

Abstract
Formation of biomolecular condensates is increasingly recognized as a mechanism employed by cells to deal with stress and to optimize enzymatic reactions. Recent studies have characterized several DNA repair foci as phase-separated condensates, behaving like liquid droplets. Concomitantly, the apparent importance of long non-coding RNAs and RNA-binding proteins for the repair of double-strand breaks has raised many questions about their exact contribution to the repair process. Here we discuss how RNA molecules can participate in condensate formation and how RNA-binding proteins can act as molecular scaffolds. We furthermore summarize our current knowledge about how properties of condensates can influence the choice of repair pathway (homologous recombination or non-homologous end joining) and identify the open questions in this field of emerging importance.
Keywords: Double-strand break repair, DNA damage foci, LLPS, lncRNA, biomolecular condensates
1. Introduction
Cells have to deal with continuous insults to their genomes. Environmental factors like radiation and chemicals as well as endogenous sources can cause a host of different types of DNA damage, impacting genome integrity [1]. DNA repair has been a topic of extensive study, which revealed the existence of multiple different pathways, each tailored toward the repair of a specific type of lesion [2]. Repair of the most severe lesion, a double strand break (DSB), starts off with a signaling cascade and the consecutive recruitment of repair factors to the damage site. Central to the DSB signaling and repair processes is the formation of a repair focus – a designated volume in which related biochemical reactions are regulated. These foci correspond to an accumulation of damage markers and repair proteins at the damage site and are considered a hallmark of DNA damage and repair, yet the regulation and inner workings of these repair centers remain elusive.
Some foci, for example those created by p53-binding protein 1 (53BP1) or RAD52, have recently been identified as phase-separated compartments in the nucleus, behaving as liquid droplets [3–5]. Condensate formation is common in cells, examples being membrane-less organelles like paraspeckles [6], Cajal bodies [7] and stress granules [8]. These compartments have dedicated functions in cell homeostasis and are not harmful to the cell per se. Certain mutations in proteins that induce condensate formation, however, can contribute to formation of pathological aggregates that are thought to underlie neurodegenerative diseases like amyotrophic lateral sclerosis (ALS) [9]. For example, mutations in the intrinsically disordered proteins FUS, EWS and TAF15 (the “FET” proteins) have been identified in ALS patients and are generally associated with an increased tendency to form solid aggregates in neurons that cannot be dissolved [10].
Over the last decades, there has been an increasing body of evidence that RNA can fulfill other roles in the cell in addition to merely being transcripts coding for proteins. RNAs can suppress gene expression by a process called RNA interference; conversely, RNAs of varying lengths can also promote gene transcription with the help of Argonaute proteins [11]. Additionally, the cellular DNA damage response involves regulation of gene expression post-transcription, in which non-coding RNAs and RNA binding proteins play a major role [12]. Recently, members of a class of RNAs known as long non-coding RNAs (lncRNAs), which are not being translated, were shown to be directly involved in DNA repair activities, interacting with repair proteins or even substituting for some [13,14].
Additionally, it was found that transcription of damage-induced lncRNAs (dilncRNAs) takes place in the immediate vicinity of DSBs, and that RNA binding proteins are recruited to the repair site [15,16]. Although transcription is in general suppressed upon detection of a DSB [17], these damage-induced lncRNAs may still be produced since they do not need a promoter [18].
The notion that many of the cell’s membrane-less organelles consist of RNA and RNA-binding proteins, together with the observation of phase-separated repair foci in the nucleus and the discovery of lncRNAs, has led to the suggestion that RNA has a more elaborate role in DNA repair. Since RNAs are complex biopolymers, differing in length, sequence, secondary structure and abundance, this role is likely not limited to a single process. Indeed, it has been shown that RNA molecules can regulate transcription of repair proteins [19], act as template for repair [20–22], aid in the formation of repair foci [3] or modulate the function of specific repair proteins by interacting with them [13,23–35].
This review will focus on the structural role of RNA and RNA-binding proteins in DNA damage foci. We will give a short overview of condensate formation and DSB repair in general, as well as highlight a couple of RNAs and RNA-binding proteins with specific roles in DNA repair. We will then provide an overview of how condensates and RNA shape the repair process. Finally, we will briefly discuss mechanisms for condensate resolution.
2. Condensate Formation in the Cell
Liquid-liquid phase separation (LLPS) has recently been implicated in the formation of a host of membrane-less organelles in cells. LLPS is the de-mixing of a solution into a dense and dilute phase [36–38]. It originates when weak multivalent interactions between related molecules become more energetically favorable than interactions of these molecules with the solvent (which is often water). In practice, this means that phase separation will occur when the concentration of a component in the cytoplasm or nucleoplasm exceeds a threshold value. Alternatively, computer simulations showed that a slowly diffusing macromolecule or “molecular scaffold” can seed phase separation at lower concentrations if it is able to retain enough other molecules in its immediate vicinity [39].
Cellular components that can phase separate are typically macromolecules without a well-defined secondary structure, such as single-stranded RNAs and intrinsically disordered domains of proteins [40]. The phase-separated compartments that they form are often referred to as “biomolecular condensates” or “liquid droplets”. Their stability is strongly dependent on environmental factors like salt concentration and pH; moreover, they can grow in size and fuse with other droplets, properties that make them highly versatile and give cells a toolbox to react to all kinds of environmental stress [38]. There are other advantages of phase separation as well: by locally increasing the concentration of certain proteins, enzymatic reactions may go faster. Condensates can also act as a sieve, providing selective access to the proteins that are needed for a specific reaction while keeping others out [41]. However, phase separation is not without risk for the cell. Biomolecular condensates can undergo a process called maturation, during which the increasing density in the core of the droplet will result in a liquid-to-solid phase transition. These can be toxic for the cell if they cannot be resolved. Indeed, mutated versions of LLPS proteins that are more prone to form aggregates are linked to neurological disorders like ALS [37]. An example is the previously mentioned FUS protein, which contains RNA-binding domains and a so-called “prion-like domain” which is intrinsically disordered [42]. The ability of the prion-like domain to establish weak multivalent interactions is thought to drive LLPS. However, pathological mutations of FUS are found in both the RNA-binding domains and the prion-like domain. These are often simple missense mutations, increasing the overall tendency of the protein to aggregate [10].
As pointed out before [43], we cannot automatically assume that formation of a certain membrane-less organelle is due to LLPS. There are more ways a cell can compartmentalize proteins, such as binding to polymeric scaffolds like DNA or RNA. In this review, we will use the overarching term “condensate” to avoid confusion.
Importantly, biomolecular condensates in cells are not homogeneous structures. Instead, they adopt some degree of organization. For example, histone protein 1a (HP1a) undergoes LLPS in Drosophila and humans to create heterochromatic domains, which were found to consist of mobile and immobile parts [44,45]. The inner immobile parts are likely caused by direct interactions between HP1a and DNA, whereas weak multivalent protein-protein interactions dominate in the outer more dynamic regions. Similarly, super-resolution imaging showed that paraspeckles, which are condensates found in the nucleus, consist of a core and shell [46]. The cores of paraspeckles consists of RNA binding proteins from the Drosophila behavior/human splicing (DBHS) protein family. In humans, this family consists of SFPQ (Splicing Factor, Proline- and Glutamine-rich), NONO (Non-POU domain-containing Octamer-binding protein) and PSPC1 (Paraspeckle Protein Component 1) [47]. DBHS proteins are often characterized as molecular scaffolds: they contain RNA binding motifs and intrinsically disorder regions, and are capable of forming heterodimers and oligomers [48,49]. Their ability to bind RNA as well as DNA provides them with a multitude of different roles in the cell, including in RNA splicing, transcriptional regulation and DNA repair, as we will see later [50]. Here, they contribute to the architecture of the paraspeckle by occupying the core, where they interact with the middle region of lncRNA NEAT1 [46,51]. Using fluorescence in situ hybridization (FISH), the authors were able to show that the 5’ and 3’ tails of NEAT1 are pointing outwards, forming the shell of the paraspeckle. Paraspeckles are thought to have a role in gene regulation and function by sequestering certain gene transcripts. They are able to retain these RNAs exactly because of their weak multivalent interactions.
In DNA damage repair, the protein 53BP1 is recruited to DSBs to form foci. These foci were recently identified as biomolecular condensates, showing droplet-like behavior [3,4]. Thus, while only a limited number of 53BP1 proteins can bind to the actual break site, there is apparently a physiological advantage to create a far larger cellular compartment around the DSB lesion. The large (1972 amino acids [52]) 53BP1 protein interacts specifically with modified histones through its Tudor and BRCT domains at sites of DNA damage, whereas its oligomerization domain is necessary to induce phase separation [4,53–55]. This results in an architecture in which interactions between the Tudor domain and the break site govern the center of the focus, while protein-protein interactions through the oligomerization domain dominate the outer shell [4].
In the nucleolus, there appear to exist “phases within a phase”, forming the sub-compartments of this membrane-less organelle. This organization originates from differences in surface tension between the individual condensates [56].
Thus, although liquid-liquid phase separation in the cell is stimulated by weak multivalent interactions between disordered domains of proteins and RNA, the resulting biomolecular condensate can adopt a structure with a well-defined architecture. Because of the multitude of factors involved in the formation of these structures in vivo, in combination with technological difficulties to visualize them at high spatial and temporal resolution, our knowledge about how that architecture contributes to function is unfortunately limited.
3. Pathways for the Repair of Double-Strand Breaks
DSBs pose an immediate threat to genomic integrity and cell viability. Extensive studies on DSB repair found a tightly regulated signaling cascade that quickly activates the repair machinery through phosphorylation and ubiquitylation of target proteins. Two main pathways for repair were identified: homologous recombination (HR) [57,58] and non-homologous end-joining (NHEJ) [59,60]. Repair of the break often proceeds through one of these two.
HR uses the homologous DNA sequence of a sister chromatid to facilitate error-free repair. This mode of repair is only possible when a sister chromatid is present, and can thus only be employed during the S or G2 phase. It adopts a mechanism in which the DNA around the break site is first resected by nuclease activity from the MRE11-RAD50-NBS1 (MRN) complex, which recognizes DNA ends, and the nucleases EXO1 and DNA2 [61–64]. The BRCA1 protein helps coordinate resection by binding to the resection factor CtIP [65]. It also antagonizes binding of 53BP1, a protein that limits end resection [66,67]. The exposed ssDNA is protected by RPA, followed by initial loading and nucleation of RAD51 recombinase, which is mediated by RAD52 [68]. This is followed by the formation of RAD51 filaments stabilized by BRCA2, to support efficient homology search and strand invasion activities. The protein SFPQ, which we described before as a molecular scaffold, was found to increase homologous pairing and strand exchange at low RAD51 concentrations, while inhibiting these activities at higher RAD51 concentrations [69]. Once a homologous sequence is found, polymerases such as Pol δ use it as a template for repair of the break, thus restoring the original sequence [70]. Repair is completed by ligation.
NHEJ, on the other hand, involves direct alignment and ligation of the broken DNA ends. It is therefore much faster and more efficient than HR and serves as the predominant cellular repair process for DSB repair; however, it is considered more error-prone than HR, often resulting in insertions or deletions. It is active throughout the cell cycle and dominant in G1.
Briefly, in NHEJ the DNA ends at the break site are recognized by the Ku heterodimer, which acts as a scaffold for other NHEJ proteins to bind to. Ku binding is followed by pairing, or synapsis, of the broken DNA ends, an essential intermediate stage enabling for the alignment and further processing of the ends and subsequent ligation. Due to its transient nature, the first study where the end synapsis step was directly measured required the use of a single-molecule FRET assay. This allowed for monitoring of the DNA ends during a reconstituted NHEJ reaction using purified human proteins [71]. In this study, as well in subsequent reports [72–74], we and others have shown that synapsis is facilitated by the scaffolding proteins XLF and XRCC4, which together with DNA ligase 4 (Lig4) were shown to form extended filaments in vitro and in cells [71,75,76]. Interestingly, it was recently found that a heterodimer of SFPQ and NONO can substitute for XLF (Figure 1A) [77,78]. When NONO was depleted, repair through NHEJ decreased while repair through HR increased [79].
Fig. 1.
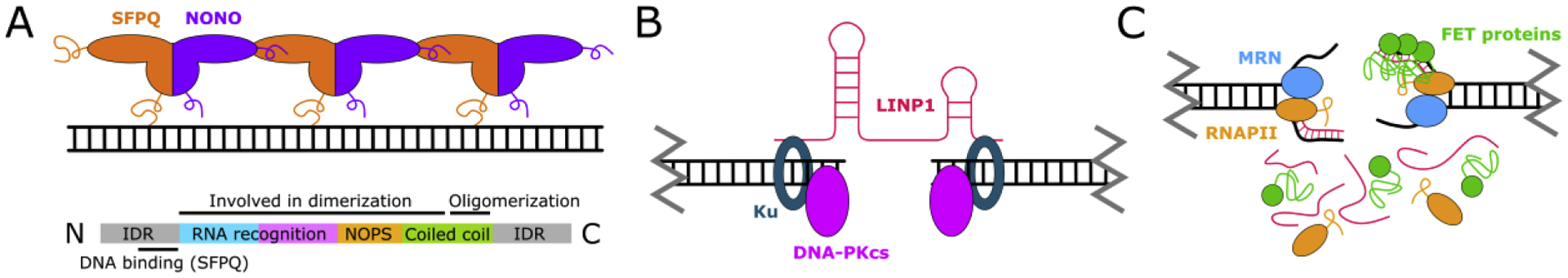
Emerging Factors in DNA Repair. A) Proteins from the DBHS family are molecular scaffolds. They contain domains that recognize nucleic acids and domains that allow them to dimerize and oligomerize. Furthermore, their intrinsically disordered N- and C-terminal domains may participate in phase separation. Here, the SFPQ-NONO dimer forms a filament along the DNA that can substitute XLF in NHEJ. IDR = intrinsically disordered region; NOPS = NonA/Paraspeckles domain (involved in dimerization). B) LncRNAs modulate repair processes. Here, LINP1 directly interacts with Ku to promote synapsis in NHEJ. C) Components of the transcription apparatus participate in condensate formation. RNAPII interacts with the MRN complex to produce dilncRNAs. The FET proteins may be involved in stimulating RNAPII activity. The intrinsically disordered domains of RNAPII and the FET proteins form weak multivalent interactions with RNA and with each other, supporting focus formation.
Another key NHEJ factor, DNA-PKcs, is a PIKKs family kinase and binds Ku to form the DNA-PK holoenzyme. While it was initially speculated that DNA-PKcs contributes to synapsis, the specific roles of DNA-PKcs in NHEJ, and especially in the mediation of end synapsis, are a matter of debate [80]. Single-molecule FRET studies using purified human proteins have consistently shown that DNA-PKcs does not contribute to the synapsis stage, and that efficient synapsis is achieved in the presence of Ku, XRCC4-Lig4 and XLF [71,74]. These findings were further supported by optical tweezers experiments, showing that complexes of XRCC4 and XLF can efficiently bridge neighboring DNA molecules [81]. In contrast, a study utilizing an Xenopus laevis egg extract system to reconstitute NHEJ measured by smFRET assays, revealed that synapsis and NHEJ in this system involves an intermediate step that heavily relies on the presence and activity of DNA-PKcs [82].
When the damage is too extensive for direct rejoining of the two ends, additional processing may take place. Proteins like Artemis (a nuclease), WRN (a helicase) and Polμ (a polymerase) are typically involved in this [83,84].
Beyond the canonical NHEJ proteins, the foci forming protein 53BP1 was also proposed to facilitate end synapsis based on observations of foci coalescence [85]. As discussed briefly before, 53BP1 is a key player in regulating DSB pathway choice by preventing excessive resection. It keeps the DNA ends mostly intact during the repair process as part of the pro-NHEJ Shieldin complex [86]. It will be important to determine how 53BP1 contributes to or affects the biochemical properties of the end synapsis step.
The repair pathways themselves are not set in stone, with a recent study showing a mechanism for NHEJ that surprisingly depends on resection of the DNA ends [87]. Other work showed how NHEJ in non-cycling cells can be error-free when homologous RNA transcripts are used as a template [20,88]. How individual enzymes steer pathway choice, however, is still unclear. Although pathway choice is mainly dependent on cell cycle, recent studies have painted a more nuanced picture with a crucial role for phase-separated domains, which we will discuss later in this review.
4. The Structural Roles of RNA Transcripts in Double-Strand Break Repair
RNA polymers are ideal candidates to participate in phase separation, because of their ability to form transient interactions with other RNAs (in a sequence-specific context) and RNA binding proteins [89]. Moreover, they can act as polymeric scaffolds to compartmentalize proteins. As discussed earlier, they are an important component of paraspeckles and stress granules [46,9] and likely support phase separation in other membrane-less organelles such as the nucleolus [56,90,91].
According to the central dogma of molecular biology, RNA serves as an “assembly guide” for proteins. However, cells contain many long transcripts that are never translated into proteins: so-called long non-coding RNAs (lncRNAs). While initially overlooked, it is now widely accepted that these lncRNAs serve dedicated functions in the cell, mostly related to chromatin architecture and transcription regulation [92,19]. The sequence-dependent secondary structure of these long RNA molecules allows them to specifically interact with target proteins or localize to a specific location in the genome.
Here we will briefly discuss the structural roles that RNA can play in DNA damage repair. We will therefore focus on the RNAs that accumulate at the damage site, rather than the lncRNAs that work at the level of transcription regulation.
4.1. LncRNAs showing direct interactions with repair factors
Over the last decade, a number of lncRNAs have been identified to interact directly with repair factors around the break, modulating their activity. We provide a brief overview of these lncRNAs, subdividing them in those that promote HR, those that promote NHEJ and those that are involved in other aspects of the DNA repair response. We also provide a short overview in tabular form (Table 1).
Table 1.
Overview of lncRNAs discussed in this article.
| IncRNA | Interactions and Functions | Promotes… | References |
|---|---|---|---|
| BGL3 | Binds PARP1 and BARD1, promoting retention of the BRCA1-BARD1 complex at DSBs | HR | [32] |
| DDSR1 | Interacts with BRCA1 and hnRNPUL1 to regulate HR | HR | [27] |
| PRLH1 | Interacts with RNF169, replacing 53BP1 from ubiquitinylated chromatin | HR | [31] |
| TERRA | Interacts with RAD51 at telomeres to form R-loops | HR (at telomeres) | [35,94] |
| HITT | Blocks the ATM binding site for the MRN complex | NHEJ | [24,100] |
| LINP1 | Binds to Ku to stabilize the synaptic complex | NHEJ | [13,97] |
| LRIK | Interacts with Ku, enhancing the efficiency of NHEJ | NHEJ | [33] |
| SNHG12 | Mediates the interaction between DNA-PK and Ku | NHEJ | [23] |
| SNHG17 |
|
NHEJ | [34] |
| GUARDIN |
|
HR and NHEJ | [26] |
| HITTERS | Promotes binding of MRE11 to RAD50 | Cell survival | [25] |
| Linc00312 | Binds to DNA-PKcs to prevent recruitment to Ku80 | Apoptosis | [28] |
| Lnc-bc060912 | Interacts with PARP1 and NPM1 | Cell survival | [29] |
| NEAT1 |
|
Genome stability | [46,51,102,103] |
| NEAT2/MALAT1 |
|
Cell survival | [30,108] |
Pro-HR
DDSR1 is a lncRNA that directly interacts with BRCA1 and hnRNPUL1, and is induced upon DNA damage in multiple different cancer cell lines [27]. hnRNPUL1 is a regulator of end resection. Indeed, upon depletion of DDSR1, less end resection is detected. Moreover, loss of DDSR1 lead to excessive accumulation of BRCA1 and repair factor RAP80 at DSBs, and thereby negatively affects the efficiency of HR.
Another lncRNA, BGL3, is recruited to DSBs by PARP1 at an early stage, interacting with its DNA-binding domain [32]. It also binds BARD1, a binding partner of the HR protein BRCA1, promoting the retention of BRCA1-BARD1 complexes at DSBs and enhancing the binding of BARD1 to other repair proteins. BGL3 therefore acts as a molecular scaffold during HR. Indeed, BGL3 deficiency led to reduced HR efficiency, while not significantly affecting NHEJ.
PRLH1 is a lncRNA expressed in p53-deficient or mutant cells (p53 is the main protein that protects genome stability in humans) [31]. PRLH1 then interacts with RNF169, displacing 53BP1 from ubiquitinylated chromatin, and paving the way for end resection and homologous recombination. Finally, telomeric repeat containing RNAs (TERRA) are a group of lncRNAs that are transcribed from regions near chromosome ends. They are targeted to short telomeres through a UUAGGG-repeat sequence motif, where they associate with RAD51 to form R-loops [35]. These R-loops trigger telomere fragility, replication stress and recombination events. HR-like repair at chromosome ends is considered undesired, since it eventually contributes to alternative lengthening of telomeres, which is a mechanism for some cancer cells to overcome telomere shortening [93]. Interestingly, NONO and SFPQ have been shown to suppress the formation of these R-loops [94], and thereby help control telomere length in cancer cells.
Pro-NHEJ
The Ku heterodimer is known to be able to interact with RNA, specifically with certain sequence motifs [95,96]. It is therefore not surprising that two lncRNAs have recently been identified that link this ability to DNA damage repair: LRIK and LINP1. The 5’ region of LRIK, which indeed contains the AATG and CATGA motifs, binds to the Ku heterodimer [33]. The interaction increases Ku’s affinity for DSBs and facilitates the efficient recruitment of downstream repair factors like XRCC4. LINP1, which is overexpressed in multiple cancers, forms condensates by itself, but adopts a filamentous structure when bound to Ku [13,97]. The Ku-LINP1 complex is still capable of interacting with XLF, but binding with another accessory protein, PAXX, is abrogated. Interestingly, Ku-LINP1 can still participate in end joining; in fact, LINP1 stabilized the synaptic complex more than PAXX did (Figure 1B). Insulin-like growth factor binding protein-3 (IGFBP3), a protein that modulates NHEJ in triple-negative breast cancer, binds NONO and SFPQ but needs LINP1 for complex formation [98,99], further cementing a role for LINP1 in promoting NHEJ.
Expression of the lncRNA HITT was found to be reduced in tissues from colon cancer, thyroid cancer and chromophobe renal cell carcinoma [24]. Subsequently, HITT was shown to prevent recruitment of ATM to sites of DNA damage [100]. ATM is a kinase involved early in the DSB response, and regulates the activity of many downstream repair factors. HITT blocks ATM’s binding site for the MRN complex, specifically the NBS1 protein, thus disfavoring HR.
Small Nucleolar RNA Host Genes (SNHG) are a subset of lncRNAs which have received attention due to their oncogenic role in cancer [101]. SNHG12 was identified as a factor that is overexpressed in atherosclerosis [23]. In this disease, cells near plaque build-up often show signs of senescence, which could be due to persistent DNA damage. SNHG12 binds to DNA-PK and seems to mediate the interaction of this repair factor with Ku. Knocking down SNHG12 leads indeed to higher levels of DNA damage in the cell, indicating a role for SNHG12 in NHEJ. Another member of this class of lncRNAs, SNHG17, was shown to be upregulated upon H. pylori infection [34]. Overabundant SNHG17 recruits NONO to DSBs and upregulates RING1, which in turn induces RAD51 breakdown. These combined effects cause a shift from HR to NHEJ. This change in DNA repair pathway is thought to contribute to the development of gastric cancer that is often associated with chronic H. pylori infection.
Other relevant repair-associated lncRNAs
There are a couple of repair-related lncRNAs that, to our current knowledge, are not necessarily pro-HR or pro-NHEJ but are more involved with cell fate decisions in general. Here, we describe a couple of lncRNAs that work at different levels to steer the cell’s response to DNA damage.
NEAT1, which we briefly discussed before as a major component of paraspeckles, is a multifunctional lncRNA that was also found to be involved in the transcriptional activation of HR genes [102]. Another study found that activation of p53 triggers the formation of paraspeckles [103]. NEAT1 further modulates ATR signaling, thereby preventing DSBs from occurring. Combined, these studies suggest a role for NEAT1 in protecting genome stability. The exact mechanisms through which NEAT1 and paraspeckles contribute to this remain unclear at the moment, but the sequestration of harmful transcripts and proteins inside biomolecular condensates is not unthinkable.
NEAT2, also known as MALAT1, was first identified in non-small cell lung cancer [104], but is upregulated in many other cancers [105–107]. In multiple myeloma, it forms a complex with PARP1 and Ligase 3 to promote alternative NHEJ, a process that involves minimal resection and subsequent alignment of microhomologies around the break site [30]. It is also reported to bind SFPQ and thereby release the oncogene PTBP2 from SFPQ/PTBP2 complexes [108]. Given the previously described role of SFPQ in DNA repair, it would be interesting to know if the interaction of NEAT2 with SFPQ has any effect on the function of SFPQ in those repair complexes.
Linc00312, a lncRNA first found in nasopharyngeal carcinoma, is associated with cell apoptosis [109]. It inhibits DNA repair by binding to DNA-PKcs and preventing its recruitment to Ku80 [28]. Overexpression of linc00312 also downregulates MRN expression, although the molecular mechanism is unknown. LncRNAs that are associated with preventing apoptosis include lnc-bc060912 and HITTERS (unrelated to HITT). lnc-bc060912 binds to nucleophosmin (NPM1), which is a nucleolar protein involved in DNA damage repair, and to PARP1 [29]. HITTERS is upregulated in certain cancer cells by endoplasmic reticulum stress [25]. It works as a scaffold to promote binding of MRE11 to RAD50, thereby supporting DNA repair.
GUARDIN is a p53-responsive lncRNA that acts on at least two different levels to protect genome integrity [26]. It prevents end-to-end fusion of chromosomes by binding to microRNA23a, which has a partially complementary sequence. microRNA23a normally suppresses TRF2 expression, which is part of the shelterin complex found at chromosome ends. GUARDIN thus indirectly helps to protect telomeres from undesired fusion events. On a different level, GUARDIN aids in BRCA1-BARD1 complex formation by directly interacting with both proteins. Depletion of GUARDIN leads to degradation of BRCA1. Additionally, the authors showed that depletion of GUARDIN also leads to impaired repair through both HR and NHEJ.
Often, only parts of the lncRNAs physically interact with the DNA repair factors. This suggests that the remainder of the RNA polymer can be used for other interactions. This may indeed include interactions with other proteins, rendering such lncRNAs molecular chaperones, or an architectural role in a condensate like the previously described NEAT1 in paraspeckles. In this context, interactions with proteins from the DBHS family are particularly interesting. As pointed out before, these proteins can complement or even substitute dedicated DNA repair proteins, and may also help with lncRNA recruitment to the break site.
4.2. Break-induced transcription
Another source of lncRNAs is break-induced transcription. Although transcription in general is downregulated upon detection of a DSB [17], it was reported that transcription in the immediate vicinity of the break still occurs [16,110]. The resulting damage-induced lncRNAs (dilncRNAs) are therefore produced from sequences directly around the break site.
A general approach to study break-induced transcription involves integrating an artificial construct with a unique cut site into the genome, which can then be used to induce a DSB site-specifically, such that the sequences flanking the break are known. The extent to which break-induced transcription occurs in genetically unperturbed cells, however, has been debated. The main criticism is that the artificial locus would not resemble the natural state of the chromatin. A single-molecule approach shows that break-induced transcription indeed depends on the chromatin landscape, with intragenic regions showing nucleosome depletion and bidirectional transcription upon induction of a DSB [111]. Another study using next-generation sequencing was not able to detect transcription when the break was induced in genic or intergenic regions, but the authors did find break-induced transcription at ribosomal DNA loci [112]. A third study could not find proof for generation of small RNAs around break sites either, but did show a role for the RNA processing enzyme DROSHA in generating DNA:RNA hybrids around the DSB [113]. Despite these concerns, de novo production of dilncRNAs was found to be crucial for formation of repair foci [3,114]. We will therefore provide a brief overview of the literature supporting break-induced transcription.
Break-induced transcription does not seem to be associated with promoters: by inducing a DSB at a specific location in the genome and using single-molecule FISH and RT-qPCR to analyze the dilncRNAs that were formed, it was shown that transcription takes place to and from the break site [114]. The polymerase involved was identified as RNAPII, which is recruited to the DSB through the MRN complex and the transcription pre-initiation complex [3]. A potential mechanism for this break-induced transcription came from the observation that the interaction between RNAPII and the MRN complex alone can stimulate RNA synthesis in vitro [18,114]. One would perhaps expect that the nuclease activity of the MRN complex would create the ideal ssDNA substrate needed for transcription. Surprisingly, however, the nuclease activity of MRN does not play a role in this. Instead, the underlying mechanism is thought to rely on the ability of MRN to melt DNA ends, creating an opportunity for RNAPII to start transcription from the break site inwards (Figure 1C). This is not difficult to imagine conceptually, since local DNA melting is always a prerequisite for transcription initiation. The transcript itself can exist in the form of an RNA:DNA hybrid, which offers a potential explanation for the observation that such hybrids exist near break sites, and are therefore not necessarily the result of interrupted transcription that occurred before the damage. Eventually, these transcripts are processed by DICER and DROSHA to create smaller DNA damage-induced RNAs, coined DDRNAs [15,115]. Their sequence allows these DDRNAs to be site-specifically recruited to break sites. DDRNAs, DICER and DROSHA were all found to be necessary for formation of repair foci [3,113,114].
4.3. Transcription-associated DSB repair
Although break-induced transcription is consistent with a repair model that involves phase separation, it is unclear if there is an additional use for the sequence information stored in RNA transcripts. For example, it has been suggested that RNA can aid in homology-based repair. In yeast, both synthetic RNAs and endogenous transcripts can indeed act as a template in DNA repair if the sequence is homologous to the break site [116,117], while RAD51 and RAD52 were shown to be able to participate in strand exchange between DNA and single-stranded RNA [21,118]. In human cells, however, that process seems to be less efficient than when DNA is used [22]. In 2015, support for transcription-associated HR came from a study that showed that in the G0/G1 phase of the cell cycle, proteins associated with HR are preferentially recruited to damage sites where transcription takes place. Additionally, this recruitment was found to rely on Cockayne Syndrome Protein B [119]. One year later, Chakraborty et al. showed that NHEJ proteins preferentially associate with transcribed genes, and can use the nascent RNA as template for error-free repair [20]. Very recently, RNA transcripts from around the break site were shown to stimulate HR in human cells [120], a process that is dependent on RAD51-associated-protein 1. Despite these findings, much remains unknown. For example, the molecular mechanisms behind RNA-templated repair are not well understood. It is also still unclear to what extent this type of repair is applied in human cells, and how it depends on cell type and phase. Considering the scope of this review, it would also be interesting to know if the increased accessibility of the transcribed genomic region plays a role in the recruitment of specific repair factors that guide pathway choice.
5. Nucleation and Development of DNA Damage Foci
The DNA damage response is a tightly organized sequence of events, starting off with a signaling cascade that helps the cell in isolating, identifying, and repairing the lesion. Although much is known about the order in which repair factors are recruited, we would like to give an overview that emphasizes the role that damage-induced lncRNAs and condensates can play in this process.
5.1. PARylation as the Initiation of Phase Separation in DNA Repair
One of the earliest responders to DNA damage is the poly (ADP-ribose) polymerase PARP1 [121]. This highly expressed protein has many functions in the cell, ranging from DNA repair to chromatin maintenance. Its enzymatic activity, the production of long and branched poly(ADP-ribose) (abbreviated as PAR) chains from NAD+, is a form of a post-translational modification and is strongly enhanced by binding to damaged DNA [122,123]. PARP1 can PARylate itself (automodification), other proteins and also DNA [124]. The resulting PAR chains are a signal for DNA repair proteins, facilitating their recruitment to the site of damage. Indeed, many DNA repair proteins contain PAR-binding motifs [123]. The importance of PARP1 activity for recruitment of downstream repair factors was illustrated by treatment with the PARP inhibitor talazoparib, which altered recruitment of many repair factors in HeLa cells, delaying the ones that are known to interact with PAR [121].
Altmeyer et al. showed that the production of PAR chains leads to phase separation around the damage site (Figure 2A,B) [125]. They made the interesting suggestion that this phase separation allows the cell to strictly control the earliest response to DNA damage by restricting the proteins that can reach the break. The negative PAR chains allow early access into the phase separated domain for certain proteins with intrinsically disorder regions (IDRs), which can then actively participate in phase separation through weak multivalent interactions with each other. Among the proteins encountered in this early phase-separated domain are the RNA-binding FET proteins. The implication of these proteins in DNA repair is intriguing, since they are known to have functions in transcription, are associated with chromosomal rearrangements in cancer and have a role in neurological disorders like ALS [10,126]. Indeed, there is a link between familial FUS mutations that promote pathological protein aggregation and increased DNA damage [127,128]. In this early stage, proteins with PAR-binding motifs are recruited early as well, as for example XRCC1 [121]. NONO, which interacts with PAR chains through its RNA recognition motif, is also recruited [79], although the timing is unclear. Interestingly, the important DNA repair factor 53BP1 is initially kept out [125].
Fig. 2.
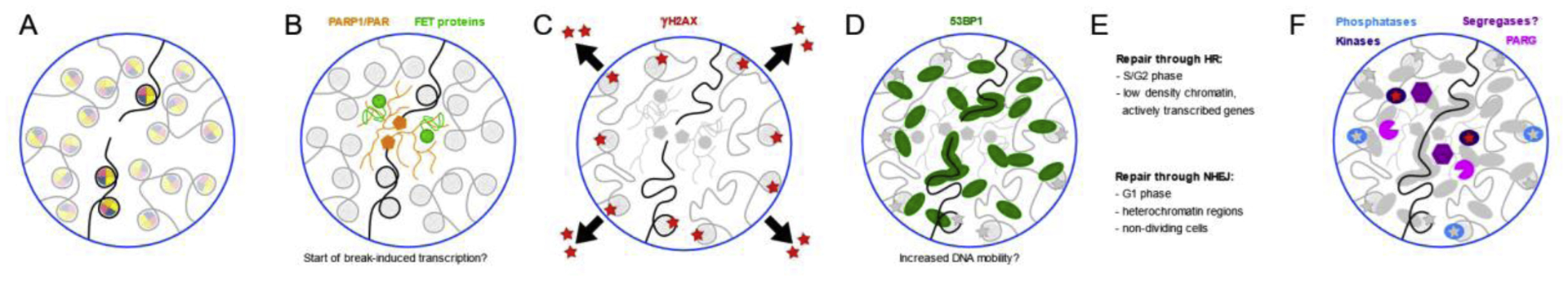
Development of DNA repair foci. A) A double strand break occurs. B) PARP1 seeds a phase-separated domain by synthesizing PAR chains. Proteins with a PAR binding motif and several RNA binding proteins (FUS, EWS and TAF15) are recruited in the phase-separated domain. Break-induced transcription may start. C) ATM phosphorylates histone H2AX in a confined volume around the break, leading to local chromatin decondensation. D) 53BP1 is recruited to the break site, interacting with the chromatin and the DNA. E) Repair proceeds through HR or NHEJ. How condensates aid in pathway choice is not fully understood, but HR seems to be preferred in low-density chromatin. F) Dissipation of the focus can occur through post-translational modifications and the breakdown of PAR chains. What role segregases like VCP play is not known exactly.
The presence of the RNA-binding FET proteins makes it tempting to speculate that transcription of damage-induced long non-coding RNAs (dilncRNAs) also occurs at this very early stage. As shown recently, members of the transcription apparatus were found to interact with the MRN complex, which is an early responder to DNA damage [3]. As discussed earlier, the MRN complex was found capable of stimulating transcription around break sites [18]. The presence of RNAPII at DSBs may be further stabilized by the ability of its C-terminal domain to participate in phase separation [129,130]. In fact, it was shown that the IDRs of the FET proteins form polymeric fibers that interact with the RNAPII C-terminal domain in a manner that correlates with transcriptional activation [131]. Further support for an early onset of transcription comes from cells in which H2AX was knocked down, halting DNA repair before γH2AX foci formation. In these cells, small non-coding RNAs localize to the break site in a sequence specific manner [114]. These RNAs could contribute to phase separation due to their negative charge and chemical resemblance to PAR chains.
It has been shown in vitro that automodification of PARP1 will promote dissociation of PARP1 from the break site, likely due to steric hindrance or electrostatic repulsion [132], making the break site available for further processing.
5.2. Chromatin Relaxation increases the Accessibility of Proteins to the Damage Site
Building further on this model for the role of phase separation in DNA repair, the next step involves ATM phosphorylating histone H2AX (Figure 2C). This spread in phosphorylation is thought to occur in 3D space, rather than in a linear fashion along the DNA, within the confines of a topologically associated domain (TAD), which can have a size in the Mbp range [133]. Phosphorylation does not extend beyond the CTCF binding sites that form the border of the TAD [134], suggesting a natural limitation exists to how large a γH2AX focus can grow. It has been shown that ATM silences transcription around DSBs [17], although ATM inhibition does not seem to affect dilncRNA production [114]. Regardless, the spread of γH2AX promotes chromatin decondensation [135,136], which fundamentally changes the phase-separated compartment around the break site, allowing more proteins in. A study that has not yet been peer-reviewed at the time of writing suggests that FUS is necessary for the organization of small γH2AX foci into larger clusters [137], suggesting FUS is still present at this point. MDC1 binds γH2AX [138], recruiting the E3 ligases RNF8 and RNF168. These two proteins then ubiquitylate the chromatin around the break site [139]. The RNA processing enzyme DROSHA is also required at the break site around the same time, preceding pathway choice [113].
5.3. 53BP1 Focus Formation
The recruitment of the large protein 53BP1 takes place downstream of ubiquitylation by RNF8 and RNF168 (Figure 2D). Its multi-domain structure allows 53BP1 to interact with the damage site in multiple different ways. First of all, the 53BP1 Tudor domain is able to bind dimethylated histone H4K20 [55]. This histone modification is also present in undamaged chromatin; however, decondensation of the chromatin around the break site is thought to make it more accessible. The Tudor domain was also shown to interact with dilncRNAs and DDRNAs in cells in which damage was induced at specific loci [114]. Additionally, the 53BP1 UDR domain interacts with ubiquitylated histone residue H2A(X)K15 [54]. Using a system in which light can trigger phase separation of a 53BP1-fusion protein, Kilic et al. showed that the oligomerization and BRCT domains of the protein are important for its phase-separating capabilities, while the disordered N-terminal domain is surprisingly dispensable [4]. Nucleation and growth of 53BP1 foci is stalled by RNAPII inhibitors, suggesting these processes are dependent on de novo transcription, which may indeed be the previously described break-induced transcription [3]. Moreover, treatment with RNase A results in dissipation of the foci [140], as does treatment with anti-sense oligonucleotides against regions around the break [114]. As pointed out above, RNAPII itself may participate in phase separation [129,130], and an active transcription apparatus could perhaps increase focus stability.
5.4. Choice of Repair Pathway and DSB mobility
A critical question to which we do not have a full answer yet, is in what way the development of a DNA repair focus contributes to the choice of repair pathway.
A study by Lemaître et al. showed that the nuclear position where the break occurs dictates pathway choice in yeast [141]. If a break occurs in the heterochromatin region of the nucleus, HR is impaired. The nuclear lamina, a dense fibrillar network, also suppresses HR. Conversely, the recruitment of NHEJ proteins to the break was not delayed, resulting in repair through an end-joining mechanism. Another example of the importance of chromatin compaction for pathway choice comes from work by Aymard et al., that shows that breaks in transcriptionally active genes (in euchromatic regions) are preferentially repaired through HR, while breaks in inactive regions are typically repaired through NHEJ [142,143]. As discussed before, Chakraborty et al. showed that NHEJ proteins are preferentially recruited to transcribed genes [20,88]. This seems at odds with the idea that active chromatin is repaired through HR. It is important to point out here that non-replicating cells, for example neurons, cannot apply classical HR due to the absence of a sister chromatid. The involvement of the NHEJ machinery and the ability to use nascent RNAs as a template can therefore be the mechanism of choice for error-free DNA repair in these cells.
In yeast, DNA damage foci that are rich in the HR protein RAD52 were found to behave like liquid droplets, with their movement and fusion mediated by nuclear filaments [5]. Clustering of these damage sites is thought to occur to facilitate repair in dedicated repair centers. The extent to which DSBs move in mammalian cells, however, has been debated before [144]. In the work by Aymard et al., discussed above, breaks in transcribed regions of the chromatin were found to be clustered in a mechanism that depends on the MRN complex, and on the actin and microtubule organizers FMN2 and the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex [143]. The involvement of FMN2 and the LINC complex suggests active transport of breaks across the nucleus; the involvement of MRN, on the other hand, suggests resection may be necessary for this to occur. The authors suggest these DSBs are sequestered to make sure they are repaired in an error-free manner in a later stage of the cell cycle.
Another possible reason for DSB mobility is that it may aid in the homology search. However, this can increase the chances of mis-rejoining if repair droplets fuse, particularly in an environment where NHEJ is the preferred pathway. A system based on dysfunctional telomeres, which are often joined together through NHEJ, showed that these telomeres exhibited increased mobility in search for other telomeres, which indeed results in mis-rejoining [145]. This behavior extended to IR-induced DSBs, and was found to depend on 53BP1, the LINC complex and dynamic microtubules. Importantly, the role of 53BP1 seems disconnected from its role in resection [146], which opens the possibility that the observed DSB mobility is due to the tendency of 53BP1 to phase separate, irrespective of whether the break requires HR or NHEJ. The authors propose that DSB mobility may be beneficial for the cell when the number of breaks is low, since that lowers the chances of mis-rejoining.
In conclusion, HR seems to be the preferred pathway if the chromatin is less condensed and therefore more mobile (particularly during S/G2), while NHEJ occurs in denser regions and non-dividing cells (Figure 2E). DNA repair proteins that are able to form condensates, like 53BP1, are able to confer mobility to the break site. Although phase separation alone can in principle explain the fusion of such repair centers, the involvement of the LINC complex and nuclear filaments suggests that the cell retains some autonomy on the formation of larger clusters.
5.5. Condensate Resolution
The importance of condensates for DNA repair and cellular function in general begs the question how the cell regulates not only their formation, but also their dissipation. Here we will briefly discuss some of the mechanisms employed by the cell to dissolve condensates, with a focus on DNA repair foci.
The PAR chains that constitute the original condensate are eventually broken down by the enzyme PAR glycohydrolase (PARG) [147] (Figure 2F). Together with the tendency of PARP1 to dissociate upon autoPARylation [132], this prevents excessive growth of the phase-separated compartment.
Another way for the cell to control the size of the condensate is by post-translational modifications, for example phosphorylation (Figure 2F). Kinases such as ATM and DNA-PK have central roles in the DNA damage signaling cascade, Indeed, phosphorylation can have a pronounced effect on the tendency of intrinsically disordered proteins to phase separate or aggregate [148]. The aggregate formation of the FUS protein has been well studied because of its role in neurological disorders. Wild-type FUS was shown to be less prone to aggregation when the protein was phosphorylated [149]. Likewise, phosphorylation of the disordered C-terminal domain of RNAPII, which produces dilncRNAs, also affects its ability to phase separate [129,130]. Moreover, one of the factors triggering chromatin condensation is thought to be the phosphorylation of HP1a [45]. Phosphorylation of histone H2AX is of course responsible for chromatin decondensation in a DNA damage context. The dephosphorylation of γH2AX, which occurs during or after repair, is performed by protein phosphatase 2A and WIP1 [150,151], and presumably helps with bringing the chromatin back to its native state, resolving the focus. Thus, a small modification like phosphorylation may be a tool for the cell to change the larger physical behavior of a repair focus.
Another post-translational modification is ubiquitylation, which can lead to the breakdown of misfolded proteins that may exist in the core of the condensate. An example of a protein that assists in this type of degradation is the segregase VCP, also known as p97 (Figure 2F). It is recruited to DNA damage sites, where it removes K48-ubiquitylated proteins to facilitate recruitment of 53BP1, BRCA1 and RAD51 [121,152]. An inhibitor of VCP was shown to induce an accumulation of ubiquitylated proteins and cell death [153,154]. It is not unthinkable that large DNA repair foci contain misfolded proteins at their core, which may be toxic if the cell cannot dissolve them. Whether such aggregates exist, and what role segregases like VCP play in resolving these, remains to be seen.
6. Concluding Remarks
Over the years it has become clear that RNA polymers can fulfill more roles than merely being an assembly guide for proteins. LncRNAs have emerged as modulators of processes like transcription and DNA repair through direct interactions with key proteins. We now know that RNA polymers, through their ability to form weak multivalent interactions and to act as scaffolds, are also ideal building blocks for biomolecular condensates. Indeed, membrane-less organelles such as stress granules, paraspeckles, nucleoli and Cajal bodies all contain large amounts of RNA.
Biomolecular condensates are dynamic yet organized structures that are highly sensitive to environmental conditions, rendering them an ideal tool for the cell to respond to stress. Although DNA damage is a major stress-inducing event, the importance of condensate formation for DNA repair has until recently received only limited attention. Canonical DNA repair pathways describe repair as a tightly orchestrated sequence of events. Condensate formation, starting with PARylation and supported by damage-induced transcription, offers a compelling explanation for the timely recruitment and spatial organization of repair factors.
The main challenge exists in determining the structure-function relationship of the biomolecular condensates of DNA repair. This starts with the identity of repair foci: are they indeed phase-separated compartments, or would another description be a better fit? This is not just semantics, since the behavior of the compartment is integral to its function and may enhance our understanding. The development of repair foci over time may hold crucial information about pathway choice. How is focus growth and eventual dissipation regulated by the cell, and what is the interplay with the different repair pathways? Not much is known about the influence of condensates on the biochemical functions of the repair enzymes. How are the kinetics of the repair process affected by the dense environment? It may well be possible that certain enzymes or substrates are excluded from the condensate. How can we best simulate those conditions in the lab?
There is a considerable amount of literature on condensate formation in artificial systems. Additionally, more and more studies are being published on other membrane-less organelles that may well behave very similar to DNA repair foci. In combination with the significant progress that has been made in imaging techniques, such as super-resolution microscopy, they should provide us with the tools to answer these intriguing questions.
Highlights.
We discuss the current knowledge about the function of biomolecular condensates in DNA repair
We describe the role of RNA and RNA binding proteins in the formation of repair foci
We highlight several long non-coding RNAs that perform dedicated tasks at the damage site
Acknowledgments
Research in the Rothenberg lab is supported by funding from the National Institute of Health (1R35GM134947-01, 1P01CA247773-01/5491; 1R01AI153040-01) American Cancer Society (RSG DMC-16-241-01-DMC), the V foundation for Cancer Research (D2018-020) and Pfizer.
We would like to thank Dipika Gupta and Huijun Xue for critically reading the manuscript. We apologize to all colleagues whose work we could not discuss due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
As stated below research in the Rothenberg lab is amongst others supported by Pfizer.
References
- [1].Tubbs A, Nussenzweig A, Endogenous DNA Damage as a Source of Genomic Instability in Cancer, Cell. 168 (2017) 644–656. 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chatterjee N, Walker GC, Mechanisms of DNA damage, repair and mutagenesis, Environ. Mol. Mutagen 58 (2017) 235–263. 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pessina F, Giavazzi F, Yin Y, Gioia U, Vitelli V, Galbiati A, Barozzi S, Garre M, Oldani A, Flaus A, Cerbino R, Parazzoli D, Rothenberg E, d’Adda di Fagagna F, Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors, Nat. Cell Biol 21 (2019) 1286–1299. 10.1038/s41556-019-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kilic S, Lezaja A, Gatti M, Bianco E, Michelena J, Imhof R, Altmeyer M, Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments, EMBO J. 38 (2019) e101379. 10.15252/embj.2018101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Oshidari R, Huang R, Medghalchi M, Tse EYW, Ashgriz N, Lee HO, Wyatt H, Mekhail K, DNA repair by Rad52 liquid droplets, Nat. Commun 11 (2020) 695. 10.1038/s41467-020-14546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fox AH, Nakagawa S, Hirose T, Bond CS, Paraspeckles: Where Long Noncoding RNA Meets Phase Separation, Trends Biochem. Sci 43 (2018) 124–135. 10.1016/j.tibs.2017.12.001. [DOI] [PubMed] [Google Scholar]
- [7].Strom AR, Brangwynne CP, The liquid nucleome – phase transitions in the nucleus at a glance, J. Cell Sci 132 (2019). 10.1242/jcs.235093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lau Y, Oamen HP, Caudron F, Protein Phase Separation during Stress Adaptation and Cellular Memory, Cells. 9 (2020). 10.3390/cells9051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wolozin B, Ivanov P, Stress granules and neurodegeneration, Nat. Rev. Neurosci 20 (2019) 649–666. 10.1038/s41583-019-0222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Svetoni F, Frisone P, Paronetto MP, Role of FET proteins in neurodegenerative disorders, RNA Biol. 13 (2016) 1089–1102. 10.1080/15476286.2016.1211225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Portnoy V, Huang V, Place RF, Li L-C, Small RNA and transcriptional upregulation, Wiley Interdiscip. Rev. RNA 2 (2011) 748–760. 10.1002/wrna.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Boucas J, Riabinska A, Jokic M, Herter-Sprie GS, Chen S, Höpker K, Reinhardt HC, Posttranscriptional regulation of gene expression—adding another layer of complexity to the DNA damage response, Front. Genet 3 (2012). 10.3389/fgene.2012.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thapar R, Wang JL, Hammel M, Ye R, Liang K, Sun C, Hnizda A, Liang S, Maw SS, Lee L, Villarreal H, Forrester I, Fang S, Tsai M-S, Blundell TL, Davis AJ, Lin C, Lees-Miller SP, Strick TR, Tainer JA, Mechanism of efficient double-strand break repair by a long non-coding RNA, Nucleic Acids Res. (n.d.) 10.1093/nar/gkaa784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Thapar R, Regulation of DNA Double-Strand Break Repair by Non-Coding RNAs, Mol. Basel Switz 23 (2018). 10.3390/molecules23112789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d’Adda di Fagagna F, Site-specific DICER and DROSHA RNA products control the DNA-damage response, Nature. 488 (2012) 231–235. 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Michalik KM, Böttcher R, Förstemann K, A small RNA response at DNA ends in Drosophila, Nucleic Acids Res. 40 (2012) 9596–9603. 10.1093/nar/gks711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA, ATM-Dependent Chromatin Changes Silence Transcription In cis to DNA Double-Strand Breaks, Cell. 141 (2010) 970–981. 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sharma S, Anand R, Zhang X, Francia S, Michelini F, Galbiati A, Williams H, Ronato DA, Masson J-Y, Rothenberg E, Cejka P, d’Adda di Fagagna F, MRE11-RAD50-NBS1 Complex Is Sufficient to Promote Transcription by RNA Polymerase II at Double-Strand Breaks by Melting DNA Ends, Cell Rep. 34 (2021) 108565. 10.1016/j.celrep.2020.108565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Durut N, Mittelsten Scheid O, The Role of Noncoding RNAs in Double-Strand Break Repair, Front. Plant Sci 10 (2019). 10.3389/fpls.2019.01155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chakraborty A, Tapryal N, Venkova T, Horikoshi N, Pandita RK, Sarker AH, Sarkar PS, Pandita TK, Hazra TK, Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes, Nat. Commun 7 (2016) 13049. 10.1038/ncomms13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mazina OM, Keskin H, Hanamshet K, Storici F, Mazin AV, Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair, Mol. Cell 67 (2017) 19–29.e3. 10.1016/j.molcel.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shen Y, Nandi P, Taylor MB, Stuckey S, Bhadsavle HP, Weiss B, Storici F, RNA-driven genetic changes in bacteria and in human cells, Mutat. Res. Mol. Mech. Mutagen 717 (2011) 91–98. 10.1016/j.mrfmmm.2011.03.016. [DOI] [PubMed] [Google Scholar]
- [23].Haemmig S, Yang D, Sun X, Das D, Ghaffari S, Molinaro R, Chen L, Deng Y, Freeman D, Moullan N, Tesmenitsky Y, Wara AKMK, Simion V, Shvartz E, Lee JF, Yang T, Sukova G, Marto JA, Stone PH, Lee WL, Auwerx J, Libby P, Feinberg MW, Long noncoding RNA SNHG12 integrates a DNA-PK–mediated DNA damage response and vascular senescence, Sci. Transl. Med 12 (2020). 10.1126/scitranslmed.aaw1868. [DOI] [PubMed] [Google Scholar]
- [24].Wang X, Li L, Zhao K, Lin Q, Li H, Xue X, Ge W, He H, Liu D, Xie H, Wu Q, Hu Y, A novel LncRNA HITT forms a regulatory loop with HIF-1α to modulate angiogenesis and tumor growth, Cell Death Differ. 27 (2020) 1431–1446. 10.1038/s41418-019-0449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wu C, Chen W, Yu F, Yuan Y, Chen Y, Hurst DR, Li Y, Li L, Liu Z, Long Noncoding RNA HITTERS Protects Oral Squamous Cell Carcinoma Cells from Endoplasmic Reticulum Stress-Induced Apoptosis via Promoting MRE11-RAD50-NBS1 Complex Formation, Adv. Sci. n/a (n.d.) 2002747. 10.1002/advs.202002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hu WL, Jin L, Xu A, Wang YF, Thorne RF, Zhang XD, Wu M, GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability, Nat. Cell Biol 20 (2018) 492–502. 10.1038/s41556-018-0066-7. [DOI] [PubMed] [Google Scholar]
- [27].Sharma V, Khurana S, Kubben N, Abdelmohsen K, Oberdoerffer P, Gorospe M, Misteli T, A BRCA1-interacting lncRNA regulates homologous recombination, EMBO Rep. 16 (2015) 1520–1534. 10.15252/embr.201540437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Guo Z, Wang Y-H, Xu H, Yuan C-S, Zhou H-H, Huang W-H, Wang H, Zhang W, LncRNA linc00312 suppresses radiotherapy resistance by targeting DNA-PKcs and impairing DNA damage repair in nasopharyngeal carcinoma, Cell Death Dis. 12 (2021) 1–15. 10.1038/s41419-020-03302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Luo H, Sun Y, Wei G, Luo J, Yang X, Liu W, Guo M, Chen R, Functional Characterization of Long Noncoding RNA Lnc_bc060912 in Human Lung Carcinoma Cells, Biochemistry. 54 (2015) 2895–2902. 10.1021/acs.biochem.5b00259. [DOI] [PubMed] [Google Scholar]
- [30].Hu Y, Lin J, Fang H, Fang J, Li C, Chen W, Liu S, Ondrejka S, Gong Z, Reu F, Maciejewski J, Yi Q, Zhao J-J, Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma, Leukemia. 32 (2018) 2250–2262. 10.1038/s41375-018-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Deng B, Xu W, Wang Z, Liu C, Lin P, Li B, Huang Q, Yang J, Zhou H, Qu L, An LTR retrotransposon-derived lncRNA interacts with RNF169 to promote homologous recombination, EMBO Rep. 20 (2019) e47650. 10.15252/embr.201847650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hu Z, Mi S, Zhao T, Peng C, Peng Y, Chen L, Zhu W, Yao Y, Song Q, Li X, Li X, Jia C, Pei H, BGL3 lncRNA mediates retention of the BRCA1/BARD1 complex at DNA damage sites, EMBO J. 39 (2020) e104133. 10.15252/embj.2019104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang D, Zhou Z, Wu E, Ouyang C, Wei G, Wang Y, He D, Cui Y, Zhang D, Chen X, Reed SH, Luo J, Chen R, LRIK interacts with the Ku70–Ku80 heterodimer enhancing the efficiency of NHEJ repair, Cell Death Differ. 27 (2020) 3337–3353. 10.1038/s41418-020-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Han T, Jing X, Bao J, Zhao L, Zhang A, Miao R, Guo H, Zhou B, Zhang S, Sun J, Shi J, pylori H infection alters repair of DNA double-strand breaks via SNHG17, J. Clin. Invest 130 (2020) 3901–3918. 10.1172/JCI125581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Feretzaki M, Pospíšilová M, Fernandes RV, Lunardi T, Krejci L, Lingner J, RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops, Nature. 587 (2020) 303–308. 10.1038/s41586-020-2815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Alberti S, Gladfelter A, Mittag T, Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates, Cell. 176 (2019) 419–434. 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pessina F, Gioia U, Brandi O, Farina S, Ceccon M, Francia S, d’Adda di Fagagna F, DNA Damage Triggers a New Phase in Neurodegeneration, Trends Genet. 0 (2020). 10.1016/j.tig.2020.09.006. [DOI] [PubMed] [Google Scholar]
- [38].Yoo H, Triandafillou C, Drummond DA, Cellular sensing by phase separation: Using the process, not just the products, J. Biol. Chem 294 (2019) 7151–7159. 10.1074/jbc.TM118.001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bracha D, Walls MT, Wei M-T, Zhu L, Kurian M, Avalos JL, Toettcher JE, Brangwynne CP, Mapping Local and Global Liquid Phase Behavior in Living Cells Using Photo-Oligomerizable Seeds, Cell. 175 (2018) 1467–1480.e13. 10.1016/j.cell.2018.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lin Y, Protter DSW, Rosen MK, Parker R, Formation and Maturation of Phase Separated Liquid Droplets by RNA Binding Proteins, Mol. Cell 60 (2015) 208–219. 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schmidt HB, Görlich D, Transport Selectivity of Nuclear Pores, Phase Separation, and Membraneless Organelles, Trends Biochem. Sci 41 (2016) 46–61. 10.1016/j.tibs.2015.11.001. [DOI] [PubMed] [Google Scholar]
- [42].Chen C, Ding X, Akram N, Xue S, Luo S-Z, Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases, Molecules. 24 (2019). 10.3390/molecules24081622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].P. A, Weber SC, Evidence for and against Liquid-Liquid Phase Separation in the Nucleus, Non-Coding RNA. 5 (2019). 10.3390/ncrna5040050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH, Phase separation drives heterochromatin domain formation, Nature. 547 (2017) 241–245. 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ, Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin, Nature. 547 (2017) 236–240. 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].West JA, Mito M, Kurosaka S, Takumi T, Tanegashima C, Chujo T, Yanaka K, Kingston RE, Hirose T, Bond C, Fox A, Nakagawa S, Structural, super-resolution microscopy analysis of paraspeckle nuclear body organization, J. Cell Biol 214 (2016) 817–830. 10.1083/jcb.201601071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Knott GJ, Bond CS, Fox AH, The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold, Nucleic Acids Res. 44 (2016) 3989–4004. 10.1093/nar/gkw271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Passon DM, Lee M, Rackham O, Stanley WA, Sadowska A, Filipovska A, Fox AH, Bond CS, Structure of the heterodimer of human NONO and paraspeckle protein component 1 and analysis of its role in subnuclear body formation, Proc. Natl. Acad. Sci 109 (2012) 4846–4850. 10.1073/pnas.1120792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lee M, Sadowska A, Bekere I, Ho D, Gully BS, Lu Y, Iyer KS, Trewhella J, Fox AH, Bond CS, The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation, Nucleic Acids Res. 43 (2015) 3826–3840. 10.1093/nar/gkv156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shav-Tal Y, Zipori D, PSF and p54nrb/NonO – multi-functional nuclear proteins, FEBS Lett. 531 (2002) 109–114. 10.1016/S0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- [51].Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, Hirose T, Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation, Mol. Cell 70 (2018) 1038–1053.e7. 10.1016/j.molcel.2018.05.019. [DOI] [PubMed] [Google Scholar]
- [52].Iwabuchi K, Li B, Massa HF, Trask BJ, Date T, Fields S, Stimulation of p53-mediated Transcriptional Activation by the p53-binding Proteins, 53BP1 and 53BP2*, J. Biol. Chem 273 (1998) 26061–26068. 10.1074/jbc.273.40.26061. [DOI] [PubMed] [Google Scholar]
- [53].Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM, Chemical proteomics reveals a γH2AX-53BP1 interaction in the DNA damage response, Nat. Chem. Biol 11 (2015) 807–814. 10.1038/nchembio.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CCY, Huang H, Landry M-C, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D, 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark, Nature. 499 (2013) 50–54. 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Botuyan MV, Lee J, Ward IM, Kim J-E, Thompson JR, Chen J, Mer G, Structural Basis for the Methylation State-Specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair, Cell. 127 (2006) 1361–1373. 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, Kriwacki RW, Pappu RV, Brangwynne CP, Coexisting Liquid Phases Underlie Nucleolar Subcompartments, Cell. 165 (2016) 1686–1697. 10.1016/j.cell.2016.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].San Filippo J, Sung P, Klein H, Mechanism of Eukaryotic Homologous Recombination, Annu. Rev. Biochem 77 (2008) 229–257. 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- [58].Heyer W-D, Ehmsen KT, Liu J, Regulation of homologous recombination in eukaryotes, Annu. Rev. Genet 44 (2010) 113–139. 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jensen RB, Rothenberg E, Preserving genome integrity in human cells via DNA double-strand break repair, Mol. Biol. Cell 31 (2020) 859–865. 10.1091/mbc.E18-10-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lieber MR, The Mechanism of Human Nonhomologous DNA End Joining, J. Biol. Chem 283 (2008) 1–5. 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- [61].Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, Burma S, Khanna KK, Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks, Nucleic Acids Res. 38 (2010) 1821–1831. 10.1093/nar/gkp1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Myler LR, Gallardo IF, Soniat MM, Deshpande RA, Gonzalez XB, Kim Y, Paull TT, Finkelstein IJ, Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair, Mol. Cell 67 (2017) 891–898.e4. 10.1016/j.molcel.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pawłowska E, Szczepanska J, Blasiak J, DNA2—An Important Player in DNA Damage Response or Just Another DNA Maintenance Protein?, Int. J. Mol. Sci 18 (2017). 10.3390/ijms18071562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC, BLM–DNA2–RPA–MRN and EXO1–BLM–RPA–MRN constitute two DNA end resection machineries for human DNA break repair, Genes Dev. 25 (2011) 350–362. 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Makharashvili N, Paull TT, CtIP: A DNA damage response protein at the intersection of DNA metabolism, DNA Repair. 32 (2015) 75–81. 10.1016/j.dnarep.2015.04.016. [DOI] [PubMed] [Google Scholar]
- [66].Bunting SF, Callén E, Wong N, Chen H-T, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng C-X, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A, 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks, Cell. 141 (2010) 243–254. 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J, 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers, Nat. Struct. Mol. Biol 17 (2010) 688–695. 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Whelan DR, Lee WTC, Yin Y, Ofri DM, Bermudez-Hernandez K, Keegan S, Fenyo D, Rothenberg E, Spatiotemporal dynamics of homologous recombination repair at single collapsed replication forks, Nat. Commun 9 (2018) 3882. 10.1038/s41467-018-06435-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Morozumi Y, Takizawa Y, Takaku M, Kurumizaka H, Human PSF binds to RAD51 and modulates its homologous-pairing and strand-exchange activities, Nucleic Acids Res. 37 (2009) 4296–4307. 10.1093/nar/gkp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, Heyer W-D, Eukaryotic DNA Polymerases in Homologous Recombination, Annu. Rev. Genet 50 (2016) 393–421. 10.1146/annurev-genet-120215-035243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Reid DA, Keegan S, Leo-Macias A, Watanabe G, Strande NT, Chang HH, Oksuz BA, Fenyo D, Lieber MR, Ramsden DA, Rothenberg E, Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair, Proc. Natl. Acad. Sci 112 (2015) E2575–E2584. 10.1073/pnas.1420115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Reid DA, Conlin MP, Yin Y, Chang HH, Watanabe G, Lieber MR, Ramsden DA, Rothenberg E, Bridging of double-stranded breaks by the nonhomologous end-joining ligation complex is modulated by DNA end chemistry, Nucleic Acids Res. 45 (2017) 1872–1878. 10.1093/nar/gkw1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Conlin MP, Reid DA, Small GW, Chang HH, Watanabe G, Lieber MR, Ramsden DA, Rothenberg E, DNA ligase IV guides end-processing choice during nonhomologous end joining, Cell Rep. 20 (2017) 2810–2819. 10.1016/j.celrep.2017.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, Lieber MR, The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis, Nat. Commun 10 (2019) 3588. 10.1038/s41467-019-11507-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney BL, Weinfeld M, Schriemer DC, Lees-Miller SP, Tainer JA, XRCC4 Protein Interactions with XRCC4-like Factor (XLF) Create an Extended Grooved Scaffold for DNA Ligation and Double Strand Break Repair♦, J. Biol. Chem 286 (2011) 32638–32650. 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ahnesorg P, Smith P, Jackson SP, XLF Interacts with the XRCC4-DNA Ligase IV Complex to Promote DNA Nonhomologous End-Joining, Cell. 124 (2006) 301–313. 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- [77].Jaafar L, Li Z, Li S, Dynan WS, SFPQ•NONO and XLF function separately and together to promote DNA double-strand break repair via canonical nonhomologous end joining, Nucleic Acids Res. 45 (2017) 1848–1859. 10.1093/nar/gkw1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Li S, Shu F, Li Z, Jaafar L, Zhao S, Dynan WS, Cell-type specific role of the RNA-binding protein, NONO, in the DNA double-strand break response in the mouse testes, DNA Repair. 51 (2017) 70–78. 10.1016/j.dnarep.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Krietsch J, Caron M-C, Gagné J-P, Ethier C, Vignard J, Vincent M, Rouleau M, Hendzel MJ, Poirier GG, Masson J-Y, PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks, Nucleic Acids Res. 40 (2012) 10287–10301. 10.1093/nar/gks798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhao B, Rothenberg E, Ramsden DA, Lieber MR, The molecular basis and disease relevance of non-homologous DNA end joining, Nat. Rev. Mol. Cell Biol 21 (2020) 765–781. 10.1038/s41580-020-00297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Brouwer I, Sitters G, Candelli A, Heerema SJ, Heller I, Melo De AJ, Zhang H, Normanno D, Modesti M, Peterman EJG, Wuite GJL, Sliding sleeves of XRCC4–XLF bridge DNA and connect fragments of broken DNA, Nature. 535 (2016) 566–569. 10.1038/nature18643. [DOI] [PubMed] [Google Scholar]
- [82].Graham TGW, Walter JC, Loparo JJ, Two-Stage Synapsis of DNA Ends during Non-homologous End Joining, Mol. Cell 61 (2016) 850–858. 10.1016/j.molcel.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lieber MR, The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End Joining Pathway, Annu. Rev. Biochem 79 (2010) 181–211. 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shamanna RA, Lu H, de Freitas JK, Tian J, Croteau DL, Bohr VA, WRN regulates pathway choice between classical and alternative non-homologous end joining, Nat. Commun 7 (2016). 10.1038/ncomms13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Difilippantonio S, Gapud E, Wong N, Huang C-Y, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, Sleckman BP, Nussenzweig A, 53BP1 facilitates long-range DNA end-joining during V(D)J recombination, Nature. 456 (2008) 529–533. 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, de Lange T, 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polα-dependent fill-in, Nature. 560 (2018) 112–116. 10.1038/s41586-018-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Biehs R, Steinlage M, Barton O, Juhász S, Künzel J, Spies J, Shibata A, Jeggo PA, Löbrich M, DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination, Mol. Cell 65 (2017) 671–684.e5. 10.1016/j.molcel.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chakraborty A, Tapryal N, Venkova T, Mitra J, Vasquez V, Sarker AH, Duarte-Silva S, Huai W, Ashizawa T, Ghosh G, Maciel P, Sarkar PS, Hegde ML, Chen X, Hazra TK, Deficiency in classical nonhomologous end-joining–mediated repair of transcribed genes is linked to SCA3 pathogenesis, Proc. Natl. Acad. Sci 117 (2020) 8154–8165. 10.1073/pnas.1917280117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Jain A, Vale RD, RNA phase transitions in repeat expansion disorders, Nature. 546 (2017) 243–247. 10.1038/nature22386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Frottin F, Schueder F, Tiwary S, Gupta R, Körner R, Schlichthaerle T, Cox J, Jungmann R, Hartl FU, Hipp MS, The nucleolus functions as a phase-separated protein quality control compartment, Science. 365 (2019) 342–347. 10.1126/science.aaw9157. [DOI] [PubMed] [Google Scholar]
- [91].Yao R-W, Xu G, Wang Y, Shan L, Luan P-F, Wang Y, Wu M, Yang L-Z, Xing Y-H, Yang L, Chen L-L, Nascent Pre-rRNA Sorting via Phase Separation Drives the Assembly of Dense Fibrillar Components in the Human Nucleolus, Mol. Cell 76 (2019) 767–783.e11. 10.1016/j.molcel.2019.08.014. [DOI] [PubMed] [Google Scholar]
- [92].Mercer TR, Dinger ME, Mattick JS, Long non-coding RNAs: insights into functions, Nat. Rev. Genet 10 (2009) 155–159. 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- [93].Cesare AJ, Reddel RR, Alternative lengthening of telomeres: models, mechanisms and implications, Nat. Rev. Genet 11 (2010) 319–330. 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- [94].Petti E, Buemi V, Zappone A, Schillaci O, Broccia PV, Dinami R, Matteoni S, Benetti R, Schoeftner S, SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability, Nat. Commun 10 (2019) 1001. 10.1038/s41467-019-08863-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dalby AB, Goodrich KJ, Pfingsten JS, Cech TR, RNA recognition by the DNA end-binding Ku heterodimer, RNA. 19 (2013) 841–851. 10.1261/rna.038703.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yoo S, Dynan WS, Characterization of the RNA Binding Properties of Ku Protein, Biochemistry. 37 (1998) 1336–1343. 10.1021/bi972100w. [DOI] [PubMed] [Google Scholar]
- [97].Zhang Y, He Q, Hu Z, Feng Y, Fan L, Tang Z, Yuan J, Shan W, Li C, Hu X, Tanyi JL, Fan Y, Huang Q, Montone K, Dang CV, Zhang L, Long noncoding RNA LINP1 regulates repair of DNA double-strand breaks in triple-negative breast cancer, Nat. Struct. Mol. Biol 23 (2016) 522–530. 10.1038/nsmb.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lin MZ, Marzec KA, Martin JL, Baxter RC, The role of insulin-like growth factor binding protein-3 in the breast cancer cell response to DNA-damaging agents, Oncogene. 33 (2014) 85–96. 10.1038/onc.2012.538. [DOI] [PubMed] [Google Scholar]
- [99].de Silva HC, Lin MZ, Phillips L, Martin JL, Baxter RC, IGFBP-3 interacts with NONO and SFPQ in PARP-dependent DNA damage repair in triple-negative breast cancer, Cell. Mol. Life Sci 76 (2019) 2015–2030. 10.1007/s00018-019-03033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhao K, Wang X, Xue X, Li L, Hu Y, A long noncoding RNA sensitizes genotoxic treatment by attenuating ATM activation and homologous recombination repair in cancers, PLOS Biol. 18 (2020) e3000666. 10.1371/journal.pbio.3000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zimta A-A, Tigu AB, Braicu C, Stefan C, Ionescu C, Berindan-Neagoe I, An Emerging Class of Long Non-coding RNA With Oncogenic Role Arises From the snoRNA Host Genes, Front. Oncol 10 (2020). 10.3389/fonc.2020.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Taiana E, Favasuli V, Ronchetti D, Todoerti K, Pelizzoni F, Manzoni M, Barbieri M, Fabris S, Silvestris I, Gallo Cantafio ME, Platonova N, Zuccalà V, Maltese L, Soncini D, Ruberti S, Cea M, Chiaramonte R, Amodio N, Tassone P, Agnelli L, Neri A, Long non-coding RNA NEAT1 targeting impairs the DNA repair machinery and triggers anti-tumor activity in multiple myeloma, Leukemia. 34 (2020) 234–244. 10.1038/s41375-019-0542-5. [DOI] [PubMed] [Google Scholar]
- [103].Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P, Boeckx B, Wijnhoven PWG, Radaelli E, Vermi W, Leucci E, Lapouge G, Beck B, van den Oord J, Nakagawa S, Hirose T, Sablina AA, Lambrechts D, Aerts S, Blanpain C, Marine J-C, p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity, Nat. Med 22 (2016) 861–868. 10.1038/nm.4135. [DOI] [PubMed] [Google Scholar]
- [104].Ji P, Diederichs S, Wang W, Böing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, Müller-Tidow C, MALAT-1, a novel noncoding RNA, and thymosin β 4 predict metastasis and survival in early-stage non-small cell lung cancer, Oncogene. 22 (2003) 8031–8041. 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- [105].Ren Shancheng, Liu Yawei, Xu Weidong, Sun Yi, Lu Ji, Wang Fubo, Wei Min, Shen Jian, Hou Jianguo, Gao Xu, Xu Chuanliang, Huang Jiaoti, Zhao Yi, Sun Yinghao, Long Noncoding RNA MALAT-1 is a New Potential Therapeutic Target for Castration Resistant Prostate Cancer, J. Urol 190 (2013) 2278–2287. 10.1016/j.juro.2013.07.001. [DOI] [PubMed] [Google Scholar]
- [106].Luo J-H, Ren B, Keryanov S, Tseng GC, Rao UNM, Monga SP, Strom S, Demetris AJ, Nalesnik M, Yu YP, Ranganathan S, Michalopoulos GK, Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas, Hepatology. 44 (2006) 1012–1024. 10.1002/hep.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Guffanti A, Iacono M, Pelucchi P, Kim N, Soldà G, Croft LJ, Taft RJ, Rizzi E, Askarian-Amiri M, Bonnal RJ, Callari M, Mignone F, Pesole G, Bertalot G, Bernardi LR, Albertini A, Lee C, Mattick JS, Zucchi I, De Bellis G, A transcriptional sketch of a primary human breast cancer by 454 deep sequencing, BMC Genomics. 10 (2009) 163. 10.1186/1471-2164-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Ji Q, Zhang L, Liu X, Zhou L, Wang W, Han Z, Sui H, Tang Y, Wang Y, Liu N, Ren J, Hou F, Li Q, Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ/PTBP2 complex, Br. J. Cancer 111 (2014) 736–748. 10.1038/bjc.2014.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zhang C, Wang M, Shi C, Shi F, Pei C, Long non-coding RNA Linc00312 modulates the sensitivity of ovarian cancer to cisplatin via the Bcl-2/Caspase-3 signaling pathway, Biosci. Trends. 12 (2018) 309–316. 10.5582/bst.2018.01052. [DOI] [PubMed] [Google Scholar]
- [110].Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang Y-G, Qi Y, A Role for Small RNAs in DNA Double-Strand Break Repair, Cell. 149 (2012) 101–112. 10.1016/j.cell.2012.03.002. [DOI] [PubMed] [Google Scholar]
- [111].Vítor AC, Sridhara SC, Sabino JC, Afonso AI, Grosso AR, Martin RM, de Almeida SF, Single-molecule imaging of transcription at damaged chromatin, Sci. Adv 5 (2019) eaau1249. 10.1126/sciadv.aau1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Bonath F, Domingo-Prim J, Tarbier M, Friedländer MR, Visa N, Next-generation sequencing reveals two populations of damage-induced small RNAs at endogenous DNA double-strand breaks, Nucleic Acids Res. 46 (2018) 11869–11882. 10.1093/nar/gky1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lu W-T, Hawley BR, Skalka GL, Baldock RA, Smith EM, Bader AS, Malewicz M, Watts FZ, Wilczynska A, Bushell M, Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair, Nat. Commun 9 (2018) 532. 10.1038/s41467-018-02893-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Michelini F, Pitchiaya S, Vitelli V, Sharma S, Gioia U, Pessina F, Cabrini M, Wang Y, Capozzo I, Iannelli F, Matti V, Francia S, Shivashankar GV, Walter NG, d’Adda di Fagagna F, Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks, Nat. Cell Biol 19 (2017) 1400–1411. 10.1038/ncb3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Francia S, Cabrini M, Matti V, Oldani A, d’Adda di Fagagna F, DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors, J. Cell Sci 129 (2016) 1468–1476. 10.1242/jcs.182188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Storici F, Bebenek K, Kunkel TA, Gordenin DA, Resnick MA, RNA-templated DNA repair, Nature. 447 (2007) 338–341. 10.1038/nature05720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F, Transcript-RNA-templated DNA recombination and repair, Nature. 515 (2014) 436–439. 10.1038/nature13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Wahba L, Gore SK, Koshland D, The homologous recombination machinery modulates the formation of RNA–DNA hybrids and associated chromosome instability, ELife. 2 (2013). 10.7554/eLife.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wei L, Nakajima S, Böhm S, Bernstein KA, Shen Z, Tsang M, Levine AS, Lan L, DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination, Proc. Natl. Acad. Sci 112 (2015) E3495–E3504. 10.1073/pnas.1507105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Ouyang J, Yadav T, Zhang J-M, Yang H, Rheinbay E, Guo H, Haber DA, Lan L, Zou L, RNA transcripts stimulate homologous recombination by forming DR-loops, Nature. (2021) 1–6. 10.1038/s41586-021-03538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Aleksandrov R, Dotchev A, Poser I, Krastev D, Georgiev G, Panova G, Babukov Y, Danovski G, Dyankova T, Hubatsch L, Ivanova A, Atemin A, Nedelcheva-Veleva MN, Hasse S, Sarov M, Buchholz F, Hyman AA, Grill SW, Stoynov SS, Protein Dynamics in Complex DNA Lesions, Mol. Cell 69 (2018) 1046–1061.e5. 10.1016/j.molcel.2018.02.016. [DOI] [PubMed] [Google Scholar]
- [122].Alemasova EE, Lavrik OI, Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins, Nucleic Acids Res. 47 (2019) 3811–3827. 10.1093/nar/gkz120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kamaletdinova T, Fanaei-Kahrani Z, Wang Z-Q, The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers, Cells. 8 (2019). 10.3390/cells8121625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Talhaoui I, Lebedeva NA, Zarkovic G, Saint-Pierre C, Kutuzov MM, Sukhanova MV, Matkarimov BT, Gasparutto D, Saparbaev MK, Lavrik OI, Ishchenko AA, Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro, Nucleic Acids Res. 44 (2016) 9279–9295. 10.1093/nar/gkw675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grøfte M, Rask M-BD, Streicher W, Jungmichel S, Nielsen ML, Lukas J, Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose), Nat. Commun 6 (2015) 8088. 10.1038/ncomms9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kovar H, Dr. Jekyll and Mr. Hyde: The Two Faces of the FUS/EWS/TAF15 Protein Family, Sarcoma. (2010). 10.1155/2011/837474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Wang W-Y, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IRA, Huang EJ, Tsai L-H, Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons, Nat. Neurosci 16 (2013) 1383–1391. 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Guerrero EN, Wang H, Mitra J, Hegde PM, Stowell SE, Liachko NF, Kraemer BC, Garruto RM, Rao KS, Hegde ML, TDP-43/FUS in motor neuron disease: Complexity and challenges, Prog. Neurobiol 145–146 (2016) 78–97. 10.1016/j.pneurobio.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Boehning M, Dugast-Darzacq C, Rankovic M, Hansen AS, Yu T, Marie-Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P, Darzacq X, Zweckstetter M, RNA polymerase II clustering through carboxy-terminal domain phase separation, Nat. Struct. Mol. Biol 25 (2018) 833–840. 10.1038/s41594-018-0112-y. [DOI] [PubMed] [Google Scholar]
- [130].Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q, Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II, Nature. 558 (2018) 318–323. 10.1038/s41586-018-0174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL, Phosphorylation-Regulated Binding of RNA Polymerase II to Fibrous Polymers of Low-Complexity Domains, Cell. 155 (2013) 1049–1060. 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Krüger A, Bürkle A, Hauser K, Mangerich A, Real-time monitoring of PARP1-dependent PARylation by ATR-FTIR spectroscopy, Nat. Commun 11 (2020) 2174. 10.1038/s41467-020-15858-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Arnould C, Legube G, The Secret Life of Chromosome Loops upon DNA Double-Strand Break, J. Mol. Biol 432 (2020) 724–736. 10.1016/j.jmb.2019.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Natale F, Rapp A, Yu W, Maiser A, Harz H, Scholl A, Grulich S, Anton T, Hörl D, Chen W, Durante M, Taucher-Scholz G, Leonhardt H, Cardoso MC, Identification of the elementary structural units of the DNA damage response, Nat. Commun 8 (2017) 15760. 10.1038/ncomms15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, Bazett-Jones DP, Nussenzweig A, Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks, J. Cell Biol 172 (2006) 823–834. 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Dellaire G, Kepkay R, Bazett-Jones DP, High resolution imaging of changes in the structure and spatial organization of chromatin, γ-H2A.X and the MRN complex within etoposide-induced DNA repair foci, Cell Cycle. 8 (2009) 3750–3769. 10.4161/cc.8.22.10065. [DOI] [PubMed] [Google Scholar]
- [137].Levone BR, Lenzken SC, Antonaci M, Maiser A, Rapp A, Conte F, Reber S, Ronchi AE, Mühlemann O, Leonhardt H, Cardoso MC, Ruepp M-D, Barabino SML, FUS-dependent liquid-liquid phase separation is an early event in double-strand break repair, BioRxiv. (2020) 798884. 10.1101/798884. [DOI] [Google Scholar]
- [138].Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP, MDC1 Directly Binds Phosphorylated Histone H2AX to Regulate Cellular Responses to DNA Double-Strand Breaks, Cell. 123 (2005) 1213–1226. 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- [139].Wilson MD, Durocher D, Reading chromatin signatures after DNA double-strand breaks, Philos. Trans. R. Soc. B Biol. Sci 372 (2017) 20160280. 10.1098/rstb.2016.0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Pryde F, Khalili S, Robertson K, Selfridge J, Ritchie A-M, Melton DW, Jullien D, Adachi Y, 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin, J. Cell Sci 118 (2005) 2043–2055. 10.1242/jcs.02336. [DOI] [PubMed] [Google Scholar]
- [141].Lemaître C, Grabarz A, Tsouroula K, Andronov L, Furst A, Pankotai T, Heyer V, Rogier M, Attwood KM, Kessler P, Dellaire G, Klaholz B, Reina-San-Martin B, Soutoglou E, Nuclear position dictates DNA repair pathway choice, Genes Dev. 28 (2014) 2450–2463. 10.1101/gad.248369.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, Legube G, Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks, Nat. Struct. Mol. Biol 21 (2014) 366–374. 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Aymard F, Aguirrebengoa M, Guillou E, Javierre BM, Bugler B, Arnould C, Rocher V, Iacovoni JS, Biernacka A, Skrzypczak M, Ginalski K, Rowicka M, Fraser P, Legube G, Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes, Nat. Struct. Mol. Biol 24 (2017) 353–361. 10.1038/nsmb.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Dion V, Gasser SM, Chromatin Movement in the Maintenance of Genome Stability, Cell. 152 (2013) 1355–1364. 10.1016/j.cell.2013.02.010. [DOI] [PubMed] [Google Scholar]
- [145].Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T, 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair, Cell. 163 (2015) 880–893. 10.1016/j.cell.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T, 53BP1 Regulates DSB Repair Using Rif1 to Control 5′ End Resection, Science. 339 (2013) 700–704. 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Harrision D, Gravells P, Thompson R, Bryant HE, Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) – Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy, Front. Mol. Biosci 7 (2020). 10.3389/fmolb.2020.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Owen I, Shewmaker F, The Role of Post-Translational Modifications in the Phase Transitions of Intrinsically Disordered Proteins, Int. J. Mol. Sci 20 (2019). 10.3390/ijms20215501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Monahan Z, Ryan VH, Janke AM, Burke KA, Rhoads SN, Zerze GH, O’Meally R, Dignon GL, Conicella AE, Zheng W, Best RB, Cole RN, Mittal J, Shewmaker F, Fawzi NL, Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity, EMBO J. 36 (2017) 2951–2967. 10.15252/embj.201696394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Chowdhury D, Keogh M-C, Ishii H, Peterson CL, Buratowski S, Lieberman J, γ-H2AX Dephosphorylation by Protein Phosphatase 2A Facilitates DNA Double-Strand Break Repair, Mol. Cell 20 (2005) 801–809. 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- [151].Moon S-H, Nguyen T-A, Darlington Y, Lu X, Donehower LA, Dephosphorylation of γ-H2AX by WIP1: An important homeostatic regulatory event in DNA repair and cell cycle control, Cell Cycle. 9 (2010) 2092–2096. 10.4161/cc.9.11.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Meerang M, Ritz D, Paliwal S, Garajova Z, Bosshard M, Mailand N, Janscak P, Hübscher U, Meyer H, Ramadan K, The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks, Nat. Cell Biol 13 (2011) 1376–1382. 10.1038/ncb2367. [DOI] [PubMed] [Google Scholar]
- [153].Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon M-K, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou H-J, Wustrow D, Rolfe M, Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis, Cancer Cell. 28 (2015) 653–665. 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Moigne RL, Aftab BT, Djakovic S, Dhimolea E, Valle E, Murnane M, King EM, Soriano F, Menon M-K, Wu ZY, Wong ST, Lee GJ, Yao B, Wiita AP, Lam C, Rice J, Wang J, Chesi M, Bergsagel PL, Kraus M, Driessen C, Soly SKV, Yakes FM, Wustrow D, Shawver L, Zhou H-J, Martin TG, Wolf JL, Mitsiades CS, Anderson DJ, Rolfe M, The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma, Mol. Cancer Ther 16 (2017) 2375–2386. 10.1158/1535-7163.MCT-17-0233. [DOI] [PubMed] [Google Scholar]