

Abstract
Most anthelmintics were discovered through in vivo screens using animal models of infection. Developing in vitro assays for parasitic worms presents several challenges. The lack of in vitro life cycle culture protocols requires harvesting worms from vertebrate hosts or vectors, limiting assay throughput. Once worms are removed from the host environment, established anthelmintics often show no obvious phenotype - raising concerns about the predictive value of many in vitro assays. However, with recent progress in understanding how anthelmintics subvert host-parasite interactions and breakthroughs in high-content imaging and machine learning, in vitro assays have the potential to discern subtle cryptic parasite phenotypes. These may prove better endpoints than conventional in vitro viability assays.
Keywords: anthelmintic, drug screening, nematode, platyhelminth, high-content imaging
A need for new approaches to screening?
Most anthelmintics (see Glossary) in current use were discovered between ~1950’s-80’s through low-throughput in vivo screens in small animal models of infection (Figure 1). While advances in genomics and breakthroughs in molecular tools are generating large amounts of data on parasite biology, this has not yet translated into the discovery of new anthelmintic classes. Anthelmintics recently approved for human use (moxidectin, triclabendazole) and macrofilaricidal leads in clinical development (oxfendazole, emodepside) belong to classes that have been in veterinary use for decades [1]. Resistance is common to classes of broad spectrum anthelmintics in agricultural settings [2], and there is a concern that mass drug administration could produce a similar outcome for human disease. Much has been written about the scarcity of new leads in the anthelmintic development pipeline [3,4]. We are interested in why this earlier era of anthelmintic drug discovery was more successful and how in vitro assays can be developed to best recapitulate past successful approaches.
Figure 1. How were existing anthelmintics discovered?

Timeline for the discovery of a selection of anthelmintics used over the past century. Early antiparasitic compounds already known to be efficacious against protozoan were used in humans harboring helminths. Drug screens were later performed on animal models of infection - either rodents (mice, rats, girds) or agricultural animals (poultry). Animal symbol denotes in vivo models used in primary screens. Petri dish indicates an in vitro assay was employed. Many of these discoveries spawned numerous derivatives, but we have restricted our summary to the first representative of the chemical series. [A] The initial benzimidazole hit was discovered using an in vitro trichostrongylid assay and an in vivo rodent model of Heligmosomoides infection. [B] In the case of praziquantel, an in vitro screen on S. mansoni complemented two in vivo murine models of flatworm infection. [C] The initial amino-acetonitrile derivative hit that led to monepantel was discovered using an in vitro Haemonchus contortus larval development assay and this series was optimized using the jird model. [D] Derquantel was developed from paraherquamide, which was discovered in rodent screens, but also incorporated structure-activity relationship data from a related hit serendipitously discovered in a C. elegans screen. A complete table of these compounds and references is found in supplementary Table S1.
While in vivo screens of infected animals capture any hit regardless of compound mechanism of action, in vitro screens are more likely to miss compounds without overt phenotypes. There is a recognition that many anthelmintics require both a significant host and parasite component to their mechanism of action. An anthelmintic could conceivably (i) act on molecular targets in the parasite but require a host component for clearance, (ii) have polypharmacological effects on both parasite and host targets, or (iii) act directly on host targets and have negligible direct antiparasitic action.
A growing body of evidence reveals that anthelmintics essential to nematode and flatworm control may fall within the middle of this spectrum (Figure 2A, Key Figure). For example, praziquantel acts directly on schistosomes causing contractile paralysis and tegument damage, which is followed by immune recognition and parasite clearance [5–7]. Praziquantel may also act on host vasculature [8] and immune cells [9]. While exhibiting potent in vitro effects against gastrointestinal (GI) nematodes, the microfilaricide ivermectin doesn’t have obvious effects on filarial worms at concentrations that are therapeutic in vivo and likely acts through dysregulation of parasite immune evasion [10–12]. Diethylcarbamazine also has subtle effects on parasites [13,14] and only exhibits in vitro filaricidal activity at high millimolar concentrations [15]. However, diethylcarbamazine is lethal to parasites in vitro if microfilariae are co-cultured with donor blood and it’s in vivo activity in an animal model is eliminated by pretreatment with an immunosuppressant [16,17]. So while the mechanisms of many anthelmintics remain poorly understood, the host immune system can clearly play an important role in drug action.
Figure 2, Key Figure. Developing in vitro assay endpoints with improved in vivo predictive value.
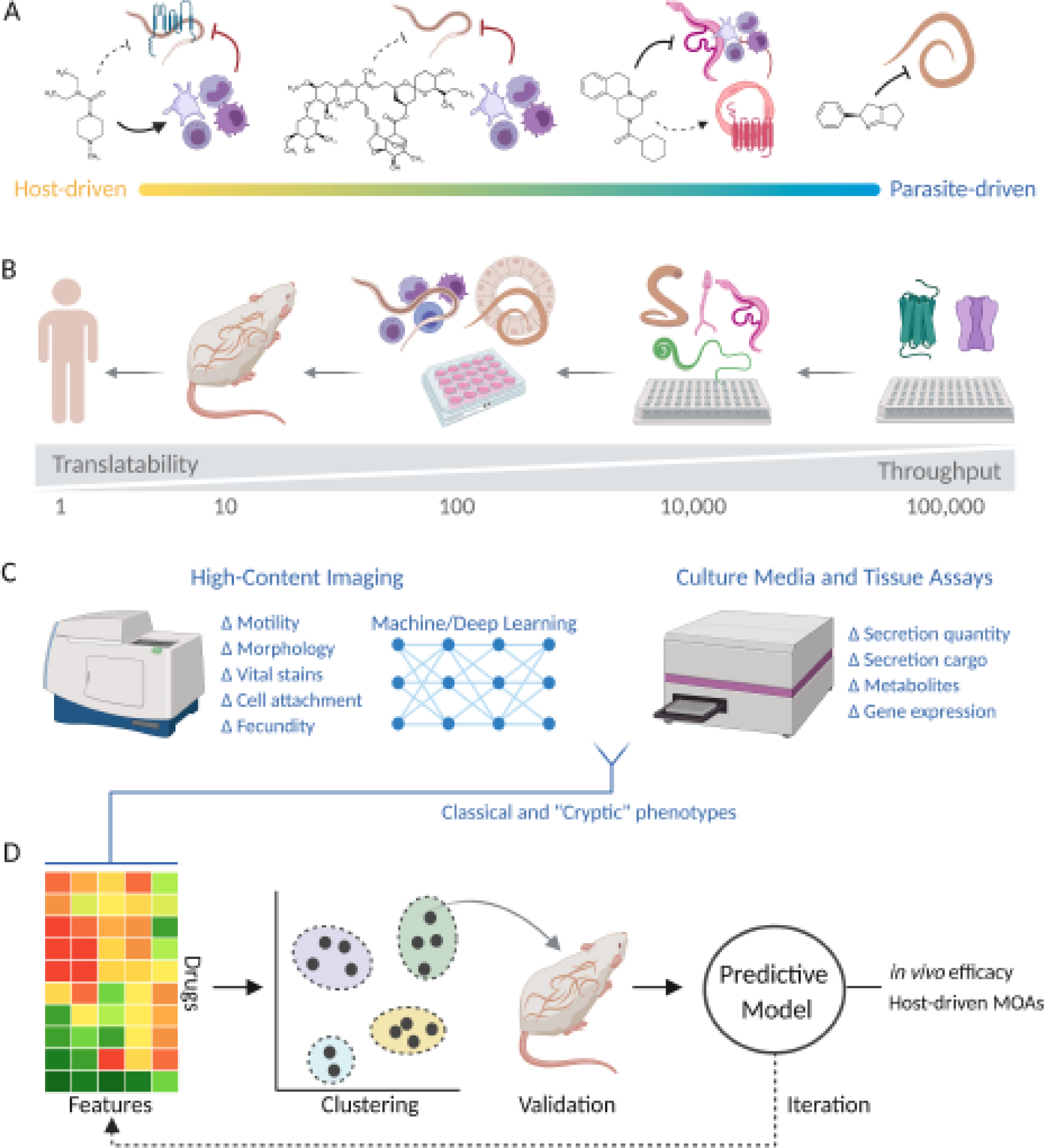
(A) Anthelmintic mechanisms of action can require varying degrees of host-involvement. Examples range from (left to right) diethylcarbamazine and ivermectin, which show little activity on filarial worms in vitro (dashed lines) but obvious immune-dependent effects in vivo (bold lines), to praziquantel (shown with schistosomes) and levamisole (shown with ascaris), which have pronounced phenotypes in vitro indicating more parasite-driven mechanisms. (B) Drug screening similarly falls along a spectrum. In vivo assays are agnostic to worm phenotype or drug target but have limited throughput. More complex in vitro culture conditions can reproduce aspects of the host environment (co-culture with host cell lines or organoids, inclusion of immune cells), although this complexity can limit throughput. Simple culture conditions are easier to scale reproducibly, especially if only one endpoint is measured in an assay, with target-based screens providing maximum throughput. This comes at the expense of host assay components and an increased likelihood of overlooking ‘true positive’ compounds that would be active in vivo. (C) High-content assays measure a greater phenotypic space and can be incorporated into machine learning pipelines for classification of drug effects. Image-derived phenotypes can be multiplexed with assays on culture media, providing additional features to define in vitro drug phenotypes. (D) Features from multivariate phenotyping can be clustered to identify distinct drug response patterns. Drugs from clusters exhibiting similar biological effects may be tested for efficacy in vivo. This data can be iteratively improved upon to identify in vitro phenotypic profiles with optimal in vivo anthelmintic predictive value.
In vivo screening: Historically productive, but limited throughput.
Screening using animal models of infection has the advantage that no prior knowledge of drug mechanism is required. Most frontline drugs were discovered screening infected animals (Figure 1) and even those that are broad spectrum may or may not evoke obvious phenotypes depending on the parasites species and developmental stage being assayed. For example, there are notable differences between filarial and GI parasites (Box 1). Would the narrow-spectrum diethylcarbamazine have been identified had it not been screened in vivo, given its lack of in vitro potency against parasites it effectively treats? Identifying the broad-spectrum macrocyclic lactones would also have required the fortuitous screening of a parasite species and developmental stage exhibiting an obvious in vitro phenotype. Similarly, artemisinins can kill schistosomes in vitro, but only following prolonged exposure to high micromolar concentrations that don’t resemble in vivo conditions [18]. Other antischistosomal hits that show no obvious phenotype in vitro include Ro 15–5458 [19] and Ro 13–1978 [20]. And even when compounds exhibit in vitro movement or morphology phenotypes, they may not be relevant to the drug’s mechanism of action. Hycanthone impairs worm movement due to action at acetylcholinesterases, separate from its therapeutic mechanism of action involving DNA binding [21]. The striking contractile phenotype of praziquantel in vitro does not strictly equate to drug efficacy in vivo; liver stage worms display the exact same contractile response to drug as adults, even though they are completely refractory to praziquantel treatment [7]. Many anthelmintics with overt in vitro phenotypes may act in vivo by damaging the parasite to facilitate immune recognition and clearance of worms (Figure 2A). In vitro approaches to screening that do not reproduce in vivo conditions will be less capable of identifying hit compounds.
Box 1. Targeting gastrointestinal versus tissue-dwelling helminths.
Chemotherapy of worms that reside within host tissues may require different considerations than targeting worms within the gastrointestinal (GI) tract.
Drug absorption and distribution.
Worms located within tissues present pharmacokinetic challenges of drug absorption and distribution that GI parasites do not. While GI parasites are endoparasites, the alimentary canal is technically ‘outside’ the body. A drug does not necessarily need to be absorbed by the host to be efficacious if it is present in the lumen and taken up by parasites via trans-cuticle or trans-tegumental route. For example, while praziquantel has near complete GI absorption and works against most tissue-dwelling and GI flatworms, the nearly identical compound epsiprantel has poor GI absorption and is only effective against cestodes. On the other hand, tissue dwelling parasites, such as adult onchocerca residing in nodules or larval tapeworms that cause cysticercosis, may be more difficult to target in vivo with exposure to sustained therapeutic concentrations of drug. Finally, certain GI helminths may also present challenges similar to tissue-dwelling parasites. Some have tissue-dwelling life cycle stages (ex. larval hookworms can enter an encysted hypobiotic state), or as adults they may be exposed to drug from both the intestinal lumen and epithelial tissue. For example, whipworm benzimidazole absorption correlates most strongly with concentrations of drug in blood plasma rather than the intestinal lumen [71].
Expulsion versus elimination.
Anthelmintics may be drive clearance of GI helminths by causing parasite paralysis resulting in expulsion. However, transient paralysis of tissue-dwelling helminths may not lead to parasite death. Worms in close proximity to rapidly absorbed drugs, such as schistosomes living in the mesenteric vasculature, can still recover after drug clearance and migrate back to their preferred locations in the body [7]. Similarly, adult filarial worms that are damaged by drug treatment can recover and microfilaria loads can rebound. Some tissue-dwelling parasites may require prolonged in vivo exposure to drug, which can be difficult to achieve in a convenient dosing form. Rather than killing these long-lived adult worms directly, drugs may trigger changes that promote immune-mediated clearance [45]. Therefore, a broad-spectrum drug may impact various parasite species differently. Ivermectin may acutely disrupt neuromuscular function of a GI nematode [12] while causing longer effects on fecundity of tissue-dwelling nematodes. Praziquantel may cause contraction of a tapeworm scolex leading to expulsion from the GI tract, while clearance of schistosomes involves host immune cells. For these reasons, different sets of in vitro phenotypes are likely to serve as reliable predictors of in vivo efficacy across tissue-dwelling and GI parasites.
While past animal screens were conducted on hundreds to thousands of compounds, the current emphasis on a reduction in animal use (replacement, reduction and refinement) makes this impractical as a primary screen. Animal screens also require synthesis of larger amounts of test compound than miniaturized in vitro screens. For example, one hit criteria for antischistosomal screening is efficacy clearing infections in mice dosed at 100 mg/kg over five days [22] - this screening paradigm would require nearly 100 mg test compound for a cohort of five mice. Therefore, in vitro screening approaches are needed, even if one limitation is that they have the potential to miss many ‘true positive’ hit compounds. We will discuss two aspects of assay development that can improve in vitro screening approaches.
Approach 1: Making assays more “in vivo like” by incorporating aspects of the host immune system.
Approach 2: Employing more subtle in vitro phenotypic endpoints that are nevertheless important predictors of in vivo anthelmintic efficacy.
Employing in vitro assays that better approximate in vivo conditions
Challenges working with parasitic worms put an inherent limit on assay throughput. Two obvious barriers to high-throughput parasite screening are sourcing parasitic worms in large quantities and the complexity of in vitro culture conditions that recapitulate the host environment.
If the goal of a high-throughput screen is viewed as simply obtaining an enriched set of bioactive compounds from a large chemically diverse library, compromises to simplify and scale an assay are probably necessary. There is an enormous chemical space available for screening, both theoretically (166 billion organic compounds in the GDB-17 database) and available for purchase (>20 million unique compounds in the Enamine Diverse REAL drug-like set). The choice of what to screen is crucial to the success of the assay (Box 2). Since most compounds will typically be inactive, it may be economical to use free-living worms that can easily be scaled to enrich for chemicals with anthelmintic activity [23]. A crude readout of viability or development may be suitable for triaging inactive compounds. The active dataset may then be explored using lower throughput assays interrogating phenotypes in more detail, as has been done with the ‘wactive’ worm bio-active compounds [24,25]. The low historical success rate of C. elegans as an anthelmintic screening model [26] is likely linked to high false negative rates and the unique biology of parasites belonging to different clades [27]. In some parasite species, juvenile worms offer comparable screening throughput and improved predictive value against disease-relevant life stages [27–29].
Box 2. What to screen? Properties of past successful anthelmintics.
Aside from assay development (‘how to screen’), there is also the problem of compound selection (‘what to screen’). Several common features can be seen in past successful anthelmintics. For example, many were derived from natural products. They also often exhibit sustained activity, either through unique pharmacokinetic or pharmacodynamic properties.
Natural Products:
Natural products have a long history as anthelmintics. Infections were treated with plant derived traditional medicines before modern pharmaceutical sciences. More recently, drugs have been developed from active compounds produced by bacteria (Streptomyces cultures yielding milbemycins and avermectins) and fungi (Mycelia sterilia cultures yielding cyclooctadepsipeptides and Penicillium and Aspergillus cultures producing paraherquamides). Natural products continue to be productive sources of new bioactive compounds to treat a range of drug resistant pathogens [72,73]. In addition to bacterial and fungal cultures, natural products produced by microfauna that interact with schistosomes are a promising area of exploration [74].
Extended pharmacokinetics:
The search for adulticidal antifilarial drugs illustrates the importance of pharmacokinetics. Suramin, although no longer clinically used, has one of the longest half lives of any drug (>1 month). Adult worm lifespan can be shortened if continuously exposed to drug (repeated ivermectin dosing against onchocerca [75]), and slower killing dynamics may be useful in avoiding inflammatory responses (Mazzotti reaction).
Unusual pharmacodynamics:
Drugs can also display prolonged action depending on their pharmacodynamic properties. Many drugs with in vivo efficacy against flatworms have a short half-life (several hours), but kill worms on a protracted timescale (artemisinin and oxamniquine ~3 days, oltipraz ~1 week (reviewed in [76])). How to reconcile this discrepancy? One explanation may be the covalent action of these drugs on their targets, uncoupling pharmacodynamics and pharmacokinetics. Numerous covalently acting antischistosomal drugs have been used in humans. Oxamniquine is activated by a parasite sulfotransferase, forming a reactive electrophilic product that binds DNA and protein [77]. Oltipraz irreversibly binds thiol groups on schistosome glutathione-S-transferase [78]. Niridazole is metabolically activated by worms and covalently binds worm proteins [79]. Dichlorvos (metabolized from metrifonate) irreversibly binds cholinesterases. Haem-activated artemisinin covalently modifies >100 malaria proteins [80], so a similar mechanism may occur in blood-feeding schistosomes. Covalent ligands come with concerns (selectivity of the reactive electrophilic functionality, or host immune response to the drug-protein conjugate), but these compounds merit consideration given the precedent of past efficacy.
When smaller numbers of compounds are screened in medium or low-throughput assays, it may be feasible to use adult parasites and incorporate aspects of the host immune system in culture conditions. This may be the case with pharmacophore-based screens centering on a small number of interesting chemical series. This approach, rather than high-throughput screening, has historically produced many of our existing anthelmintics and consists of screening a limited number (~several hundred) of structurally related compounds that display interesting bioactivity. At this stage, in vitro assays that more closely reflect the disease state should better predict in vivo efficacy, as the assay endpoint is a better approximation of the disease endpoint [30].
Culture protocols have been developed that mimic the host environment in attempts to derive adult parasites from juvenile stages. Whole blood culture systems promote the in vitro development of schistosomes from juvenile schistosomula [5]. Co-culture systems with human leukocytes and endothelial cells [31] or 3-D skin models [32] promote the in vitro development of Onchocerca volvulus, which has no laboratory animal screening model to generate adult worms. And larval hookworms co-cultured with human intestinal epithelial cells exhibit feeding behavor and gene expression profiles that more closely match similar stage parasites harvested from animal hosts [33]. Conceivably, worm co-culture within organoids may eventually provide a route to study host-parasite interactions [34].
In addition to promoting parasite development, these host cell types are likely crucial for drug mechanism of action in vivo (Figure 2). Ivermectin and diethylcarbamazine are examples of anthelmintics that do not elicit obvious in vitro phenotypes in filarial worms at therapeutically relevant concentrations. Some of these drugs promote immune-cell adhesion to parasites in vitro [16,35], an outcome crucial for parasite killing. Of the antischistosomal drugs, artemisinins are relatively inactive on cultured schistosomes unless media is supplemented with haemin or red blood cells [5,18], and praziquantel-mediated killing likely has a large immune component [5–7]. Including blood cells in parasite cultures may make it easier to detect compounds whose mechanisms involve the host immune system, particularly for tissue-dwelling or blood-consuming life cycle stages.
Identifying in vitro phenotypes with in vivo predictive value
Another strategy to improve in vitro assays is to develop methods to detect subtle drug-evoked parasite changes that are nonetheless crucial to anthelmintic efficacy. Often, in vitro screens on roundworms [27,36,37] and flatworms [38–40] use changes in movement or morphology as a phenotypic readout, with the understandable assumption that dead worms don’t move. But an immobile worm may not be a dead worm. Studies on C. elegans have shown movement inhibition may only be transient and may vary based on the developmental stage being screened [41,42]. Brugia parasites show recovery after transient inhibition of movement in response to diethylcarbamazine and levamisole [14,43,44]. And while the premise that compounds which kill worms in vitro should be prioritized for in vivo screening seems reasonable, it is possible that compounds that are efficacious in vivo will not cause obvious movement or morphology changes in vitro. However, this is not to say that drugs do not have nuanced phenotypes that may be important predictors of in vivo efficacy. For example, staining antigens on the schistosome surface after in vitro drug treatment reveals damage to the tegument that is likely crucial for immune recognition and parasite clearance [5], and drug-evoked changes in secretion of parasite-derived molecules may disrupt host-parasite signaling. As Moreno et al. note, adult parasites do not show obvious acute changes in mobility when treated with benzimidazoles or macrocyclic lactones, but these drugs do impact secretion of parasite derived molecules that may well alter host-parasite cross-talk [45]. So what types of assays may be employed to achieve higher content and more informative phenotyping of test compounds?
High-content imaging is an approach using automated image acquisition and analysis to explore a larger phenotypic space than conventional high-throughput screens, which often look at just a single endpoint. Images of samples from various treatment conditions are analyzed to detect features, and combinations of features define a profile that distinguishes one phenotype from another. These assays could look at developmental outcomes (for example, larval development or molting assays [46]), fecundity (for example, adult female filarial release of microfilaria), or discern subtle changes in movement and morphology that segregate compounds based on mechanism of action [47,48]. Use of fluorescent dyes or molecular probes can also allow for multiplexing assays on numerous tissues or quantification of subtle tissue / cell type changes (Figure 2C), including observations of endosymbiotic Wolbachia [49]. Imaging may also be performed tracking worms across a time-course following drug treatment. Fixed timepoints can limit the mechanistic profile of the resulting hits, and protozoa screens have found that capturing both ‘fast acting’ and ‘slow acting’ hits increases the diversity of hit mechanisms [50,51]. Machine learning allows for phenotypes to be detected within these large datasets [47,48]. In supervised learning strategies, features collected from imaging data may be combined with manually annotated labels to generate training sets for future hit classification. While this allows screening of visually obvious phenotypes at increased scale, it is low resolution in that it builds in our inability to distinguish subtle, cryptic phenotypes that may be valuable indicators of drug action. Unsupervised learning and deep learning approaches can be used to identify and discriminate new phenotypic categories from image-extracted features in an automated manner [52].
Multivariate phenotyping doesn’t need to be limited to imaging data. This workflow can be multiplexed to include changes in gene expression, as well as biochemical endpoints measuring changes to assay media (Figure 2C). For example, while in vitro ivermectin treatment may cause only subtle changes in morphology or movement, it causes measurable changes in secreted products (proteins and extracellular vesicles) secreted into the assay media [10,44,53]. Media can also be used to measure metabolic changes [54,55]. These assays don’t always correspond to visual scoring of worm viability [45,56,57], indicating there are interesting differences in underlying mechanisms.
Features from imaging and biochemical data can be used to define profiles that cluster based on compounds’ mechanism of action. These clusters can then be screened in animal models to iteratively refine in vitro endpoints for optimal predictive value in vivo (Figure 2D). It is clear from other anti-parasitic drug screening efforts that diverse assay endpoints are needed to ensure mechanistically diverse leads in the development pipeline [50,58], which is crucial given the widespread emergence of resistance to many anthelmintics [2].
Screening of new and existing compounds using quantitative endpoints may allow for combinatorial testing of drugs in a systematic way that can identify instances of synergy or additive effects. For example, synergistic drug combinations may exhibit increased potency and allow for decreased dosing and off target effects and this can be investigated by generating isobolograms from plates consisting of a matrix of drug combinations across a series of concentrations. Additive effects can also be studied when multiplexing assay endpoints. Drug combinations that overlay independent phenotypes may indicate independent mechanisms of action, which is desirable for slowing the emergence of resistance. These studies would be useful not just in screening new libraries, but also gaining a better mechanistic understanding of existing anthelmintics.
Identifying targets that underpin in vitro phenotypes
A complete understanding of how anthelmintics work will require insight into the molecular mechanisms underpinning clusters of phenotypic profiles. The therapeutic targets of many anthelmintics have yet to be deorphanized, although recent functional data has proposed candidates for praziquantel [59,60] and diethylcarbamazine [14]. This is also important since truly high-throughput assays may require target-based rather than phenotypic screens, and historically ‘best in class’ drugs have come from target-based screens on deorphanized receptors [61]. There is currently a wealth of parasite genomic information [62] which can inform screening by predicting essential drug targets and metabolic chokepoints [63]. However, tools to functionally annotate this genome, such as CRISPR and transgenesis protocols [64–66], are not routinely employed in parasitic nematodes. Similarly, schistosome studies have reported editing of parasite eggs [67], but hatched miracidia need to survive propagation through snail and vertebrate hosts to allow routine interrogation of gene function in intra-mammalian parasite stages. Most species can’t be cryopreserved and there is a lack of any in vitro ‘egg-to-egg’ parasite culture system - meaning maintenance and propagation of edited lines will not be trivial for large scale genomic screens. These obstacles will need to be addressed in order to fully understand the genetic basis for phenotypic profiles of either existing anthelmintics or novel leads.
Concluding remarks
Recent decades have seen a scarcity of new leads in the anthelmintic pipeline and few new drug classes, and resistance to many broad spectrum anthelmintics is common in veterinary settings. Advances in genomics have allowed us to better understand the mechanisms of existing anthelmintics (often aided by resistant strains [68–70]), but progress in the search for drugs with new mechanisms has been slow. Over the past century, most anthelmintics were discovered using in vivo screens on animal models of infection. We argue that the efficiency of in vitro assays can be increased by designing phenotypic screens to better recapitulate the in vivo environment.
Drugs may conceivably act across a spectrum of mechanisms ranging from indirect antiparasitic action, subtly impacting the ability of worms to reside undetected within the host, to direct parasite killing. Assays looking only at obvious outcomes such as gross changes in movement or viability may neglect compounds that would be efficacious in vivo but lack obvious effects in vitro. In high-content imaging, an analogy has been made to the psychological phenomenon of inattentional blindness. In this case, a focus on an obvious outcome that we expect to see when looking for anthelmintics (perhaps changes in movement or viability) may actually distract us from subtle phenotypes that are nonetheless valuable for predicting anti-parasitic effects.
Advances in automated imaging and the computational frameworks for analysis of these data have made it possible to profile worm phenotypes in unprecedented detail. By exploring this expanded phenotypic space and screening more subtle features of drug action, we may not only improve upon existing anthelmintics but also have the opportunity to identify new classes of compounds. While resolving the exact mode of action for these compounds may require advances in functional genomics or the use of free-living models [70] (see Outstanding Questions), leads that fall under distinct phenotypic clusters from existing anthelmintic classes would merit prioritization as potentially acting via unique mechanisms.
OUTSTANDING QUESTIONS.
Before embarking on screening new compounds, do we have a complete understanding of the phenotypic profile for existing frontline anthelmintics against target parasite species and stages?
Will high-content whole organism screens using new multivariate and ‘cryptic’ endpoints reveal anthelmintics with novel mechanisms of action? Will deep phenotypic profiling better resolve anthelmintic interactions and inform combinatorial strategies to mitigate the spread of resistance?
How can we experimentally validate the in vivo efficacy of test compounds against parasites that are not amenable to animal models of infection? Are small animal hosts made permissive through immunodeficiency less useful in capturing drug mechanisms of action that require a significant immune component?
How will the development of more routine parasite transgenesis and genome editing tools alter in vitro and in vivo approaches to drug screening?
How can we associate specific genes to parasite drug response phenotypes, given sparse evidence-based genomic annotations and a lack of scalable functional genomic tools in many species?
Supplementary Material
HIGHLIGHTS.
Many current anthelmintics were discovered screening relatively low-throughput animal models of infection. Several frontline anthelmintics lack obvious in vitro phenotypes and would not be detected using common in vitro screens.
Anthelmintics that lack overt phenotypes may have ‘cryptic phenotypes’, disrupting subtle processes that are nonetheless crucial to parasite survival within the host.
Provision of parasite material may be an inherent limit on assay throughput, but improved profiling of existing anthelmintics and expanding the scope of phenotypes observed can maximize the productivity of parasite screening.
Advances in high content imaging allow for in-depth profiling of anthelmintic phenotypes, and advances in parasite culture can more closely approximate the in vivo environment.
Acknowledgements.
Funding support includes R21AI153545 (JDC and MZ), R21AI146540 (JDC) and R01AI151171 (MZ). The authors would like to thank Sebastian Raschka for helpful comments on this manuscript.
Glossary
- Anthelmintic
medication for human or veterinary use to treat infection with parasitic worms.
- Deep learning
class of machine learning algorithms that rely on multilayer neural networks that are particularly well-suited for learning from large and unstructured datasets.
- High-content imaging
automated microscopy of drug-parasite interactions to enable the collection of complex spatial and morphological readouts of cell and organismal health.
- Machine learning
algorithms capable of learning from structured data to make determinations and predictions without explicit programming.
- Mechanism of action
how a compound interacts with its target and, in the case of anthelmintics, triggers parasite elimination from the host.
- Supervised learning
machine learning technique that relies on the use of labeled data to train a predictive model.
- Unsupervised learning
machine learning technique to deduce structures present in unlabeled data.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Moraes J and Geary TG (2020) FDA-Approved Antiparasitic Drugs in the 21st Century: A Success for Helminthiasis? Trends Parasitol. 36, 573–575 [DOI] [PubMed] [Google Scholar]
- 2.Wolstenholme AJ et al. (2004) Drug resistance in veterinary helminths. Trends Parasitol. 20, 469–476 [DOI] [PubMed] [Google Scholar]
- 3.Geary TG et al. (2015) Anthelmintic drug discovery: into the future. J. Parasitol 101, 125–133 [DOI] [PubMed] [Google Scholar]
- 4.Nixon SA et al. (2020) Where are all the anthelmintics? Challenges and opportunities on the path to new anthelmintics. Int. J. Parasitol. Drugs Drug Resist 14, 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reimers N et al. (2015) Drug-induced exposure of Schistosoma mansoni antigens SmCD59a and SmKK7. PLoS Negl. Trop. Dis. 9, e0003593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panic G et al. (2017) Immunohistochemical Investigations of Treatment with Ro 13–3978, Praziquantel, Oxamniquine, and Mefloquine in Schistosoma mansoni-Infected Mice. Antimicrob. Agents Chemother. 61, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCusker P et al. (2021) Schistosoma mansoni alter transcription of immunomodulatory gene products following in vivo praziquantel exposure. PLoS Negl. Trop. Dis 15, e0009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan JD et al. (2017) The anthelmintic praziquantel is a human serotoninergic G-protein-coupled receptor ligand. Nat. Commun 8, 1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eyoh E et al. (2019) The anthelmintic drug praziquantel promotes human Tr1 differentiation. Immunol. Cell Biol 97, 512–518 [DOI] [PubMed] [Google Scholar]
- 10.Moreno Y et al. (2010) Ivermectin disrupts the function of the excretory-secretory apparatus in microfilariae of Brugia malayi. Proc. Natl. Acad. Sci. U. S. A 107, 20120–20125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berrafato T et al. (2019) Macrocyclic lactone anthelmintic-induced leukocyte binding to Dirofilaria immitis microfilariae: Influence of the drug resistance status of the parasite. Int. J. Parasitol. Drugs Drug Resist 10, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin RJ et al. (2021) Ivermectin: An Anthelmintic, an Insecticide, and Much More. Trends Parasitol. 37, 48–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawking F et al. (1948) Mode of action of hetrazan in filariasis. Lancet 2, 730. [DOI] [PubMed] [Google Scholar]
- 14.Verma S et al. Diethylcarbamazine activates TRP channels including TRP-2 in filaria, Brugia malayi. , Communications Biology, 3. (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denham DA et al. (1978) Studies with Brugia pahangi 17. The anthelmintic effects of diethylcarbamazine. J. Parasitol 64, 463–468 [PubMed] [Google Scholar]
- 16.Cesbron JY et al. (1987) Platelets mediate the action of diethylcarbamazine on microfilariae. Nature 325, 533–536 [DOI] [PubMed] [Google Scholar]
- 17.McGarry HF et al. (2005) Diethylcarbamazine activity against Brugia malayi microfilariae is dependent on inducible nitric-oxide synthase and the cyclooxygenase pathway. Filaria J. 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao S et al. (2001) Artemether administered together with haemin damages schistosomes in vitro. Trans. R. Soc. Trop. Med. Hyg 95, 67–71 [DOI] [PubMed] [Google Scholar]
- 19.Probst A et al. (2020) Efficacy, metabolism and pharmacokinetics of Ro 15–5458, a forgotten schistosomicidal 9-acridanone hydrazone. J. Antimicrob. Chemother DOI: 10.1093/jac/dkaa247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keiser J et al. (2015) Aryl hydantoin Ro 13–3978, a broad-spectrum antischistosomal. J. Antimicrob. Chemother 70, 1788–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pica-Mattoccia L et al. (1989) Binding of oxamniquine to the DNA of schistosomes. Trans. R. Soc. Trop. Med. Hyg 83, 373–376 [DOI] [PubMed] [Google Scholar]
- 22.Nwaka S and Hudson A (2006) Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discov 5, 941–955 [DOI] [PubMed] [Google Scholar]
- 23.Burns AR et al. (2015) Caenorhabditis elegans is a useful model for anthelmintic discovery. Nat. Commun 6, 7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burns AR et al. (2017) The novel nematicide wact-86 interacts with aldicarb to kill nematodes. PLoS Negl. Trop. Dis 11, e0005502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamal M et al. (2019) The marginal cells of the Caenorhabditis elegans pharynx scavenge cholesterol and other hydrophobic small molecules. Nat. Commun 10, 3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geary TG et al. (1999) Mechanism-based screening: discovery of the next generation of anthelmintics depends upon more basic research. Int. J. Parasitol 29, 105–12; discussion 113–4 [DOI] [PubMed] [Google Scholar]
- 27.Elfawal MA et al. (2019) Drug Screening for Discovery of Broad-spectrum Agents for Soil-transmitted Nematodes. Scientific Reports, 9(1):12347. doi: 10.1038/s41598-019-48720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mansour NR et al. (2016) High Throughput Screening Identifies Novel Lead Compounds with Activity against Larval, Juvenile and Adult Schistosoma mansoni. PLoS Negl. Trop. Dis 10, e0004659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guidi A et al. (2017) Discovery by organism based high-throughput screening of new multi-stage compounds affecting Schistosoma mansoni viability, egg formation and production. PLoS Negl. Trop. Dis 11, e0005994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moffat JG et al. (2017) Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov 16, 531–543 [DOI] [PubMed] [Google Scholar]
- 31.Voronin D et al. (2019) Development of a preliminary in vitro drug screening assay based on a newly established culturing system for pre-adult fifth-stage Onchocerca volvulus worms. PLoS Negl. Trop. Dis 13, e0007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malkmus C et al. (2020) Preliminary evaluations of 3-dimensional human skin models for their ability to facilitate in vitro the long-term development of the debilitating obligatory human parasite Onchocerca volvulus. PLoS Negl. Trop. Dis 14, e0008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feather CM et al. (2017) Ancylostoma ceylanicum infective third-stage larvae are activated by co-culture with HT-29-MTX intestinal epithelial cells. Parasit. Vectors 10, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duque-Correa MA et al. (2020) Organoids - new models for host-helminth interactions. Trends Parasitol. 36, 170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vatta AF et al. (2014) Ivermectin-dependent attachment of neutrophils and peripheral blood mononuclear cells to Dirofilaria immitis microfilariae in vitro. Vet 334 Parasitol. 1–2, 38–42. [DOI] [PubMed] [Google Scholar]
- 36.Bulman CA et al. (2015) Repurposing Auranofin as a Lead Candidate for Treatment of Lymphatic Filariasis and Onchocerciasis. PLoS Negl. Trop. Dis 9, e0003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keiser J et al. (2016) Evaluation of an FDA approved library against laboratory models of human intestinal nematode infections. Parasit. Vectors 9, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdulla MH et al. (2009) Drug discovery for schistosomiasis: hit and lead compounds identified in a library of known drugs by medium-throughput phenotypic screening. PLoS Negl. Trop. Dis 3, e478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panic G et al. (2015) Activity Profile of an FDA-Approved Compound Library against Schistosoma mansoni. PLoS Negl. Trop. Dis 9, e0003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duguet TB et al. (2020) Identification of annotated bioactive molecules that impair motility of the blood fluke Schistosoma mansoni. Int. J. Parasitol. Drugs Drug Resist 13, 73–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spensley M et al. (2018) Acute Effects of Drugs on Caenorhabditis elegans Movement Reveal Complex Responses and Plasticity. G3 8, 2941–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Del Borrello S et al. (2019) Rhodoquinone biosynthesis in C. elegans requires precursors generated by the kynurenine pathway. Elife 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mostafa E et al. (2015) Transient effects of levamisole on Brugia malayi microfilariae. Invert. Neurosci 15, 5. [DOI] [PubMed] [Google Scholar]
- 44.Loghry HJ et al. (2020) Ivermectin inhibits extracellular vesicle secretion from parasitic nematodes. J Extracell Vesicles 10, e12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moreno Y et al. (2021) When secretomes meet anthelmintics: Lessons for therapeutic interventions. Trends Parasitol DOI: 10.1016/j.pt.2021.01.007 [DOI] [PubMed] [Google Scholar]
- 46.Jawahar S et al. (2021) Drugs that target early stages of Onchocerca volvulus: A revisited means to facilitate the elimination goals for onchocerciasis. PLoS Negl. Trop. Dis 15, e0009064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen S et al. (2020) A multi-dimensional, time-lapse, high content screening platform applied to schistosomiasis drug discovery. Commun Biol 3, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDermott-Rouse A et al. (2021) Behavioral fingerprints predict insecticide and anthelmintic mode of action. bioRxiv. doi: 10.1101/2021.01.27.428391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gunderson EL et al. (2020) The endosymbiont Wolbachia rebounds following antibiotic treatment. PLoS Pathog. 16, e1008623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jumani RS et al. (2019) A suite of phenotypic assays to ensure pipeline diversity when prioritizing drug-like Cryptosporidium growth inhibitors. Nat. Commun. 10, 1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abraham M et al. (2020) Probing the Open Global Health Chemical Diversity Library for Multistage-Active Starting Points for Next-Generation Antimalarials. ACS Infect Dis 6, 613–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chandrasekaran SN et al. (2021) Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat. Rev. Drug Discov 20, 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harischandra H et al. (2018) Profiling extracellular vesicle release by the filarial nematode Brugia malayi reveals sex-specific differences in cargo and a sensitivity to ivermectin. PLoS Negl. Trop. Dis 12, e0006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.James CE and Davey MW (2007) A rapid colorimetric assay for the quantitation of the viability of free-living larvae of nematodes in vitro. Parasitol. Res 101, 975–980 [DOI] [PubMed] [Google Scholar]
- 55.Howe S et al. (2015) Lactate as a novel quantitative measure of viability in Schistosoma mansoni drug sensitivity assays. Antimicrob. Agents Chemother 59, 1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lalli C et al. (2015) Development and validation of a luminescence-based, medium-throughput assay for drug screening in Schistosoma mansoni. PLoS Negl. Trop. Dis 9, e0003484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aguiar PHN et al. (2017) A high-throughput colorimetric assay for detection of Schistosoma mansoni viability based on the tetrazolium salt XTT. Parasit. Vectors 10, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hovlid ML and Winzeler EA (2016) Phenotypic Screens in Antimalarial Drug Discovery. Trends Parasitol. 32, 697–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Babes RM et al. (2017) The anthelminthic drug praziquantel is a selective agonist of the sensory transient receptor potential melastatin type 8 channel. Toxicol. Appl. Pharmacol 336, 55–65 [DOI] [PubMed] [Google Scholar]
- 60.Park SK et al. (2019) The anthelmintic drug praziquantel activates a schistosome transient receptor potential channel. J. Biol. Chem 294, 18873–18880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swinney DC and Anthony J (2011) How were new medicines discovered? Nat. Rev. Drug Discov 10, 507–519 [DOI] [PubMed] [Google Scholar]
- 62.International Helminth Genomes Consortium, (2019) Comparative genomics of the major parasitic worms. Nat. Genet 51, 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Curran DM et al. (2020) Modeling the metabolic interplay between a parasitic worm and its bacterial endosymbiont allows the identification of novel drug targets. Elife 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryant AS et al. (2018) A Critical Role for Thermosensation in Host Seeking by Skin-Penetrating Nematodes. Curr. Biol 28, 2338–2347.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lok JB (2019) CRISPR/Cas9 Mutagenesis and Expression of Dominant Mutant Transgenes as Functional Genomic Approaches in Parasitic Nematodes. Front. Genet 10, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu C et al. (2020) In vivo imaging of transgenic Brugia malayi. PLoS Negl. Trop. Dis 14, e0008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ittiprasert W et al. (2019) Programmed genome editing of the omega-1 ribonuclease of the blood fluke, Schistosoma mansoni. Elife 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anderson TJC et al. (2018) Genetic crosses and linkage mapping in schistosome parasites. Trends Parasitol. 34, 982–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doyle SR and Cotton JA (2019) Genome-wide approaches to investigate anthelmintic resistance. Trends Parasitol. 35, 289–301 [DOI] [PubMed] [Google Scholar]
- 70.Wit J et al. (2021) Complementary approaches with free-living and parasitic nematodes to understanding anthelmintic resistance. Trends Parasitol. 37, 240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hansen TVA et al. (2017) Pathway of oxfendazole from the host into the worm: Trichuris suis in pigs. Int J Parasitol Drugs Drug Resist. 7(3):416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rottmann M et al. (2010) Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang F et al. (2020) A marine microbiome antifungal targets urgent-threat drug-resistant fungi. Science 370, 974–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao J et al. (2019) A rotifer-derived paralytic compound prevents transmission of schistosomiasis to a mammalian host. PLoS Biol. 17, e3000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walker M et al. (2017) Macrofilaricidal Efficacy of Repeated Doses of Ivermectin for the Treatment of River Blindness. Clin. Infect. Dis 65, 2026–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Archer S (1985) The chemotherapy of schistosomiasis. Annu. Rev. Pharmacol. Toxicol 25, 485–508 [DOI] [PubMed] [Google Scholar]
- 77.Valentim CL et al. (2013) Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science 342, 1385–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nare B et al. (1992) Mechanisms of inactivation of Schistosoma mansoni and mammalian glutathione S-transferase activity by the antischistosomal drug oltipraz. Biochem. Pharmacol 43, 1345–1351 [DOI] [PubMed] [Google Scholar]
- 79.Tracy JW et al. (1983) Reductive metabolism of niridazole by adult Schistosoma mansoni. Correlation with covalent drug binding to parasite macromolecules. Mol. Pharmacol 24, 291–299 [PubMed] [Google Scholar]
- 80.Wang J et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. , Nature Communications, 6. (2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.