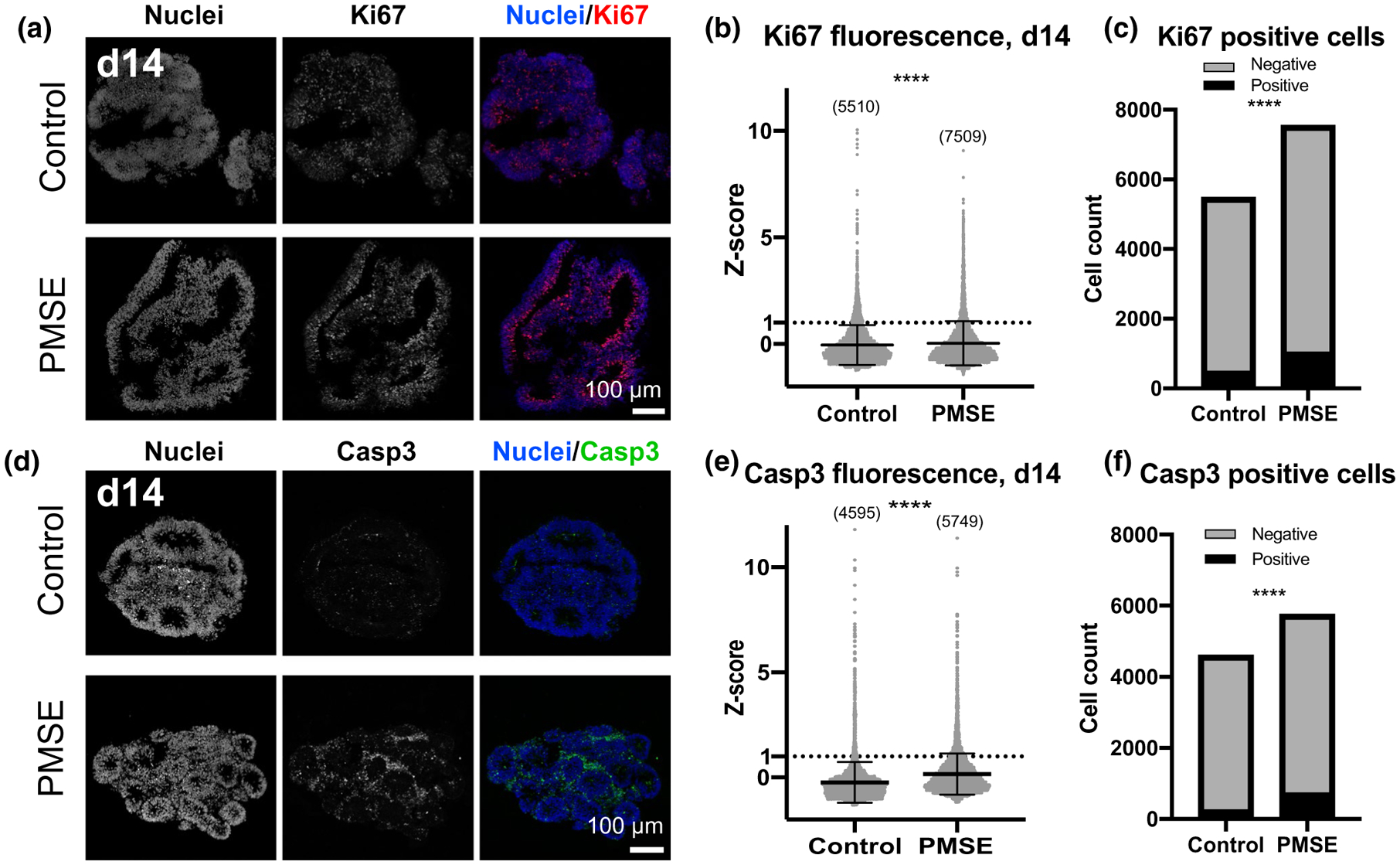
Increased proliferation and apoptosis in PMSE organoids. (a) At day 14, Ki67 immunoreactivity was present in the neural rosettes, often near the apical lumens, and more strongly in PMSE. (b) Scatter plots of Ki67 relative fluorescence demonstrated increased proliferation in PMSE hCOs. Each dot represents one cell, and data were compiled from four separate differentiations. (c) The percentage of Ki67-positive cells, defined as expression greater than one standard deviation above the mean, was significantly increased in PMSE (13.7%) compared to controls (8.9%). (d) Active Caspase-3 staining was also increased in day 14 hCOs, and the staining was most prominent outside the neural rosettes. (e) Scatter plots of active Caspase-3 relative fluorescence demonstrated increased apoptosis in PMSE hCOs. Analysis was done as described in panel b. (f) The percentage of active Caspase-3-positive cells, defined as expression greater than one standard deviation above the mean, was significantly increased in PMSE (12.6%) compared to controls (5.4%). Mean ± SD is displayed. ****p < .0001. Cell numbers in panels b and e are noted in parentheses. Scale bars = 100 μm
To assess for apoptotic cells, we labeled with active Caspase-3 and found positive cells surrounding the neural rosettes in both controls and PMSE hCOs (Figure 4d). Similar to the Ki67 analysis, we measured the active Caspase-3 fluorescence of individual cells and found increased expression in PMSE (Figure 4e). We then determined the proportion of active Caspase-3 fluorescence by using one standard deviation above the mean as a threshold. Unexpectedly, there was a higher proportion of positive cells in PMSE hCOs (12.6%) compared to controls (5.4%) (Figure 4f, p < .0001, Fisher’s exact test). Together, these findings suggest that in PMSE the increased proliferation is not entirely offset by the increase in cell death, leading to early hCO expansion, and that the decreased neurogenesis is secondary to delayed neural stem cell differentiation perhaps combined with death of cells that would have become TBR2+ SVZ progenitors.
3.4 |. Increased neuroepithelial budding in PMSE hCOs was dependent on a Wnt pathway agonist
The mTOR and Wnt signaling pathways have significant interactions (Shimobayashi & Hall, 2014) and Wnt pathway activation has been found to increase neuroepithelial buds in hCOs modeling lissencephaly (Iefremova et al., 2017). We hypothesized that the presence of CHIR-99021, a GSK3-β inhibitor/Wnt pathway agonist used in the hCO differentiation protocol from day 5–14, would have an additive effect with mTOR pathway activation to generate the increase in neuroepithelial buds. We differentiated a control line and a PMSE line from subject 2 into hCOs using two different media conditions: one that included CHIR-99021 according to the usual protocol, and one that omitted CHIR-99021. In two replicate differentiations, we found that CHIR-99021 was necessary for the increase in neuroepithelial budding in PMSE, but that its presence did not significantly affect the PMSE hCO size (Figure 5). The absence of CHIR-99021 did not affect the size or neuroepithelial budding of the control hCOs.
FIGURE 5.
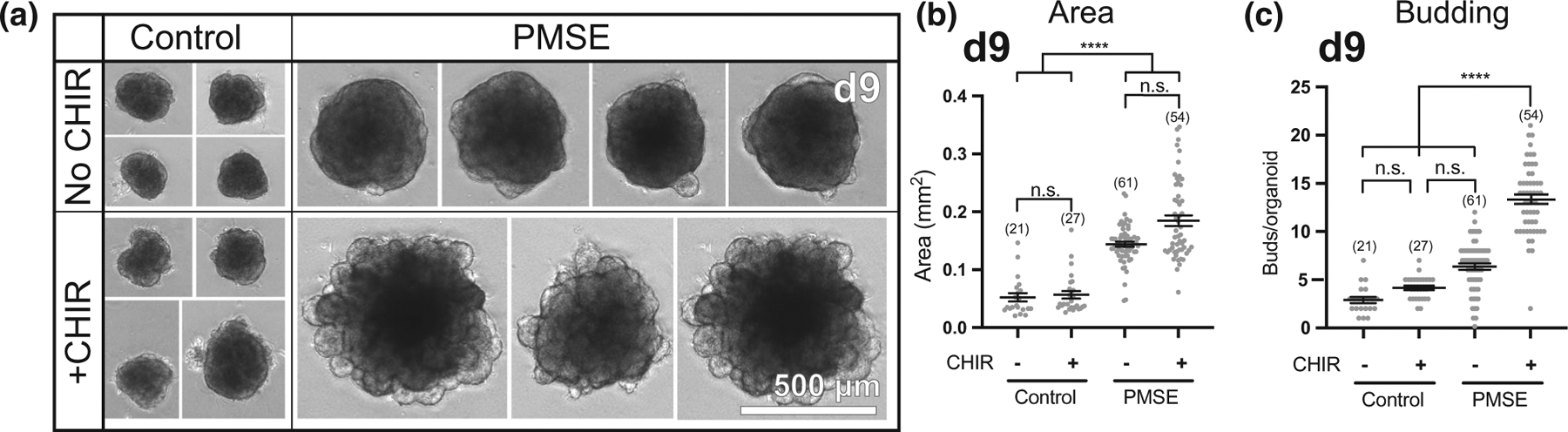
Increased neuroepithelial budding in PMSE hCOs was dependent on exposure to a Wnt pathway agonist. (a) When CHIR-99021 was not included in the differentiation media (top), day 9 PMSE hCOs did not display robust neuroepithelial budding while control hCOs were not affected. (b) Quantitation of hCO area, with and without CHIR-99021, redemonstrated that PMSE hCOs were larger than controls, but this increased size was not dependent on the presence of CHIR-99021. (c) Quantification of neuroepithelial buds around the perimeter of hCOs demonstrated that the increase in budding in PMSE hCOs is dependent on CHIR-99021. The presence or absence of CHIR-99021 did not affect budding in control hCOs. Each dot represents a single hCO. ****p < .0001. Mean ± SEM is displayed. Organoid numbers are noted in parentheses. Data were compiled from two independent experiments. Scale bars = 500 μm
3.5 |. PMSE organoids displayed increased extraluminal primary cilia
Primary cilia are specialized organelles that are found on the apical surface of radial glia as well as other neural progenitors and neurons, and they have a reciprocal regulatory role with several developmental pathways, including Wnt and mTOR (Lancaster et al., 2011; Lepanto et al., 2016). In mTORopathies, abnormal neuronal ciliogenesis likely contributes to cortical dyslamination (Park et al., 2018). To determine whether radial glial ciliogenesis is abnormal in PMSE, primary cilia in day 14 hCOs were labeled with Arl13b, a small GTPase found in cilia. Both control and PMSE hCOs had robust expression of Arl13b in the lumens of neural rosettes (Figure 6a,b). However, in PMSE hCOs, scattered Arl13b expression was also found outside the rosette lumens, on cells that comprised the rosettes and also on cells that were not incorporated into a rosette. Image quantitation demonstrated a significant increase in extraluminal Arl13b expression in PMSE hCOs (Figure 6c). This finding suggests that PMSE hCOs exhibit a disorganized structure with neural progenitors that have disrupted apical-basal polarity or impaired ability to arrange into normally structured neural rosettes.
FIGURE 6.
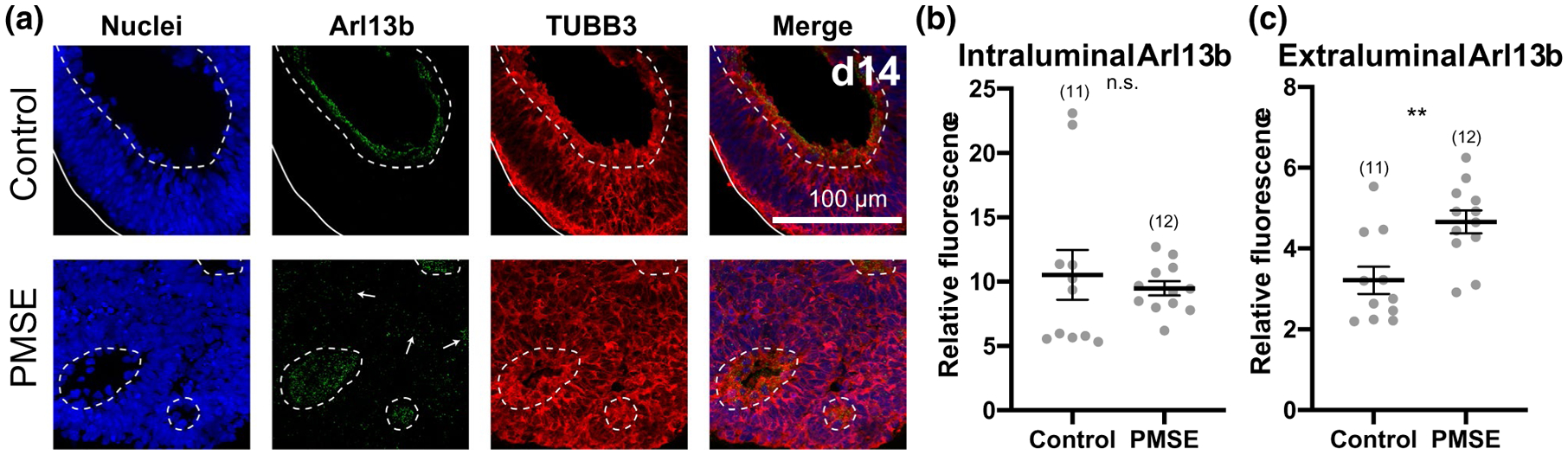
Primary cilia outside the lumens of neural rosettes were present in greater quantity in PMSE organoids at day 14. (a) Confocal images of representative Arl13b immunostained hCOs show Arl13b-positive primary cilia were present in neural rosette lumens in both control and PMSE organoids. In PMSE hCOs, more Arl13b expression was apparent outside the lumens of neural rosettes (arrows; lumens outlined by dashed lines) compared to control hCOs. (b) Intraluminal expression of Arl13b was not significantly different between control and PMSE hCOs. (c) Extraluminal expression of Arl13b was increased in PMSE hCOs compared to controls. **p < .01. Mean ± SEM is displayed. Each dot represents a separate image of an organoid. Data were compiled from three independent differentiations. hCO numbers are noted in parentheses. Scale bars = 100 μm
3.6 |. Neurogenesis recovered in PMSE organoids, but they exhibited increased outer radial glia
To determine whether neurogenesis recovers at later time points, and whether a bias exists toward early- or late-born neurons in PMSE hCOs, week 8 and week 12 hCOs were labeled for PAX6+ neural progenitors, CTIP2 (BCL11B)+ early-born neurons, and SATB2+ later-born neurons (Figure 7). By week 8, neural rosette structures appeared indistinguishable between PMSE and control organoids, but PMSE hCOs displayed significantly decreased CTIP2 expression with similar expression of PAX6 and SATB2, the latter at very low levels, compared to controls (Figure 7a,b). By week 12, the expression of PAX6, CTIP2, and SATB2 was not significantly different between PMSE and control hCOs (Figure 7c,d). Moreover, expression of the neuronal marker TUBB3 in 12-week hCOs did not differ significantly between control and PMSE groups (Figure 8a,c). These findings support a transient delay in neuronal differentiation in PMSE organoids that eventually recovers.
FIGURE 7.
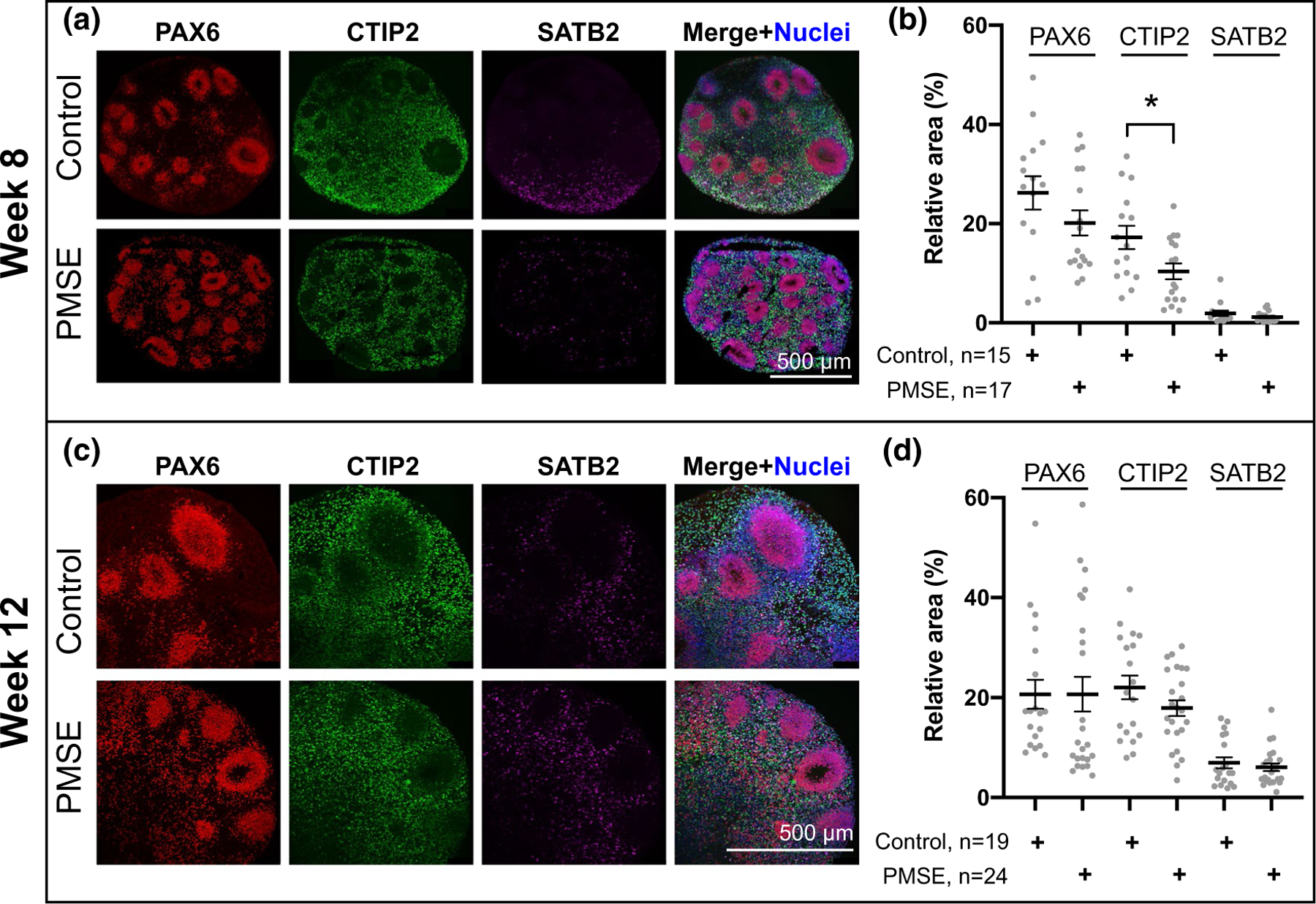
Progenitor and cortical neuronal layer markers at week 8 and 12 of differentiation. (a) At week 8, PAX6+ progenitors, CTIP2+ deep-layer neurons, and SATB2+ mid-upper-layer neurons were present in control and PMSE hCOs. (b) Quantitation of these markers demonstrated a significant decrease in CTIP2 expression in PMSE hCOs, *p < .05. (c) At week 12, there was increased SATB2 compared to week 8 (a), and both control and PMSE hCOs had similar numbers of PAX6-, CTIP2- and SATB2-positive cell numbers. (d) Quantitation of cell markers demonstrated no significant differences. Each dot represents a separate image of an organoid. Mean ± SEM is displayed. Data were compiled from three independent differentiations. Scale bars = 500 μm
FIGURE 8.
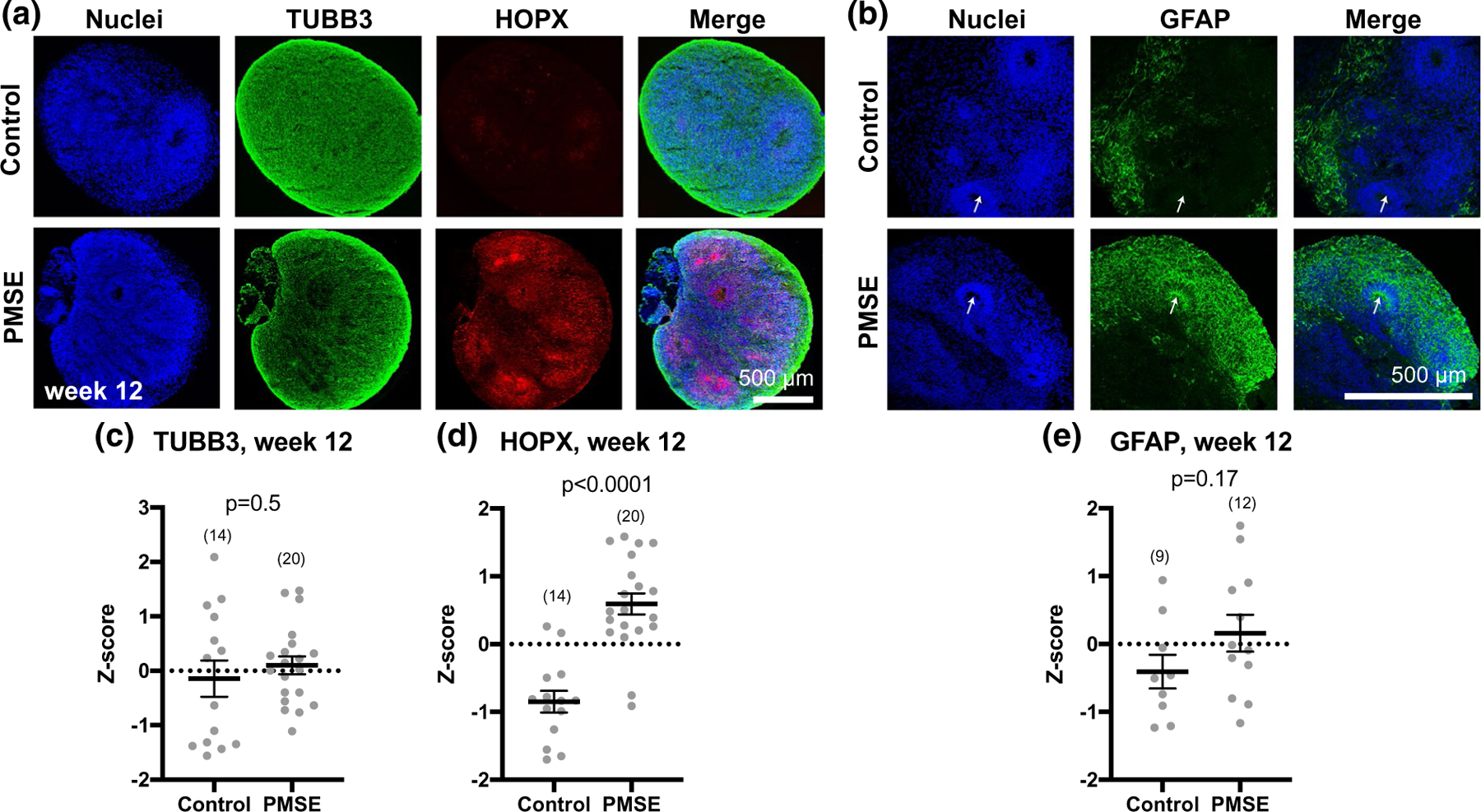
PMSE organoids at week 12 had increased outer radial glial, but no significant difference in neuronal or astroglial markers. (a) PMSE hCOs had significantly increased numbers of HOPX+ outer radial glia compared to controls, but similar numbers of TUBB3+ neurons. (b) GFAP expression was not significantly different in PMSE versus control hCOs, although the pattern of expression was different, with more GFAP+ staining in neural rosettes of PMSE hCOs (arrows). (c–e) Quantification of cell markers revealed a significant increase in HOPX expression in PMSE hCOs, with no significant differences in neurons or astrocytes. Mean ± SEM is displayed. Each dot represents a separate image of an organoid (total numbers in parentheses). Data were compiled from three independent differentiations. Scale bars = 500 μm
The mTOR pathway is important in the regulation of oRG (Andrews et al., 2020; Nowakowski et al., 2017), and oRG are a major contributor to the expansion of the human cortex (Hansen et al., 2010). At week 12, we found that there was a significant increase in HOPX+ oRG cells in PMSE hCOs compared to controls (Figure 8a,d). The glial marker GFAP was not significantly different between controls and PMSE (Figure 8b,e). Interestingly, while there were clusters of GFAP-expressing cells near the hCO surface in both PMSE and controls, prominent GFAP expression appeared in PMSE neural rosette structures but not in those of control hCOs (Figure 8b, arrows). The increase in oRG suggests another potential mechanism for megalencephaly in PMSE.
4 |. DISCUSSION
mTORopathies typically result from somatic mutations or heterozygous germline mutations, for example, TSC1, TSC2, or DEPDC5, combined with a “second hit” somatic mutation, to cause focal MCD (Ye et al., 2019). The developmental stage at which these somatic mutations occur determines how cortical development is affected. Our study of hCOs derived from PMSE patients with germline homozygous STRADA loss of function (Dang et al., 2020) provides a disease-relevant hCO model of how mTORC1 hyperactivity affects the earliest stages of corticogenesis. Our results suggest that loss of STRADA transiently promotes maintenance of a neural stem cell fate and delayed neurogenesis, based on decreased TUBB3 and TBR2 expression at day 14, and decreased CTIP2 expression at week 8. Combined with increased proliferation within the VZ, the retention of a neural stem cell fate expands the neural stem cell pool, reflected in the increased cell proliferation, size, and neuroepithelial budding observed in the second week of differentiation. The increase in apoptotic cells in PMSE was mostly outside of the VZ, suggesting that these cells were destined to become TBR2+ SVZ progenitors or neurons had they survived. The increased presence of primary cilia outside of the lumens of neural rosettes in PMSE hCOs at day 14 suggests structural disorganization of the hCOs or aberrant apical/basal polarity of radial glial cells and is consistent with cortical disorganization seen with mTORopathies.
Previous studies of mTOR pathway hyperactivation in hCOs demonstrated some similarities to the findings in the present study. Heterozygous Phosphatase and Tensin Homolog (PTEN) mutations are associated with PTEN Hamartoma Tumor Syndrome, and autism spectrum disorder with macrocephaly (Leslie & Longy, 2016). Human embryonic stem cells (hESCs) were genetically edited to have biallelic loss-of-function mutations in PTEN, an upstream regulator of mTORC1 via PI3K and AKT3, to investigate the effect on cortical development (Li et al., 2017). Similar to the STRADA-mutant hCOs described here, PTEN−/− hCOs showed a transient delay in neurogenesis and increased proliferation of neural stem cells. PTEN−/− hCOs also displayed morphological changes, with expansion and folding of the neuroepithelium, observed at 4–8 weeks in vitro, a significantly later time point compared to our findings of altered structure in week 2 STRADA-mutant hCOs. These temporal differences may reflect the use of different protocols to generate hCOs or differences in mTOR-independent consequences of PTEN and STRADA loss of function.
Another group used genetically engineered heterozygous and homozygous truncating mutations in TSC1 and TSC2 to study the effect of mTOR pathway hyperactivation on human cortical development (Blair et al., 2018). No differences were seen in cell type composition, measured by PAX6, SOX2, MAP2, and GFAP, or in proliferation assayed by Ki67 immunolabeling in TSC1 and TSC2-mutant hCOs at day 20, the earliest time point analyzed. It is possible that a transient delay in neurogenesis and increased proliferation was present earlier, and a more detailed examination of TBR2+ SVZ progenitors or neuronal layer markers was not performed. TSC1/TSC2 mutant hCOs exhibited impaired neuronal and increased glial differentiation at later time points (day 100 and later). While we did not find significant changes in TUBB3 or GFAP at week 12 in PMSE hCOs, future experiments examining later time points may show differences.
The transient nature of delayed neurogenesis in PMSE hCOs suggests the presence of compensatory mechanisms that counteract the effects of mTOR hyperactivation and implies that mTOR pathway activity is just one of many factors that regulate the complex neurodevelopmental program of neural stem cell maintenance and neurogenesis. Another pathway known to regulate early corticogenesis is the Wnt pathway. The emergence of the increased neuroepithelial budding observed here was dependent on the inclusion of CHIR-99021, a GSK3-β inhibitor/Wnt pathway agonist, in the media. Normally, in the absence of the Wnt ligand, GSK3-β phosphorylates β-catenin, resulting in ubiquitination and degradation of β-catenin. With Wnt stimulation, Disheveled is activated and blocks GSK3-β from phosphorylating β-catenin. Both Wnt and mTOR pathways play an important role in early neurogenesis, and there is crosstalk between these two pathways (Shimobayashi & Hall, 2014). GSK3-β inhibition can result in decreased phosphorylation of TSC2 and disinhibition of mTORC1 (Inoki et al., 2006). Phosphorylation of TSC1/TSC2 results in inhibition of the Wnt pathway through degradation of β-catenin (Mak et al., 2003). Therefore, mTOR and Wnt pathway activation may have a reciprocally reinforcing effect and combine to increase the formation of neuroepithelial buds. Our results are complementary to findings in iPSC-derived brain organoids from individuals with Miller-Dieker syndrome (MDS) (Iefremova et al., 2017). In that study, MDS hCOs had impairment in β-catenin signaling, decreased hCO size, and impaired neural rosette formation. Adding CHIR-99021 to the differentiation media did not significantly affect control hCOs but rescued the phenotypic abnormalities in MDS hCOs. Enhanced Wnt/β-catenin signaling has been previously reported in hemimegalencephaly, an MCD also associated with enhanced mTOR activation (Yu et al., 2005). The dependence of the increased budding in PMSE on a Wnt pathway agonist underscores the reciprocal and reinforcing actions of the mTOR and Wnt pathways and shows that their concerted action promotes the formation of neuroepithelium in early corticogenesis. Although inclusion of CHIR-99021 in the differentiation protocol was necessary for the budding phenotype to manifest in PMSE hCOs, the use of CHIR-99021 may also mask Wnt-related phenotypes.
At week 12 of differentiation, the increased HOPX expression found in PMSE hCOs suggests increased production of oRG and reinforces the finding that mTOR pathway signaling is active and important in the development of oRG (Andrews et al., 2020; Nowakowski et al., 2017). This finding also underscores the importance of human-based neurodevelopmental disease models, as oRG are rare in the developing rodent brain. Our results in PMSE hCOs are concordant with that of PTEN−/− hCOs, which also demonstrated increased HOPX expression at week 12 of differentiation (Li et al., 2017). One testable prediction from this finding is that PMSE (and PTEN mutant) hCOs will develop expanded superficial cortical layers with prolonged culture.
There are several caveats to our study. While the loss of STRADA has been shown to cause mTOR pathway activation in mouse models, human brain specimens (Orlova et al., 2010), and iPSC-derived PMSE neurons (Dang et al., 2020), numerous substrates of AMPK exist aside from TSC1/TSC2 that could be dysregulated due to loss of STRADA. In addition, STRADA is a pseudokinase that augments LKB1 kinase activity when bound to LKB1; in the absence of STRADA, LKB1 has minimal kinase activity. While variants in LKB1 and MO25 are not linked to human MCD or epilepsy, knockout of Lkb1 in mice leads to abnormal brain development (Asada et al., 2007; Barnes et al., 2007). LKB1 has several targets aside from AMPK, and thus other effects of STRADA on LKB1 signaling could also contribute to our results. Therefore, our PMSE hCO model may not directly reflect abnormalities solely due to mTOR pathway hyperactivity. However, the likely explanation for the phenotypic overlap between STRADA-mutant hCOs and PTEN−/− hCOs is that the shared features result from the common downstream effect of mTORC1 hyperactivation.
Another caveat is that our PMSE iPSC lines in this study each had one sub-karyotypic chromosomal abnormality that was discovered on SNP chip analysis. Because these abnormalities were different for the two PMSE lines and both lines demonstrated the same phenotype, it is unlikely that these different chromosomal microduplications were responsible for the observed phenotypes in PMSE hCOs. Chromosomal analysis of iPSC lines at the resolution of SNP chip analysis is often not reported in publications, and it is unclear how to interpret small, non-recurrent, copy number variants in iPSCs (Assou et al., 2018). The 20q11.21 amplification is a recurrent chromosomal abnormality that has been described in at least one brain organoid publication (Blair et al., 2018). In that study, the usage of several different stem cell lines with consistent results and the presence of the chromosomal abnormality in both control and mutant hESC lines helped to allay concerns that the observed phenotypes were secondary to the 20q11.21 amplification.
In summary, our results demonstrate how the loss of STRADA affects early corticogenesis and suggest that megalencephaly in PMSE may be secondary to an increased neural stem cell pool during early corticogenesis, as well as increased oRG at later gestational ages. The PMSE hCO model allows the interrogation of the effect of mTOR hyperactivity at the earliest stages of human corticogenesis using a disease-relevant homozygous germline mutation. Future avenues of research include determining the time-dependent effect of mTOR pathway inhibition on PMSE hCOs, examining the effect of the expanded oRG cells at later time points, and assessing for abnormal cell types that are seen in mTORopathies, such as balloon cells and cytomegalic dysmorphic neurons. Another important future consideration will be to perform functional analyses of mature PMSE hCOs using electrophysiological recordings. Such recordings may detect network hyperexcitability that reflects the in vivo seizure phenotype and may result in improved understanding of the pathogenic mechanisms for mTOR-related epilepsies. The PMSE hCO model will allow for mechanistic probing of the many dynamic changes uncovered in this study, deepening our understanding of corticogenesis in mTORopathies, and potentially revealing new avenues for therapeutic development.
ACKNOWLEDGEMENTS
This work was supported by NIH NS109289 and by a Ravitz Advancement Award from the Department of Pediatrics at Michigan Medicine (LTD). None of the authors has any conflict of interest related to the contents of this article.
Footnotes
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Andrews MG, Subramanian L, & Kriegstein AR (2020). MTOR signaling regulates the morphology and migration of outer radial glia in developing human cortex. Elife, 9, e58737. 10.7554/eLife.58737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada N, Sanada K, & Fukada Y (2007). LKB1 regulates neuronal migration and neuronal differentiation in the developing neocortex through centrosomal positioning. Journal of Neuroscience, 27(43), 11769–11775. 10.1523/JNEUROSCI.1938-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assou S, Bouckenheimer J, & Vos JD (2018). Concise review: Assessing the genome integrity of human induced pluripotent stem cells: What quality control metrics? Stem Cells, 36(6), 814–821. 10.1002/stem.2797 [DOI] [PubMed] [Google Scholar]
- Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, & Clevers HC (2003). Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. The EMBO Journal, 22(12), 3062–3072. 10.1093/emboj/cdg292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, & Polleux F (2007). LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell, 129(3), 549–563. 10.1016/j.cell.2007.03.025 [DOI] [PubMed] [Google Scholar]
- Bi W, Glass IA, Muzny DM, Gibbs RA, Eng CM, Yang Y, & Sun A (2016). Whole exome sequencing identifies the first STRADA point mutation in a patient with polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE). American Journal of Medical Genetics, Part A, 170(8), 2181–2185. 10.1002/ajmg.a.37727 [DOI] [PubMed] [Google Scholar]
- Blair JD, Hockemeyer D, & Bateup HS (2018). Genetically engineered human cortical spheroid models of tuberous sclerosis. Nature Medicine, 24(10), 1568–1578. 10.1038/s41591-018-0139-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, & Alessi DR (2003). MO25α/β interact with STRADα/β enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. The EMBO Journal, 22(19), 5102–5114. 10.1093/emboj/cdg490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB (2016). The mTOR signalling cascade: Paving new roads to cure neurological disease. Nature Reviews Neurology, 12(7), 379–392. 10.1038/nrneurol.2016.81 [DOI] [PubMed] [Google Scholar]
- Dang LT, Glanowska KM, Iffland PH II, Barnes AE, Baybis M, Liu Y, Patino G, Vaid S, Streicher AM, Parker WE, Kim S, Moon UY, Henry FE, Murphy GG, Sutton M, Parent JM, & Crino PB (2020). Multimodal analysis of STRADA function in brain development. Frontiers in Cellular Neuroscience, 14, 122. 10.3389/fncel.2020.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lullo E, & Kriegstein AR (2017). The use of brain organoids to investigate neural development and disease. Nature Reviews Neuroscience, 18(10), 573–584. 10.1038/nrn.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers C, Staufner C, Granzow M, Paramasivam N, Hinderhofer K, Kaufmann L, Fischer C, Thiel C, Opladen T, Kotzaeridou U, Wiemann S, Schlesner M, Eils R, Kölker S, Bartram CR, Hoffmann GF, & Moog U (2017). Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Molecular Genetics and Metabolism, 121(4), 297–307. 10.1016/j.ymgme.2017.06.014 [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PRL, & Kriegstein AR (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature, 464(7288), 554–561. 10.1038/nature08845 [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, Alessi DR, & Hardie DG (2003). Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. Journal of Biology, 2(4), 28. 10.1186/1475-4924-2-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iefremova V, Manikakis G, Krefft O, Jabali A, Weynans K, Wilkens R, Marsoner F, Brändl B, Müller F-J, Koch P, & Ladewig J (2017). An organoid-based model of cortical development identifies non-cell-autonomous defects in Wnt signaling contributing to Miller-Dieker Syndrome. Cell Reports, 19(1), 50–59. 10.1016/j.celrep.2017.03.047 [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang C, He X, MacDougald OA, You M, Williams BO, & Guan K-L (2006). TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell, 126(5), 955–968. 10.1016/j.cell.2006.06.055 [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Schroth J, & Gleeson JG (2011). Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nature Cell Biology, 13(6), 700–707. 10.1038/ncb2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepanto P, Badano JL, & Zolessi FR (2016). Neuron’s little helper: The role of primary cilia in neurogenesis. Neurogenesis, 3(1), e1253363. 10.1080/23262133.2016.1253363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, & Longy M (2016). Inherited PTEN mutations and the prediction of phenotype. Seminars in Cell & Developmental Biology, 52, 30–38. 10.1016/j.semcdb.2016.01.030 [DOI] [PubMed] [Google Scholar]
- Li Y, Muffat J, Omer A, Bosch I, Lancaster MA, Sur M, Gehrke L, Knoblich JA, & Jaenisch R (2017). Induction of expansion and folding in human cerebral organoids. Cell Stem Cell, 20(3), 385–396. 10.1016/j.stem.2016.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O’Malley HA, Patino GA, O’Brien JE, Rusconi R, Gupta A, Thompson RC, Natowicz MR, Meisler MH, Isom LL, & Parent JM (2013). Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Annals of Neurology, 74(1), 128–139. 10.1002/ana.23897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, & Kriegstein AR (2011). Development and evolution of the human neocortex. Cell, 146(1), 18–36. 10.1016/j.cell.2011.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak BC, Takemaru K-I, Kenerson HL, Moon RT, & Yeung RS (2003). The tuberin-hamartin complex negatively regulates β-catenin signaling activity. Journal of Biological Chemistry, 278(8), 5947–5951. 10.1074/jbc.C200473200 [DOI] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, Ounadjela JR, Shuga J, Wang X, Lim DA, West JA, Leyrat AA, Kent WJ, & Kriegstein AR (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science (New York, N.Y.), 358(6368), 1318–1323. 10.1126/science.aap8809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, & Crino PB (2010). STRADα deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. The Journal of Clinical Investigation, 120(5), 1591–1602. 10.1172/JCI41592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Lim JS, Ramakrishina S, Kim SH, Kim WK, Lee J, Kang H-C, Reiter JF, Kim DS, Kim H. (Henry), & Lee JH (2018). Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron, 99(1), 83–97.e7. 10.1016/j.neuron.2018.05.039 [DOI] [PubMed] [Google Scholar]
- Parker WE, Orlova KA, Parker WH, Birnbaum JF, Krymskaya VP, Goncharov DA, Baybis M, Helfferich J, Okochi K, Strauss KA, & Crino PB (2013). Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Science Translational Medicine, 5(182), 182ra53. 10.1126/scitranslmed.3005271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenberger EG, Strauss KA, Ramsey KE, Craig DW, Stephan DA, Robinson DL, Hendrickson CL, Gottlieb S, Ramsay DA, Siu VM, Heuer GG, Crino PB, & Morton DH (2007). Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain, 130(7), 1929–1941. 10.1093/brain/awm100 [DOI] [PubMed] [Google Scholar]
- Qian X, Jacob F, Song MM, Nguyen HN, Song H, & Ming G (2018). Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nature Protocols, 13(3), 565–580. 10.1038/nprot.2017.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, Yoon K-J, Jeang W, Lin LI, Li Y, Thakor J, Berg DA, Zhang CE, Kang E, Chickering M, … Ming G-L (2016). Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell, 165(5), 1238–1254. 10.1016/j.cell.2016.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, & Hall MN (2014). Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nature Reviews Molecular Cell Biology, 15(3), 155–162. 10.1038/nrm3757 [DOI] [PubMed] [Google Scholar]
- Tidball AM, Dang LT, Glenn TW, Kilbane EG, Klarr DJ, Margolis JL, Uhler MD, & Parent JM (2017). Rapid generation of human genetic loss-of-function iPSC lines by simultaneous reprogramming and gene editing. Stem Cell Reports, 9(3), 725–731. 10.1016/j.stemcr.2017.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball AM, Neely MD, Chamberlin R, Aboud AA, Kumar KK, Han B, Bryan MR, Aschner M, Ess KC, & Bowman AB (2016). Genomic instability associated with p53 knockdown in the generation of Huntington’s disease human induced pluripotent stem cells. PLoS ONE, 11(3), e0150372. 10.1371/journal.pone.0150372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, McQuillan L, Poduri A, Green TE, Matsumoto N, Mefford HC, Scheffer IE, Berkovic SF, & Hildebrand MS (2019). Somatic mutation: The hidden genetics of brain malformations and focal epilepsies. Epilepsy Research, 155, 106161. 10.1016/j.eplepsyres.2019.106161 [DOI] [PubMed] [Google Scholar]
- Yu J, Baybis M, Lee A, McKhann G, Chugani D, Kupsky WJ, Aronica E, & Crino PB (2005). Targeted gene expression analysis in hemimegalencephaly: Activation of β-catenin signaling. Brain Pathology, 15(3), 179–186. 10.1111/j.1750-3639.2005.tb00518.x [DOI] [PMC free article] [PubMed] [Google Scholar]