

Abstract
Over the past decade, pharmacogenetic testing has emerged in clinical practice to guide selected cardiovascular therapies. The most common implementation in practice is CYP2C19 genotyping to predict clopidogrel response and assist in selecting antiplatelet therapy after percutaneous coronary intervention. Additional examples include genotyping to guide warfarin dosing and statin prescribing. Increasing evidence exists on outcomes with genotype-guided cardiovascular therapies from multiple randomized controlled trials and observational studies. Pharmacogenetic evidence is accumulating for additional cardiovascular medications. However, data for many of these medications are not yet sufficient to support the use of genotyping for drug prescribing. Ultimately, pharmacogenetics might provide a means to individualize drug regimens for complex diseases such as heart failure, in which the treatment armamentarium includes a growing list of medications shown to reduce morbidity and mortality. However, sophisticated analytical approaches are likely to be necessary to dissect the genetic underpinnings of responses to drug combinations. In this Review, we examine the evidence supporting pharmacogenetic testing in cardiovascular medicine, including that available from several clinical trials. In addition, we describe guidelines that support the use of cardiovascular pharmacogenetics, provide examples of clinical implementation of genotype-guided cardiovascular therapies and discuss opportunities for future growth of the field.
ToC blurb
In this Review, Cavallari and colleagues examine the evidence supporting pharmacogenetic testing in cardiovascular medicine, describe guidelines for the use of cardiovascular pharmacogenetics and provide examples of the clinical implementation of genotype-guided therapies.
Introduction
Precision medicine involves tailoring disease treatment and prevention strategies on the basis of genotype, environment, lifestyle and other patient-specific factors. Pharmacogenetics is an important component of precision medicine and aims to minimize the traditional trial-and-error approach to drug therapy by considering an individual’s genetic code, in addition to other patient-specific information, to select optimal drug therapy. Decades of pharmacogenetic research have identified associations between genetics and the safety and effectiveness of numerous therapies that are now being translated to clinical practice.1
Variations in genes encoding drug-metabolizing enzymes are currently the most commonly tested in the clinical setting to inform pharmacotherapy2. Subsequent drug responses depend on whether the enzyme is responsible for metabolizing the drug to a more or less active form. For prodrugs requiring bioactivation, such as clopidogrel, an inherited enzyme deficiency can render the drug ineffective owing to minimal conversion to the active metabolite, which elicits the effects of the drug. For drugs such as warfarin that are administered in their pharmacologically active form, enzyme deficiency might lead to increased drug exposure and serious adverse effects (Fig. 1). Limited examples also exist in which clinical testing for genes encoding a drug transporter (for example, SLCO1B1 for statins) or drug target (for example, VKORC1 for warfarin) guides therapy3–6.
Fig. 1 |. Mechanisms by which variation in genes encoding drug-metabolizing enzymes can affect cardiovascular drug pharmacokinetics.
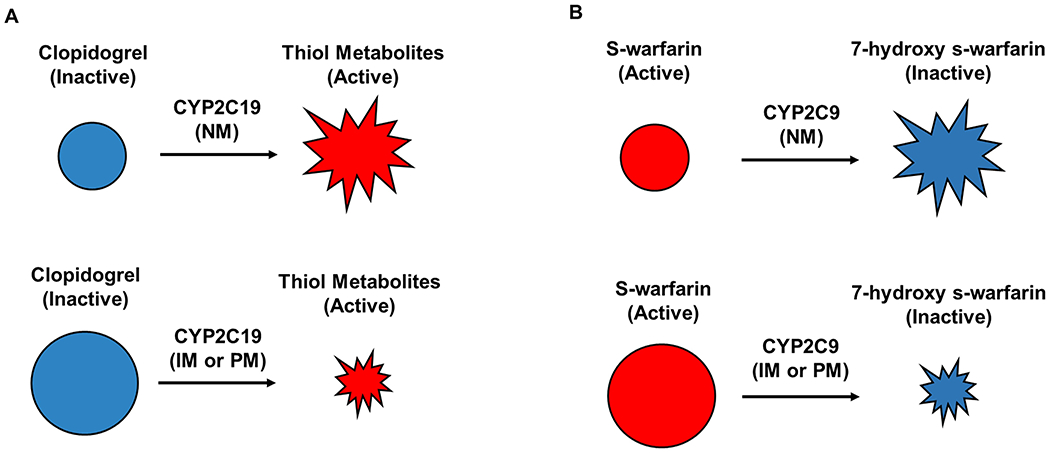
a | For prodrugs, such as clopidogrel, CYP2C19 genotypes associated with poor or intermediate metabolism lead to lower formation of the active metabolite and can result in treatment failure. b | For active drugs, such as warfarin, CYP2C9 genotypes associated with poor or intermediate metabolism lead to higher concentrations of the active form of the drug and can result in toxicity. CYP2C19, cytochrome P450 2C19; IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer.
To facilitate pharmacogenetic testing in practice, the Clinical Pharmacogenetics Implementation Consortium (CPIC) provides guidelines for gene–drug pairs that have sufficient evidence to support the consideration of genotype data in prescribing decisions.7 Similar guidance is provided by the Dutch Pharmacogenetics Working Group (DPWG), Canadian Pharmacogenomics Network for Drug Safety (CPNDS) and other professional societies8,9. These guidelines are all annotated in the Pharmacogenomics Knowledgebase (PharmGKB)10. These guidelines generally do not address whether or not to order pharmacogenetic testing, leaving that to the discretion of the physician. Instead, the guidelines provide recommendations on how to apply existing test results to optimize pharmacotherapy, with the assumption that genotype data will be increasingly available through initiatives at the health system level or through direct-to-consumer testing.
PharmGKB ranks the evidence underlying their clinical annotations for gene–drug pairs, with level 1 indicating the strongest level of evidence and level 4 the lowest. CPIC similarly categorizes gene–drug pairs from level A, signifying that genetic information should be considered in drug prescribing, to level D, indicating that the evidence is weak or conflicting. These rankings can assist clinicians when assessing the merits of pharmacogenetic evidence to support clinical implementation. As of early 2021, 25 CPIC guidelines have been published, all for gene–drug pairs ranked as level A. These guidelines are freely available through the CPIC website (CPIC). Three guidelines address cardiovascular drugs: clopidogrel, warfarin and simvastatin. The DPWG also provides guidance for the use of these drugs and for acenocoumarol, atorvastatin, flecainide, metoprolol and propafenone. Clopidogrel and warfarin are also included in a table of gene–drug pairs for which the FDA believes that the data support therapeutic management recommendations11. Simvastatin is included in a separate table of genes that might affect drug response. Pharmacogenetic evidence is accumulating for additional cardiovascular medications such as hydralazine12 and β-blockers13. However, for many drugs, the data are insufficient to support the use of pharmacogenetics in clinical practice to guide prescribing decisions.
Similarly, while much research remains to be done, pharmacogenetics might potentially be a means to guide the treatment of cardiovascular diseases in which multiple drug regimens are required, but the optimal drug combination for a given patient is unknown. In this Review, we summarize the evidence supporting pharmacogenetic testing in cardiovascular medicine, describe guidelines for the use of cardiovascular pharmacogenetics and provide examples of the clinical implementation of genotype-guided therapies. We also discuss opportunities for the growth of cardiovascular pharmacogenetics.
Clopidogrel
Dual antiplatelet therapy with aspirin plus a P2Y purinoceptor 12 (P2Y12 receptor) inhibitor (clopidogrel, ticagrelor or prasugrel) is the standard of care after percutaneous coronary intervention (PCI) to reduce the risk of major adverse cardiovascular events (MACE).14 In patients presenting with acute coronary syndrome (ACS), guidelines give preference to prasugrel or ticagrelor over clopidogrel on the basis of their superior efficacy in clinical trials15,16. Although the use of these newer agents is therefore increasing, these drugs are associated with higher cost and bleeding risks15–17. These risk factors, in addition to the dyspnoea that occurs with the use of ticagrelor, contribute to higher discontinuation rates of prasugrel and ticagrelor than of clopidogrel and limit their universal use17,18. Furthermore, clinical trials did not account for the fact that approximately 30% of individuals inherit a deficiency in the cytochrome P450 (CYP) 2C19 enzyme, which leads to reduced clopidogrel effectiveness. Data from a 2019 study suggested that clopidogrel is as effective as an alternative agent after PCI in patients with full enzyme activity19.
Pharmacogenetics
Clopidogrel is a prodrug that undergoes a two-step metabolism to its active form that irreversibly inhibits platelet activation20 (Fig. 2). Multiple CYP enzymes are involved in activating clopidogrel. However, CYP2C19 is involved in both steps and has a crucial function in the bioactivation process of clopidogrel21. The gene encoding CYP2C19 is highly polymorphic, with *1 denoting the allele associated with normal enzyme function and *2 and *3 denoting alleles associated with no enzyme function22,23. Individuals with two no-function (also called loss-of-function) alleles (for example, *2/*2 genotype) have no CYP2C19 enzyme activity and are poor metabolizers (PMs). Intermediate metabolizers (IMs) have a single no-function allele (for example, *1/*2 genotype) and markedly reduced enzyme activity. Conversely, the *17 allele is an allele associated with increased enzyme function that confers the rapid metabolizer (RM; *1/*17 genotype) and ultra-rapid metabolizer (UM; *17/*17 genotype) phenotypes. Approximately 30% of individuals are PMs or IMs, and another 30% are RMs or UMs, but phenotype frequencies vary by ancestry (Table 1). Although other less common no-function alleles have been described, the *2, *3, and *17 alleles are the only alleles that are considered essential for testing by the US Association of Molecular Pathology24,25.
Fig. 2 |. Clopidogrel metabolism.
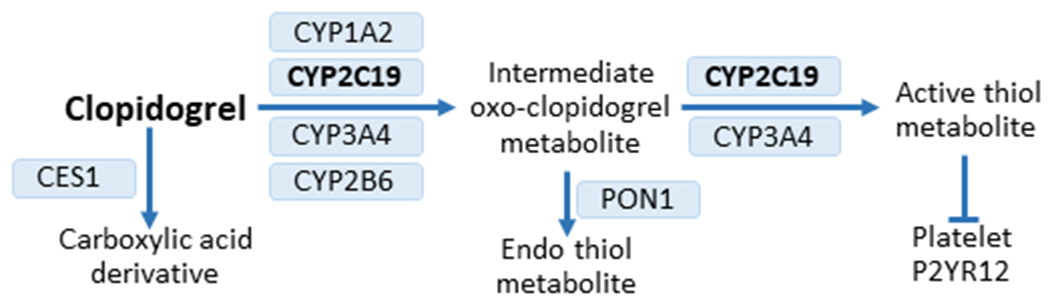
Clopidogrel is administered as a prodrug. Approximately 85% of the prodrug is hydrolysed by carboxyl esterase 1 (CES1) to an inactive carboxylic acid derivative. The remaining drug undergoes metabolism to the oxo-clopidogrel intermediate metabolite, which is then metabolized to either the active thiol metabolite or to an inactive endo thiol metabolite via paraoxonase 1 (PON1). The active thiol metabolite inhibits platelet activation through irreversible binding to the platelet P2Y purinoceptor 12 (P2Y12 receptor). Multiple cytochrome P450 (CYP) enzymes are involved in the two-step bioactivation process21. CYP2C19 is involved in both steps of the process and is essential for clopidogrel activation.
Table 1 |.
Prevalence of phenotypes that influence response to cardiovascular pharmacotherapy
| Phenotype | Prevalence | ||||||
|---|---|---|---|---|---|---|---|
| European | African | East Asian | Latino or Native American | Central or South Asian | Middle Eastern | Oceanian | |
| CYP2C19 | |||||||
| Ultra-rapid metabolizer | 0.05 | 0.04 | 0.00 | 0.02 | 0.03 | 0.04 | 0.00 |
| Rapid metabolizer | 0.27 | 0.21 | 0.03 | 0.19 | 0.19 | 0.26 | 0.02 |
| Normal metabolizer | 0.40 | 0.31 | 0.38 | 0.58 | 0.30 | 0.45 | 0.04 |
| Intermediate metabolizer | 0.26 | 0.34 | 0.46 | 0.20 | 0.41 | 0.24 | 0.37 |
| Poor metabolizer | 0.02 | 0.05 | 0.13 | 0.01 | 0.08 | 0.02 | 0.57 |
| CYP2C9 | |||||||
| Normal metabolizer | 0.63 | 0.74 | 0.84 | 0.79 | 0.60 | 0.61 | 0.91 |
| Intermediate metabolizer | 0.35 | 0.25 | 0.15 | 0.20 | 0.36 | 0.36 | 0.09 |
| Poor metabolizer | 0.03 | 0.01 | 0.01 | 0.01 | 0.04 | 0.03 | 0.00 |
| VKORC1 (−1639G>A or rs9923231) | |||||||
| Warfarin-sensitive (A/A) | 0.19 | 0.03 | 0.84 | 0.46 | NA | NA | NA |
| Intermediate warfarin sensitivity (A/G) | 0.41 | 0.06 | 0.16 | 0.42 | NA | NA | NA |
| Warfarin-resistant (G/G) | 0.40 | 0.91 | 0.00 | 0.12 | NA | NA | NA |
| SLCO1B1 (rs4149056) | |||||||
| Normal function (T/T) | 0.71 | 0.98 | 0.76 | 0.89 | NA | NA | NA |
| Intermediate function (C/T) | 0.27 | 0.02 | 0.22 | 0.10 | NA | NA | NA |
| Low function (C/C) | 0.01 | 0 | 0.02 | 0 | NA | NA | NA |
CYP2C19 and clopidogrel exposure.
The CYP2C19 PM and IM phenotypes are associated with lower exposure to the active clopidogrel metabolite and higher on-treatment platelet reactivity than the normal metabolizer (NM) phenotype (that is,*1/*1 genotype).26–29 Use of higher doses of clopidogrel has been examined as a strategy to overcome the reduced antiplatelet effects in IMs and PMs. Tripling the dose to 225 mg per day can produce a similar level of platelet inhibition in IMs as a 75 mg per day dose in NMs, whereas a dose as high as 300 mg per day is insufficient for PMs30–32. Given that CYP2C19 genotype does not affect the pharmacokinetics or pharmacodynamics of prasugrel or ticagrelor, these drugs are recommended over clopidogrel dose escalation in IMs and PMs in the absence of contraindications (such as a high risk of bleeding)33,34.
The effect of the *17 allele on clopidogrel response is unclear. An early study showed that this allele improved the inhibition of platelet aggregation and increased the risk of bleeding with clopidogrel, but the analysis did not adjust for the *2 allele35. The *2 and *17 alleles rarely occur on the same haplotype and, consequently, whether the effects observed were truly owing to the presence of the *17 allele or instead the absence of the *2 allele is unclear.
CYP2C19 and clopidogrel effectiveness.
Multiple studies have shown an increased risk of MACE (generally defined as cardiovascular death, myocardial infarction or stroke) after PCI in clopidogrel-treated IMs and PMs compared with similarly-treated patients without a no-function allele26,36–38. In a meta-analysis of nine studies, including 9,685 clopidogrel-treated patients (55% with an ACS and 91% with PCI), the risk of MACE was significantly higher in IMs than in NMs (HR 1.55, 95% CI 1.11–2.17), with an even greater risk in PMs (HR 1.76, 95% CI 1.24–2.50)39. This allele dose-dependent effect further supports the biological plausibility that each no-function allele causes decreased CYP2C19 metabolic function and therefore an increased risk of MACE in patients receiving clopidogrel. Several additional meta-analyses have also shown higher rates of cardiovascular events in clopidogrel-treated PMs and IMs than in individuals without a no-function allele40–43. However, studies of lower-risk populations, such as patients receiving clopidogrel for atrial fibrillation or with a medically managed ACS, did not show an increased risk of MACE in clopidogrel-treated PMs and IMs44,45. Nevertheless, the CYP2C19 no-function genotype remained associated with stent thrombosis45. Taken together, the data strongly demonstrate reduced clopidogrel effectiveness in IMs and PMs after PCI, but the relationship between CYP2C19 genotype and clopidogrel response in lower-risk populations is less clear25,42.
Clinical trial data.
Two large, multicentre trials (POPular-Genetics and TAILOR PCI) and several smaller trials or interventional studies have examined outcomes with a CYP2C19-guided approach to antiplatelet therapy selection after PCI (Table 2, Box 1)19,46–50. The genotype-guided groups of the POPular-Genetics and TAILOR-PCI trials19,49 consisted of prasugrel or ticagrelor for IMs and PMs and clopidogrel for patients with other phenotypes (such as NMs, RMs or UMs). The POPular-Genetics trial19 showed that among patients undergoing PCI after an ST-segment elevation myocardial infarction, a CYP2C19-guided approach was non-inferior to treatment with prasugrel or ticagrelor in preventing atherothrombotic events but was superior in reducing bleeding risk. These data suggest that in patients without a CYP2C19 no-function allele, clopidogrel is as effective at preventing MACE as an alternative P2Y12 inhibitor. The TAILOR-PCI trial49 included patients with either stable coronary disease or ACS undergoing PCI. In contrast to the POPular-Genetics trial, patients in the comparator group of the TAILOR-PCI trial received clopidogrel. Event rates were lower than anticipated, requiring revision of the power analysis during the trial such that a 50% reduction in the primary end point of major atherothrombotic events was necessary to show statistical significance. The primary analysis was conducted in a subset of participants in each group with the IM or PM phenotype and showed a 34% lower occurrence of events with genotype-guided therapy, which narrowly missed the threshold for statistical significance (P = 0.06). Risk reduction for the secondary end point of stent thrombosis was also close to the threshold for significance (P = 0.05). In addition, there was a reduction in the prespecified end point of total number of events per patient (P = 0.01) with genotype-guided therapy compared with conventional therapy. The event-free survival curves separated early, reflecting the high risk of events in the early period after PCI. A post-hoc analysis revealed a reduction in the rate of events in the first 90 days (P = 0.001) in favour of genotype-guided therapy compared with conventional therapy. These data suggest that genotype-guided therapy has the greatest benefit in the early period (for example, 3 months) following PCI.
Table 2 |.
Studies of outcomes with CYP2C19-guided antiplatelet therapy after percutaneous coronary intervention
| Study (year) | Design | Intervention | Patient population | Primary end points and results | Ref. |
|---|---|---|---|---|---|
| TAILOR-PCI (2020) | Randomized controlled trial | Genotype-guided therapy (n = 903) versus conventional therapy with clopidogrel (n = 946) in IMs and PMs | ACS or stable CAD and PCI | Cardiovascular death, MI, stroke, stent thrombosis or severe recurrent ischaemia occurred in 4.0% of the genotype-guided group and 5.9% of the conventional group (HR 0.66, 95% CI 0.43–1.02, P = 0.06) | 49 |
| Hulot et al. (2020) | Multi-site observational study | Clinical genotyping with recommendations for alternative therapy in slow (for example, *1/*2) and very slow (*2/*2) metabolizers (n = 1,445 total) | STEMI and PCI | Rate of death, MI or stent thrombosis was 3.04% in slow and very slow metabolizers who received optimized therapy versus 3.31% in normal and rapid metabolizers (P = 0.82) and 15.6% in slow or very slow metabolizers without treatment adjustment (P < 0.05) | 64 |
| POPular Genetics (2019) | Randomized controlled trial | Genotype-guided therapy (n = 1,242) versus standard treatment with either ticagrelor or prasugrel (n = 1,246) | STEMI and PCI | All-cause death, MI, stent thrombosis, stroke or major bleeding occurred in 5.1% of the genotype-guided group and 5.9% of the standard-treatment group (P < 0.001 for non-inferiority); major or minor bleeding occurred in 9.8% of the genotype group and 12.5% of the standard-treatment group (HR 0.78, 95% CI 0.61–0.98, P = 0.04) | 19 |
| PHARMCLO (2018) | Randomized controlled trial | Genotype-guided therapy (n = 448) versus standard treatment (n = 440) | STEMI or non-STEMI and PCI (62% had PCI) | Cardiovascular death, MI, stroke or major bleeding occurred in 15.9% of the genotype group and 25.9% of the standard-treatment group (HR 0.58, 95% CI 0.43–0.78, P < 0.001) | 46 |
| Cavallari et al. (2018) | Multi-site observational study | Clinical genotyping with recommendations for alternative therapy in IMs and PMs (n = 1,815 total) | ACS or stable CAD and PCI | Rate of death, MI or ischaemic stroke was 13.5 per 100 patient-years in IMs or PMs prescribed clopidogrel versus 8.7 per 100 patient-years in IMs or PMs prescribed alternative therapy (propensity score adjusted HR 2.26, 95% CI 1.18–4.32, P = 0.013) | 62 |
| Deiman et al. (2016) | Single-centre, observational study | Clinical genotyping with recommendations for alternative therapy in PMs (n = 3,260 total) | Elective PCI | Occurrence of death from cardiovascular causes, MI, stent thrombosis, repeat revascularization or stroke in PMs was 31% with clopidogrel and 5% with prasugrel (P = 0.003) | 63 |
| Sanchez-Ramos et al. (2016) | Intervention study versus historical controls | Genotype-guided strategy (n = 317) versus non-tailored strategy (n = 402) | PCI with stent implantation (86% with ACS) | Cardiovascular death, ACS or stroke occurred in 10% of patients in the genotype group and 14% of controls (P = 0.037) | 50 |
| Shen et al. (2016) | Intervention study versus historical controls | Genotype-guided group (n = 309) versus usual care with clopidogrel (n = 319) | CAD and PCI | All-cause death, MI or target-vessel revascularization occurred at rate of 4.2% in the genotype group and 9.4% in the usual-care group (P = 0.010) | 47 |
| IAC-PCI (2013) | Randomized controlled trial | Personalized therapy (n = 301) versus conventional therapy with clopidogrel (n = 299) | CAD and PCI with stent implantation | Cumulative rate of all-cause death, MI, stroke or target-vessel revascularization at 6 months was 2.6% in the genotype group and 9.0% in the conventional treatment group (P < 0.01) | 48 |
ACS, acute coronary syndrome; CAD, coronary artery disease; IM, intermediate metabolizer; MI, myocardial infarction; PCI, percutaneous coronary intervention; PM, poor metabolizer; STEMI, ST-segment elevation myocardial infarction.
Box 1 |. Limitations of RCT data.
Although data from randomized controlled trials (RCTs) are available for clopidogrel and warfarin and forthcoming for statins, their interpretation is challenging given limitations inherent to the field of pharmacogenetics. Namely, for existing medications, most patients have a genotype associated with safe and effective therapy. Otherwise, the drug would not have reached the market, at least in the absence of having a companion diagnostic device to identify patients expected to benefit. Therefore, large sample sizes are needed to identify a sufficient number of patients with variant genotypes of interest in whom a genotype-guided approach is hypothesized to be of benefit. For example, the TAILOR PCI trial49 enrolled >5,000 patients to identify the subset (approximately 1,850) with a CYP2C19 no-function allele, in whom the primary end point was tested. Even with the large overall study population, the study was powered to detect only a relatively large difference (50% relative risk reduction) between the groups49.
Concerns also exist about the generalizability of RCT data to real-world patient populations. In particular, POPular-Genetics and TAILOR-PCI participants seem to have a higher prevalence of risk factors for cardiovascular events (for example, diabetes and kidney disease) and bleeding (for example, oral anticoagulant use and history of stroke) than those who received CYP2C19 testing in clinical practice (see the table)19,49,62. Therefore, whether trial results can be translated to higher-risk populations is unclear.
Differences in allele frequencies according to ancestry create another challenge, in which the variants tested must be appropriate for the patient populations included. This challenge is most clearly demonstrated with the warfarin pharmacogenetic trials, all of which limited genotyping to variants important for non-African ancestry populations, and while trials enrolling predominately non-African ancestry populations showed positive outcomes89,121, the COAG trial122 with a large African ancestry population did not. Indeed, genotype-guided dosing was worse in this population, more often resulting in over-anticoagulation. These data further point to the limited generalizability of RCT data to populations underrepresented in the trial. Had the COAG trial not been conducted and the results from the EU-PACT and GIFT trials been translated into clinical practice, with genotyping limited to the variants tested in the trials, this approach could have resulted in over-dosing and harm to patients of African ancestry.
| Study (year) | Diabetes (%) | Hypertension (%) | Dyslipidaemia (%) | Chronic kidney disease (%) | Oral anticoagulation (%) | History of stroke or transient ischaemic attack (%) | Ref. |
|---|---|---|---|---|---|---|---|
| IGNITE (2018) | 38 | 80 | 68 | 30 | 8 | 11 | 62 |
| POPular-Genetics (2019) | 12 | 42 | 21 | 9 | 4 | Not reported | 19 |
| TAILOR-PCI (2020) | 27 | 63 | 52 | 10 | Excluded | 3 | 49 |
Clinical use
In 2010, the FDA approved a revision to add a boxed warning to the label for clopidogrel about reduced clopidogrel effectiveness in PMs (but not IMs), in whom alternative P2Y12 inhibitors are recommended51. Similar language exists on the clopidogrel label approved by the European Medicines Agency, Japanese Pharmaceutical and Medical Devices Agency and other regulatory bodies. The FDA addresses both IMs and PMs in their table of gene–drug pairs in which the data support therapeutic management recommendations11. CPIC guidelines for the use of clopidogrel also address both IMs and PMs and focus recommendations on patients with an ACS and PCI, in whom substantial data exist, whereas the DPWG extends recommendations to patients with stroke9,25. The guidelines recommend prasugrel or ticagrelor in CYP2C19 IMs or PMs in the absence of a contraindication, but make no recommendations on whether genetic testing should be performed. Joint PCI guidelines from 2016 by the ACC/AHA recommend against routine genotyping for all patients undergoing PCI, citing a lack of randomized controlled trial (RCT) data at the time the guidelines were written14. However, these organizations state that testing might be considered in high-risk patients and, similar to CPIC, either prasugrel or ticagrelor is recommended for patients with a no-function allele.
CYP2C19 testing to guide the use of antiplatelet therapy after PCI is one of the most common examples of pharmacogenetic testing in clinical practice2,52. Some sites reserve testing for patients at high risk of MACE on the basis of clinical presentation or coronary anatomy, in line with ACC/AHA guidelines, whereas other sites genotype the majority of patients undergoing PCI52. Most sites provide recommendations for alternative P2Y12 inhibitor therapy for both IMs and PMs via clinical consult notes or automated decision support. Testing strategies vary between sites, with rapid genotyping in place at some institutions, allowing results to be available soon after sample collection (Box 2). At other sites where rapid genotyping is not available, results are usually returned within a week, and patients with a no-function CYP2C19 allele who are receiving clopidogrel can be switched to alternative therapy at that time. However, this strategy might place patients at an elevated risk of MACE in the vulnerable, high-risk period immediately after PCI. Therefore, another approach in the absence of rapid genotyping is to treat patients with prasugrel or ticagrelor until the genotype results are obtained. When the genotyping results arrive, IMs and PMs might be continued on the newer agents, whereas those without a no-function allele might be switched to clopidogrel in a so-called ‘de-escalation’ approach. This approach has the potential to maximize benefit and reduce bleeding risk given the high risk of atherothrombotic events early after ACS and PCI and high risk of bleeding with newer agents during long-term therapy.53–56 The 2020 ESC guidelines address genotype-guided de-escalation as a strategy for patients who are unsuitable for long-term therapy with more potent agents after PCI because of bleeding risk or other factors, citing the POPular Genetics trial as the basis for this recommendation.19,57,58
Box 2 |. Challenges with pharmacogenetic implementation.
Despite substantial evidence supporting genetic associations with the response to clopidogrel, warfarin and simvastatin and existing examples of genotyping in practice, pharmacogenetics has not been widely implemented in clinical practice. A major barrier to implementation is the demand by many stakeholders, including physicians and third-party payers, for randomized controlled trial data demonstrating that testing improves clinical outcomes. However, some argue that pharmacogenetics is held to a higher standard than that required for other laboratory tests (such as renal function and blood counts) that are routinely ordered (and reimbursed) to inform drug therapy216. Multiple factors influence the drug response, and a pharmacogenetic test result provides just one, albeit important, piece of information that contributes to a patient’s full clinical picture, as is the case with many other laboratory results.
Another major challenge with pharmacogenetic testing in practice is genotype turnaround time. Pharmacogenetic results are most useful when available at the time of prescribing to avoid the need to delay prescribing decisions until after results become available. However, many current implementations follow a reactive testing model, in which testing is ordered at the time a drug therapy change can be considered. Challenges with turnaround time might be overcome with rapid, point-of-care testing, as described previously6, but this approach might not be feasible in all settings. This challenge is particularly true in countries such as the USA, where genotyping is considered to be high-complexity testing, requiring that it be done by specially certified personnel127. Pre-emptive pharmacogenetic testing (where patients are tested before any prescribing decision is made) and the eventual promise of integrating genome sequence data into the electronic health record could greatly facilitate pharmacogenetic adoption. Then the questions of who or when to genotype become moot and are replaced by the question of how to use existing genetic information in prescribing decisions, for which guidance exists from the Clinical Pharmacogenetics Implementation Consortium and other pharmacogenetic groups. However, pre-emptive testing creates additional challenges, including the need to integrate results into the electronic health record, ideally in a life-time results section, to be readily accessible when needed. For optimal use of pre-emptive test results, there should also be a means to alert the provider of test results when applicable and provide recommendations for therapy based on the results. This approach might be best provided through automated alerts in the electronic health record, triggered when a relevant drug (such as clopidogrel) is prescribed to a patient with a genotype associated with reduced effectiveness (for example, CYP2C19*1/*2) or increased risk of toxicity. Ideally, there should also be a mechanism for test results to accompany patients who move from one health-care institution to another.
Pre-emptive genotyping of patients at high risk of cardiovascular events has also been described59 and allows for results to be readily available if the patient requires PCI in the future. Given the lack of urgency with pre-emptive testing, samples can be batched and processed together, substantially reducing the cost of testing. Given that the CYP2C19 genotype has implications for multiple drugs other than clopidogrel, such as proton-pump inhibitors60 and selective serotonin reuptake inhibitors61, the results could be valuable to guide other therapies even if the patient never requires PCI. However, because many third-party payers are reluctant to reimburse for pre-emptive testing, such a model might require an investment from the health system to cover the cost of testing.
Real-world outcome data.
Adding to the RCT evidence on outcomes with CYP2C19-guided antiplatelet therapy are data from real-world observations of patients receiving genetic testing as part of clinical care62–64. A Dutch study of >3,000 patients undergoing elective PCI showed a significant reduction in the risk of MACE among PMs prescribed prasugrel compared with PMs treated with clopidogrel63. However, the outcomes in IMs were not examined in this study. In a US-based study, we examined outcomes with CYP2C19 testing in 1,815 patients undergoing either emergent or elective PCI across seven institutions in the NHGRI-funded Implementing GeNomics In pracTicE (IGNITE) Network62. In line with CPIC guidelines, all sites recommended alternative therapy for IMs and PMs, with the ultimate prescribing decision left to physician discretion. No recommendations were provided for patients without a non-functional allele. Among the 31.5% of patients with the IM or PM phenotype, 61% received alternative therapy (most commonly prasugrel) in line with recommendations, and the remainder received clopidogrel. The risk of MACE was significantly higher in IMs and PMs treated with clopidogrel than with alternative therapy (Table 2). These differences were most pronounced in patients with ACS and remained significant when limiting the analysis to IMs. No significant difference was observed in outcomes between patients without a no-function allele (85% receiving clopidogrel) and IMs and PMs treated with prasugrel or ticagrelor, providing further data that clopidogrel might be as effective as newer agents in those without a no-function allele. Similar results were shown in an observational study of CYP2C19-guided antiplatelet therapy after STEMI and PCI across 57 centres in France64.
The IGNITE dataset was expanded to include two additional sites and >1,500 additional patients. An initial study with the larger data set aimed to examine the effect of the increased-function CYP2C19*17 allele on clopidogrel effectiveness and safety65. The study specifically compared outcomes between clopidogrel-treated RMs or UMs (for example, those with the *1/*17 or *17/*17 genotype) versus NMs and showed no significant difference between the groups in the risk of atherothrombotic or bleeding events after PCI. These data contrast with previous studies comparing outcomes between *17 allele carriers and non-carriers, in which PMs and IMs were included in the non-carrier group, which might have confounded results35,66,67.
Emerging evidence
In addition to PCI, clopidogrel is prescribed for stroke prevention and peripheral arterial disease, and data show that the CYP2C19 genotype influences clopidogrel effectiveness in these settings. In particular, a large RCT of patients with a minor ischaemic stroke or transient ischaemic attack showed that the combination of clopidogrel plus aspirin was more effective than aspirin alone in preventing recurrent stroke, but only in those without a CYP2C19 no-function allele68. In a subsequent meta-analysis of 15 studies and nearly 5,000 clopidogrel-treated patients with ischaemic stroke or transient ischaemic attack, the risk of new stroke was significantly higher in IMs and PMs than in individuals with other phenotypes69. Similarly, after endovascular treatment for peripheral arterial disease, reduced rates of stent patency have been observed with clopidogrel treatment in IMs and PMs70,71. Concerns about reduced clopidogrel effectiveness on the basis of CYP2C19 genotype prompted investigators to exclude PMs from the large, multi-site EUCLID trial72 of ticagrelor versus clopidogrel in peripheral artery disease.
Additional clopidogrel-response modifiers
Genetic factors.
A genome-wide association study (GWAS) confirmed that the CYP2C19 gene was the major genetic contributor to the inter-patient variability that mediates the antiplatelet effects of clopidogrel73. Researchers have sought to identify additional genetic contributions to the clopidogrel response. Other genes examined include ABCB1, CES1 and PON1. Clopidogrel is a substrate for P-glycoprotein, an intestinal efflux transporter encoded by the ABCB1 gene. Some studies have suggested an association between the c.3435C>T (rs1045642) polymorphism and the effectiveness of clopidogrel after PCI74,75, whereas other studies have not shown an association76. Similarly, PON1, which encodes esterase paraoxonase 1, an enzyme involved in clopidogrel bioactivation, was initially linked to the risk of stent thrombosis during clopidogrel treatment after PCI.77 However, multiple studies have not replicated this finding78–80. The CES1 genotype data are perhaps the most promising. Carboxylesterase 1 (CES1) hydrolyses clopidogrel into an inactive carboxylic acid metabolite, influencing the amount of drug available for conversion to its active form via CYP2C19. The CES1 variant Gly143Glu (rs71647871) reduces CES1 activity and has been associated with reduced platelet reactivity after clopidogrel administration81,82. Members of the International Clopidogrel Pharmacogenomics Consortium reported that a polygenic risk score, including variants in CYP2C19, CES1 and CYP2B6, predicted an increased risk of cardiovascular events and increased cardiovascular mortality in patients receiving clopidogrel82. Ultimately, a polygenic risk score, combined with clinical factors (described below), might provide superior prediction of the clopidogrel response than any single factor alone.
Clinical variables.
Clinical factors that can modify the clopidogrel response include age, body size, renal function and diabetes mellitus. Lower exposure to the active clopidogrel metabolite and higher residual platelet reactivity have been observed in clopidogrel-treated patients with diabetes83,84. The presence of chronic kidney disease, age >75 years or obesity further impairs the antiplatelet effects of clopidogrel85,86. The ABCD-GENE (age, BMI, chronic kidney disease, diabetes mellitus and CYP2C19 genotype) score was developed and validated as a predictor of cardiovascular events after PCI86. Multiple studies have also examined whether proton-pump inhibitors can reduce the clopidogrel response through inhibitory effects on CYP2C19 activity, but the data are inconsistent87.
Oral anticoagulants
The vitamin K antagonist warfarin and the direct-acting oral anticoagulants (DOACs) apixaban, dabigatran, edoxaban and rivaroxaban are indicated for the treatment and prevention of thromboembolic disorders. Warfarin has a narrow therapeutic index and requires frequent monitoring of the international normalized ratio (INR) to ensure optimal anticoagulation, defined as an INR of 2–3 for most indications. Warfarin doses necessary to attain an INR of 2–3 vary from <1 mg per day to >10 mg per day among patients88. Genotype is a major contributor to this dose variability but is rarely considered in clinical practice. Instead, warfarin treatment is typically initiated at a similar dose (for example, 5 mg per day) in all patients. Loading doses (for example, a 10 mg dose) are sometimes used for the initial 1–3 days of therapy, particularly in Europe, with subsequent dose adjustment on the basis of an INR response89,90. However, these ‘fixed-dose’ approaches can result in over-anticoagulation in individuals with genotypes associated with reduced warfarin metabolism or increased sensitivity. Conversely, delays can occur in attaining therapeutic anticoagulation in those with warfarin-resistance genotypes, especially in the absence of a loading dose.
DOACs have a wider therapeutic index than warfarin, do not require regular monitoring, and are associated with greater reductions in the risk of stroke or systemic embolic events and a lower risk of intracranial haemorrhage compared with warfarin91,92. Although genetic variation, specifically in the CES1 gene, has been linked to plasma levels of dabigatran and risk of bleeding, no consistent evidence exists on genetic associations with DOAC effectiveness93. Owing to the favourable profile of DOACs compared with warfarin, DOAC use has steadily increased over the past 10 years. By 2015, DOAC use comprised approximately 30% of oral anticoagulation in the USA and 50% of prescriptions for patients with atrial fibrillation in Norway94,95. However, because of the higher cost, lower adherence rates and more limited indications for DOACs than for warfarin, in addition to the high cost of reversal agents for DOACs in the event of over-anticoagulation, warfarin remains commonly prescribed — particularly in older patients and those at increased risk of bleeding or with substantial comorbidity96,97.
Pharmacogenetics
Genetic associations.
The major genes influencing the response to warfarin are CYP2C9 and VKORC1, with a minor contribution from CYP4F2 (Fig. 3). The CYP2C9*2, *3, *5, *6, *8 and *11 alleles reduce S-warfarin (the more active enantiomer) clearance and therefore patient dose requirements98–101. Dose reductions of approximately 5–7 mg per week have been reported with the *2, *8 and *11 alleles, with reductions approaching 14 mg per week reported for the *3 and *5 alleles102,103. These variants are also associated with increased risk of over-anticoagulation and bleeding, with a high risk of bleeding persisting throughout warfarin therapy104,105. The *2 and *3 alleles are the most common CYP2C9 variants in individuals of European ancestry, whereas the *5, *6, *8 and *11 alleles occur almost exclusively in populations with African ancestry.
Fig. 3 |. Sites of action for pharmacogenes involved in the response to warfarin.
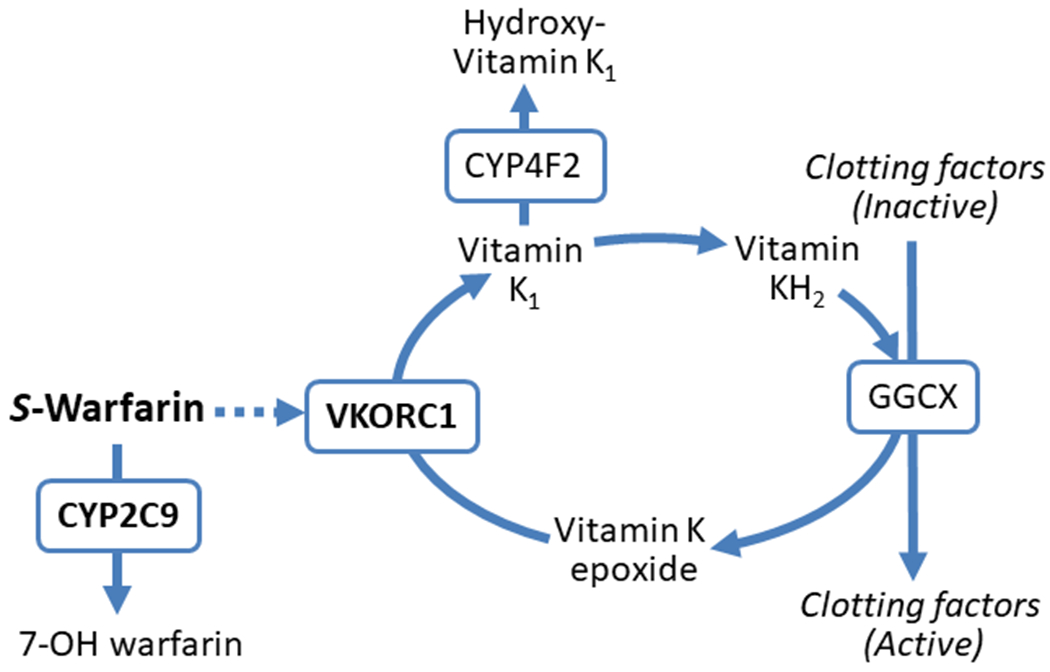
The more potent S-enantiomer of warfarin is metabolized by the cytochrome P450 (CYP) 2C9 enzyme to the inactive 7-OH warfarin protein. Warfarin exerts its anticoagulation effects by inhibiting vitamin K oxidoreductase complex 1 (VKORC1), thereby preventing reduction of vitamin K epoxide to vitamin K1, which is subsequently reduced to vitamin KH2, a necessary cofactor for γ-carboxylation and activation of clotting factors II, VII, IX and X. The CYP4F2 enzyme metabolizes vitamin K1 to an inactive hydroxyl-vitamin K1 metabolite so that less is available for reduction to vitamin KH2. Polymorphisms in the genes encoding the CYP2C9, VKORC1 and CYP4F2 proteins can affect warfarin metabolism, sensitivity to warfarin and vitamin K availability, respectively, thereby influencing warfarin dose requirements and bleeding risk. GGCX, γ-glutamyl carboxylase.
Vitamin K epoxide reductase complex 1 (VKORC1) is the protein target of warfarin, and rare variants contribute to warfarin resistance and the need for very high doses (such as ≥20 mg per day) to attain therapeutic anticoagulation106. A common variant, c.–1639G>A (rs9923231) in the regulatory region of VKORC1 is associated with reduced VKORC1 expression and lower warfarin dose requirements.107,108 Compared with individuals of European ancestry, the –1639 AA (highly sensitive) genotype is more prevalent in Asian individuals, and the –1639 GG (reduced sensitivity) genotype is more common in populations with African ancestry (Table 1). This variation accounts for the lower doses generally observed in Asian individuals and higher doses observed in individuals of African ancestry than in European individuals109.
The CYP4F2 enzyme metabolizes vitamin K1 to hydroxyl-vitamin K1. CYP4F2 activity is reduced in the presence of the *3 allele (rs2108622), resulting in higher concentrations of vitamin K1 being available for reduction to vitamin K hydroquinone and activation of clotting factors110. The *3 allele is associated with higher warfarin dose requirements than the *1 allele in European and Asian cohorts111. However, no association has been identified in populations with African ancestry111,112.
GWAS have confirmed that VKORC1 and CYP2C9 are the primary genetic contributors to warfarin dose requirements in populations with European or Asian ancestry.113–115 A GWAS in a population with African ancestry revealed an additional variant near the CYP2C cluster on chromosome 10, rs12777823, associated with lower dose requirements for warfarin.116 This single-nucleotide polymorphism (SNP) is also correlated with reduced S-warfarin clearance, and although this SNP is common across the general population, its association with the warfarin response seems to be limited to persons of African ancestry116. Therefore, rs12777823 is not thought to be a functional polymorphism, but instead to be in linkage disequilibrium with a functional variant(s) found in populations of African ancestry. A subsequent GWAS identified a variant on chromosome 6 upstream of EPHA7 that was associated with the risk of major bleeding in warfarin-treated African American individuals with an INR of <4117. This variant occurs exclusively in persons of African ancestry, with an allele frequency of 7%, and its association with bleeding risk reached genome-wide significance when the discovery and replication cohorts were combined. The SNP also seems to increase bleeding risk prediction when included in the HAS-BLED score, but this finding requires validation117,118.
Dosing algorithms.
Dosing algorithms including genotype (VKORC1 –1639G>A, CYP2C9*2 and CYP2C9*3 alleles) and clinical variables that influence the response to warfarin (for example, age, height, weight and use of CYP2C9 inhibitors) have been developed by the International Warfarin Pharmacogenetics Consortium (IWPC)119 and Gage and colleagues120 to assist with estimating initial warfarin doses (both available at Warfarin Dosing). The IWPC algorithm also includes the use of CYP2C9 inducers (such as phenytoin or rifampin) and is preferred for patients receiving these drugs. Otherwise, both algorithms provide similar dose estimations. Importantly, neither published algorithm includes the CYP2C9*5, *6, *8, *11 or rs12777823 variant, although the *5 and *6 alleles are incorporated into the online version of the Gage algorithm, as is the CYP4F2*3 allele.
Clinical trial data.
Three large multi-site RCTs have evaluated the efficacy of genotype-guided warfarin dosing89,121,122 (Table 3, Box 1). All three trials used a pharmacogenetic algorithm that included the VKORC1 –1639G>A and CYP2C9*2 and *3 variants. The GIFT trial121 additionally genotyped for CYP4F2. The primary end point for the EU-PACT and COAG trials89,122 was time in therapeutic range, whereas the GIFT trial121 was powered to detect differences in clinical outcomes. Both the EU-PACT and GIFT trials, but not the COAG trial, showed significant improvement in their primary end points with pharmacogenetic dosing.
Table 3 |.
Comparison of warfarin pharmacogenomic trials
| Trial | Participants | Comparator group | Genetic polymorphisms tested | Loading dose | Primary end point | Outcome | Ref. |
|---|---|---|---|---|---|---|---|
| EU-PACT (2013) | Adults from Sweden or the UK (n = 455; 98.5% white) who were newly starting warfarin for atrial fibrillation or venous thromboembolism | Fixed dose of 10 mg on day 1 (or 5 mg if aged >75 years), then 5 mg on days 2 and 3, then dose guided by INR | CYP2C9*2 and *3, VKORC1 −1639G>A | Yes, loading dose algorithm used on days 1–3 in the genotype-guided group; 10 mg dose given on day 1 for patients aged ≤75 years in the control group | Percent of time with an INR of 2.0–3.0 during the initial 12 weeks of warfarin therapy | Mean percent time in INR range was 67.4% in the genotype-guided group and 60.3% in the control group (P < 0.001) | 89 |
| COAG (2013) | Adults from the USA newly initiating warfarin treatment with a target INR of 2.0–3.0 (n = 1,015; 27% Black, 73% non-Black) | Clinical algorithm-guided dosing | CYP2C9*2 and *3, VKORC1 −1639G>A | No, but CYP2C9 variants were ignored for the initial dose in the genotype-guided group | Percent of time with an INR of 2.0–3.0 from day 4 or 5 through to day 28 of warfarin therapy | All patients: mean percent time in INR range was 45.2% in the genotype-guided group and 45.4% in the clinically-guided group (P = 0.91); Black patients: mean percent time in INR range was 35.2% in the genotype-guided group and 43.5% in the clinically guided group (P = 0.01) | 122 |
| GIFT (2017) | Patients aged ≥65 years undergoing elective total-hip or total-knee arthroplasty (n = 1,650; 91% white, 6% Black) | Clinical algorithm-guided dosing | CYP2C9*2 and *3, VKORC1 −1639G>A, CYP4F2*3 | No, but CYP2C9 variants were ignored for the first 2 days of therapy in the genotype-guided group | Composite of major bleeding, INR ≥4, venous thromboembolism or death | 10.8% in the genotype-guided group and 14.7% in the clinically guided group had at least one end point (relative rate 0.73, 95% CI 0.56–0.95, P = 0.02) | 121 |
INR, international normalized ratio.
Important differences between trials might have contributed to the disparate results (Table 3). Of note, both the EU-PACT and GIFT trials were conducted in predominately European ancestry populations, whereas the COAG trial included a more diverse population, with 27% of participants being African American. None of the trials genotyped for the African-specific variants, and failure to account for these variants is associated with substantial warfarin overdosing among African Americans because individuals with a CYP2C9*5, *6, *8 or *11 allele (approximately 15% of patients with African ancestry) or rs12777823 A allele (>40% patients) would be misclassified as NMs (for example, *1/*1) and dosed accordingly123. Indeed, among COAG participants of African ancestry, genotype-guided warfarin dosing resulted in worse anticoagulation control than dosing on the basis of clinical factors alone.122
Another important difference between the trials was that loading doses were used in the EU-PACT but not in the COAG trial. We have reported that loading doses are needed in most patients starting warfarin to reduce the amount of time required to achieve therapeutic anticoagulation and that loading doses should be genotype-based124. Specifically, rapid INR increases and supratherapeutic anticoagulation can occur with warfarin initiation in patients with two or more CYP2C9 or VKORC1 variants (such as the VKORC1 –1639AA genotype or a combination of VKORC1 –1639AG and CYP2C19 *1/*3 genotypes). We reported substantial delays in time to the first therapeutic INR for individuals without a CYP2C9 or VKORC1 variant or with a single variant124. These data suggest that cautious dosing is warranted in individuals with two or more variants to avoid over anticoagulation. Conversely, aggressive loading doses might be needed in patients without a variant or with a single variant, which includes most patients of European or African ancestry, to reduce the time needed to achieve therapeutic anticoagulation. This aggressive loading dose schedule is supported by results from the EU-PACT trial, in which genotype-guided loading doses were used in the genotype group. The genotype-guided group had a higher percentage of time spent in the therapeutic range than the control group in which patients received a standard loading dose regimen89. Given that the primary end point of the COAG trial was time in therapeutic range at 1 month, the absence of loading doses, and especially loading doses individualized on the basis of genotype results, might have delayed the time required to reach therapeutic anticoagulation and affected trial results.
Clinical use
Pharmacogenetic guidance.
The US Association for Molecular Pathology has defined the CYP2C9*2, *3, *5, *6, *8, *11 and VKORC1 –1639G>A alleles as the minimum set of variants that should be included in clinical warfarin genotyping assays125. The FDA-approved warfarin labelling and guidelines by CPIC and DPWG provide dosing recommendations on the basis of genotype. However, both the FDA labelling and DPWG guidelines limit recommendations to the VKORC1 –1639G>A and CYP2C9*2 and *3 alleles108,126. By contrast, CPIC addresses other CYP2C9 variants and provides separate guidance for patients of African ancestry and non-African ancestry108. For populations of non-African ancestry, CPIC recommends estimating the dose on the basis of CYP2C9*2 and *3 and VKORC1 variants with the use of one of the available dosing algorithms. Genotyping for CYP4F2*3 is considered optional, but if detected, a dose increase of 5–10% is recommended. For those of African ancestry, genotype-guided dosing is recommended only if the information on the CYP2C9*5, *6, *8 and *11 genotypes is available, with genotyping for rs12777823 considered to be optional. If this additional genotype information is available, the warfarin dose should first be estimated with the use of a warfarin-dosing algorithm and then reduced by 15–30% for each CYP2C9*5, *6, *8 or *11 allele, with an additional 15–30% reduction if the rs12777823 variant is detected.
Clinical implementation.
Several examples exist of genotype-guided warfarin dosing in practice4–6. Genotype-guided dosing, with the use of a point-of-care testing device and the same dosing algorithm used in the EU-PACT trial, was implemented in three anticoagulation clinics in northern England6. The genotype-guided approach resulted in greater time in the therapeutic range in the initial 3 months of therapy than standard (non-genotype-guided) dosing. These data demonstrate the feasibility of implementing genotype-guided warfarin dosing in practice and show that improved outcomes can be achieved with this approach outside of the controlled clinical trial setting.
Similarly, we have described the implementation of genotype-guided dosing as the standard of care for patients starting warfarin during hospitalization3. In contrast to Europe, where point-of-care testing is acceptable, genotyping in the USA is considered to be ‘high complexity’ and cannot be conducted as a point-of-care test127 (Box 2). Therefore, to allow time for genotyping to be done, patients received an initial warfarin dose guided by clinical factors, calculated via a dosing algorithm embedded in the electronic health record, with subsequent dosing guided by genotype results. This approach requires an efficient turnaround of genotyping data so that results could be available before the second warfarin dose. This approach is labour intensive and probably not practical at most medical centres. An alternative approach taken by other centres is to genotype pre-emptively, with results entered into the electronic health record, so that genotyping data are available if warfarin is later prescribed128–131. Ideally, automated clinical decision support should be available to provide dose estimates on the basis of genotype and clinical factors at the point of initial warfarin prescribing.
Genetic information might also assist in the selection of oral anticoagulant therapy and specifically in decisions about initiating warfarin treatment versus a DOAC. Data to support this approach come from genetic substudies of trials comparing the efficacy of DOACs versus warfarin in stroke prevention132. In an ENGAGE AF TIMI-48 substudy132, warfarin-treated participants with a sensitive or highly sensitive genotype (for example, VKORC1 –1639AA or CYP2C9*1/*3) had greater proportions of time with an INR in the supratherapeutic range (that is, INR >4) and had higher rates of bleeding in the initial 90 days of treatment than those with non-sensitive genotypes. The reduction in bleeding risk with edoxaban compared with warfarin was greatest among patients with the sensitive and highly sensitive genotypes, suggesting these patients might be good candidates for DOAC therapy. In a genetic substudy of the RE-LY trial133, carriers of the CES1 rs2244613 minor allele had a reduced risk of bleeding with dabigatran than with warfarin, whereas there was no significant difference in bleeding risk between treatment groups in non-carriers. Although the functional effects of this variant are unclear,133 these data further support the use of genotype data in the selection of oral anticoagulant therapy.
Statins
Pharmacogenetics
Although still one of the most commonly prescribed HMG-CoA reductase inhibitors (or statins), simvastatin use has declined in the past decade — in part because simvastatin use has been associated with an increased risk of myopathy or myopathy-like symptoms, which is estimated to occur in 1–5% of simvastatin-treated patients134,135. The risk of myopathy with simvastatin treatment increases in a dose-dependent manner, which prompted the FDA in 2011 to recommend restricting the use of simvastatin 80 mg per day, the maximum approved dose136. Simvastatin use is associated with a wide spectrum of muscle-related symptoms, ranging from mild myalgia to severe, life-threatening rhabdomyolysis, and at least a portion of myopathy risk can be explained by genetic variation in the SLCO1B1 gene137. A less consistent genetic association exists with more general muscle-related symptoms, which might be due to patient and physician expectations related to this well-known adverse effect associated with statins (the so-called nocebo effect).138 SLCO1B1 encodes the organic anion transporting polypeptide 1B1 (OATP1B1), a transporter mediating the hepatic uptake of endogenous compounds and many drugs (such as some statins) for metabolism139. In the SLCO1B1 gene, the rs4149056 SNP has, by far, the most evidence supporting its influence on OATP1B1 function. The rs4149056 variant C allele is associated with decreased OATP1B1 transporter function, with homozygous patients having notably reduced function140. This reduced drug transport to the liver can lead to accumulation of circulating levels of simvastatin acid, the active form of simvastatin141,142. The rs4149056 variant has the greatest effect on exposure to simvastatin acid, followed by pitavastatin, lovastatin and atorvastatin143,144. Owing to increased drug exposure generally increasing the risk of adverse effects, patients with the rs4149056 C allele receiving simvastatin have an increased risk of myopathy, with the highest risk in homozygous patients.135,145,146
Genetic associations with statin-induced myopathy.
Two genome-wide association studies of atorvastatin response have been published147,148. One study tested associations with plasma levels of atorvastatin and its major metabolites in patients with ACS147. In addition to rs4149056 (which was strongly associated, but not at a genome-wide significant level), the researchers identified two strong associations with atorvastatin-metabolite ratios: rs45446698 just upstream of CYP3A7 and rs887829 located in multiple overlapping UGT1A genes. Atorvastatin is partially metabolized by the CYP3A family. Although CYP3A7 is predominantly expressed in the fetal liver, expression continues into adulthood in approximately 10% of adults149. Multiple UDP-glucuronosyltransferase 1A1 enzymes are also probably involved in atorvastatin metabolism, so these associations seem biologically plausible.
The other GWAS was a case–control analysis of statin-induced myopathy, in which >50% of patients received simvastatin and approximately 30% received atorvastatin148. Only rs4149056 reached the threshold of genome-wide significance, being strongly associated with severe myopathy. This association was consistent across the replication and additional validation cohorts, with a combined meta-analysis of all cohorts showing a threefold increased risk with the presence of each C allele. Another SNP, rs4149000 (in SLCO1A2), was associated with a similar risk profile in the meta-analysis but was no longer significant after adjusting for the rs4149056 genotype. Also, while solute carrier organic anion transporter family member 1A2 (SLCO1A2) might transport other statins, no evidence currently exists that SLCO1A2 transports simvastatin or atorvastatin.
Other genetic variations have also been associated with statin-associated myopathy, but not with the consistency observed with SLCO1B1. Polymorphisms in genes encoding proteins involved in statin transport and metabolism, such as CYP3A4 and ABCB1, have shown conflicting associations150–153. Similarly, variants in genes encoding proteins associated with muscle physiology, such as COQ2 (involved in coenzyme Q10 synthesis) and GATM (involved in creatine synthesis), have also shown limited or conflicting associations146,154,155.
Genetic associations with statin effectiveness.
Polygenic risk scores that are predictive of an increased risk of atherosclerotic cardiovascular disease have been described156, and, in at least one case, the score was also predictive of risk reduction with statin therapy156,157. The success of a polygenic risk score in predicting the effectiveness of statin therapy contrasts with earlier efforts that focused on single gene variants as predictors of statin effectiveness158. The variants included in the polygenic risk score are not obviously related to statin pharmacokinetics or pharmacodynamics, so whether the score specifically predicts statin response or reflects a more severe phenotype that would be likely to respond better to most risk-lowering therapies for coronary artery disease is unclear. Indeed, a report showed that the same risk score that predicted statin-associated cardiovascular risk reduction also predicted risk reduction with the proprotein convertase subtilisin kexin type 9 inhibitor alirocumab159.
Clinical trial data.
Unlike with clopidogrel and warfarin, large RCTs testing genotype-guided statin prescribing have yet to be completed. A pragmatic trial randomly assigned 159 patients who discontinued statins because of myalgia to either restarting statin therapy informed by their SLCO1B1 genotype or according to standard non-genetic recommendations sent to the patients’ primary providers through the electronic health record160. Among patients who restarted statin therapy, the primary end point of self-reported statin adherence was similar between groups. However, the genotype group had more patients who restarted statin therapy and greater reductions in plasma LDL-cholesterol level than the group given non-genetic recommendations. Patients with myopathy associated with any statin were included, but because SLCO1B1 variation does not seem to clinically influence all statins similarly, these data do not provide clarity for specific genotype-guided simvastatin (and potentially atorvastatin) therapy.
The I-PICC trial161 is an ongoing, pseudo-cluster RCT comparing pre-emptive SLCO1B1-informed statin therapy with usual care in approximately 400 statin-naive patients who meet guideline recommendations for statin therapy. Patients are enrolled when a lipid profile is ordered, with the leftover blood sample used for genotyping to allow for pharmacogenetic data to be available in time to inform statin prescribing. Genotype-based recommendations seem to mirror CPIC simvastatin guidelines. The primary outcome is a change in plasma LDL-cholesterol level at 1 year.
Clinical use
CPIC provides guidance for the use of the SLCO1B1 genotype to guide simvastatin therapy143 on the basis of the evidence linking rs4149056 and simvastatin-associated myopathy. These guidelines recommend lowering the dose of simvastatin to no more than 20 mg per day or prescribing another statin in patients who carry at least one reduced-function rs4149056 C allele. Rosuvastatin or pravastatin are recommended as alternatives to simvastatin because secondary analyses of clinical trials of these drugs showed no association between the SLCO1B1 genotype and increased risk of myopathies145,162. Other pharmacogenetic consortia similarly recommend reducing the simvastatin dose or choosing an alternative statin for C allele carriers163,164. The French National Network of Pharmacogenetics further recommends rs4149056 genotyping before starting simvastatin treatment in patients with risk factors for myopathy164.
Owing to the increased generic availability of statins associated with a lower risk of associated myopathy, the clinical applicability of SLCO1B1 genotyping to inform simvastatin dosing has declined. However, whether simvastatin is the only statin in which rs4149056 can be clinically used to inform therapy remains unclear. Evidence has shown associations between the variant C allele and increased rates of atorvastatin intolerance and muscle-associated adverse effects147,165,166. The DPWG guidelines extend their recommendations to atorvastatin, specifically advising the avoidance of atorvastatin in C allele carriers with additional substantial risk factors for myopathy163. CPIC is currently in the process of updating its SLCO1B1-simvastatin guidelines, taking into account published research from the past 7 years in this field.
Emerging opportunities
In addition to existing guidelines for clopidogrel, warfarin and statins, CPIC has plans to review the evidence for additional cardiovascular gene–drug pairs for potential guideline creation. Each pair is supported by less evidence than the drugs already discussed, but as new data are published, the evidence might rise to a level that would support clinical implementation.
β-Blockers
CYP2D6 genotype.
β-Adrenergic receptor antagonists, or β-blockers, are indicated for a variety of cardiovascular diseases, including heart failure, hypertension and secondary prevention of myocardial infarction. Carvedilol, metoprolol, nebivolol, propranolol and timolol are primarily metabolized by the highly polymorphic CYP2D6 enzyme, with common CYP2D6 variants producing an array of phenotypes from increased enzyme function (via gene duplication) to complete loss of enzyme function (via gene deletion or splicing defect). A reasonable amount of evidence exists to support genetic polymorphisms in CYP2D6 that affect the pharmacokinetics of these β-blockers167–169. However, the evidence supporting the use of genetic information in prescribing β-blockers is weak (PharmGKB level 2–3, CPIC level B/C). Although some data have shown that CYP2D6 variants affect heart rate response to these β-blockers, whether CYP2D6 genotype notably affects blood pressure response or influences β-blocker-associated reductions in cardiovascular risk remains unclear170–174.
ADRB1, ADRB2 and GRK5 genotypes.
Three other genes have been studied in relation to β-blocker response: ADRB1, ADRB2 and GRK5. ADRB1 encodes the β1-adrenergic receptor, which is expressed primarily in the myocardium. This G protein-coupled receptor is antagonized by all β-blockers and is thought to be the primary site of action mediating the cardiovascular benefits of this drug. Two common variants in ADRB1, Ser49Gly (rs1801252) and Arg389Gly (rs1801253), are by far the most studied and are associated with decreased receptor activity through receptor downregulation and decreased signal transduction, respectively175. These variants have been associated with the decreased diastolic blood pressure response to β-blockers but do not seem to influence systolic blood pressure or heart rate response176–178. Multiple groups have reported that patients with the Ser49Ser and Arg389Arg genotypes (particularly Arg389Arg) have a greater risk reduction from β-blockers than individuals with other genotypes for indications including heart failure, atrial fibrillation and hypertension178–180.
In contrast to data on ADRB1, multiple reports indicate that the ADRB2 genotype is not associated with differential risk profiles in β-blocker-treated patients181–183. ADRB2 encodes the β2-adrenergic receptor, which is antagonized by non-selective β-blockers such as bucindolol, carvedilol, labetalol and propranolol. Two variants in ADRB2, Arg16Gly (rs1042713) and Gln27Glu (rs1042714) are associated with greater β-blocker-associated reductions in heart rate and diastolic blood pressure178,184. However, conflicting evidence exists on whether these variants influence clinical outcomes in β-blocker-treated patients178,185,186.
GRK5 encodes the G protein-coupled receptor kinase 5 (GRK5), which works intracellularly to blunt signalling from the β1 and β2 adrenergic receptors. The Gln41Leu (rs2230345) variant increases GRK5 function by mimicking the effect of a β-blocker187. On the basis of its β-blocker-like effect, the GRK5 41Leu variant is associated with reduced mortality in patients with heart failure or hypertension, regardless of β-blocker usage187,188. Indeed, β-blockers might not provide benefit to these variant carriers187, which if confirmed, might identify a patient population in which other drug classes might be preferred over β-blockers, depending on the indication. However, additional studies are warranted to determine the clinical utility of these variants to inform β-blocker therapy.
Hydralazine
Hydralazine is a direct vasodilator recommended by clinical guidelines as a secondary agent for the treatment of hypertension189. Hydralazine is metabolized primarily by acetylation, which is mostly accomplished in the liver by N-acetyltransferase type 2 (NAT2). Genetic variation in NAT2 has been associated with the acetylation rate of NAT2 in humans, with NAT2*4 defining the common allele, which is associated with a ‘rapid acetylator’ phenotype. Alleles such as NAT2*5, *6 and *7 are associated with a ‘slow acetylator’ phenotype, and individuals with one of each allele (for example, *4/*5 genotype) are defined as ‘intermediate acetylators’. Among healthy volunteers given hydralazine, rapid acetylators have decreased hydralazine exposure compared with slow acetylators190,191. This pharmacokinetic effect seems to correlate with the blood-pressure response to hydralazine, given that one small study reported that, in a population of patients with resistant hypertension, only slow acetylators had notable blood-pressure reductions192.
Acetylator status might also influence the risk of adverse effects. Overall, hydralazine use has been associated with a rare occurrence of lupus-like symptoms, although this relationship is not well established193. Indirect evidence suggests that slow acetylators might be at a higher risk of developing these lupus-like symptoms if exposed to hydralazine, but these data are far from conclusive194–196. Therefore, slow acetylators might have the greatest antihypertensive response to hydralazine, probably due to increased drug exposure. However, slow acetylators might also incur an increased risk of rare, but potentially severe, adverse effects. Further evidence is needed to support the use of NAT2 genotyping to predict the safety and effectiveness of hydralazine treatment.
Antiarrhythmic drugs
Similar to β-blockers described above, the class 1 antiarrhythmic drugs flecainide and propafenone undergo CYP2D6-mediated metabolism. Although the evidence is limited for these drugs (PharmGKB level 2A, CPIC level B/C), CYP2D6 variation seems to affect the pharmacokinetics of these drugs197–199. Studies have shown differences in QTc interval response with variations in the CYP2D6 genotype200,201. The FDA-approved labelling for propafenone warns that patients with a CYP2D6 deficiency (or inhibition secondary to concomitant drug therapy), when combined with CYP3A4 inhibition, are at a greater risk of increased propafenone exposure and associated proarrhythmia or other adverse events. However, the majority of the data supporting pharmacogenetic associations with these drugs are from studies in healthy volunteers, so additional studies in patients requiring rhythm control are warranted. Although no CPIC guidelines exist to address flecainide or propafenone, the DPWG recommends reducing the flecainide and propafenone doses to 50% and 30% of the standard dose, respectively, in CYP2D6 PMs163.
A small pilot study showed a significant correlation between a polygenic risk score (including 61 common variants outside of CYP2D6) and quinidine-induced or dofetilide-induced QT prolongation in individuals of European descent202,203. The score was also associated with drug-induced torsades de pointe in an independent cohort,203 suggesting that a multi-variant panel might potentially be used to predict the risk of drug-induced arrhythmias.
Additional pharmacogenetic opportunities
Complex disease management.
Pharmacogenetics might eventually prove valuable to inform the treatment of chronic, heterogeneous diseases, such as heart failure, where prescribing multiple medications that target various pathogenic disease pathways is the norm. For decades, β-blockers and angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers have been considered the cornerstone therapy for heart failure with reduced ejection fraction (HFrEF) regardless of aetiology, on the basis of substantial RCT evidence that these drugs reduce morbidity and mortality204. Evidence later emerged showing benefit of mineralocorticoid-receptor antagonists and hydralazine plus nitrates as ‘add-on’ therapy for additional reduction of morbidity and mortality. Then, after a decade with minimal progress in the field, angiotensin receptor–neprilysin inhibitors, sodium–glucose cotransporter 2 inhibitors, soluble guanylate cyclase stimulators and other therapies are emerging as additional therapies to improve outcomes205. This situation creates a challenge when selecting the optimal drug combination for an individual patient, with little data to inform the selection of one medication over another. Pharmacogenomics holds potential promise as a tool to individualize HFrEF therapy, with some limited data showing genetic associations in response to individual agents206,207. However, research in this field will require widening the focus beyond a single drug or gene to examine data across the genome that predicts response to drug combinations. Advances in machine learning and artificial intelligence (AI) increase the feasibility of large-scale analytical tasks.
Unlike in HFrEF, no drug therapies have been shown to increase survival in patients with heart failure with preserved ejection fraction (HFpEF). The aetiology of HFpEF might be even more complex than that of HFrEF owing to the involvement of multiple biological pathways in HFpEF. Machine learning techniques have been used to derive sub-phenotypes associated with the development of HFpEF208,209. Targeting these sub-phenotypes could be crucial to identifying successful therapies, with each sub-phenotype requiring different therapeutic strategies. Here, pharmacogenomics might help to classify patients more accurately to a sub-phenotype for treatment decision-making. Similar to its use in the discovery of sub-phenotypes in HFpEF, advanced methods such as machine learning and AI would assist both the discovery of novel pharmacogenetic variants and allow the synthesis of data from multiple loci across the genome to create a patient’s ‘predicted clinical response phenotype’ for medications. This work is already beginning in other fields outside of cardiovascular disease210.
Multi-omics.
Integration of genetic data with other data types, such as transcriptomics, proteomics and metabolomics, might help to elucidate the biological processes underlying individual responses to cardiovascular drugs. Use of these ‘multi-omics’ approaches are evident in relation to the mechanisms of cardiovascular disease211–213. However, the use of multi-omic data to characterize the drug response is sparse, possibly because data related to medication exposure over time might not be readily available in large cohort and clinical trial populations. The use of high-precision surrogate markers, such as cardiac imaging data, might reduce the population sample size needed to assess drug response and the time and complexity required for these data-intense analyses. Owing to the vast amounts of data that multi-omics (and cardiac imaging) analyses generate, these data provide additional opportunities for machine learning or AI. AI might be beneficial in integrating multiple types of omics data with the use of biological knowledge or for efficiently identifying even minor features from cardiac imaging files that would be impractical, or even impossible, for a human to detect and document reliably. AI might also facilitate the integration of genomic data with other ‘big data’, such as electronic health record data, perhaps with the use of natural language processing to improve the resolution of phenome-wide association studies to detect drug-response phenotypes214.
Conclusions
Strong and consistent evidence exists of genetic associations with clopidogrel effectiveness after PCI, warfarin dose requirements and simvastatin tolerability. Pharmacogenetic testing for each of these examples has been successfully implemented into clinical practice in multiple institutions. Although the data are not entirely consistent, clinical trial and observational evidence support improved outcomes with genotype-guided antiplatelet therapy and warfarin dosing. Investigation into the efficacy of pharmacogenetic approaches to guide statin selection is now underway. Ongoing investigations will help to elucidate the value of pharmacogenetic testing to guide other cardiovascular therapies. Although the logistical challenges associated with obtaining genotype results within the initial days of therapy might limit the clinical application of pharmacogenetic testing, pre-emptive testing or, where allowed, point-of-care testing might overcome these challenges. The addition of genetic testing strategies to clinical practice guidelines57 and expanded coverage of testing215 might signal that the broader adoption of testing is on the horizon. Future genomic discovery efforts integrating AI and machine learning techniques could be crucial to elucidate polygenetic predictors of response to therapies for heart failure and other complex diseases.
Key points.
Substantial evidence supports CYP2C19 genotyping to predict the effectiveness of clopidogrel after percutaneous coronary intervention.
Data strongly support genetic associations with warfarin dose requirements and risk of simvastatin-induced myopathy, but the use of genotyping in clinical practice to guide prescribing of these drugs is limited.
Limited evidence exists to support genetic testing to guide the use of other cardiovascular drugs at present.
Modern computational approaches can harness large multi-origin data to elucidate genetic predictors of the response to drug treatment for diseases requiring multiple medications targeting various pathogenic pathways.
Acknowledgements
J.D.D. and L.H.C. are supported by NIH grant NIH/NHGRI U01 HG00729. L.H.C. is supported by grants NIH/NHLBI R01 HL149752 and NIH/NCATS UL1TR001427.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Cardiology thanks J. W. Jukema and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Related links
CPIC: https://cpicpgx.org/
PharmGKB: https://www.pharmgkb.org/
Warfarin Dosing: http://www.warfarindosing.org/Source/Home.aspx
References
- 1.Roden DM et al. Pharmacogenomics. Lancet 394, 521–532 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volpi S et al. Research Directions in the Clinical Implementation of Pharmacogenomics: An Overview of US Programs and Projects. Clin. Pharmacol. Ther. 103, 778–786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nutescu EA et al. Feasibility of implementing a comprehensive warfarin pharmacogenetics service. Pharmacotherapy 33, 1156–1164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavallari LH et al. The IGNITE Pharmacogenetics Working Group: An Opportunity for Building Evidence with Pharmacogenetic Implementation in a Real-World Setting. Clin. Transl. Sci. 10, 143–146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavallari LH et al. Implementation of inpatient models of pharmacogenetics programs. Am. J. Health. Syst. Pharm. 73, 1944–1954 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorgensen AL et al. Implementation of genotype-guided dosing of warfarin with point-of-care genetic testing in three UK clinics: a matched cohort study. BMC Med. 17, 76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Relling MV & Klein TE CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 89, 464–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross CJ et al. The Canadian Pharmacogenomics Network for Drug Safety: a model for safety pharmacology. Thyroid 20, 681–687 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Swen JJ et al. Pharmacogenetics: from bench to byte--an update of guidelines. Clin Pharmacol. Ther. 89, 662–673 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Klein TE et al. Integrating genotype and phenotype information: an overview of the PharmGKB project. Pharmacogenetics Research Network and Knowledge Base. Pharmacogenomics J. 1, 167–170 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Food and Drug Administration. Table of Pharmacogenetic Associations. https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations.
- 12.Collins KS et al. Genotype-Guided Hydralazine Therapy. Am. J. Nephrol. 51, 764–776 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas CD & Johnson JA Pharmacogenetic factors affecting beta-blocker metabolism and response. Expert Opin. Drug. Metab. Toxicol. 16, 953–964 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine GN et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Thorac. Cardiovasc. Surg. 152, 1243–1275 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Wallentin L et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 361, 1045–1057 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Wiviott SD et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 357, 2001–2015 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Dayoub EJ et al. Trends in platelet adenosine diphosphate P2Y12 receptor inhibitor use and adherence among antiplatelet-naive patients after percutaneous coronary intervention, 2008-2016. JAMA Intern. Med. 178, 943–950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lombardi N et al. Ticagrelor-related late-onset dyspnea as cause of emergency department visit: a 3-year outpatient study. J. Cardiovasc. Med. (Hagerstown) 19, 284–289 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Claassens DMF et al. A Genotype-Guided Strategy for Oral P2Y12 Inhibitors in Primary PCI. N. Engl. J. Med. 381, 1621–1631 (2019). [DOI] [PubMed] [Google Scholar]; A major clinical trial of genotype-guided antiplatelet therapy showing similar net adverse clinical outcomes (composite of MACE and major bleeding) but decreased risk of major or minor bleeding with genotype-guided antiplatelet therapy after PCI compared with prasugrel or ticagrelor treatment.
- 20.Kazui M et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 38, 92–99 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Sangkuhl K, Klein TE & Altman RB Clopidogrel pathway. Pharmacogenet. Genomics 20, 463–465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caudle KE et al. Standardizing terms for clinical pharmacogenetic test results: consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet. Med. 19, 215–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaedigk A et al. The Pharmacogene Variation (PharmVar) Consortium: Incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin. Pharmacol. Ther. 103, 399–401 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pratt VM et al. Recommendations for Clinical CYP2C19 Genotyping Allele Selection: A Report of the Association for Molecular Pathology. J. Mol. Diagn. 20, 269–276 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Scott SA et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin. Pharmacol. Ther. 94, 317–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mega JL et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 360, 354–362 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Brandt JT et al. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J. Thromb. Haemost. 5, 2429–2436 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Umemura K, Furuta T & Kondo K The common gene variants of CYP2C19 affect pharmacokinetics and pharmacodynamics in an active metabolite of clopidogrel in healthy subjects. J. Thromb. Haemost. 6, 1439–1441 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Varenhorst C et al. Genetic variation of CYP2C19 affects both pharmacokinetic and pharmacodynamic responses to clopidogrel but not prasugrel in aspirin-treated patients with coronary artery disease. Eur. Heart J. 30, 1744–1752 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carreras ET et al. Diabetes mellitus, CYP2C19 genotype, and response to escalating doses of clopidogrel. Insights from the ELEVATE-TIMI 56 Trial. Thromb. Haemost. 116, 69–77 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Mega JL et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 306, 2221–2228 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Price MJ et al. Influence of genetic polymorphisms on the effect of high- and standard-dose clopidogrel after percutaneous coronary intervention: the GIFT (Genotype Information and Functional Testing) study. J. Am. Coll. Cardiol. 59, 1928–1937 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Mega JL et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation 119, 2553–2560 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Wallentin L et al. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet 376, 1320–1328 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Sibbing D et al. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 121, 512–518 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Collet JP et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 373, 309–317 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Trenk D et al. Cytochrome P450 2C19 681G>A polymorphism and high on-clopidogrel platelet reactivity associated with adverse 1-year clinical outcome of elective percutaneous coronary intervention with drug-eluting or bare-metal stents. J. Am. Coll. Cardiol. 51, 1925–1934 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Giusti B et al. Relation of cytochrome P450 2C19 loss-of-function polymorphism to occurrence of drug-eluting coronary stent thrombosis. Am. J. Cardiol. 103, 806–811 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Mega JL et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA 304, 1821–1830 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Combescure C et al. Clinical implications of clopidogrel non-response in cardiovascular patients: a systematic review and meta-analysis. J. Thromb. Haemost. 8, 923–933 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Hulot JS et al. Cardiovascular risk in clopidogrel-treated patients according to cytochrome P450 2C19*2 loss-of-function allele or proton pump inhibitor coadministration: a systematic meta-analysis. J. Am. Coll. Cardiol. 56, 134–143 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Sorich MJ, Rowland A, McKinnon RA & Wiese MD CYP2C19 genotype has a greater effect on adverse cardiovascular outcomes following percutaneous coronary intervention and in Asian populations treated with clopidogrel: a meta-analysis. Circ. Cardiovasc. Genet. 7, 895–902 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Zabalza M et al. Meta-analyses of the association between cytochrome CYP2C19 loss- and gain-of-function polymorphisms and cardiovascular outcomes in patients with coronary artery disease treated with clopidogrel. Heart 98, 100–108 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Bauer T et al. Impact of CYP2C19 variant genotypes on clinical efficacy of antiplatelet treatment with clopidogrel: systematic review and meta-analysis. BMJ 343, d4588 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmes MV, Perel P, Shah T, Hingorani AD & Casas JP CYP2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events: a systematic review and meta-analysis. JAMA 306, 2704–2714 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Notarangelo FM et al. Pharmacogenomic Approach to Selecting Antiplatelet Therapy in Patients With Acute Coronary Syndromes: The PHARMCLO Trial. J Am Coll Cardiol 71, 1869–1877 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Shen DL et al. Clinical Value of CYP2C19 Genetic Testing for Guiding the Antiplatelet Therapy in a Chinese Population. J. Cardiovasc. Pharmacol. 67, 232–236 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Xie X et al. Personalized antiplatelet therapy according to CYP2C19 genotype after percutaneous coronary intervention: a randomized control trial. Int. J. Cardiol. 168, 3736–3740 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Pereira NL et al. Effect of Genotype-Guided Oral P2Y12 Inhibitor Selection vs Conventional Clopidogrel Therapy on Ischemic Outcomes After Percutaneous Coronary Intervention: The TAILOR-PCI Randomized Clinical Trial. JAMA 324, 761–771 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; A major clinical trial of genotype-guided antiplatelet therapy showing a trend towards reduced risk of MACE and a significant reduction in the risk of multiple recurrent events with genotype-guided therapy after PCI compared with clopidogrel treatment.
- 50.Sanchez-Ramos J et al. Results of genotype-guided antiplatelet therapy in patients who undergone percutaneous coronary intervention with stent. Int. J. Cardiol. 225, 289–295 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Holmes DR Jr. et al. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on clinical expert consensus documents and the American Heart Association endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 56, 321–341 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Empey PE et al. Multisite Investigation of Strategies for the Implementation of CYP2C19 Genotype-Guided Antiplatelet Therapy. Clin. Pharmacol. Ther. 104, 664–674 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Angiolillo DJ et al. International Expert Consensus on Switching Platelet P2Y12 Receptor-Inhibiting Therapies. Circulation 136, 1955–1975 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Antman EM et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J. Am. Coll. Cardiol. 51, 2028–2033 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Becker RC et al. Bleeding complications with the P2Y12 receptor antagonists clopidogrel and ticagrelor in the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur. Heart J. 32, 2933–2944 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Rollini F, Franchi F & Angiolillo DJ Switching P2Y12-receptor inhibitors in patients with coronary artery disease. Nat. Rev. Cardiol. 13, 11–27 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Collet JP et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart. J. (2020). doi: 10.1093/eurheartj/ehaa575. [DOI] [PubMed] [Google Scholar]
- 58.Claassens DM & Sibbing D De-Escalation of Antiplatelet Treatment in Patients with Myocardial Infarction Who Underwent Percutaneous Coronary Intervention: A Review of the Current Literature. J. Clin. Med 9, 2983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peterson JF et al. Physician response to implementation of genotype-tailored antiplatelet therapy. Clin. Pharmacol. Ther. 100, 67–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lima JJ et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2C19 and Proton Pump Inhibitor Dosing. Clin. Pharmacol. Ther. (2020). doi: 10.1002/cpt.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hicks JK et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin. Pharmacol. Ther. 98, 127–134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cavallari LH et al. Multisite Investigation of Outcomes With Implementation of CYP2C19 Genotype-Guided Antiplatelet Therapy After Percutaneous Coronary Intervention. JACC Cardiovasc. Interv. 11, 181–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; A multicentre collaboration providing CYP2C19 genotyping to guide antiplatelet therapy after PCI as part of clinical care showing an increased risk of MACE in CYP2C19 IMs and PMs treated with clopidogrel compared with other antiplatelet therapy.
- 63.Deiman BA et al. Reduced number of cardiovascular events and increased cost-effectiveness by genotype-guided antiplatelet therapy in patients undergoing percutaneous coronary interventions in the Netherlands. Neth. Heart J. 24, 589–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hulot JS et al. Routine CYP2C19 Genotyping to Adjust Thienopyridine Treatment After Primary PCI for STEMI: Results of the GIANT Study. JACC Cardiovasc. Interv. 13, 621–630 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Lee CR et al. Impact of the CYP2C19*17 allele on outcomes in patients receiving genotype-guided antiplatelet therapy after percutaneous coronary intervention. Clin. Pharmacol. Ther. 109, 705–715 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tiroch KA et al. Protective effect of the CYP2C19 *17 polymorphism with increased activation of clopidogrel on cardiovascular events. Am. Heart J. 160, 506–512 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Gross L et al. Genotype-Phenotype Association and Impact on Outcomes following Guided De-Escalation of Anti-Platelet Treatment in Acute Coronary Syndrome Patients: The TROPICAL-ACS Genotyping Substudy. Thromb. Haemost. 118, 1656–1667 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Wang Y et al. Association Between CYP2C19 Loss-of-Function Allele Status and Efficacy of Clopidogrel for Risk Reduction Among Patients With Minor Stroke or Transient Ischemic Attack. JAMA 316, 70–78 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Pan Y et al. Genetic Polymorphisms and Clopidogrel Efficacy for Acute Ischemic Stroke or Transient Ischemic Attack: A Systematic Review and Meta-Analysis. Circulation 135, 21–33 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Diaz-Villamarin X et al. Genetic polymorphisms influence on the response to clopidogrel in peripheral artery disease patients following percutaneous transluminal angioplasty. Pharmacogenomics 17, 1327–1338 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Guo B et al. Patients carrying CYP2C19 loss of function alleles have a reduced response to clopidogrel therapy and a greater risk of in-stent restenosis after endovascular treatment of lower extremity peripheral arterial disease. J. Vasc. Surg. 60, 993–1001 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Hiatt WR et al. Ticagrelor versus Clopidogrel in Symptomatic Peripheral Artery Disease. N. Engl. J. Med. 376, 32–40 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Shuldiner AR et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 302, 849–857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mega JL et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet 376, 1312–1319 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simon T et al. Genetic determinants of response to clopidogrel and cardiovascular events. N. Engl. J. Med. 360, 363–375 (2009). [DOI] [PubMed] [Google Scholar]
- 76.Zhai Y et al. Meta-analysis of effects of ABCB1 polymorphisms on clopidogrel response among patients with coronary artery disease. Eur. J. Clin. Pharmacol. 73, 843–854 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Bouman HJ et al. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat. Med. 17, 110–116 (2011). [DOI] [PubMed] [Google Scholar]
- 78.Mega JL et al. PON1 Q192R genetic variant and response to clopidogrel and prasugrel: pharmacokinetics, pharmacodynamics, and a meta-analysis of clinical outcomes. J. Thromb. Thrombolysis 41, 374–383 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Hulot JS et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ. Cardiovasc. Interv 4, 422–428 (2011). [DOI] [PubMed] [Google Scholar]
- 80.Lewis JP et al. Paraoxonase 1 (PON1) gene variants are not associated with clopidogrel response. Clin. Pharmacol. Ther. 90, 568–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lewis JP et al. The functional G143E variant of carboxylesterase 1 is associated with increased clopidogrel active metabolite levels and greater clopidogrel response. Pharmacogenet. Genomics 23, 1–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lewis JP et al. Pharmacogenomic polygenic response score predicts ischaemic events and cardiovascular mortality in clopidogrel-treated patients. Eur. Heart J. Cardiovasc. Pharmacother 6, 203–210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Franchi F, Rollini F & Angiolillo DJ Defining the link between chronic kidney disease, high platelet reactivity, and clinical outcomes in clopidogrel-treated patients undergoing percutaneous coronary intervention. Circ. Cardiovasc. Interv 8, e002760 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Angiolillo DJ et al. Impaired responsiveness to the platelet P2Y12 receptor antagonist clopidogrel in patients with type 2 diabetes and coronary artery disease. J. Am. Coll. Cardiol. 64, 1005–1014 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Angiolillo DJ et al. Impact of chronic kidney disease on platelet function profiles in diabetes mellitus patients with coronary artery disease taking dual antiplatelet therapy. J. Am. Coll. Cardiol. 55, 1139–1146 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Angiolillo DJ et al. Derivation, Validation, and Prognostic Utility of a Prediction Rule for Nonresponse to Clopidogrel: The ABCD-GENE Score. JACC Cardiovasc. Interv. 13, 606–617 (2020). [DOI] [PubMed] [Google Scholar]
- 87.Bouziana SD & Tziomalos K Clinical relevance of clopidogrel-proton pump inhibitors interaction. World J. Gastrointest. Pharmacol. Ther. 6, 17–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wadelius M et al. The largest prospective warfarin-treated cohort supports genetic forecasting. Blood 113, 784–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pirmohamed M et al. A randomized trial of genotype-guided dosing of warfarin. N. Engl. J. Med. 369, 2294–2303 (2013). [DOI] [PubMed] [Google Scholar]
- 90.Stewart A, Ganguli A, FitzGerald R & Pirmohamed M Variation in warfarin prescribing and dosing in the UK: a national survey of anticoagulation clinics. J. Clin. Pharm. Ther. 40, 466–471 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Proietti M, Romanazzi I, Romiti GF, Farcomeni A & Lip GYH Real-World Use of Apixaban for Stroke Prevention in Atrial Fibrillation: A Systematic Review and Meta-Analysis. Stroke 49, 98–106 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Ntaios G et al. Real-World Setting Comparison of Nonvitamin-K Antagonist Oral Anticoagulants Versus Vitamin-K Antagonists for Stroke Prevention in Atrial Fibrillation: A Systematic Review and Meta-Analysis. Stroke 48, 2494–2503 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Pare G et al. Genetic determinants of dabigatran plasma levels and their relation to bleeding. Circulation 127, 1404–1412 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Ziakas PD, Kourbeti IS, Poulou LS, Vlachogeorgos GS & Mylonakis E Medicare part D prescribing for direct oral anticoagulants in the United States: Cost, use and the “rubber effect”. PLoS One 13, e0198674 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kjerpeseth LJ et al. Trends in use of warfarin and direct oral anticoagulants in atrial fibrillation in Norway, 2010 to 2015. Eur. J. Clin. Pharmacol. 73, 1417–1425 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Ho KH, van Hove M & Leng G Trends in anticoagulant prescribing: a review of local policies in English primary care. BMC Health Serv. Res. 20, 279 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhu J, Alexander GC, Nazarian S, Segal JB & Wu AW Trends and Variation in Oral Anticoagulant Choice in Patients with Atrial Fibrillation, 2010-2017. Pharmacotherapy 38, 907–920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Allabi AC et al. Functional impact of CYP2C95, CYP2C96, CYP2C98, and CYP2C911 in vivo among black Africans. Clin. Pharmacol. Ther. 76, 113–118 (2004). [DOI] [PubMed] [Google Scholar]
- 99.Dickmann LJ et al. Identification and functional characterization of a new CYP2C9 variant (CYP2C9*5) expressed among African Americans. Mol. Pharmacol. 60, 382–387 (2001). [DOI] [PubMed] [Google Scholar]
- 100.Liu Y et al. Decreased warfarin clearance associated with the CYP2C9 R150H (*8) polymorphism. Clin. Pharmacol. Ther. 91, 660–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scordo MG et al. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on warfarin maintenance dose and metabolic clearance. Clin. Pharmacol. Ther. 72, 702–710 (2002). [DOI] [PubMed] [Google Scholar]
- 102.Asiimwe IG et al. Genetic Factors Influencing Warfarin Dose in Black-African Patients: A Systematic Review and Meta-Analysis. Clin. Pharmacol. Ther. 107, 1420–1433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cavallari LH et al. Genetic and clinical predictors of warfarin dose requirements in African Americans. Clin. Pharmacol. Ther. 87, 459–464 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Higashi MK et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 287, 1690–1698 (2002). [DOI] [PubMed] [Google Scholar]
- 105.Limdi NA et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin. Pharmacol. Ther. 83, 312–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rost S et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 427, 537–541 (2004). [DOI] [PubMed] [Google Scholar]
- 107.Wang D et al. Regulatory polymorphism in vitamin K epoxide reductase complex subunit 1 (VKORC1) affects gene expression and warfarin dose requirement. Blood 112, 1013–1021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Johnson JA et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Pharmacogenetics-Guided Warfarin Dosing: 2017 Update. Clin. Pharmacol. Ther. 102, 397–404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Limdi NA et al. Warfarin pharmacogenetics: a single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood 115, 3827–3834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McDonald MG, Rieder MJ, Nakano M, Hsia CK & Rettie AE CYP4F2 is a vitamin K1 oxidase: An explanation for altered warfarin dose in carriers of the V433M variant. Mol. Pharmacol. 75, 1337–1346 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Danese E et al. Effect of CYP4F2, VKORC1, and CYP2C9 in Influencing Coumarin Dose: A Single-Patient Data Meta-Analysis in More Than 15,000 Individuals. Clin. Pharmacol. Ther. 105, 1477–1491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bress A et al. Effect of NQO1 and CYP4F2 genotypes on warfarin dose requirements in Hispanic-Americans and African-Americans. Pharmacogenomics 13, 1925–1935 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cooper GM et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 112, 1022–1027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Takeuchi F et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 5, e1000433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cha PC et al. Genome-wide association study identifies genetic determinants of warfarin responsiveness for Japanese. Hum. Mol. Genet. 19, 4735–4744 (2010). [DOI] [PubMed] [Google Scholar]
- 116.Perera MA et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. Lancet 382, 790–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.De T et al. Association of Genetic Variants With Warfarin-Associated Bleeding Among Patients of African Descent. JAMA 320, 1670–1677 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pisters R et al. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 138, 1093–1100 (2010). [DOI] [PubMed] [Google Scholar]
- 119.International Warfarin Pharmacogenetics, C. et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N. Engl. J. Med. 360, 753–764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gage BF et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin. Pharmacol. Ther. 84, 326–331 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gage BF et al. Effect of Genotype-Guided Warfarin Dosing on Clinical Events and Anticoagulation Control Among Patients Undergoing Hip or Knee Arthroplasty: The GIFT Randomized Clinical Trial. JAMA 318, 1115–1124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; A randomized clinical trial that demonstrated a reduction in the composite of major bleeding, INR ≥4, venous thromboembolism or death in the genotype-guided group compared with the clinically-guided dosing group after total joint arthroplasty.
- 122.Kimmel SE et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 369, 2283–2293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Drozda K et al. Poor warfarin dose prediction with pharmacogenetic algorithms that exclude genotypes important for African Americans. Pharmacogenet. Genomics 25, 73–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arwood MJ et al. Anticoagulation endpoints with clinical implementation of warfarin pharmacogenetic dosing in a real-world setting: A proposal for a new pharmacogenetic dosing approach. Clin. Pharmacol. Ther. 101, 675–683 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pratt VM et al. Recommendations for Clinical Warfarin Genotyping Allele Selection: A Report of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 22, 847–859 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.U.S. Food & Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling. https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling.
- 127.Kaul KL, Leonard DG, Gonzalez A & Garrett CT Oversight of genetic testing: an update. J. Mol. Diagn. 3, 85–91 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee YM et al. Analysis of comprehensive pharmacogenomic profiling to impact in-hospital prescribing. Pharmacogenet. Genomics 29, 23–30 (2019). [DOI] [PubMed] [Google Scholar]
- 129.Leary E, Brilliant M, Peissig P & Griesbach S Preliminary outcomes of preemptive warfarin pharmacogenetic testing at a large rural healthcare center. Am. J. Health Syst. Pharm. 76, 387–397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roden DM et al. Benefit of Preemptive Pharmacogenetic Information on Clinical Outcome. Clin. Pharmacol. Ther. 103, 787–794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marrero RJ et al. How to Transition from Single-Gene Pharmacogenetic Testing to Preemptive Panel-Based Testing: A Tutorial. Clin. Pharmacol. Ther. 108, 557–565 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mega JL et al. Genetics and the clinical response to warfarin and edoxaban: findings from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet 385, 2280–2287 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Shi J et al. Dabigatran etexilate activation is affected by the CES1 genetic polymorphism G143E (rs71647871) and gender. Biochem. Pharmacol. 119, 76–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Buettner C et al. Statin use and musculoskeletal pain among adults with and without arthritis. Am. J. Med. 125, 176–182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Link E et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N. Engl. J. Med. 359, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 136.Food and Drug Administration. Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-new-restrictions-contraindications-and-dose-limitations-zocor.
- 137.Hopewell JC et al. Independent risk factors for simvastatin-related myopathy and relevance to different types of muscle symptom. Eur. Heart J. 41, 3336–3342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gupta A et al. Adverse events associated with unblinded, but not with blinded, statin therapy in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid-Lowering Arm (ASCOT-LLA): a randomised double-blind placebo-controlled trial and its non-randomised non-blind extension phase. Lancet 389, 2473–2481 (2017). [DOI] [PubMed] [Google Scholar]
- 139.Shitara Y Clinical importance of OATP1B1 and OATP1B3 in drug-drug interactions. Drug Metab. Pharmacokinet. 26, 220–227 (2011). [DOI] [PubMed] [Google Scholar]
- 140.Tirona RG, Leake BF, Merino G & Kim RB Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J. Biol. Chem. 276, 35669–35675 (2001). [DOI] [PubMed] [Google Scholar]
- 141.Choi HY et al. Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 gene polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid. Pharmacogenet. Genomics 25, 595–608 (2015). [DOI] [PubMed] [Google Scholar]
- 142.Pasanen MK, Neuvonen M, Neuvonen PJ & Niemi M SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet. Genomics 16, 873–879 (2006). [DOI] [PubMed] [Google Scholar]
- 143.Ramsey LB et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther. 96, 423–428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tornio A et al. SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharmacogenet. Genomics 25, 382–387 (2015). [DOI] [PubMed] [Google Scholar]
- 145.Voora D et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J. Am. Coll. Cardiol. 54, 1609–1616 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carr DF et al. SLCO1B1 genetic variant associated with statin-induced myopathy: a proof-of-concept study using the clinical practice research datalink. Clin. Pharmacol. Ther. 94, 695–701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Turner RM et al. A Genome-wide Association Study of Circulating Levels of Atorvastatin and Its Major Metabolites. Clin. Pharmacol. Ther. 108, 287–297 (2020). [DOI] [PubMed] [Google Scholar]; A genome-wide association study showing strong associations between rs4149056 and both circulating atorvastatin levels and muscle-related adverse effects in patients who had been hospitalized with a non-ST-segment elevation ACS and treated with atorvastatin.
- 148.Carr DF et al. Genomewide Association Study of Statin-Induced Myopathy in Patients Recruited Using the UK Clinical Practice Research Datalink. Clin. Pharmacol. Ther. 106, 1353–1361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sim SC, Edwards RJ, Boobis AR & Ingelman-Sundberg M CYP3A7 protein expression is high in a fraction of adult human livers and partially associated with the CYP3A7*1C allele. Pharmacogenet. Genomics 15, 625–631 (2005). [DOI] [PubMed] [Google Scholar]
- 150.Becker ML et al. Influence of genetic variation in CYP3A4 and ABCB1 on dose decrease or switching during simvastatin and atorvastatin therapy. Pharmacoepidemiol. Drug Saf 19, 75–81 (2010). [DOI] [PubMed] [Google Scholar]
- 151.Fiegenbaum M et al. The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin. Pharmacol. Ther. 78, 551–558 (2005). [DOI] [PubMed] [Google Scholar]
- 152.Hoenig MR, Walker PJ, Gurnsey C, Beadle K & Johnson L The C3435T polymorphism in ABCB1 influences atorvastatin efficacy and muscle symptoms in a high-risk vascular cohort. J. Clin. Lipidol. 5, 91–96 (2011). [DOI] [PubMed] [Google Scholar]
- 153.Wilke RA, Moore JH & Burmester JK Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage. Pharmacogenet. Genomics 15, 415–421 (2005). [DOI] [PubMed] [Google Scholar]
- 154.Ruano G et al. Mechanisms of statin-induced myalgia assessed by physiogenomic associations. Atherosclerosis 218, 451–456 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mangravite LM et al. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature 502, 377–380 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mega JL et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 385, 2264–2271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Natarajan P et al. Polygenic Risk Score Identifies Subgroup With Higher Burden of Atherosclerosis and Greater Relative Benefit From Statin Therapy in the Primary Prevention Setting. Circulation 135, 2091–2101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Postmus I et al. In search for genetic determinants of clinically meaningful differential cardiovascular event reduction by pravastatin in the PHArmacogenetic study of Statins in the Elderly at risk (PHASE)/PROSPER study. Atherosclerosis 235, 58–64 (2014). [DOI] [PubMed] [Google Scholar]
- 159.Damask A et al. Patients With High Genome-Wide Polygenic Risk Scores for Coronary Artery Disease May Receive Greater Clinical Benefit From Alirocumab Treatment in the ODYSSEY OUTCOMES Trial. Circulation 141, 624–636 (2020). [DOI] [PubMed] [Google Scholar]
- 160.Peyser B et al. Effects of Delivering SLCO1B1 Pharmacogenetic Information in Randomized Trial and Observational Settings. Circ. Genom. Precis. Med. 11, e002228 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Vassy JL et al. The Integrating Pharmacogenetics in Clinical Care (I-PICC) Study: Protocol for a point-of-care randomized controlled trial of statin pharmacogenetics in primary care. Contemp. Clin. Trials 75, 40–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Danik JS et al. Lack of association between SLCO1B1 polymorphisms and clinical myalgia following rosuvastatin therapy. Am. Heart J. 165, 1008–1014 (2013). [DOI] [PubMed] [Google Scholar]
- 163.Dutch guidelines August 2020 update. https://www.knmp.nl/downloads/pharmacogenetic-recommendations-august-2020.pdf
- 164.Lamoureux F, Duflot T & French Network of P Pharmacogenetics in cardiovascular diseases: State of the art and implementation-recommendations of the French National Network of Pharmacogenetics (RNPGx). Therapie 72, 257–267 (2017). [DOI] [PubMed] [Google Scholar]
- 165.de Keyser CE et al. The SLCO1B1 c.521T>C polymorphism is associated with dose decrease or switching during statin therapy in the Rotterdam Study. Pharmacogenet. Genomics 24, 43–51 (2014). [DOI] [PubMed] [Google Scholar]
- 166.Linskey DW et al. Association of SLCO1B1 c.521T>C (rs4149056) with discontinuation of atorvastatin due to statin-associated muscle symptoms. Pharmacogenet. Genomics 30, 208–211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Blake CM, Kharasch ED, Schwab M & Nagele P A meta-analysis of CYP2D6 metabolizer phenotype and metoprolol pharmacokinetics. Clin. Pharmacol. Ther. 94, 394–399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Baudhuin LM et al. Relation of ADRB1, CYP2D6, and UGT1A1 polymorphisms with dose of, and response to, carvedilol or metoprolol therapy in patients with chronic heart failure. Am. J. Cardiol. 106, 402–408 (2010). [DOI] [PubMed] [Google Scholar]
- 169.Vieira CP, Neves DV, Coelho EB & Lanchote VL Effect of CYP2D6 Poor Metabolizer Phenotype on Stereoselective Nebivolol Pharmacokinetics. Drug Metab. Lett. 12, 68–70 (2018). [DOI] [PubMed] [Google Scholar]
- 170.Lefebvre J, Poirier L, Poirier P, Turgeon J & Lacourciere Y The influence of CYP2D6 phenotype on the clinical response of nebivolol in patients with essential hypertension. Br. J. Clin. Pharmacol. 63, 575–582 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Meloche M et al. CYP2D6 polymorphism and its impact on the clinical response to metoprolol: A systematic review and meta-analysis. Br. J. Clin. Pharmacol. 86, 1015–1033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zineh I et al. Pharmacokinetics and CYP2D6 genotypes do not predict metoprolol adverse events or efficacy in hypertension. Clin. Pharmacol. Ther. 76, 536–544 (2004). [DOI] [PubMed] [Google Scholar]
- 173.Anstensrud AK et al. Impact of genotype-predicted CYP2D6 metabolism on clinical effects and tolerability of metoprolol in patients after myocardial infarction - a prospective observational study. Eur. J. Clin. Pharmacol. 76, 673–683 (2020). [DOI] [PubMed] [Google Scholar]
- 174.Batty JA et al. An investigation of CYP2D6 genotype and response to metoprolol CR/XL during dose titration in patients with heart failure: a MERIT-HF substudy. Clin. Pharmacol. Ther. 95, 321–330 (2014). [DOI] [PubMed] [Google Scholar]
- 175.Johnson JA & Liggett SB Cardiovascular pharmacogenomics of adrenergic receptor signaling: clinical implications and future directions. Clin. Pharmacol. Ther. 89, 366–378 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Johnson JA et al. Beta 1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin. Pharmacol. Ther. 74, 44–52 (2003). [DOI] [PubMed] [Google Scholar]
- 177.Liu J et al. beta1-Adrenergic receptor polymorphisms influence the response to metoprolol monotherapy in patients with essential hypertension. Clin. Pharmacol. Ther. 80, 23–32 (2006). [DOI] [PubMed] [Google Scholar]
- 178.Pacanowski MA et al. beta-adrenergic receptor gene polymorphisms and beta-blocker treatment outcomes in hypertension. Clin. Pharmacol. Ther. 84, 715–721 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kao DP et al. Effect of bucindolol on heart failure outcomes and heart rate response in patients with reduced ejection fraction heart failure and atrial fibrillation. Eur. J. Heart. Fail 15, 324–333 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Liggett SB et al. A polymorphism within a conserved beta(1)-adrenergic receptor motif alters cardiac function and beta-blocker response in human heart failure. Proc. Natl Acad. Sci. USA 103, 11288–11293 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cresci S et al. Clinical and genetic modifiers of long-term survival in heart failure. J. Am. Coll. Cardiol. 54, 432–444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Magvanjav O et al. Pharmacogenetic Associations of beta1-Adrenergic Receptor Polymorphisms With Cardiovascular Outcomes in the SPS3 Trial (Secondary Prevention of Small Subcortical Strokes). Stroke 48, 1337–1343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.White HL et al. An evaluation of the beta-1 adrenergic receptor Arg389Gly polymorphism in individuals with heart failure: a MERIT-HF sub-study. Eur. J. Heart Fail 5, 463–468 (2003). [DOI] [PubMed] [Google Scholar]
- 184.Sehrt D, Meineke I, Tzvetkov M, Gultepe S & Brockmoller J Carvedilol pharmacokinetics and pharmacodynamics in relation to CYP2D6 and ADRB pharmacogenetics. Pharmacogenomics 12, 783–795 (2011). [DOI] [PubMed] [Google Scholar]
- 185.Huang J et al. ADRB2 polymorphism Arg16Gly modifies the natural outcome of heart failure and dictates therapeutic response to beta-blockers in patients with heart failure. Cell Discov. 4, 57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Terra SG et al. beta-Adrenergic receptor polymorphisms and responses during titration of metoprolol controlled release/extended release in heart failure. Clin. Pharmacol. Ther. 77, 127–137 (2005). [DOI] [PubMed] [Google Scholar]
- 187.Liggett SB et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat. Med. 14, 510–517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lobmeyer MT et al. Polymorphisms in genes coding for GRK2 and GRK5 and response differences in antihypertensive-treated patients. Pharmacogenet. Genomics 21, 42–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Whelton PK et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 71, e127–e248 (2018). [DOI] [PubMed] [Google Scholar]
- 190.Gonzalez-Fierro A et al. Pharmacokinetics of hydralazine, an antihypertensive and DNA-demethylating agent, using controlled-release formulations designed for use in dosing schedules based on the acetylator phenotype. Int. J. Clin. Pharmacol. Ther. 49, 519–524 (2011). [DOI] [PubMed] [Google Scholar]
- 191.Han LW et al. Effect of N-Acetyltransferase 2 Genotype on the Pharmacokinetics of Hydralazine During Pregnancy. J. Clin. Pharmacol. 59, 1678–1689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Spinasse LB, Santos AR, Suffys PN, Muxfeldt ES & Salles GF Different phenotypes of the NAT2 gene influences hydralazine antihypertensive response in patients with resistant hypertension. Pharmacogenomics 15, 169–178 (2014). [DOI] [PubMed] [Google Scholar]
- 193.Schoonen WM et al. Do selected drugs increase the risk of lupus? A matched case-control study. Br. J. Clin. Pharmacol. 70, 588–596 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Marsden JR, Mason GG, Coburn PR, Rawlins MD & Shuster S Drug acetylation and expression of lupus erythematosus. Eur. J. Clin. Pharmacol. 28, 387–390 (1985). [DOI] [PubMed] [Google Scholar]
- 195.Mazari L, Ouarzane M & Zouali M Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc. Natl. Acad. Sci. U.S.A 104, 6317–6322 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Weber WW & Hein DW N-acetylation pharmacogenetics. Pharmacol. Rev. 37, 25–79 (1985). [PubMed] [Google Scholar]
- 197.Doki K et al. Effect of CYP2D6 genotype on flecainide pharmacokinetics in Japanese patients with supraventricular tachyarrhythmia. Eur. J. Clin. Pharmacol. 62, 919–926 (2006). [DOI] [PubMed] [Google Scholar]
- 198.Doki K, Sekiguchi Y, Kuga K, Aonuma K & Homma M Serum flecainide S/R ratio reflects the CYP2D6 genotype and changes in CYP2D6 activity. Drug Metab. Pharmacokinet. 30, 257–262 (2015). [DOI] [PubMed] [Google Scholar]
- 199.Rouini MR & Afshar M Effect of CYP2D6 polymorphisms on the pharmacokinetics of propafenone and its two main metabolites. Therapie 72, 373–382 (2017). [DOI] [PubMed] [Google Scholar]
- 200.Jazwinska-Tarnawska E et al. The influence of CYP2D6 polymorphism on the antiarrhythmic efficacy of propafenone in patients with paroxysmal atrial fibrillation during 3 months propafenone prophylactic treatment. Int. J. Clin. Pharmacol. Ther. 39, 288–292 (2001). [DOI] [PubMed] [Google Scholar]
- 201.Lim KS et al. Changes in the QTc interval after administration of flecainide acetate, with and without coadministered paroxetine, in relation to cytochrome P450 2D6 genotype: data from an open-label, two-period, single-sequence crossover study in healthy Korean male subjects. Clin. Ther 32, 659–666 (2010). [DOI] [PubMed] [Google Scholar]
- 202.Arking DE et al. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat. Genet. 46, 826–836 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Strauss DG et al. Common Genetic Variant Risk Score Is Associated With Drug-Induced QT Prolongation and Torsade de Pointes Risk: A Pilot Study. Circulation 135, 1300–1310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yancy CW et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 136, e137–e161 (2017). [DOI] [PubMed] [Google Scholar]
- 205.Murphy SP, Ibrahim NE & Januzzi JL Jr. Heart Failure With Reduced Ejection Fraction: A Review. JAMA 324, 488–504 (2020). [DOI] [PubMed] [Google Scholar]
- 206.McNamara DM et al. Endothelial nitric oxide synthase (NOS3) polymorphisms in African Americans with heart failure: results from the A-HeFT trial. J. Card. Fail 15, 191–198 (2009). [DOI] [PubMed] [Google Scholar]
- 207.McNamara DM et al. Pharmacogenetic interactions between angiotensin-converting enzyme inhibitor therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. J. Am. Coll. Cardiol. 44, 2019–2026 (2004). [DOI] [PubMed] [Google Scholar]
- 208.Segar MW et al. Phenomapping of patients with heart failure with preserved ejection fraction using machine learning-based unsupervised cluster analysis. Eur. J. Heart Fail. 22, 148–158 (2020). [DOI] [PubMed] [Google Scholar]
- 209.Shah SJ et al. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 131, 269–279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Athreya AP et al. Pharmacogenomics-Driven Prediction of Antidepressant Treatment Outcomes: A Machine-Learning Approach With Multi-trial Replication. Clin. Pharmacol. Ther. 106, 855–865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Schlotter F et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 138, 377–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schussler-Fiorenza Rose SM et al. A longitudinal big data approach for precision health. Nat. Med. 25, 792–804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Infante T et al. Network Medicine: A Clinical Approach for Precision Medicine and Personalized Therapy in Coronary Heart Disease. J. Atheroscler. Thromb. 27, 279–302 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Denny JC, Bastarache L & Roden DM Phenome-Wide Association Studies as a Tool to Advance Precision Medicine. Annu. Rev. Genomics Hum. Genet. 17, 353–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Centers for Medicare & Medicaid Services. Local Coverage Determination (LCD): MolDX: Pharmacogenomics Testing (L38294). https://www.cms.gov/medicare-coverage-database/details/lcd-details.aspx?LCDId=38294&ver=16&Cntrctr=All&UpdatePeriod=889&bc=AAAACAAAAAAA&. [PubMed]
- 216.Relling MV, Altman RB, Goetz MP & Evans WE Clinical implementation of pharmacogenomics: overcoming genetic exceptionalism. Lancet Oncol. 11, 507–509 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]