

Abstract
Angiogenesis is essential for the sustained growth of solid tumors. Hypoxia-inducible factor 1 (HIF-1) is a master regulator of angiogenesis and constitutive activation of HIF-1 is frequently observed in human cancers. Therefore, understanding the mechanisms governing the activation of HIF-1 is critical for successful therapeutic targeting of tumor angiogenesis. Herein, we establish a new regulatory mechanism responsible for the constitutive activation of HIF-1α in cancer, irrespective of oxygen tension. PIM1 kinase directly phosphorylates HIF-1α at threonine 455, a previously uncharacterized site within its oxygen-dependent degradation domain. This phosphorylation event disrupts the ability of prolyl hydroxylases to bind and hydroxylate HIF-1α, interrupting its canonical degradation pathway and promoting constitutive transcription of HIF-1 target genes. Moreover, phosphorylation of the analogous site in HIF-2α (S435) stabilizes the protein through the same mechanism, indicating post-translational modification within the oxygen-dependent degradation domain as a mechanism of regulating the HIF-α subunits. In vitro and in vivo models demonstrate that expression of PIM1 is sufficient to stabilize HIF-1α and HIF-2α in normoxia and stimulate angiogenesis in a HIF-1-dependent manner. CRISPR mutants of HIF-1α (Thr455D) promoted increased tumor growth, proliferation and angiogenesis. Moreover, HIF-1α-T455D xenograft tumors were refractory to the anti-angiogenic and cytotoxic effects of PIM inhibitors. These data identify a new signaling axis responsible for hypoxia-independent activation of HIF-1 and expand our understanding of the tumorigenic role of PIM1 in solid tumors.
Keywords: PIM kinase, HIF-1, hypoxia, angiogenesis
Introduction:
Angiogenesis, or the branching of blood vessels, is a rate-limiting step in the development of solid tumors [1]. Hypoxia-inducible factor 1 (HIF-1) is a basic helix-loop-helix-PAS domain transcription factor that is a critical mediator of the cellular response to oxygen deprivation and a key driver of tumor angiogenesis [2]. HIF-1 is a heterodimer that consists of HIF-1β, a constitutively expressed subunit, HIF-1α and HIF-2α, subunits whose expression is tightly regulated in an oxygen-dependent manner. In the presence of oxygen, cytoplasmic HIF-α subunits are rapidly hydroxylated on prolines 402 (Pro402) and 564 (Pro564), located within its oxygen-dependent degradation domain (ODDD), by prolyl hydroxylase-domain proteins (PHDs) 1–3 [3, 4]. Hydroxylation causes the von Hippel-Lindau (VHL) tumor suppressor protein to recognize HIF-α and recruit a ubiquitin-protein ligase complex that leads to the ubiquitination and rapid degradation of HIF-α by the 26S proteasome [5]. In low oxygen, PHDs become less active and hydroxylation of HIF-α is reduced, allowing it to accumulate in the cell and translocate to the nucleus, where it binds to hypoxia-response elements (HREs) to promote the transcription of target genes [6]. Both HIF-1α and HIF-2α subunits are widely expressed in human cancers, and HIF-1α and HIF-2α have been shown to promote tumor progression. It was proposed that the tumorigenic effects of HIF-1α and HIF-2α were largely through overlapping functions, more recent studies indicate that these subunits have differential effects that can promote distinct outcomes through independent regulation of distinct target genes, as well as through direct and indirect interactions with complexes containing important oncoproteins and tumor suppressors [7].
Constitutive expression of HIF-1α and HIF-2α is common in human cancers, regardless of oxygen tension. Stabilization of HIF-α subunits in normoxia has been attributed to genetic alterations, such as loss of VHL, as well as transcriptional upregulation due to the activation of oncogenic signaling pathways [8–10]. Post-translational modification also plays a critical role in controlling HIF-1α expression and function. Phosphorylation of HIF-1α and HIF-2α by various kinases has been described to positively and negatively impact protein stability [11–16]. Regardless of the mechanism, stabilization of HIF-1α HIF-2α in normoxia upregulates genes that initiate and sustain angiogenesis during tumor growth [17]. Hence, the identification of novel, oxygen-independent mechanisms that regulate HIF-1α expression is valuable in the effort to understand tumor progression and effectively target angiogenesis as a therapeutic strategy.
The Proviral integration site for Moloney murine leukemia virus (PIM) kinases are a family of serine-threonine kinases that are known to promote tumorigenesis by impacting cell cycle progression, survival, and proliferation [18, 19]. PIM kinases are elevated in a host of other solid tumors, including colon, breast, and lung cancer, and their overexpression is associated with higher staging, increased metastasis, and diminished overall survival [20–27]. As a result, several small-molecule PIM kinase inhibitors are actively being tested as anti-cancer agents in clinical trials. We recently reported that PIM inhibitors display synergistic anti-tumor effects in combination with anti-angiogenic agents in prostate and colon cancers that were characterized by a dramatic loss of vasculature [19, 28]. Here, we establish PIM1 as an important factor responsible for driving tumor angiogenesis. Mechanistically, we show that PIM1 promotes angiogenesis through a novel signaling axis directly linking PIM1 to HIF-1 via a previously uncharacterized direct phosphorylation event that disrupts the canonical HIF-1α and HIF-2α degradation pathway. Our results suggest that the ability of PIM1 to induce angiogenesis and tumor growth is dependent on the stabilization of HIF-1 and that the anti-tumor effects of PIM inhibitors are largely due to their anti-angiogenic properties.
Results
PIM1 expression is correlated with angiogenesis in human cancers:
We recently discovered that overexpression of PIM1 kinase was sufficient to sustain vasculature during treatment with anti-vascular endothelial growth factor (VEGF) therapy [28]. Therefore, we sought to determine whether PIM1 expression was correlated with tumor angiogenesis. To this end, we performed immunohistochemistry (IHC) to quantify PIM1 and CD31 (an endothelial cell marker) levels in three prostate cancer tissue microarrays (TMAs) (#2 n = 36, #5 n = 44, and #13 n = 29). These TMAs were obtained from diagnostic cores of radical prostatectomies of patients prior to treatment at the University of Arizona, and samples ranged in disease severity (Gleason score 6–9). We observed a statistically significant correlation between PIM1 expression and microvessel density (Fig. 1A and B), indicating that PIM1 expression is significantly correlated with vascularization in human tumor samples. Moreover, publicly available data from The Cancer Genome Atlas database showed a statistically significant correlation between PIM1 and PECAM1 (an endotheilial cell marker) in human prostate, colon, and lung cancer tumors (Fig. S1A). We next sought to determine whether PIM1 is sufficient to promote angiogenesis in vivo using preclinical imaging modalities. To this end, 1 × 106 PC3 prostate cancer cells were stably transfected with PIM1 or a vector control (VEC) and implanted subcutaneously into the flanks of male NSG mice. Verifying previous results, PIM1-overexpressing tumors grew significantly faster than control tumors (Fig. 1C, [28]). To assess angiogenesis, a paramagnetic contrast agent (CA) was injected intravenously, and magnetic resonance imaging (MRI) was performed to visualize uptake of the CA in the tumor and reference tissue and generate a time-concentration curve. DCE-MRI data from size-matched PC3/VEC and PC3/PIM1 tumors were analyzed using the Linear Reference Region Model [29]. The resulting time-concentration curve revealed a dramatic increase in the uptake of the CA in PIM1-overexpressing tumors compared to control tumors, indicating that PIM1 expression significantly increases perfusion (Fig. 1D), and the average Rktrans was significantly greater in PIM1-overexpressing tumors than in control tumors (Fig. 1E). Together, these analyses indicate that PIM1-overexpressing tumors have more blood flow and a greater number of mature vessels than control tumors. This correlation was confirmed with endpoint staining of CD31, which demonstrated that microvessel density was significantly higher in PIM1-overexpressing tumors than in control tumors (Fig. S1B and C). Taken together, these data demonstrate that PIM1 expression is sufficient to enhance tumor angiogenesis.
Figure 1. PIM1 correlates with angiogenesis in human cancer samples.

A) Representative PIM1 and CD31 staining of three human prostate cancer TMAs, n=109. B) Scoring of PIM1 and CD31 were graphed to determine the association between PIM1 and vasculature by Pearson correlation in each of the indicated TMAs. C) Mice (n=4/group) were injected with PC3-VEC or PC3-PIM1 cells and tumor volume was measured via caliper over time. D) Representative DCE-MRI trace from size-matched PC3-PIM1 and PC3-VEC tumors and E) average vascular perfusion (Ktrans), n=4/group. Scale bar = 50 μm. *p < 0.05, error bars = SEM.
PIM1 induces angiogenesis in a HIF-1-dependent manner:
To determine whether the pro-angiogenic effect of PIM1 is dependent on HIF-1, HIF-1/2α were knocked down using siRNA in PC3 cells stably expressing a doxycycline (Dox)-inducible lentiviral vector encoding PIM1 (Dox-PIM1) [25], and in vitro angiogenesis assays were performed. Forty-eight hours after knockdown, Dox-PIM1 cells were treated with Dox, and cell lysates and conditioned media (CM) were collected after 24 h. Then, human umbilical vein endothelial cells (HUVECs) were suspended in CM from each experimental condition and plated on reduced growth factor basement membrane, and tube formation was assessed over time. Immunoblotting verified that PIM1 stabilized HIF-1/2α, and siRNA effectively reduced HIF-1/2α levels (Fig. 2A). CM from Dox-PIM1 cells substantially enhanced tube formation compared to CM from Dox-VEC cells (Fig. 2B). Strikingly, the pro-angiogenic effect of PIM1 expression was abolished in cells lacking HIF-1/2α. PIM1 overexpression significantly increased the average tube length and number of branch points, and HIF-1/2α knockdown restored tube formation to basal levels (Fig. 2C and D). Conditioned media from PIM1-overexpressing cells contained nearly 5 times more VEGF than CM from control cells, and knockdown of HIF-1/2α restored VEGF levels to basal levels (Fig. 2E). To show that the pro-angiogenic effect of PIM1 requires HIF-1α in vivo, 5 × 106 RKO cells were injected subcutaneously into the flanks of SCID mice (n=3/group). PIM1-overexpressing tumors grew significantly faster than control tumors, whereas knockdown of HIF-1α abolished the growth advantage conferred by PIM1 overexpression (Fig. 2F). To monitor angiogenesis, mice from each group were injected with Angiosense 750EX, a fluorescent probe that remains localized in the vasculature to allow for in vivo imaging of angiogenesis. To calculate the relative amount of functional vasculature in each cohort, Angiosense signal was normalized to differences in tumor volume to obtain a vascular index for each group. RKO tumors overexpressing PIM1 were significantly more vascular than control tumors, whereas no increase in angiogenesis was observed with PIM1 overexpression in tumors lacking HIF-1α (Fig. 2G). CD31 staining of tumors from each group confirmed that the pro-angiogenic effect of PIM1 was lost in tumors with knockdown of HIF-1α (Fig. 2H and I). Similarly, the expression of HIF-1 target genes (VEGF and HK2) was increased by PIM1 expression in vivo, but not in HIF-1α-knockdown tumor tissue (Fig. S2F). The growth advantage associated with PIM1 overexpression was attributable to increased survival, not proliferation, as PIM1 significantly reduced cell death (CC3 staining) but did not change Ki67 staining (Fig. 2H). Interstingly, tumors lacking HIF-1α had significantly more cell death compared to control tumors, regardless of PIM1 overexpression (Fig. 2I). Taken together, these data indicate that PIM1 promotes angiogenesis in a HIF-1-dependent manner and suggests that the pro-tumorigenic effect of PIM1 can largely be attributed to its ability to promote angiogenesis and prevent cell death.
Figure 2. PIM1 induces angiogenesis in vivo and in vitro.

A) Dox-PIM1 PC3 cells were transfected with siHIF-1/2α prior to treatment with dox for 24 h, and lysates were collected for immunoblotting and conditioned media (CM) was harvested for in vitro angiogenesis assay. B) Representative images of tube formation at 1 and 6 hours after plating HUVEC cells in CM. C) Quantification of mean tube length and D) total branch points as measured with Image J Angiogenesis Analyzer plug-in. E) VEGF-A levels in CM from the indicated conditions were measured by ELISA. F) Twelve mice (n=3/group) were injected with the indicated RKO cell lines, and tumor volume was measured over time. G) Mice were injected with 2 nmol of Angiosense 750EX 24 hours prior to imaging for fluorescence intensity. The vascular index was calculated by normalizing the bioluminescence signal to the tumor volume. H) Tumors derived from each cell line were harvested and immunostained with CD31, PIM1, HIF-1α, and CC3. I) Quantification of IHC. Scale bar = 50 μm. *p < 0.05, n.s. = not significant, error bars = SEM.
PIM1 promotes pro-angiogenic gene expression through HIF-1:
Because the pro-angiogenic effect of PIM1 is dependent on HIF-1, we hypothesized that elevated PIM1 expression is sufficient to activate HIF-1 in the absence of hypoxia. Immunoblotting was used to evaluate HIF-1/2α protein levels in control and PIM1-overexpressing colon (RKO), prostate (PC3), and lung (A549) cell lines. Strikingly, PIM1 expression was sufficient to increase HIF-1α and HIF-2α in all cell lines tested (Fig. 3A–C). Importantly, treatment with chemically distinct pan-PIM kinase inhibitors (PIM447 and AZD1208) blocked the ability of PIM1 to increase HIF-1/2α protein levels (Fig. 3A and C). To ensure that the levels of HIF-1/2α observed after PIM1 induction were sufficient to activate HIF-dependent transcription, Dox-VEC or Dox-PIM1 cells were co-transfected with Renilla-luciferase and a previously described HIF-1 reporter that drives luciferase expression (HRE-Luc) [30], treated for 24 h with Dox to induce PIM1, and then treated with DMSO or AZD1208 for 4 h. PIM1 expression increased HIF-1 activity by approximately 2-fold in normoxia, and this effect was reversed by treatment with AZD1208 (Fig. 3D). To assess the effect of PIM1 on HIF-1 target genes, we used a semi-high-throughput qPCR assay to measure a panel of 84 hypoxia-inducible genes (Qiagen RT Profiler). PC3 Dox-PIM1 cells were cultured in normoxic conditions with or without 20 ng/mL Dox for 24 h to induce PIM1 and treated with or without AZD1208 for 8 h, at which point mRNA was collected for subsequent gene expression analysis. PIM1-overexpression altered the transcript levels of several classes of hypoxia-responsive genes (Fig. 3E). To identify genes whose induction was specific to PIM1, we focused on genes that were upregulated at least 3-fold by PIM1 expression in normoxia and significantly reduced by treatment with AZD1208. Of the eleven genes that fit these criteria, seven are known to promote angiogenesis, and all are established targets of HIF-1 (Fig. 3F). We validated that PIM1 increased the expression of several well-known HIF-1 target genes (VEGF-A, ANGPTL4, and HK2) by qRT-PCR, and treatment with PIM447 restored the expression of each to basal levels (Fig. 3G). To compare the amplitudes of HIF-1-mediated gene expression induced by hypoxia and PIM1 overexpression, PC3-LN4 and HCT116 cell were transfected with vector or PIM1 or cultured in 1.0% O2 for 8 h, and the expression of HIF-1 target genes was assessed by qPCR. In both cell lines, hypoxia increased HIF-1α protein levels to a greater extent than PIM1 overexpression, but the induction of HIF-1 target genes was similar, indicating that overexpression of PIM1 in normoxia is sufficient for full activation of HIF-1-mediated transcription (Fig. S3A and B). To confirm that PIM1 alters gene expression in a HIF-1-dependent manner, we assessed HIF-1 target genes in the previously described RKO cell line with stable knockdown of HIF-1α. PIM1 overexpression significantly increased the transcript level of all three genes compared to that of control cells, whereas no increase was observed in RKO cells lacking HIF-1α (Fig. 3H). Thus, PIM1 expression is sufficient to stabilize HIF-1/2α in normoxic conditions and increase the transcription of HIF-1 target genes.
Figure 3. PIM1 is sufficient to stabilize HIF-1α and activate HIF-1 in normoxia.

A) RKO colon cancer cells ± PIM1 were treated with DMSO or PIM447 (1 μM) for 6 h. B) A549 and H460 lung cancer cells were stably infected with lentiviral constructs expressing Vector (VEC) or PIM1. C) Dox-VEC or Dox-PIM1 PC3 cells were treated with Dox for 24 h prior to DMSO or AZD1208 (3 μM) for 6 h. D) PC3 Dox-PIM1 cells expressing HRE-Luc were treated with Dox for 24 h prior to DMSO or AZD1208 for 6 h, and bioluminescence was measured. E and F) PC3 Dox-PIM1 cells were treated with Dox for 24 h prior to DMSO or AZD1208 for 6 h and RNA was harvested to measure the expression of hypoxia-inducible genes. F) HIF-1 target genes upregulated 3-fold by PIM1 and reduced by AZD1208. G) PC3 Dox-PIM1 cells were treated with Dox for 24 h prior to DMSO or AZD1208 for 6 h and RNA was harvested to measure gene expression by qRT-PCR. H) RNA was harvested from the indicated cell lines and gene expression was measured by qRT-PCR. *p < 0.05, n.s. = not significant, error bars = SEM.
PIM1 phosphorylates HIF-1α at threonine 455:
Because PIM1 is a serine-threonine kinase, we hypothesized that PIM1 may directly phosphorylate HIF-1α to alter its protein stability. To test whether these proteins interact in cells, HA-PIM1 was overexpressed in 293T cells and treated for 6 h with MG132 in normoxia or hypoxia to stabilize HIF-1α prior to harvest. Immunoprecipitated PIM1 pulled down endogenous HIF-1α in both normoxia and hypoxia, demonstrating that these proteins complex in cells (Fig. 4A). Next, in vitro kinase assays using recombinant PIM1 and HIF-1α revealed that PIM1 was able to phosphorylate HIF-1α (Fig. 4B). To identify PIM-mediated phosphorylation sites, mass spectrometry analysis was performed to identify post-translational modifications on HIF-1α. PIM1 phosphorylated HIF-1α at two sites in vitro. One site, Ser643, has been previously described as an ERK target site that enhances the nuclear localization of HIF-1α but does not alter its protein stability [31]. The second site, Thr455, is a previously uncharacterized site located within the ODDD of HIF-1α between Pro402 and Pro564, which are hydroxylated by PHDs as a signal initiating the proteasomal degradation of HIF-1α (Fig. 4C). Thr455 is evolutionarily conserved among mammals, suggesting its importance as a regulatory site (Fig. 4D). Based on its localization within the ODDD, we chose to further investigate the effect of T455 phosphorylation on HIF-1α stability. To verify that this site is phosphorylated in cells, PIM1 and HA-HIF-1α were co-transfected into 293T cells, HIF-1α was immunoprecipitated, and mass spectrometry was used to detect phosphorylation. HIF-1α was robustly phosphorylated at T455 in cells overexpressing PIM1 (Fig. S4A). Next, we generated a phospho-specific antibody against HIF-1α Thr455. To verify the specificity of this antibody, we used site-directed mutagenesis to create a T455A mutant of HIF-1α. Wild-type or T455A HA-HIF-1α were immunoprecipitated from cells and incubated with recombinant PIM1 in the same conditions used for in vitro kinase assays. Wild-type HIF-1α displayed robust phosphorylation at Thr455 by PIM1, which was blocked by AZD1208, whereas the T455A mutant was not recognized by the phospho-antibody (Fig. 4E). Additionally, staining of tumor sections showed increased phosphorylation of HIF-1α (T455) in tumor tissue over expressing PIM1 compared to control, and no staining was observed in HIF-1α knockdown tumors (Fig. S4B and C). These results confirm that PIM1 directly phosphorylates HIF-1α at T455 and our antibody recognizes phosphorylation specific to this site. To ensure that Thr455 phosphorylation correlated with changes in endogenous PIM1 expression, C42-B and PC3-LN4 cell lines were transfected with PIM1 siRNA prior to exposure to hypoxia. As expected, hypoxia induced PIM1, total HIF-1α, and phosphorylation of T455. Notably, silencing of PIM1 significantly reduced the accumulation of total HIF-1α in hypoxia in C42-B (and to a lesser extent in PC3-LN4) and nearly abolished T455 phosphorylation in both cell lines in normoxia and hypoxia, demonstrating that physiological levels of PIM1 correlate with T455 phosphorylation (Fig 4F). Taken together, these studies establish that PIM1 directly phosphorylates HIF-1α at Thr455.
Figure 4. PIM1 phosphorylates HIF-1α at Thr455.
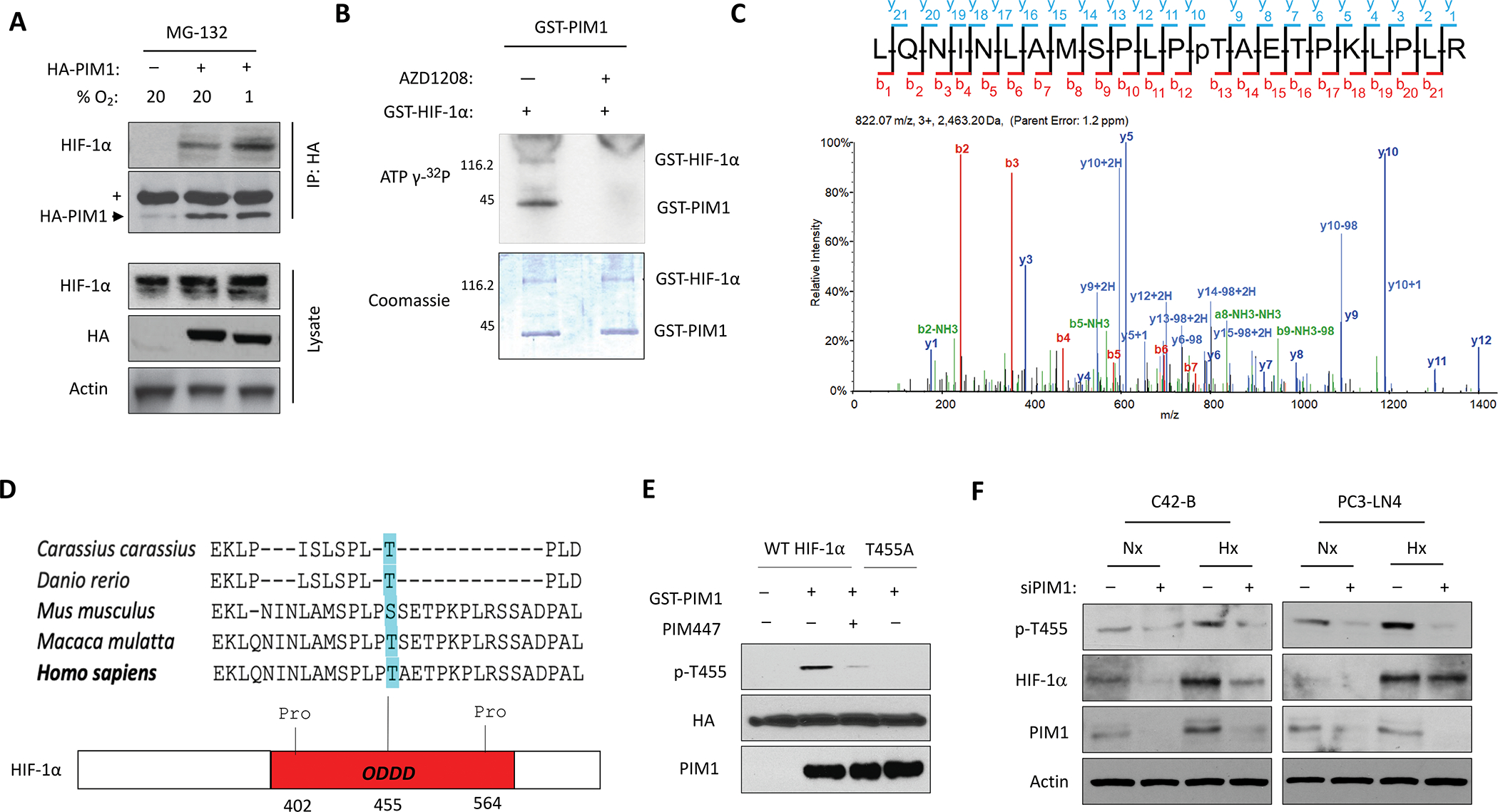
A) 293T cells were transfected with HA-PIM1 and treated with MG-132 in normoxia or hypoxia for 6 h. HA-PIM1 was immunoprecipitated and interaction with endogenous HIF-1α was assessed by western blotting. (+ = Heavy chain IgG). B) Images of Coomassie and autoradiography of in vitro kinase assays using recombinant PIM1 and HIF-1α. C) Spectra from mass spectrometry analysis of HIF-1α from an in vitro kinase assay showing phosphorylation of HIF-1α at Thr455 by PIM1. D) Sequence alignment across species and schematic of HIF-1α ODDD (Thr455 highlighted in blue) E) Immunoblot of in vitro kinase assay combining recombinant PIM1 with immunoprecipitated HIF-1α constructs. F) C42-B and PC3-LN4 cells were transfected with scrambled or PIM1 siRNA and cultured in normoxia (Nx) or hypoxia (Hx) for 6 hours prior to harvest.
PIM-mediated phosphorylation of HIF-1α at Thr455 increases its protein stability:
Next, we characterized the effect of T455 phosphorylation on HIF-1α protein levels. Parental and RKO cells stably expressing PIM1 were cultured in hypoxia (1% O2) for 4 hours to stabilize HIF-1α and then returned to normoxia (20% O2), and lysates were collected over a 30-min time course. The half-life of HIF-1α was significantly longer in PIM1-overexpressing cells than in parental cells (30.1 ± 1.2 min vs. 9.8 ± 0.5 min) (Fig. 5A). To directly assess the effect of Thr455 phosphorylation on HIF-1α protein stability, we used site-directed mutagenesis to generate HIF-1α T455D (phosphomimetic) and T455A (phospho-null) constructs. Following transfection of WT, T455D, or T455A HIF-1α, HEK293T cells were treated with cycloheximide and lysates were collected over time to determine the rate of protein degradation. The half-life of the phospho-null mutant (T455A) was significantly shorter than that of WT HIF-1α (1.4 ± 0.2 h vs. 2.1 ± 0.2 h), whereas the half-life of the phospho-mimetic (T455D) was significantly longer, showing little degradation over the 4 h time course (Fig. 5B). Next, we tested whether PIM1 decreased HIF-1α ubiquitination. HEK293T cells stably expressing PIM1 or a vector control were transfected with HA-HIF-1α and treated with DMSO or PIM447 for 30 min, followed by MG-132 treatment for 4 h to allow for the accumulation of ubiquitinated HIF-1α. HIF-1α was immunoprecipitated and ubiquitin was detected by immunoblotting. PIM1 expression decreased the amount of ubiquitin bound to HIF-1α by approximately 3-fold, whereas PIM inhibition significantly increased the amount of ubiquitination (Fig. 5C). To assess the effect of Thr455 phosphorylation on ubiquitination, 293T cells expressing VEC or PIM1 were transfected with HA-HIF-1α WT, T455D, or T455A and ubiquitination assays were performed. Overexpression of PIM1 significantly decreased WT HIF-1α ubiquitination compared to VEC, whereas PIM1-overexpression did not alter the ubiquitination of T455D or T455A HA-HIF-1α (Fig. 5D).
Figure 5. Phosphorylation of HIF-1α at T455 disrupts PHD2 binding and increases HIF-1α stability.

A) RKO ± PIM1 cells were incubated in hypoxia (1.0% O2) for 1 h then lysed at different time points after restoring normal oxygen (20% O2). B) 293T cells were transfected with HA-HIF-1α, T455A or T455D and incubated in hypoxia for 4 h prior to treatment with cycloheximide (CHX, 10 μM). Densitometry was used to determine the rate of protein decay. C) 293T ± PIM1 cells were transfected with HA-HIF-1α and treated with MG-132 (10 μM) and DMSO or AZD1208 (3 μM) for 4 h. HIF-1α constructs were immunoprecipitated and ubiquitination was measured by immunoblotting and quantified by densitometry. D) 293T cells ± PIM1 were transfected with HA- HIF-1α, T455D or T455A and treated with MG-132 for 4 h. HIF-1α constructs were immunoprecipitated and ubiquitination was measured by immunoblotting and quantified by densitometry. E) SW620 and PC3 cells were transfected with HA-PIM1 and lysates were collected. Relative HIF-OH (Pro564) is graphed. F) HA-HIF-1α WT, T455D or T455A. HA-HIF-1α constructs were immunoprecipitated and blotted for HIF-OH (Pro564). The ratio of hydroxylated to total HIF-1α is graphed. G) 293T ± PIM1 cells were transfected with HA-HIF-1α and treated with DMSO or PIM447 (3 μM) for 4 h. HA-HIF1α constructs were immunoprecipitated and PHD2 was probed by western blotting; relative abundance was calculated by densitometry. H) HA-HIF1α constructs were immunoprecipitated and PHD2 was probed by western blotting; relative abundance quantified below. *p < 0.05, n.s. = not significant, error bars = SEM.
To assess whether PIM alters HIF-1α hydroxylation at Pro564, SW620 and PC3 cells were treated for 4 h with MG132 to allow for the accumulation of HIF-1α and hydroxylation was measured by western blotting. Despite similar levels of total HIF-1α, hydroxylation of HIF-1α at Pro564 was significantly reduced by PIM1-overexpression (Fig. 5E). In a parallel assay, HIF-1α T455D showed no visible hydroxylation, whereas hydroxylation of the T455A mutant was significantly more than WT HIF-1α (Fig. 5F). Because Thr455 is located within the ODDD, we hypothesized that phosphorylation at this site may disrupt PHD binding to HIF-1α. To test this, HA-HIF-1α was transfected into 293T cells stably expressing PIM1 or vector control and ubiquitination assays were performed. Significantly less PHD2 was bound to HIF-1α in cells overexpressing PIM1 compared to VEC cells (Fig. 5G). Similarly, co-immunoprecipitation assays using HA-HIF-1α WT, T455D, or T455A revealed significantly more PHD2 was bound to T455A and significantly less was bound to T455D compared to HIF-1α WT (Fig. 5H). These data indicate that PIM1-mediated phosphorylation of HIF-1α at Thr455 increases protein stability by blocking PHD binding, hydroxylation, and subsequent proteasomal degradation of HIF-1α. Because we observed that PIM1 expression also increased HIF-2α levels, we asked whether an analogous site exists in HIF-2α that could disrupt proteasomal degradation. Alignment of the amino acid sequences of HIF-1α and HIF-2α showed that Ser435 of HIF-2α aligns with Thr455 of HIF-1α (Fig 6A). To test whether this site alters HIF-2α protein degradation, we generated a S435D mutant and compared its stability to that of wild-type HIF-2α using cycloheximide chase. The half-life of the phospho-mimetic mutant (T435D) was significantly longer than that of WT HIF-1α (5.1 ± 0.4 h vs. 2.6 ± 0.2 h) (Fig. 6B). Next, we determined whether phosphorylation at this S435 site would block the ability of PIM inhibition to reduce HIF-2α levels. Wild-type and S435D mutants were transfected into 293T cells prior to 4 h incubation in hypoxia to stabilize HIF-2α and treatment with vehicle or PIM447. PIM inhibition reduced WT HIF-2α levels, whereas levels of the S435D mutant were higher basally and refractory to PIM447 treatment, suggesting that this site is critical for PIM-mediated regulation of HIF-2α levels (Fig. 6C). Next, we asked whether phosphorylation at S435 disrupts PHD2 binding and ubiquitination of HIF-2α. Co-immunoprecipitation assays showed reduced PHD2 binding to the S435D mutant compared to wild-type HIF-2α (Fig. 6D), and there was significantly less ubiquitination of mutant HIF-2α than wild-type HIF-2α after 4 h treatment with MG-132 (Fig 6E).
Figure 6. Phosphorylation of HIF-2α at S435 increases protein stability.

A) Sequence alignment of HIF-1α and HIF-2α ODDDs (Thr455 and Ser435 highlighted) B) 293T cells were transfected with HA-HIF-2α or T455D and incubated in hypoxia for 4 h prior to treatment with cycloheximide (CHX, 10 μM). Densitometry was used to determine the rate of protein decay. C) 293T cells were transfected with the indicated HA-HIF-2α constructs and treated with DMSO or PIM447 (3 μM) for 4 h. D) The indicated HA-HIF-2α constructs were immunoprecipitated and PHD2 was probed by western blotting; relative abundance was calculated by densitometry. E) The indicated HA-HIF-2α constructs were immunoprecipitated after 4 h treatment with MG-132 and ubiquitin was probed by western blotting. *p < 0.05, n.s. = not significant, error bars = SEM.
The anti-tumor effects of PIM inhibitors depend on downregulation of HIF-1:
To confirm the significance of Thr455 phosphorylation in vivo and in vitro, we created two homozygous HIF-1α T455D mutant SW620 colon cancer cell lines using CRISPR site-directed mutagenesis. We validated these lines by Sanger sequencing (Fig. S5A). Both of these cell lines showed stable HIF-1α protein in normoxic conditions (Fig. 7A) and had significantly increased expression of HIF-1 target genes (Fig. S5B). To confirm that HIF-1α was refractory to PIM inhibition in the T455D mutant cell lines, cells were treated with PIM447 (1 μM) for 4 h. Immunoblotting revealed HIF-1α levels were significantly higher and refractory to PIM inhibition in the T455D mutant cell lines (Fig. 6A). To determine whether Thr455 phosphorylation also impacted angiogenesis, CM was harvested from parental and B24 and C34 SW620 cells treated with or without PIM447 for 24 h and used for in vitro tube formation assays. CM from both B24 and C34 cells significantly increased both the total tube length and number of branch points compared to parental SW620 CM. Treatment with PIM447 significantly reduced tube formation resulting from SW620 CM compared, whereas the mutant cell lines were refractory to the anti-angiogenic effect of PIM inhibitors (Fig. 7B and C). Next, we assessed the tumorigenicity of these cell lines and sensitivity to PIM inhibition in vivo. Five million parental SW620, C34, or B24 cells were injected into the flanks of 12 (n=4/group) NSG mice. Tumors were allowed to grow to an average size of 100 mm3, and then the mice were randomly segregated into vehicle or AZD1208 (30 mg/kg) treatment groups. Both B24 and C34 tumors grew significantly faster than parental SW620 tumors (Fig. 7D and Fig. S6A). Strikingly, treatment with AZD1208 significantly reduced the volume of SW620 tumors but was unable to slow the growth of either the B24 or C34 mutant xenografts (Fig. 7D and Fig. S6A). Hematoxylin and eosin (H&E) staining revealed that AZD1208 disrupted tumor vasculature in the SW620 xenografts, resulting in necrotic tissue, whereas C34 and B24 tumors were highly vascular, regardless of PIM inhibition (Fig. 7E and Fig. S6A and B). Importantly, AZD1208 significantly decreased microvessel density (CD31 staining) in SW620 tumors, whereas no significant decrease in vasculature was observed in mutant tumors after treatment with AZD1208 (Fig. 7F). AZD1208 significantly increased apoptosis (cleaved caspase-3) in SW620 tumors, but not in HIF-1α T455D mutant tumors (Fig. 7G). Strikingly, HIF-1α levels were significantly higher in C34 tumors than in parental SW620 tumors, and AZD1208 significantly reduced HIF-1α levels in the SW620 tumors, but not in the C34 tumors (Fig 7H). RT-PCR analysis of tumor tissue confirmed that PIM inhibition reduced the expression of pro-angiogenic genes VEGF-A and ANGPTL4 in SW620 tumors, whereas PIM inhibition had no effect in B24 and C34 tumors (Fig. 7I). These data indicate that phosphorylation of Thr455 is sufficient to drive angiogenesis and increase tumor growth.
Figure 7. HIF-1α-T455D CRISPR mutants increase tumor growth and are resistant to PIM inhibition.
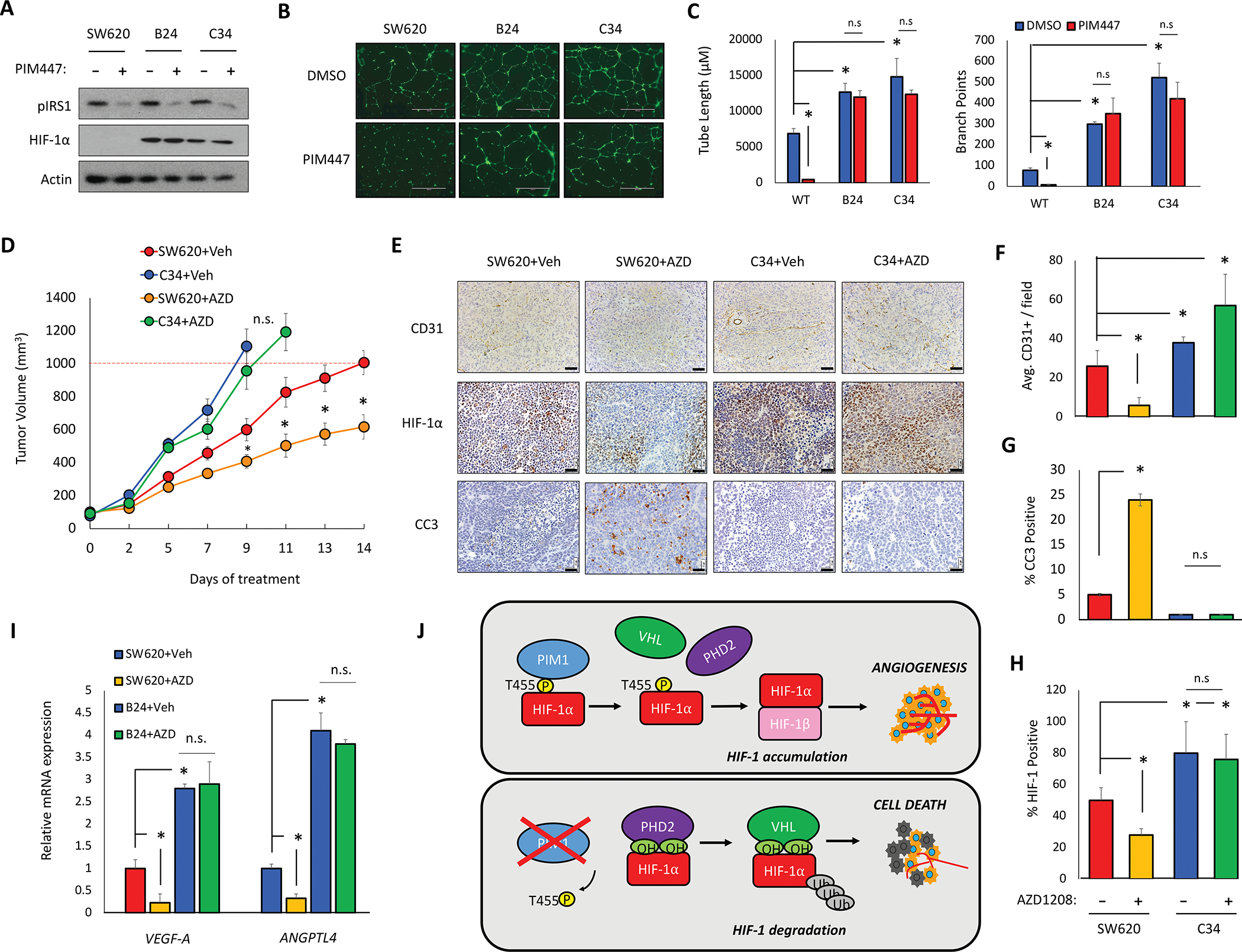
Two homozygous HIF-1α-T455D SW620 cell lines were generated via CRISPR site directed mutagenesis (B24 and C34). A) SW620, B24, and C34 cells were treated with PIM447 (3 μM) for 6 h. B) Representative images of tube formation after 24 h incubation in the indicated CM. C) Mean tube length and total branch points were quantified. D) Mice (n=4/group) were injected with 5 × 106 SW620 or C34 cells and treated with vehicle or AZD1208 (30 mg/kg); tumor volume was measured over time. E-H) IHC staining and quantification of the indicated markers in tumors from each cohort. I) RNA was harvested from tumor tissue and mRNA expression was measured by qRT-PCR. J) Model depicting the mechanism and physiological outcome of PIM1-mediated phosphorylation of HIF-1α. *p < 0.05, Scale bar = 50 μm, n.s. = not significant, error bars = SEM.
Discussion:
HIF-1 activation is arguably the most important driver of angiogenesis in solid tumors. In clinical settings, IHC analysis of patient’s biopsy specimens indicate dramatic HIF-1 overexpression in common human cancers, which leads to increased metastasis and mortality rates [32–35]. We are the first to demonstrate that PIM1 is sufficient to activate HIF-1 in normoxic conditions and promote angiogenesis (Fig. 2), establishing a new mechanism by which PIM kinases promote tumor progression. Moreover, elevated PIM1 provides a novel explanation for the constitutive activation of HIF-1 in tumors that is frequently observed in the absence of hypoxia. We identified a novel post-translational modification that controls the proteasomal degradation of HIF-1α (Fig. 3 and 4). Mechanistically, phosphorylation of HIF-1α by PIM1 at Thr455 prevents the binding of PHDs and subsequent hydroxylation of HIF-1α, ultimately reducing its degradation by the 26S proteasome (Fig. 7L). Interestingly, alignment with HIF-2α revealed an analogous site (Ser435) within the ODDD that regulates its stability through a similar mechanism (Fig. S5). While we were unable to confirm direct phosphorylation of this site by PIM1 using in vitro kinase assays, a phospho-mimetic mutation at this site was sufficient to block the ability of PIM inhibitors to reduce HIF-2α levels. Hence, it is possible that PIM2 or PIM3 could preferentially target HIF-2α (S435) or that PIM1 regulates this site through an indirect mechanism. Development of a phospho-specific antibody to HIF-1α (Thr455) confirmed that this site is phosphorylated in human cancer cell lines and that it is sensitive to changes in PIM1 activity. However, there is evidence that this site may be regulated by other kinases as well. For example, PIM3 is correlated with increased VEGF expression in pancreatic cancer, suggesting that other PIM isoforms could phosphorylate HIF-1α at Thr455 [36]. Moreover, protein kinase A has been reported to phosphorylate HIF-1α at Thr455 [16], and a proteomic-based screen searching for targets of aurora kinase inhibitors found significantly decreased phospho-Thr455 after treatment with aurora kinase inhibitors (AZD1152 and ZM447439) [37]. Therefore, phosphorylation of Thr455 appears to be a widely used mechanism through which multiple kinases regulate HIF-1α protein levels and HIF-1 activation. Unfortunately, HIF-1α itself has yet to be crystallized, so we are unable to speculate the consequences of T455 phosphorylation to HIF-1α structure. The sequence surrounding HIF-1α at Thr455 does not closely resemble the phosphorylation consensus motif predicted for PIM kinases (RXRHXS). Several established PIM1 substrates also have atypical phosphorylation sites, making it unclear how complete the described consensus sequences are for predicting PIM substrates. Other recent sites have identified PIM phospho-sites despite no canonical sequence being present, including, but not limited to, PIM1 autophosphorylation [38]. Future studies warrant further understanding of how PIM kinases recognize alternative substrate sequences and what determines the specificity of these targets.
PIM1 is elevated in many solid tumors and feeds into several pro-tumorigenic signaling pathways [39–41]. Here, we establish that heightened expression of PIM1 was sufficient to initiate and sustain angiogenesis in solid tumors (Fig. 1 and 2). Importantly, analysis of prostate cancer TMAs revealed a significant correlation between PIM1 and microvessel density, confirming that our in vitro and in vivo data translate to human cancers. Tumor vasculature is also the primary route for tumor cell dissemination [42, 43]. Several preclinical and clinical studies in various solid tumor models show that upregulation of PIM isoforms is associated with increased metastasis and PIM inhibitors alone prevent metastasis [28, 44, 45]. Specifically, a recent clinical study revealed that PIM1 is overexpressed at a high frequency in circulating tumor cells from patients with castrate-resistant prostate cancer [46]. Our results suggest that the pro-metastatic role for PIM described in the literature could be due to increased tumor vascularization. In contrast to control tumors, overexpression of PIM1 in xenograft tumors lacking HIF-1α did not increase tumor growth, proliferation, or angiogenesis, indicating that the pro-tumorigenic effects of PIM1 are largely dependent on stabilization of HIF-1α and heightened angiogenesis (Fig 2). In addition, CRISPR cell lines containing a phospho-mimetic mutation at Thr455 were completely refractory to the anti-tumor effects of PIM inhibitors in vivo. (Fig 7). Thus, the anti-tumor effect of PIM inhibitors is dependent on their ability to downregulate HIF-1 and reduce angiogenesis (Fig. 7). Synergistic anti-tumor effects have been reported when combining PIM inhibitors with a plethora of targeted therapies and chemotherapies, regardless of their mechanism of action [28, 47–50]. Our results suggest that the wide-ranging efficacy observed when combining PIM inhibitors and cytotoxic therapies could be attributed to their anti-angiogenic effects. We previously reported that PIM inhibitors selectively kill hypoxic tumor cells by increasing cellular reactive oxygen species in a HIF-1-independent manner [51]. This research expands our working model to include HIF-1; PIM inhibitors reduce angiogenesis by inhibiting HIF-1, which exacerbates the effects of hypoxia and renders tumor cells more susceptible to their cytotoxic effects (Fig. 7J).
Successful therapeutic targeting of transcription factors has proven challenging. However, reducing the expression of key transcription factors through post-translational modifications represents a promising approach [52–54]. Ongoing efforts to block angiogenesis as a treatment strategy have been fraught with disappointment, in many cases due to an increase in hypoxia associated with more aggressive disease. While many small molecules have been reported as HIF-1α inhibitors, no clinically approved selective HIF-1α inhibitor has been reported to date. Those that have been tested in the clinic have shown modest benefit [55], suggesting that alternative, HIF-independent pro-survival pathways are able to compensate or overcome HIF-1 inhibition. Taken in the context of our results and those of others, PIM is poised to fill this role. Blocking PIM represents a promising approach to overcome hypoxia-mediated therapeutic resistance due to both HIF-dependent and HIF-independent effects. Taken together with our previous studies showing a synergistic anti-tumor effect of inhibiting PIM1 and VEGF [28, 56], this work provides rationale for how this strategy works at the molecular level and identifies HIF-1α phosphorylation at Thr455 as a potential biomarker for the efficacy of PIM inhibitors in human tumors. Therefore, further translation of PIM inhibitors in combination with approved therapies is warranted in solid tumors as a new strategy to inhibit HIF-1 and angiogenesis while simultaneously targeting the hypoxic tumor cell population that is commonly associated with acquired resistance to therapy and treatment failure.
Materials and Methods:
Cell lines:
Parental and genetically modified A549, H460, HEK293T, RKO, and SW620 cells were maintained in DMEM medium containing 10% FBS. RKO vector and shHIF-1α cell lines were provided by Dr. Nicolas Denko (OSU) and have been previously described [57]. SW620 HIF-1α T455D cell lines were generated by CRISPR-Cas9 mediated mutagenesis using ssODN AAAATTACAGAATATAAATTTGGCAATGTCTCCATTACCCGATGCTGAAACGCCAAAGCCACTTCGAAGTAGTGCTGACCCTG and CRISPR sgRNA 5’GGCTTTGGCGTTTCAGCGGT – 3’. Cells were sequenced for the presence of mutations (Fig. S4A), and positive clones were expanded for use (B24 and C34). PC3/VEC and PC3/PIM1 cell lines were maintained in RPMI medium containing 10% FBS. HUVECs from Gibco were cultured in complete Med-200 containing 1X LVES media supplement (Invitrogen), and HUVEC cells from Lonza were cultured in EGM-2 medium with kit supplements added (Lonza). All cells were cultured at 37°C in 5% CO2, routinely screened for mycoplasma, and authenticated by short tandem repeat DNA profiling performed by the University of Arizona Genetics Core Facility and were used for fewer than 50 passages. When appropriate, cells were cultured in a hypoxic environment (1% O2, 5% CO2, 94% N2) using an InVivo2 400 hypoxia workstation (Baker Ruskinn).
Plasmids:
HA-HIF-1α (18949) and HRE-Luc were purchased from Addgene. HA-HIF-1α-T455A and HA-HIF-1α-T455D mutant plasmids were generated using site directed mutagenesis (Clontech). HA-PIM1 and HA-PIM1-K67M were gifts from Dr. Andrew S Kraft (UA). hPMI1 (short) was cloned into pCIP (lentiviral backbone) for generation of the hPIM1 cell lines, and PIM1 in FUCRW (RFP-lentiviral backbone) for generation of RFP-PIM1 cell lines. HA-Ub was a gift from Dr. Alexandra Newton (UCSD).
siRNA and Antibodies:
siHIF-1α experiments were performed using a forward transfection protocol (Lipofectamine 3000 Invitrogen). HIF-1α and siRNA was purchased from Santa Cruz Biotechnology (sc-44225 and sc-35316, respectively). PIM1 siRNA sequences were previously reported [58]. The following western blot antibodies were used for western blot: actin (BD Biosciences); and HA, HIF-1α (cat #: 3724S), HIF-1α-OH [P564] (cat #: 3434S), p-IRS1 [S1101] (cat #: 2385S), PHD2 (cat #: 4835S), PIM1 (cat #: 4835S), and ubiquitin (cat #: 3933S) (Cell Signaling Technologies). The p-HIF-1α [T455] antibody was generated by Pacific Immunology for western blot and IHC in this manuscript. The following antibodies were also used for IHC: CC3 (cat #: ab4051), CD31 (cat #: ab124432), HIF-1α (cat #: ab16066) and PIM1 (cat #: ab245417)(Abcam).
Supplementary Material
Acknowledgments:
We would like to thank the following shared resources at the University of Arizona Cancer Center for their help and support: EMSR, TACMASR, Gene Editing, and Biostatistics. We would also like to thank Dr. Dan Buster for performing in vitro kinase assays and Brenda Baggett and Dr. Marty Pagel for assisting with in vivo imaging. Studies were supported by funding from the National Cancer Institute (T32CA009213) on behalf of A.L.C., American Cancer Society (RSG-16-159-01-CDD) to N.A.W., and Department of Defense PCRP (W81XWH-19-1-0455) to N.A.W. A Cancer Center Support grant from the National Institute of Health (P30CA023074) also supported this research.
Footnotes
Conflict of Interest: The authors declare no competing interests.
References
- 1.Folkman J Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182–1186. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Perspectives on oxygen sensing. Cell (Review) 1999; 98: 281–284. [DOI] [PubMed] [Google Scholar]
- 3.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001; 294: 1337–1340. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) (Review) 2004; 19: 176–182. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature (Research Support, Non-U.S. Gov’t) 1999; 399: 271–275. [DOI] [PubMed] [Google Scholar]
- 6.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.) 2001; 292: 464–468. [DOI] [PubMed] [Google Scholar]
- 7.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 2011; 12: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de Bruine AP. VHL and HIF signalling in renal cell carcinogenesis. J Pathol 2010; 221: 125–138. [DOI] [PubMed] [Google Scholar]
- 9.Hua Zhong KC, David Feldser, Erik Laughner, Colleen Hanrahan, Maria-Magdalena Georgescu, Jonathan W. Simons, and Gregg L Semenza Modulation of Hypoxia-inducible Factor 1α Expression by the Epidermal Growth Factor/Phosphatidylinositol 3-Kinase/PTEN/AKT/FRAP Pathway in Human Prostate Cancer Cells: Implications for Tumor Angiogenesis and Therapeutics 2000; 60: 1541–1545. [PubMed] [Google Scholar]
- 10.Richard DE BE, Gothié E, Roux D, Pouysségur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 1999; 274: 32631–32637. [DOI] [PubMed] [Google Scholar]
- 11.Flügel DGA, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol 2007; 27: 3253–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem 2002; 277: 29936–29944. [DOI] [PubMed] [Google Scholar]
- 13.Xu D YY, Lu L, Costa M, Dai W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J Biol Chem 2010: 38944–38950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warfel NA, Dolloff NG, Dicker DT, Malysz J, El-Deiry WS. CDK1 stabilizes HIF-1alpha via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t) 2013; 12: 3689–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol Cell 2010; 40: 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bullen JW, Tchernyshyov I, Holewinski RJ, DeVine L, Wu F, Venkatraman V et al. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci Signal 2016; 9: ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartwich J, Orr WS, Ng CY, Spence Y, Morton C, Davidoff AM. HIF-1alpha activation mediates resistance to anti-angiogenic therapy in neuroblastoma xenografts. J Pediatr Surg (Evaluation Studies) 2013; 48: 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer (Review) 2011; 11: 23–34. [DOI] [PubMed] [Google Scholar]
- 19.Malone T, Schafer L, Simon N, Heavey S, Cuffe S, Finn S et al. Current perspectives on targeting PIM kinases to overcome mechanisms of drug resistance and immune evasion in cancer. Pharmacol Ther 2020; 207: 107454. [DOI] [PubMed] [Google Scholar]
- 20.Chen WW, Chan DC, Donald C, Lilly MB, Kraft AS. Pim family kinases enhance tumor growth of prostate cancer cells. Mol Cancer Res 2005; 3: 443–451. [DOI] [PubMed] [Google Scholar]
- 21.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K et al. Delineation of prognostic biomarkers in prostate cancer. Nature (Research Support, Non-U.S. Gov’t) 2001; 412: 822–826. [DOI] [PubMed] [Google Scholar]
- 22.Xie YXK, Dai B, Guo Z, Jiang T, Chen H, Qiu Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene 2006. [DOI] [PubMed] [Google Scholar]
- 23.Chauhan SS, Toth RK, Jensen CC, Casillas AL, Kashatus DF, Warfel NA. PIM kinases alter mitochondrial dynamics and chemosensitivity in lung cancer. Oncogene 2020; 39: 2597–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M, Liu T, Sun H, Weng W, Zhang Q, Liu C et al. Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. Cancer Sci 2018; 109: 1468–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao X, Liu X, Lu Y, Wang Y, Cao W, Liu X et al. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer 2019; 26: 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braso-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E et al. Erratum: PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nature medicine 2017; 23: 526. [DOI] [PubMed] [Google Scholar]
- 27.Zhao W, Qiu R, Li P, Yang J. PIM1: a promising target in patients with triple-negative breast cancer. Med Oncol 2017; 34: 142. [DOI] [PubMed] [Google Scholar]
- 28.Casillas AL, Toth RK, Sainz AG, Singh N, Desai AA, Kraft AS et al. Hypoxia-Inducible PIM Kinase Expression Promotes Resistance to Antiangiogenic Agents. Clinical cancer research : an official journal of the American Association for Cancer Research 2018; 24: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardenas-Rodriguez J, Howison CM, Pagel MD. A linear algorithm of the reference region model for DCE-MRI is robust and relaxes requirements for temporal resolution. Magn Reson Imaging (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.) 2013; 31: 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res (Research Support, U.S. Gov’t, P.H.S.) 2002; 62: 4316–4324. [PubMed] [Google Scholar]
- 31.Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem 2006; 281: 33095–33106. [DOI] [PubMed] [Google Scholar]
- 32.Baba Y, Nosho K, Shima K, Irahara N, Chan AT, Meyerhardt JA et al. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am J Pathol 2010; 176: 2292–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong D, Liu Q, Liu G, Yuan W, Wang L, Guo Y et al. The HIF/PHF8/AR axis promotes prostate cancer progression. Oncogenesis 2016; 5: e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu ZJ, Semenza GL, Zhang HF. Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang Univ Sci B 2015; 16: 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, Garner EI, Welch WR, Berkowitz RS, Mok SC. Over-expression of hypoxia-inducible factor 1 alpha in ovarian clear cell carcinoma. Gynecol Oncol 2007; 106: 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C, Li HY, Liu B, Huang S, Wu L, Li YY. Pim-3 promotes the growth of human pancreatic cancer in the orthotopic nude mouse model through vascular endothelium growth factor. J Surg Res 2013; 185: 595–604. [DOI] [PubMed] [Google Scholar]
- 37.Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 2011; 4: rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bullock AN, Debreczeni J, Amos AL, Knapp S, Turk BE. Structure and substrate specificity of the Pim-1 kinase. The Journal of biological chemistry 2005; 280: 41675–41682. [DOI] [PubMed] [Google Scholar]
- 39.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 2010; 95: 1004–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warfel NA, Kraft AS. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol Ther 2015; 151: 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rebello RJ, Huglo AV, Furic L. PIM activity in tumours: A key node of therapy resistance. Adv Biol Regul 2018; 67: 163–169. [DOI] [PubMed] [Google Scholar]
- 42.Bielenberg DR, Zetter BR. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J 2015; 21: 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tannock IF. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer 1968; 22: 258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santio NM, Eerola SK, Paatero I, Yli-Kauhaluoma J, Anizon F, Moreau P et al. Pim Kinases Promote Migration and Metastatic Growth of Prostate Cancer Xenografts. PloS one 2015; 10: e0130340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu Y, Zhang C, Du E, Wang A, Yang Y, Guo J et al. Pim-3 is a Critical Risk Factor in Development and Prognosis of Prostate Cancer. Med Sci Monit 2016; 22: 4254–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markou A, Tzanikou E, Strati A, Zavridou M, Mastoraki S, Bournakis E et al. PIM-1 Is Overexpressed at a High Frequency in Circulating Tumor Cells from Metastatic Castration-Resistant Prostate Cancer Patients. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurmasheva RT, Huang S, Houghton PJ. Predicted mechanisms of resistance to mTOR inhibitors. Br J Cancer 2006; 95: 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cen B, Xiong Y, Song JH, Mahajan S, DuPont R, McEachern K et al. The Pim-1 protein kinase is an important regulator of MET receptor tyrosine kinase levels and signaling. Mol Cell Biol 2014; 34: 2517–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 2005; 105: 4477–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moody SE, Schinzel AC, Singh S, Izzo F, Strickland MR, Luo L et al. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene 2015; 34: 2061–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warfel NA, Sainz AG, Song JH, Kraft AS. PIM Kinase Inhibitors Kill Hypoxic Tumor Cells by Reducing Nrf2 Signaling and Increasing Reactive Oxygen Species. Mol Cancer Ther 2016; 15: 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filtz TM, Vogel WK, Leid M. Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol Sci 2014; 35: 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer 2019; 19: 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang CY, Chan CY, Chou IT, Lien CH, Hung HC, Lee MF. Quercetin induces growth arrest through activation of FOXO1 transcription factor in EGFR-overexpressing oral cancer cells. J Nutr Biochem 2013; 24: 1596–1603. [DOI] [PubMed] [Google Scholar]
- 55.Fallah J, Rini BI. HIF Inhibitors: Status of Current Clinical Development. Curr Oncol Rep 2019; 21: 6. [DOI] [PubMed] [Google Scholar]
- 56.Chauhan SS, Warfel NA. Targeting PIM kinases to oppose hypoxia-mediated therapeutic resistance. Oncoscience 2018; 5: 254–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3: 187–197. [DOI] [PubMed] [Google Scholar]
- 58.Warfel NA, Sainz AG, Song JH, Kraft AS. PIM Kinase Inhibitors Kill Hypoxic Tumor Cells by Reducing Nrf2 Signaling and Increasing Reactive Oxygen Species. Mol Cancer Ther 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.