

Abstract
There is increasing interest in targeting CD33 in malignant and non-malignant disorders. In acute myeloid leukemia, longer survival with the CD33 antibody-drug conjugate gemtuzumab ozogamicin (GO) validates this strategy. Still, GO benefits only some patients, prompting efforts to develop more potent CD33-directed therapeutics. As one limitation, CD33 antibodies typically recognize the membrane-distal V-set domain. Using various artificial CD33 proteins, in which this domain was differentially positioned within the extracellular portion of the molecule, we tested whether targeting membrane-proximal targeting epitopes enhances the effector functions of CD33 antibody-based therapeutics. Consistent with this idea, a CD33V-set/CD3 bispecific antibody (BsAb) and CD33V-set-directed chimeric antigen receptor (CAR)-modified T cells elicited substantially greater cytotoxicity against cells expressing a CD33 variant lacking the entire C2-set domain than cells expressing full-length CD33, whereas cytotoxic effects induced by GO were independent of the position of the V-set domain. We therefore raised murine and human antibodies against the C2-set domain of human CD33 and identified antibodies that bound CD33 regardless of the presence/absence of the V-set domain (“CD33PAN antibodies”). These antibodies internalized when bound to CD33 and, as CD33PAN/CD3 BsAb, had potent cytolytic effects against CD33+ cells. Together, our data provide rationale for further development of CD33PAN antibody-based therapeutics.
INTRODUCTION
CD33 (Siglec-3) is a differentiation antigen that is primarily displayed on maturing and mature myeloid cells and their neoplastic cell counterparts.1,2 With this expression pattern, there have been long-standing efforts in therapeutically targeting CD33+ cells, first and foremost in acute myeloid leukemia (AML)1,3,4 but also CD33+ tumor cells in other malignancies, CD33+ myeloid-derived suppressor cells, and normal CD33+ microglial cells.5 In AML, longer survival of some patients treated with the antibody-drug conjugate gemtuzumab ozogamicin (GO) validates CD33 as drug target.6
The success and limitations of GO have fueled ongoing work to develop more effective CD33-directed therapeutics.7 However, targeting CD33 has proven difficult, and several drugs failed clinically because of lack of efficacy.7 Efforts have therefore centered around developing more potent anti-CD33 treatment modalities, including T cell engaging bispecific antibodies (BsAbs) and chimeric antigen receptor (CAR)-modified T cells.7 As one important limitation of these efforts, existing and investigational therapeutics, including GO, almost exclusively recognize immune-dominant epitope(s) within the exon 2-encoded membrane-distal V-set domain of CD33.7 Since membrane-proximal binding of antibodies can increase their effector functions,8–11 we reasoned targeting CD33 with antibodies against the membrane-proximal C2-set domain might optimize CD33-directed therapy that engage immune effector cells. Here, we test this concept experimentally and describe the generation of a series of C2-set domain-directed CD33 antibodies and derived therapeutics.
MATERIALS AND METHODS
Generation of artificial CD33 proteins and chimeras.
“CD33FL [full-length CD33] + CD22 4D” was generated using the endogenous CD33 signal peptide (amino acids [aa] 1-17), a 6-histidine tag, 3x glycine linker, the human CD33 extracellular domain (ECD, aa 18-259), a portion of the human CD22 ECD comprising C2-type domains 3-6 (aa 331-683), the CD33 transmembrane domain, and the CD33 intracellular domain (aa 260-364). Codons were optimized for human translation and cDNA synthesized as gBlock (Integrated DNA Technologies, Coralville, Iowa, USA) for cloning into pRRLsin.cPPT.MSCV lentivirus constructs containing an IRES-Enhanced Green Fluorescent Protein (EGFP) cassette. The “CD33FL + CD22 2D” construct used CD33FL + CD22 4D as template and Gibson assembly to splice out CD22 aa 331-504, removing C2-type domains 3 and 4. A CD33FL construct has been described.12–15 A truncated CD33 construct lacking the exon 3/4-encoded C2-set domain (CD33ΔE3-4) was engineered using site-directed mutagenesis to splice out CD33 ECD aa 140-232. All lentiviral constructs were confirmed by Sanger sequencing.
Parental and engineered human acute leukemia cell lines.
Human myeloid K562 and MOLM-13 cells were grown in RPMI-1640 medium with 10% fetal bovine serum and penicillin/streptomycin. Growth conditions for all other cell lines have been described.15 Lentivirally-transduced sublines overexpressing various CD33 proteins were generated at multiplicities of infection (MOI) of 0.25-25.12–14,16 EGFP-positive cells were isolated by FACS and re-cultured for further analysis/use. All cell lines were routinely tested for mycoplasma contamination (MycoAlert™ Mycoplasma Detection Kit; Lonza, Basel, Switzerland) and were authenticated using standard STR CODIS typing.
Primary AML patient specimens.
Frozen aliquots of primary AML patient specimens were obtained from an institutional repository under protocols approved by the Fred Hutchinson Cancer Research Center Institutional Review Board and cultured as described.15,17 All patients provided written informed consent for the collection and use of their biospecimens for research purposes.
Genetic deletion of CD33.
Clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-editing was carried out by electroporating purified Cas9 protein (TrueCut Cas9 V2; ThermoFisher Scientific) complexed with synthetic guide RNA (sgRNA) targeting exon 1 of CD33 (sequence 5’- CTGCTGCCCCTGCTGTGGGC-3’) using the ECM 380 Square Wave Electroporation system (Harvard Apparatus, Cambridge, MA) as described.15,18 CD33− single cells were isolated via FACS, and genomic DNA from individual clones analyzed by Sanger sequencing to confirm disruption or frame-shift mutation at all CD33 alleles.
Generation of murine and human CD33PAN antibodies.
Peptide immunogens consisting of the ECD of human CD33FL (aa 1-256), human CD33ΔE2 ECD (lacking aa 13-139 of CD33FL), or mouse/human chimeric ECD, all fused to the mouse IgG1 Fc domain, were generated, expressed in Freestyle™ 293-F cells, purified, and characterized biochemically as described.15,19 Mouse 3T3 cells were used to generate cell-based immunogens via lentiviral transduction, using cDNA for human CD33 or mouse/human chimeric CD33. BALB/c, CD1, F1, and humanized (Trianni mouse®) mice were immunized with a mixture of immunogens. Hybridoma screening was done by flow cytometry using beads coupled to peptide immunogens or with parental human lymphoid cells and sublines overexpressing CD33FL or CD33ΔE2.15 Hybridomas with reactivity against CD33ΔE2 and CD33FL were subcloned, and antibodies isotyped and sequenced as described.15
Expression and purification of recombinant murine CD33PAN antibodies.
As described previously,15,19 protein sequences were reverse-translated using human codons for cloning into a modified pCVL lentiviral vector, lentivirus particles used to transduce Freestyle™ 293-F cells, and secreted antibodies purified from conditioned media. Fractions corresponding to monomeric proteins were pooled, quantitated, and analyzed by SDS-PAGE.15
Surface plasmon resonance
Surface plasmon resonance (SPR) experiments were performed at 25°C on a Biacore T100 (Cytiva) using a running buffer of 10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P20 with 0.1 mg/mL bovine serum albumin. Rabbit anti-mouse IgG (Biacore BR100838) was amine coupled to 2 flow cells of a Series S CM5 chip (~7300 RUs). 1H7 at 0.2 μg/mL was injected at 10 μL/min over 1 flow cell of immobilized anti-mouse IgG for either 2 mins or 5 mins to capture ~94 or ~163 RUs of antibody for the CD33FL and CD33ΔE2 binding experiments, respectively. Purified ectodomains for CD33FL and CD33ΔE2 (engineered with C-terminal His and Avi tags) were run as concentration series at 35 μL/min over both the captured antibody and anti-mouse IgG alone (reference) surfaces. CD33FL was injected for 10 mins and allowed to dissociate for 15 mins. CD33ΔE2 was injected for 7 mins and dissociated for 7 mins. Ectodomain concentrations (serial 2-fold dilutions starting at 75 nM for CD33FL and 2 μM for CD33ΔE2) were run in duplicate, randomized, and included a buffer blank every 4th injection. The CM5 chip was regenerated with 10 mM glycine, pH 1.7 for 3 minutes at 20 μL/min and 1H7 recaptured prior to each CD33 injection. Data was double referenced and analyzed in BiaEval 2.0.4. CD33FL was fit with a 1:1 kinetic binding model to give a ka of 1.09(1) x 105 M−1s−1 and kd of 5.81(1) x 10−4s−1. CD33ΔE2 was fit with a 1:1 steady-state affinity model.
Construction, expression, and purification of CD33/CD3-directed BsAbs.
An anti-V-set domain-directed CD33/CD3 BsAb was constructed in the scFv-scFv format using published sequences (United States patent application publication US 2016/0317657 A1, November 3, 2016; SEQ ID NO 227).20 1H7/CD3 and 1E6/CD3 BsAbs were generated by replacing the CD33 scFv in the anti-V-set domain construct. An additional 1H7/CD3 BsAb was generated in an IgG-scFv format21 composed of the entire 1H7 antibody and scFvs against CD3. Protein sequences were reverse-translated using human codons for cloning into a modified pCVL lentiviral vector to produce purified BsAb similar to the approach detailed above.
CD33 single nucleotide polymorphism (SNP) rs12459419 genotyping.
CD33 rs12459419 genotyping was done as described.22
Quantification of CD33 expression.
Expression of CD33 variants and artificial constructs on human leukemia cell lines and primary AML cells was quantified by flow cytometry either using a directly labeled CD33 antibody (clone P67.6; BD Biosciences)12,13 or unlabeled P67.6 or 1H7 followed by APC-conjugated goat anti-mouse Ig (Multiple Adsorption, ThermoFisher Scientific). To identify non-viable cells, samples were stained with 4′,6-diamidino-2-phenylindole (DAPI). 10,000 events were acquired on a BD FACSCanto II flow cytometer (BD Biosciences), and DAPI-negative cells analyzed using FlowJo version 10 (BD Biosciences).
Quantification of CD33 internalization and modulation.
To quantify CD33 internalization, CD33+ AML cells were incubated with 2 μg/mL unlabeled CD33 antibody at 37°C and aliquots were removed at multiple time points.12,23,24 Samples were then stained with APC-conjugated goat anti-mouse Ig to identify remaining antibody on the cell surface and fluorescence quantified by flow cytometry as described above. To quantify CD33 modulation, CD33+ AML cells were incubated with 2 μg/mL unlabeled CD33 antibody at 37°C for 24 hours, after which additional CD33 antibody was added at 2 μg/mL. Secondary antibody and subsequent analysis by flow cytometry was performed as described above.
Quantification of BsAb- and GO-induced cytotoxicity.
Cytotoxicity induced by BsAb, using healthy donor T cells enriched from unstimulated peripheral blood mononuclear cells collected from healthy adult volunteers, or GO (Pfizer, New York, NY, USA) was determined flow cytometrically as described.13,16,17,20,22,25–27 For cell line experiments, cytotoxicity was quantified as a change in the percentage of dead cells as measured by DAPI staining. For primary cell experiments, drug-specific cytotoxicity was quantified as described.17,22
CAR-T cell generation
CAR T cells were generated through lentiviral transduction as previously described.28 Briefly, healthy donor negative selected human CD8+ T cells with a epHIV7 lentivirus encoding the scFv from the CD33V-set/CD3 BsAb described above linked to a IgG4 CH3 domain spacer, CD28-transmembrane domain, CD3zeta and 4–1BB intracellular signaling domain and truncated CD19 (tCD19) transduction marker. tCD19 CD8+ CAR-T cells were sorted and expanded in IL-7 and IL-15 (10ng/mL; Peprotech, Rocky Hill, NJ, USA) each for 14 days with media and cytokine changes every other day.
Chromium51 release cytotoxicity assay
CAR-T cell cytotoxicity was assessed following incubation with chromium51 labelled targets for 4 hours as described.28
Statistical analysis.
Comparisons of CD33 expression levels and drug-induced cytotoxicity were performed using paired Student’s t-test or repeat measure one-way ANOVA with multiple comparison testing, as appropriate, using Prism 8 (GraphPad Software, San Diego, CA, USA).
RESULTS
Binding distance from cell membrane correlates with immune effector functions of CD33 antibodies
To examine whether the distance between target epitope and the cell membrane influences the efficacy of T cell-engaging immunotherapies, we generated a series of artificial proteins in which the V-set domain of human CD33 was held at different distances from the cell membrane to allow targeting with a V-set domain-directed CD33 antibody-based therapeutic such as a CD33V-set/CD3 BsAb or CD33V-set-directed CAR T cells. Specifically, to bring the CD33 target epitope closer to the cell membrane, we generated an artificial CD33 protein that lacked the entire C2-set domain by removing exons 3 and 4 (CD33ΔE3-4; Figure 1A). Engineered human CD33+ AML cell lines in which endogenous CD33 was deleted via CRISPR/Cas918 were used to express either CD33FL or CD33ΔE3-4. In a first series of experiments, sublines expressing relatively similar levels of target molecules were subjected to short-term in vitro cytotoxicity assays with various doses of a CD33V-set/CD3 BsAb and healthy donor T cells as immune effector cells. As comparator, we used GO, which entirely depends on the toxic effects induced by the calicheamicin-γ1 payload for anti-tumor effects.1,4,6 As shown in Figure 1B–D, CD33V-set/CD3 BsAbs exerted greater cytotoxicity against AML and ALL cells expressing CD33ΔE3-4 than cells expressing CD33FL, whereas cytotoxic effects induced by GO were similar. Similar effects were seen in REH and RS4;11 cells (human CD33− B-acute lymphoblastic leukemia [B-ALL] cell lines) expressing these same CD33 constructs when treated with CD33V-set/CD3 BsAbs (data not shown). To further demonstrate the importance of target epitope membrane distance for efficacy of CD33-directed therapies engaging T cells, we also generated chimeric proteins using various portions of human CD22 to extend the distance between CD33 target epitope and the cell membrane (Figure 1A). As summarized in Figure 1E, the cytotoxic effects of CD33V-set/CD3 BsAbs were lower against AML cells expressing CD22/CD33FL chimeric proteins than paired cells expressing CD33FL. To confirm that the importance of membrane distance modulation effect was not isolated to BsAbs targeting CD33, we conducted a second series of similar experiments for which we generated CAR T cells directed against the V-set domain of CD33 using a CAR construct with known clinical activity.28 Indeed, as shown in Figure 1F, CD33V-set-directed CAR T cells showed significantly enhanced cytotoxicity against engineered K562 cells expressing CD33ΔE3-4 as compared to cells expressing matched levels of CD33FL, consistent with our findings with CD33V-set/CD3 BsAbs. Together, these data demonstrated that altering the position of the CD33 antibody binding epitope changes the effector functions of the CD33 antibody-derived therapies and suggested that membrane-proximal targeting of CD33 via C2-set domain-specific therapeutics could improve the efficacy of CD33-targeted T cell immunotherapy.
Figure 1. Membrane proximity of the target epitope modulates the anti-tumor efficacy of CD33/CD3 BsAbs and CAR T cells.
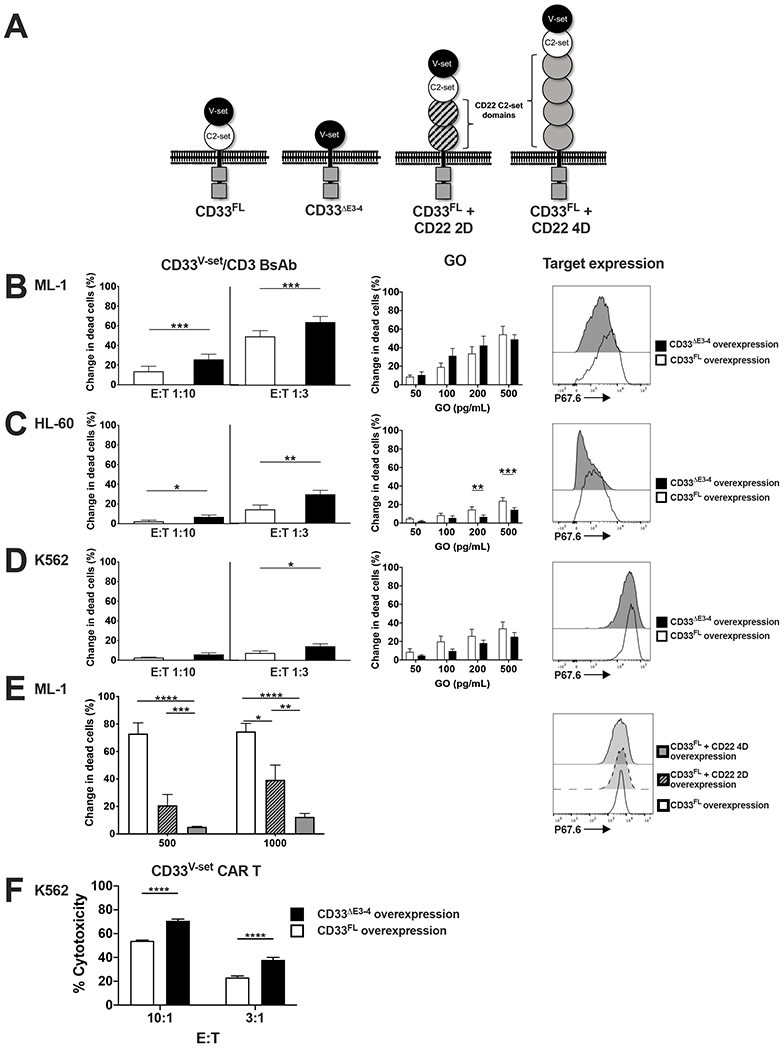
(A) Schematic of full-length CD33 (CD33FL) and artificial CD33 molecules with deletion of exons 3 and 4, resulting in membrane proximal relocation of the V-set domain (CD33ΔE3-4), or insertion of of either 2 C2-set domains of CD22 (CD33FL + CD22 2D) or 4 C2-set domains of CD22 (CD33FL + CD22 4D). (B-D) AML cell lines with CRISPR/Cas9-mediated deletion of the endogenous CD33 locus were engineered to overexpress either CD33FL or CD33ΔE3-4 via lentiviral gene transfer. Relative expression of the target proteins was flow cytometrically assessed via V-set domain CD33 antibody, P67.6, with representative histograms shown in the far-right panel. Cells were then treated with a V-set domain-targeting CD33/CD33 BsAb at a concentration of 1000 pg/mL and at the effector:target (E:T) cell ratios shown (left panel). Myeloid cells were also treated with gemtuzumab ozogamicin (GO) at the concentrations shown (middle panel). (E) The AML cell line ML-1 with CRISPR/Cas9-mediated deletion of the endogenous CD33 locus was engineered to overexpress CD33FL, CD33FL + CD22 2D or CD33FL + CD22 4D via lentiviral gene transfer. Relative expression of the CD33 constructs was flow cytometrically assessed using the V-set domain CD33 antibody, P67.6 (right panel). Cells were then treated with a V-set domain-targeting CD33/CD33 BsAb at the concentrations shown at an E:T cell ratio of 1:1. (F) V-set domain-directed CAR T cells were assessed for cytotoxicity in a chromium51 release assay against the CD33FL and CD33ΔE3-4 expressing K562 sublines from (D). Mean+SEM from a minimum of three separate experiments is shown. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Production of first-generation murine antibodies recognizing the membrane-proximal Ig-like C2-set domain of human CD33 regardless of presence of V-set domain (“CD33PAN antibodies”)
Since well-characterized antibodies recognizing the C2-set domain of human CD33 currently do not exist, we raised antibodies with this specificity in BALB/c, CD1, and F1 mice injected with immunogens consisting of the murine IgG1 Fc domain linked to the entire ECD of human CD33FL or the entire ECD of human CD33ΔE2. In screening assays in which we used human acute leukemia cell lines engineered to express either CD33ΔE2 or CD33FL to confirm epitope specificity, we identified several hybridomas showing binding to both CD33ΔE2 and CD33FL, demonstrating that the epitope recognized by these antibodies is located in the C2-set domain of CD33 and is accessible to the antibody regardless of whether or not the V-set domain is expressed (see examples in Figure 2A). Experiments with human CD33+ AML cells (ML-1) and an ML-1 subline in which we removed CD33 via CRISPR/Cas9 confirmed binding specificity to human CD33. This binding pattern is consistent with a CD33PAN antibody since all naturally occurring variants of human CD33 so far identified contain the C2-set domain (whereas some variants, e.g. CD33ΔE2, lack the V-set domain).
Figure 2. Biophysical characterization of the CD33PAN antibody, 1H7.
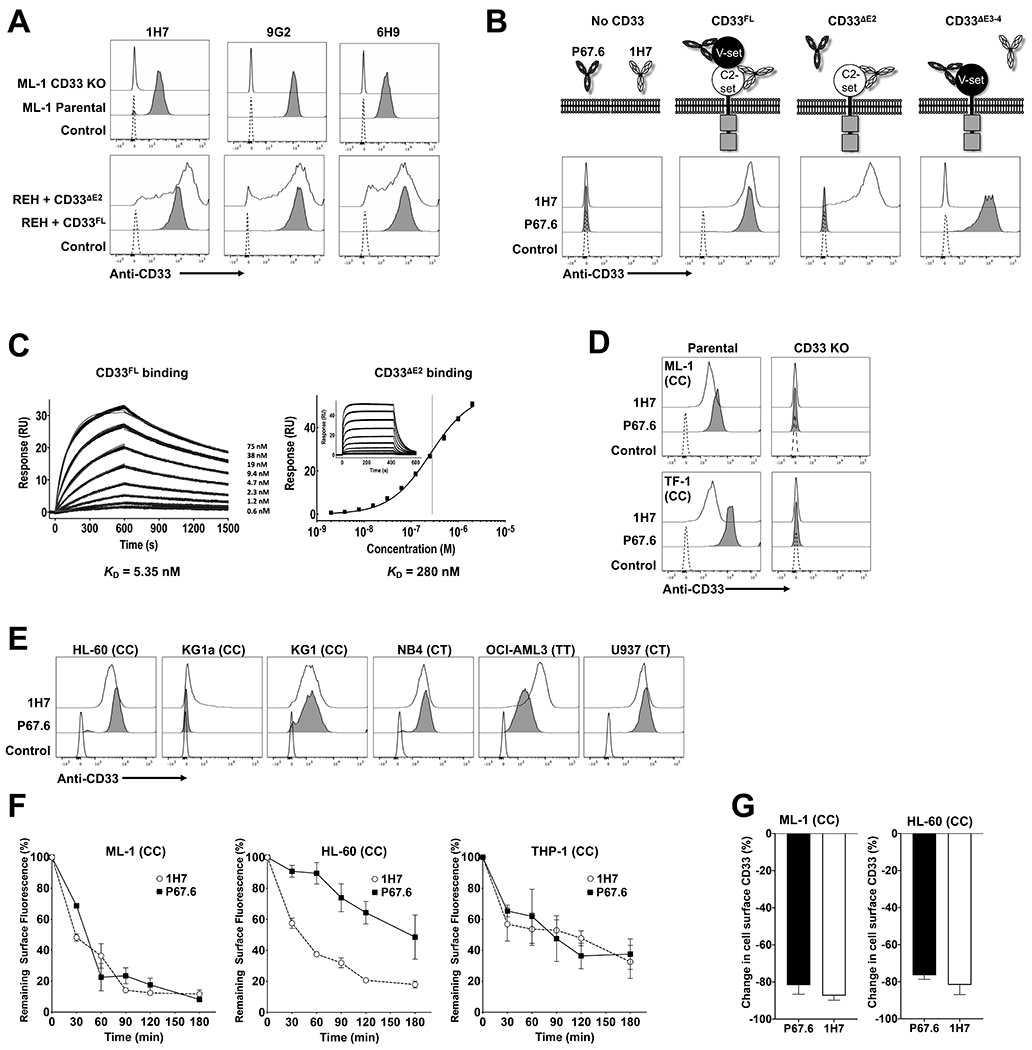
(A) Murine CD33PAN antibodies (clones 1H7, 9G2, 6H9) were tested flow cytometrically against parental ML-1 cells as well as ML-1 cells with CRISPR/Cas9-mediated deletion of CD33 (“CD33 KO”), as well as CD33neg REH sublines engineered to express CD33FL or CD33yyyyyyyΔE2, as indicated. A control without primary antibody was included. (B) Binding of recombinant 1H7 and anti-V-set domain antibody P67.6 was compared flow cytometrically against parental CD33neg REH cells and subclones engineered via lentiviral gene transfer to overexpress full-length CD33 (CD33FL), a CD33 variant with deletion of exon 2 resulting in loss of the V-set domain (CD33ΔE2), and an artificial CD33 molecule with deletion of exons 3 and 4 resulting in loss of the C2-set domain (CD33ΔE3-4). (C) Surface plasmon resonance assessment of purified extracellular domains from CD33FL or CD33ΔE2 binding to captured 1H7. Black lines are data and gray lines are the model fits. (D) Binding of 1H7 and P67.6 to parental ML-1 and TF-1 cells and cells with CRISPR/Cas9-mediated knockout of the CD33 locus (CD33 KO) was assessed flow cytometrically. Secondary antibody only negative control is shown. (E) Binding of 1H7 and P67.6 to parental AML cell lines was assessed flow cytometrically. Secondary antibody only negative control is shown. (F) Internalization of 1H7 and P67.6 s. AML cell lines were incubated with CD33 antibody at 37°C for the time indicated. Fluorescently labeled secondary antibody was then added to quantify remaining CD33 antibody on the cell surface. Results are presented as a percentage of the fluorescence signal present at time 0. Mean±SEM of 3 separate experiments is shown. (G) Modulation of CD33 expression by continuous exposure to anti-CD33 antibodies 1H7 and P67.6. ML-1 and HL-60 cells were exposed continuously to 1H7 or P67.6 for 24 hours. Primary antibody was then added again in excess to bind all available CD33 molecules, and fluorescently labeled secondary antibody was added for flow cytometric quantification. Results are presented as percentage change in fluorescence compared to control cells untreated with antibody. Mean±SEM of 3 separate experiments are shown. For all AML cell line experiments, CD33 rs12459419 genotype is shown in parentheses.
Biophysical characterization of CD33PAN antibodies
One hybridoma we identified early on (1H7) was sequenced and generated as a recombinant antibody. Using REH (human CD33− B-ALL) cells and sublines engineered to express CD33ΔE2 or CD33FL, we showed recombinant 1H7 indeed binds CD33ΔE2 and CD33FL, i.e. recognizes the C2-set domain even in the presence of the V-set domain, the defining characteristic of a CD33PAN antibody; Figure 2B). Consistent with C2-set domain binding, 1H7 did not recognize human acute leukemia cells engineered to express CD33ΔE3-4 as the only CD33 variant, whereas a V-set domain-specific antibody (clone P67.6), did (Figure 2B). Biophysical characterization via surface plasmon resonance (SPR) showed a KD of 5.35 nM and 280 nM for binding to CD33FL and CD33ΔE2 (Figure 2C). 1H7 showed appropriate negative binding to AML cells with CRISPR/Cas9-mediated deletion of CD33 (Figure 2D) and bound a range of AML cell lines encompassing all CD33 rs12459419 genotypes (Figure 2E). For further characterization, we subjected 1H7 to antibody internalization and CD33 antigen modulation experiments. As shown in Figure 2F, 1H7 was internalized in ML-1 and THP-1 cells with similar kinetics as P67.6, the parent murine CD33V-set antibody used in GO, while 1H7 was more rapidly internalized than P67.6 in HL-60 cells.1,4,6 Also similar was the extent to which cell surface density of CD33 was reduced upon 24 hours exposure to either 1H7 or P67.6 (Figure 2G). Together, these data indicate that CD33PAN antibodies are internalized, rendering them suitable for delivery of toxic payloads.
Development and characterization of CD33PAN/CD3 BsAb
To determine whether CD33PAN antibody-based therapeutics have cytotoxic properties, we built a CD33PAN/CD3 BsAb with 1H7 sequences in the canonical scFv-scFv format.20 We then conducted in vitro short-term cytotoxicity assays, using peripheral blood-derived T cells from healthy adult donors as effector cells. This 1H7/CD3 BsAb showed activity against AML cell lines including OCI-AML3 cells with rs12459419 genotypes TT (Figure 3A) but no cytolytic effect on ML-1 cells with CRISPR/Cas9-mediated deletion of CD33 (Figure 3B). Furthermore, in a bivalent IgG-scFv format, 1H7 was effective against REH cells and ML-1 cells overexpressing CD33ΔE2, confirming the C2-set domain specificity of this BsAb (Figure 3C). To assess the activity of the 1H7/CD3 BsAb more broadly, we tested it in vitro against a panel of primary patient AML specimens. We tested a total of 20 specimens, with 11 meeting our pre-defined criteria for inclusion based on viability at 48 hours (threshold >35%, a threshold similar to that used in previous studies17). Characteristics of these patient specimens are summarized in Supplementary Table 1; although several specimens from patients with TT rs12459419 genotype were tested, none met our inclusion criteria. As shown in Figure 3D, the 1H7/CD3 BsAb induced dose-dependent cytotoxicity against primary blasts from AML patients in these studies. Together, these data indicate that CD33PAN antibodies can serve as basis for therapeutics that engage immune effector cells such as T cells as mechanism of action.
Figure 3. A murine CD33PAN/CD3 BsAb redirects T cell-mediated cytotoxicity against AML cells in a CD33- and epitope-specific manner.
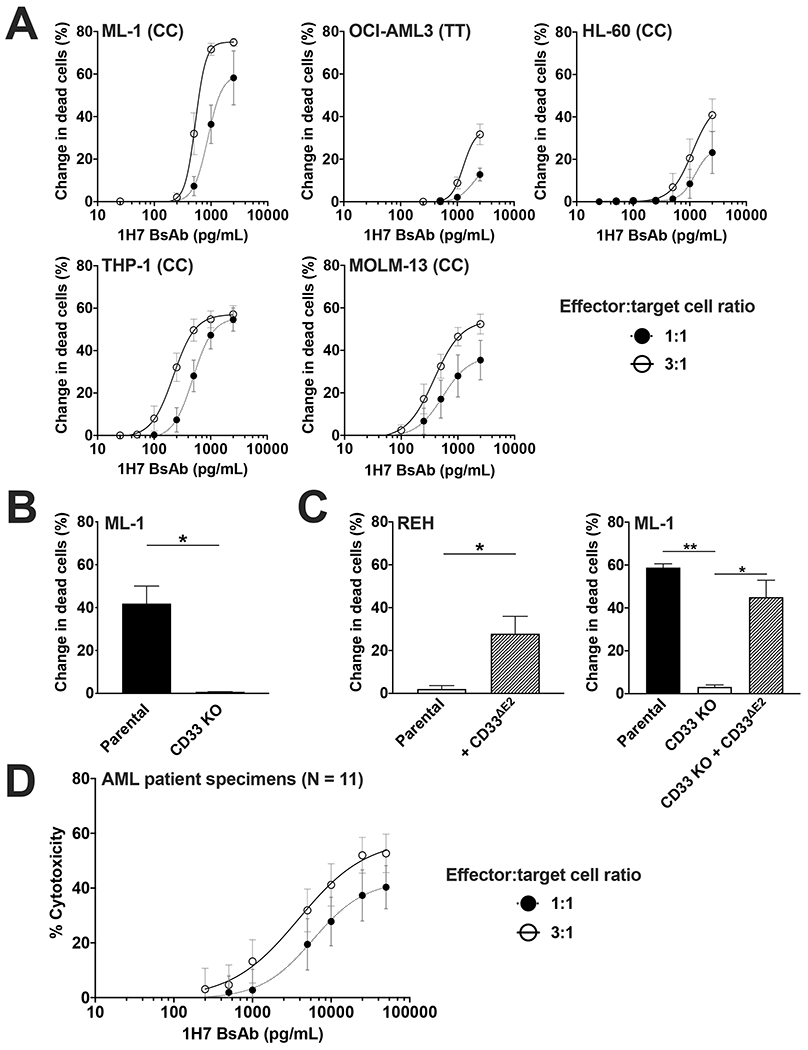
(A) Parental AML cell lines were treated with healthy donor T cells at the effector:target (E:T) cell ratios shown and 1H7/CD3 BsAb. (B) Parental ML-1 cells and a subline with CRISPR/Cas9-mediated knockout (KO) of CD33 were treated with 1H7 scFv-scFv BsAb at 500 pg/mL and healthy donor T cells at an E:T of 1:1. (C) Parental REH cell or a subline engineered to overexpress CD33ΔE2 (left panel), and parental ML-1 cells, ML-1 CD33 KO cells, and CD33 KO cells with overexpression of CD33ΔE2 (CD33 KO + CD33ΔE2) were treated with 1H7/CD3 IgG-scFv BsAb at a concentration of 500 pg/mL and healthy donor T cells at an E:T of 1:1. In cell line experiments, dead leukemic cells were enumerated after 48 hours via flow cytometry, and change in dead cells compared to no BsAb treatment is shown. CD33 rs12459419 genotype is shown in parentheses. Mean±SEM of 3 separate experiments are shown. *p<0.05; **p<0.01. (D) A panel of 11 primary AML patient samples was treated with the 1H7/CD3 BsAb and healthy donor T-cells at the E:T ratios shown. Mean±SEM across the 11 patient samples is shown. Cytotoxicity was determined enumerating both dead cells and total cell number as described.17,22
Second-generation CD33PAN antibodies with fully human variable domain sequences and derived therapeutics
Because the immunogenicity of murine amino acid sequences is a potential clinical concern, we conducted a second immunization campaign in which the same CD33 immunogens were used in humanized mice to yield antibodies with fully human variable domain sequences. As shown as examples in Figure 4A, we identified several hybridomas with binding to both CD33ΔE2 and CD33FL (i.e. CD33PAN antibody specificity). Experiments with CD33+ ML-1 cells and an ML-1 subline in which we removed CD33 via CRISPR/Cas9-mediated gene editing confirmed binding specificity of these antibodies to human CD33. For proof-of-principle studies, we sequenced one hybridoma (1E6) and then built a 1E6/CD3 BsAb in the scFv-scFv format. Similar to what we found for the 1H7/CD3 BsAbs, the 1E6/CD3 BsAb was highly potent against CD33+ human acute leukemia cells (Figure 4B) but lacked activity against CD33 knockout cells (Figure 4C). The 1E6/CD3 BsAb also potently killed REH cells overexpressing both CD33FL and CD33ΔE2, whereas the CD33V-set/CD3 BsAb had activity only against CD33FL-expressing cells (Figure 4D). Finally, we found that the 1E6/CD3 BsAb also had robust activity against a variety of primary AML patient specimens in vitro (Figure 4E).
Figure 4. A human CD33PAN/CD3 BsAb redirects T cell-mediated cytotoxicity against AML cells in a CD33- and epitope-specific manner.
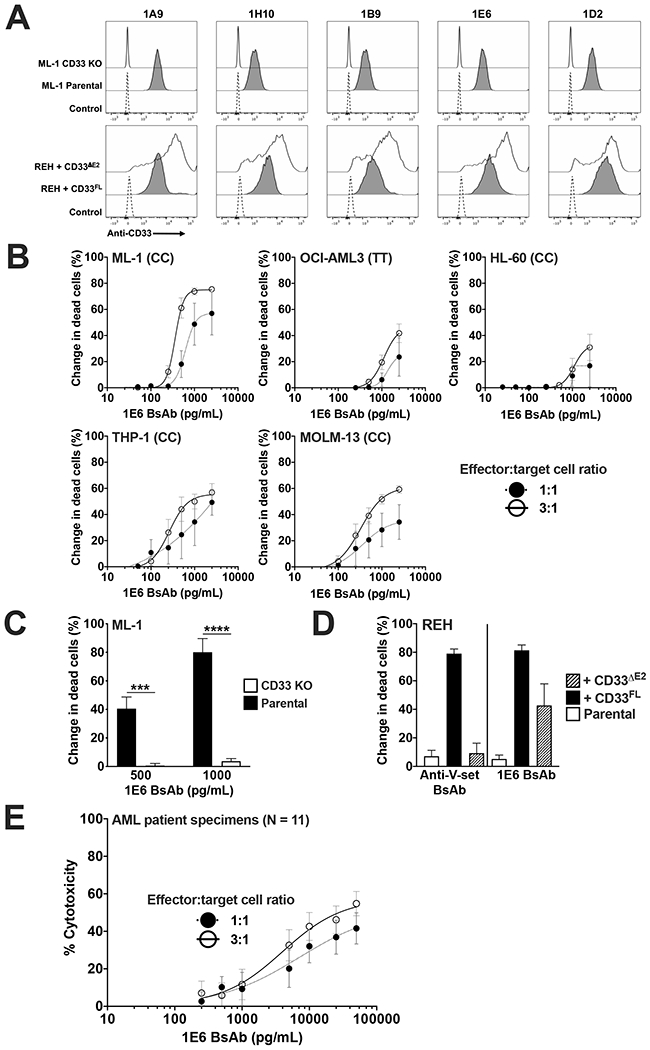
(A) Human CD33PAN antibody clones (clones 1A9, 1H10, 1B9, 1E6, and 1D2) were tested flow cytometrically against parental ML-1 cells as well as ML-1 cells with CRISPR/Cas9-mediated deletion of CD33 (“CD33 KO”), as well as CD33neg REH sublines engineered to express CD33FL or CD33ΔE2, as indicated. A control without primary antibody was included. (B) Parental AML cell lines were treated with healthy donor T cells at the effector:target (E:T) cell ratios shown and various doses of 1E6/CD3 BsAb. (C) Parental ML-1 cells and a subline with CRISPR/Cas9-mediated knockout (KO) of CD33 were treated with 1E6 BsAb at the indicated concentrations and healthy donor T cells at an E:T of 1:1. (D) Parental CD33neg REH cells or sublines engineered to overexpress CD33FL or CD33ΔE2 were treated with anti-V-set CD33/CD3 BsAb or 1E6 BsAb at a dose of 1000 pg/mL and an E:T of 3:1. In cell line experiments, dead leukemic cells were enumerated after 48 hours via flow cytometry, and change in dead cells compared to no BsAb treatment is shown. CD33 rs12459419 genotype is shown in parentheses. Mean±SEM of three separate experiments are shown. ***p<0.001; ****p<0.0001. (E) A panel of 11 primary AML patient samples was treated with the 1E6/CD3 BsAb and healthy donor T-cells at the E:T ratios shown. Mean cytotoxicity±SEM across the 11 patient samples is shown. Cytotoxicity was determined enumerating both dead cells and total cell number as described.17,22
DISCUSSION
For several cell surface proteins, including CD20, CD22, CD52, and melanoma chondroitin sulfate proteoglycan, binding membrane proximal epitopes was shown to enhance the efficacy of monoclonal antibodies as well as CD3-directed BsAbs or CAR T cells.8–11 To what degree the distance from the cell membrane influences the efficacy of CD33 antibodies has so far not been studied, perhaps partly because immune-dominant epitope(s) are located in the membrane-distal V-set domain of CD33 and CD33 antibodies therefore almost exclusively recognize this domain. In fact, only very recently, a CD33/CD3 BsAb, JNJ-67571244, has been described that is capable of binding to the C2-set domain of CD33.29
To determine the role of membrane distance for the anti-tumor efficacy of CD33 antibodies, we employed a strategy described previously,11 namely the use of a series of artificial proteins in which the V-set domain of CD33 was held at different distances from the cell membrane. While this approach can be criticized for its reliance on non-natural antigens and a test system that requires careful control of antigen expression levels, it avoids the potential issues arising from the use of antibodies with different affinities and/or the targeting of subtly different binding epitopes.11 These caveats are even more relevant when relying on modalities such as small BsAbs and CAR T cells for which efficacy and potency are affected by many additional parameters. Given the current interest in T cell engaging therapies, we used a CD33V-set/CD3 BsAb as well as CD33V-set CAR T cells as clinically relevant examples of CD33-directed immunotherapies. Overall, the findings from our well-controlled studies support the notion that membrane-proximal targeting of CD33 enhances the ability of CD33 antibodies to engage immune effector cells such as T cells for optimal cytolytic activity. These data suggest that the use of optimized therapeutics that are based on antibodies against the membrane-proximal C2-set domain could form one strategy to increase the efficacy of CD33-directed immunotherapy. Importantly, GO was similarly effective against cells expressing a CD33 protein lacking the C2-set domain (to allow membrane-proximal targeting) than against CD33FL-expressing cells. This argues against the possibility that these CD33 proteins differed significantly with regard to internalization properties, which otherwise could have accounted for some of the differences in observed efficacy of BsAb and, perhaps, CAR T cells.
Besides improved immune effector function, a second conceptual reason to pursue C2-set domain-directed CD33 antibodies as therapeutics is the existence of CD33 variants that lack the V-set domain. Recent studies have shown splicing of shorter isoforms of CD33 in AML cells, with CD33ΔE2 currently being of particular interest.14 However, while CD33ΔE2 is uniformly present in human AML cells at the mRNA level,14,15 it is unclear to what degree CD33ΔE2 is a clinically relevant target. This uncertainty is based on the observation that we were unable to detect CD33ΔE2, using CD33ΔE2-specific antibodies we newly developed, on human AML cell lines or primary blast cells from a smaller cohort of AML patients with CC, CT, and TT rs12459419 genotypes.15 Whether CD33ΔE2 is expressed as a cell surface protein in other disorders of interest for CD33-targeted immunotherapy has yet to be determined.
Our mechanistic studies showing superior immune effector engagement with membrane-proximal binding of CD33 provided the direct impetus to raise murine and human antibodies against the C2-set domain of human CD33. In detailed screening studies with engineered human acute leukemia cell lines, we identified a series of molecules that bound CD33 regardless of the presence/absence of the V-set domain. Since all known naturally occurring CD33 variants contain the C2 domain, we nicknamed these antibodies CD33PAN antibodies. Functional studies demonstrated these antibodies internalize with similar kinetics as CD33V-set antibodies, indicating they could deliver toxic payloads, e.g. small molecule drugs or radionuclides, and such studies are underway. Perhaps more interestingly, CD33PAN antibodies could serve as basis for immune effector cell engaging therapeutics such as BsAbs and CAR T cells. Our proof-of-concept studies with CD33PAN/CD3 BsAbs indeed demonstrate potent cytolytic effects against human CD33+ target cell lines and primary blasts from AML patients. As predicted, these BsAbs were also cytotoxic against human leukemia cells engineered to exclusively express the CD33ΔE2 protein variant. Together, these data provide the rationale for the further preclinical development of agents that are based on binding sequences from CD33PAN antibodies as we have initiated. Further studies on the protein expression patterns of CD33FL and CD33ΔE2 in normal and abnormal tissues could be helpful for the prediction of the toxicities of CD33PAN antibody-derived therapeutics. The currently available data suggest that such therapies exclusively (or, at the minimum, highly preferentially) engage CD33FL and that CD33ΔE2 does not play any (or, at the minimum, any significant) role as target antigen. While our mechanistic studies support superiority of membrane-proximal over targeting membrane-distal targeting of CD33, they do not provide any reason to believe this superiority would be restricted to abnormal target cells. It is therefore plausible the efficacy of CD33PAN antibody-derived therapeutics is increased against normal cells to the same degree as against abnormal cells – a possibility that suggests careful evaluation for unwanted on-target toxicities will be important during the drug development process.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Christopher Mehlin, Dr. James M. Olson, Jane Carter, and other members of the Molecular Design and Therapeutics (MDT) core facility at Fred Hutchinson Cancer Research Center for help with the generation of CD33 antibodies, and Dr. Della J. Friend for conducting the SPR experiments. Research reported in this publication was supported by the Leukemia & Lymphoma Society (Translational Research Program, grant 6489-16) and the National Institutes of Health/National Cancer Institute (NIH/NCI) (R21 CA223409, R21-CA234203, R21-CA245594, P30-CA015704, and P50-CA100632 [MD Anderson Cancer Center Leukemia SPORE]). C.D.G. is supported by a fellowship training grant from the NIH/National Heart, Lung, and Blood Institute (NHLBI; T32-HL007093), an institutional K12 grant from the NIH/NCI (K12-CA076930) an American Society of Clinical Oncology/Conquer Cancer Foundation Young Investigator Award and an Alex’s Lemonade Stand Young Investigator Grant. The Fred Hutchinson Cancer Research Center Antibody Technology Resource received support from the M.J. Murdock Charitable Trust.
Conflict of interest: C.D.G received research funding from Immunogen and Pfizer. H.P.K. is a consultant to and has ownership interests with Rocket Pharma and Homology Medicines and is a consultant to CSL Behring and Magenta Therapeutics. C.J.T. received research funding from AstraZeneca, Juno Therapeutics/Bristol Myers Squibb, Minerva, Nektar Therapeutics, and TCR2 Therapeutics; has ownership interests with ArsenalBio, Caribou Biosciences, Eureka Therapeutics, Myeloid Therapeutics, Precision Biosciences; is an inventor on a patent licensed to Juno Therapeutics; and is (or has been) a consultant to Amgen, ArsenalBio, Astra Zeneca, Caribou Biosciences, Century Therapeutics, Eureka Therapeutics, Myeloid Therapeutics, Nektar Therapeutics, PACT Pharma, Precision Biosciences, and T-CURX. R.B.W. received laboratory research grants and/or clinical trial support from Amgen, Aptevo, Celgene, Immunogen, Macrogenics, Jazz, Pfizer, and Selvita; has ownership interests with Amphivena; and is (or has been) a consultant to Agios, Amphivena, Aptevo, Astellas, Bristol Myers Squibb, Celgene, Genentech, Janssen, Kite, Macrogenics, and Pfizer. The other authors declare no competing financial interests.
REFERENCES
- 1.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012; 119(26): 6198–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duan S, Paulson JC. Siglecs as immune cell checkpoints in disease. Annu Rev Immunol 2020; 38: 365–395. [DOI] [PubMed] [Google Scholar]
- 3.Grossbard ML, Press OW, Appelbaum FR, Bernstein ID, Nadler LM. Monoclonal antibody-based therapies of leukemia and lymphoma. Blood 1992; 80(4): 863–878. [PubMed] [Google Scholar]
- 4.Laszlo GS, Estey EH, Walter RB. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev 2014; 28(4): 143–153. [DOI] [PubMed] [Google Scholar]
- 5.Walter RB. Expanding use of CD33-directed immunotherapy. Expert Opin Biol Ther 2020; 20(9): 955–958. [DOI] [PubMed] [Google Scholar]
- 6.Godwin CD, Gale RP, Walter RB. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017; 31(9): 1855–1868. [DOI] [PubMed] [Google Scholar]
- 7.Walter RB. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expert Opin Investig Drugs 2018; 27(4): 339–348. [DOI] [PubMed] [Google Scholar]
- 8.Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, et al. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother 2010; 59(8): 1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin TS. Ofatumumab: a novel monoclonal anti-CD20 antibody. Pharmgenomics Pers Med 2010; 3: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013; 121(7): 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleary KLS, Chan HTC, James S, Glennie MJ, Cragg MS. Antibody distance from the cell membrane regulates antibody effector mechanisms. J Immunol 2017; 198(10): 3999–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood 2005; 105(3): 1295–1302. [DOI] [PubMed] [Google Scholar]
- 13.Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014; 123(4): 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laszlo GS, Harrington KH, Gudgeon CJ, Beddoe ME, Fitzgibbon MP, Ries RE, et al. Expression and functional characterization of CD33 transcript variants in human acute myeloid leukemia. Oncotarget 2016; 7(28): 43281–43294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Godwin CD, Laszlo GS, Wood BL, Correnti CE, Bates OM, Garling EE, et al. The CD33 splice isoform lacking exon 2 as therapeutic target in human acute myeloid leukemia. Leukemia 2020; 34(9): 2479–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laszlo GS, Gudgeon CJ, Harrington KH, Walter RB. T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood Cancer J 2015; 5: e340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington KH, Gudgeon CJ, Laszlo GS, Newhall KJ, Sinclair AM, Frankel SR, et al. The broad anti-AML activity of the CD33/CD3 BiTE antibody construct, AMG 330, is impacted by disease stage and risk. PLoS One 2015; 10(8): e0135945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humbert O, Laszlo GS, Sichel S, Ironside C, Haworth KG, Bates OM, et al. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia 2019; 33(3): 762–808. [DOI] [PubMed] [Google Scholar]
- 19.Bandaranayake AD, Correnti C, Ryu BY, Brault M, Strong RK, Rawlings DJ. Daedalus: a robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res 2011; 39(21): e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godwin CD, Bates OM, Garling EE, Beddoe ME, Laszlo GS, Walter RB. The Bruton’s tyrosine kinase inhibitor ibrutinib abrogates bispecific antibody-mediated T-cell cytotoxicity. Br J Haematol 2020; 189(1): e9–e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orcutt KD, Ackerman ME, Cieslewicz M, Quiroz E, Slusarczyk AL, Frangioni JV, et al. A modular IgG-scFv bispecific antibody topology. Protein Eng Des Sel 2010; 23(4): 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laszlo GS, Beddoe ME, Godwin CD, Bates OM, Gudgeon CJ, Harrington KH, et al. Relationship between CD33 expression, splicing polymorphism, and in vitro cytotoxicity of gemtuzumab ozogamicin and the CD33/CD3 BiTE(R) AMG 330. Haematologica 2019; 104(2): e59–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter RB, Raden BW, Zeng R, Häusermann P, Bernstein ID, Cooper JA. ITIM-dependent endocytosis of CD33-related Siglecs: role of intracellular domain, tyrosine phosphorylation, and the tyrosine phosphatases, Shp1 and Shp2. J Leukoc Biol 2008; 83(1): 200–211. [DOI] [PubMed] [Google Scholar]
- 24.Walter RB, Häusermann P, Raden BW, Teckchandani AM, Kamikura DM, Bernstein ID, et al. Phosphorylated ITIMs enable ubiquitylation of an inhibitory cell surface receptor. Traffic 2008; 9(2): 267–279. [DOI] [PubMed] [Google Scholar]
- 25.Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res 2016; 22(23): 5829–5838. [DOI] [PubMed] [Google Scholar]
- 26.Correnti CE, Laszlo GS, Schueren WJ de van der, Godwin CD, Bandaranayake A, Busch MA, et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018; 32(5): 1239–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klupsch K, Baeriswyl V, Scholz R, Dannenberg J, Santimaria R, Senn D, et al. COVA4231, a potent CD3/CD33 bispecific FynomAb with IgG-like pharmacokinetics for the treatment of acute myeloid leukemia. Leukemia 2019; 33(3): 805–808. [DOI] [PubMed] [Google Scholar]
- 28.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 2016; 8(355): 355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nair-Gupta P, Diem M, Reeves D, Wang W, Schulingkamp R, Sproesser K, et al. A novel C2 domain binding CD33xCD3 bispecific antibody with potent T-cell redirection activity against acute myeloid leukemia. Blood Adv 2020; 4(5): 906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.