

Abstract
To maintain an adequate iron supply for hemoglobin synthesis and essential metabolic functions while counteracting iron toxicity, humans and other vertebrates have evolved effective mechanisms to conserve and finely regulate iron concentration, storage, and distribution to tissues. At the systemic level, the iron-regulatory hormone hepcidin is secreted by the liver in response to serum iron levels and inflammation. Hepcidin regulates the expression of the sole known mammalian iron exporter, ferroportin, to control dietary absorption, storage and tissue distribution of iron. At the cellular level, iron regulatory proteins 1 and 2 (IRP1 and IRP2) register cytosolic iron concentrations and post-transcriptionally regulate the expression of iron metabolism genes to optimize iron availability for essential cellular processes, including heme biosynthesis and iron-sulfur cluster biogenesis. Genetic malfunctions affecting the iron sensing mechanisms or the main pathways that utilize iron in the cell cause a broad range of human diseases, some of which are characterized by mitochondrial iron accumulation. This review will discuss the mechanisms of systemic and cellular iron sensing with a focus on the main iron utilization pathways in the cell, and on human conditions that arise from compromised function of the regulatory axes that control iron homeostasis.
Keywords: iron metabolism, IRP1, ACO1, IRP2, IREB2, ferroportin, iron-sulfur clusters, ALAD, heme biosynthesis, HSCB, HSC20
Introduction
Iron is one of the most abundant trace elements in the human body. It plays a crucial role in vivo by participating in many essential biological functions, including but not limited to, oxygen transport, nucleotide synthesis, and ATP production. Iron acts as a catalyst for these processes by serving as a ligand in heme-, iron-sulfur cluster-, and iron-containing proteins. Iron deficiency compromises red blood cell (RBC) production resulting in anemia, a primary manifestation of iron deficiency. On the other hand, iron overload is equally detrimental by promoting oxidative stress that causes damage to lipids, proteins, and DNA molecules, which can ultimately lead to cell death and tissue damage. Iron levels must therefore be tightly regulated in vivo to maintain iron availability while avoiding iron overload.
Most iron is utilized during erythropoiesis to generate heme in erythroid cells, accounting for about 90% of daily iron consumption. Therefore, erythroid cells have the highest iron content within the body, accumulating as many as 1.2 billion iron atoms per cell. Thus, iron metabolism and erythropoiesis are intertwined with one another via regulatory networks that coordinate systemic iron homeostasis and RBC production. Intestinal absorption accounts for only 10% of the daily iron requirement, whereas 90% is recycled through erythrophagocytosis of senescent RBCs in the spleen, liver, and bone marrow by reticuloendothelial macrophages [1].
Dietary iron is absorbed by the intestinal mucosa through a highly regulated process. On the apical membrane, divalent iron transporter 1 (DMT1) transports iron into the cells with the facilitation of a ferrireductase duodenal cytochrome b561 (DcytB) [2, 3]. On the basolateral membrane, ferroportin (FPN) transports iron across to apotransferrin facilitated by the ferroxidase hephaestin or ceruloplasmin[4–6]. Diferric transferrin (Tf) then transports iron throughout the body via the circulation to cells that import iron via Tf/TFR1 (transferrin receptor) cycles. Excess cellular iron is either exported by FPN or stored into ferritin, a 24-mer composed of L- and H- subunits that assemble into a sphere that can store as many as 4500 iron atoms [7]. The details of the process can be found in excellent reviews [8, 9] and in papers of this same issue. FPN is highly expressed in cells that are important for iron trafficking, including splenic macrophages that phagocytose senescent red cells, enterocytes that mediate gastrointestinal iron uptake, and hepatocytes, which store iron in ferritin and constitute the main iron storage depot in the body. In addition, FPN was recently found to be present at high levels in erythroblasts and mature RBCs [10, 11], where it accounts for up to 1% of the total human and murine RBC membrane proteins. Erythroid-specific FPN deficiency was found to significantly decrease blood iron levels, suggesting that iron released by erythroid cells is a major previously unrecognized source of iron that replenishes circulating diferric transferrin and contributes to maintenance of systemic iron homeostasis[10]. As more iron is released into the circulation and transferrin saturation levels increase, hepatocytes secrete hepcidin, a 25 amino acid peptide hormone, which decreases FPN activity, thereby reducing the iron flux across membranes[12]. When systemic iron levels are low, hepcidin transcription is downregulated to allow increased iron flux from storage and diet. While the hepcidin-FPN axis controls iron homeostasis systemically, Iron Regulatory Proteins 1 and 2 (IRP1 and IRP2) regulate iron homeostasis at the cellular level, as outlined in the following sections.
Regulation of cellular iron homeostasis by Iron Regulatory Proteins 1 and 2
IRP1 and IRP2 bind to the cis-regulatory iron-responsive elements (IREs) [13] in the mRNAs of iron metabolism genes to regulate their expression and thereby optimize cellular iron availability. IREs are RNA secondary structures of about 30 base pairs, consisting of a conserved “CAGUGN” loop (N could be A, T, C, but not G), followed by a stem composed of base-pairs interrupted by a conserved C-bulge 5 nucleotide upstream of the loop (Fig. 1). Under iron deficiency, IRPs bind to IREs in order to decrease iron storage and export, and increase iron uptake, thus restoring cellular iron balance. Binding of IRPs to IREs inhibits the translation of mRNAs that have IREs in the 5’-untranslated region (5’UTR), such as H- and L-ferritin [14, 15], FPN [5, 6], mitochondrial aconitase (ACO2)[16], the erythroid-specific 5-aminolevulinate synthase (eALAS, aka ALAS2)[17], hypoxia-inducible factor 2 alpha (HIF2α)[18], and Drosophila succinate dehydrogenase B (dSDH)[19] mRNAs, and stabilizes, by preventing endoribonuclease digestion, the mRNAs that have IREs in their 3’UTR, such as TFR1 [20], DMT1[3], cell division cycle 14A (CDC14A)[21], myotonic dystrophy kinase-related Cdc42-binding kinase α (MRCKα)[22], and profilin 2 (Pfn2)[23]. Some of the genes that encode mRNAs with IRE elements also express non-IRE-containing isoforms, such as FPN, DMT1, and CDC14A, which might provide a mechanism for bypassing regulation by IRPs under specific conditions, as in the case of FPN transcripts, which will be the focus of a dedicated section. By IRP/IRE complex immunoprecipitation and microarray analysis, 35 putative IRE-containing mRNAs were reported[24]. However, the significance of these IREs remains to be elucidated. Mutations of the L-ferritin IRE cause hereditary hyperferritinemia-cataract syndrome [25–27], and a mutation in the ALAS2 IRE was reported to contribute to the severity of the erythropoietic protoporphyria associated with loss of function mutations in the ATP-dependent Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit X, CLPX [28], emphasizing the essential function of IREs in the regulation of these target genes and of systemic iron homeostasis.
Figure 1. IRP1, a protein with dual function.
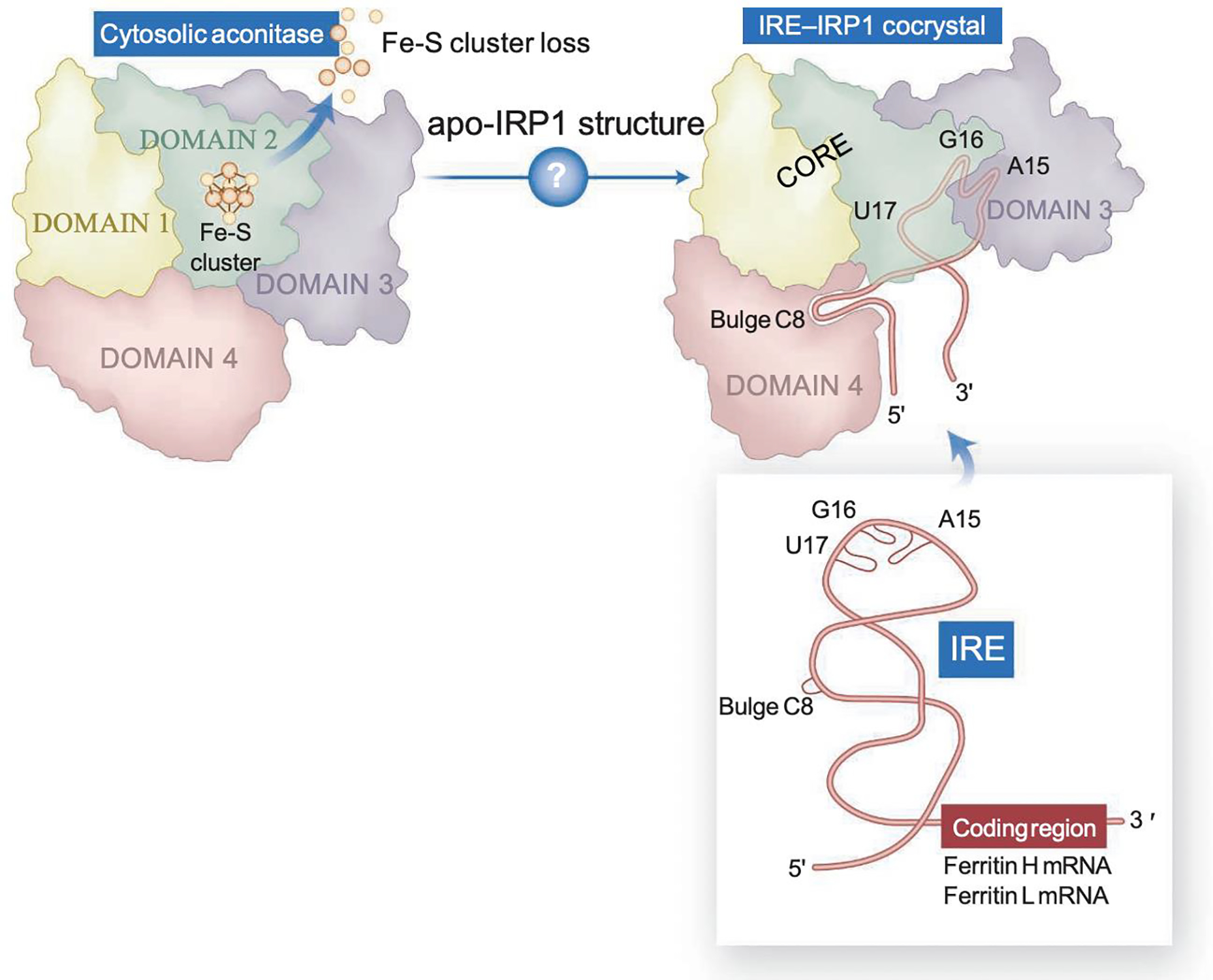
IRP1 alternates between function as a cytosolic aconitase, which contains a [4Fe-4S] cluster in the active site cleft, to an apoprotein form that lacks the cluster and binds to IRE stem-loop structures present in several transcripts encoding iron metabolism proteins. Upon binding, IRP1 represses translation of transcripts that contain IREs near the 5’-end and stabilizes from endonucleolytic degradation mRNAs that contain IREs at the 3’-UTR. Apo-IRP1 undergoes a large conformational change that creates a complex IRE-binding pocket, in which the bulge C binds to domain 4, and three residues of the loop make finger-like projections into newly accessible regions of domain 3. The length of the upper stem of the IRE optimizes the distance between its two main IRP contact points, resulting in high binding affinity (From[167]).
Human IRP1 and IRP2 have 889 and 963 amino acids, respectively, and share ~70% sequence similarity. Nevertheless, they are regulated differently. IRP1 is a bifunctional enzyme with a [4Fe-4S] cluster that functions as iron and oxygen sensor (Fig. 1). Under iron-replete conditions, IRP1 coordinates an iron-sulfur cluster (holo-form) and functions as cytosolic aconitase (ACO1), whereas under iron deficiency, IRP1 loses the cluster and binds to IREs to regulate cytosolic iron availability (Fig. 1). By contrast, IRP2 does not ligate an iron-sulfur cluster, and the protein levels are regulated by the F-box and leucine-rich repeat protein 5 (FBXL5)-mediated ubiquitination and proteasomal degradation in an iron- and oxygen-dependent manner[29–31] (see section entitled “Iron sulfur cluster cofactors are required at several steps in mammalian heme biosynthesis“).
Physiological significance of iron regulatory proteins
The physiological significance of IRPs is evident from the fact that mouse embryos with deletions of both Irp1 and Irp2 do not survive through the blastocyst stage[32]. Blastocysts from crosses between Irp1−/−Irp2+/− mice, from which 25% of the embryos are expected to harbor an Irp1−/−Irp2−/− genotype, displayed abnormal morphology and brown color likely due to ferritin overexpression. The lack of viability of Irp1−/−Irp2−/− mice further establishes the indispensable role of IRPs in iron homeostasis and overall physiology in mammals [32]. Mice with deletion of either Irp1 or Irp2 are viable indicating that IRPs are, at least partially, functionally redundant and each IRP can compensate for the loss of the other[33, 34]. Regulation of transcription of each IRP has not been thoroughly investigated, and it is not clear why the ratios of expression of the two IRPs differ among cells and tissues. However, differences in relative expression levels have been observed in RNA seq atlases, likely explaining why complete loss of one of the two Irps results in different phenotypic manifestations. For instance, transcript levels of IRP2 are approximately three times higher than expression of IRP1 in the cerebral cortex, throughout the central nervous system, and in erythroid and immune cells (Human Protein Atlas available from http://www.proteinatlas.org). In contrast, IRP1 transcript levels are relatively high in kidneys, liver, and adipose tissue, approaching 10-fold higher expression levels in proximal renal tubule cells (Human Protein Atlas available from http://www.proteinatlas.org).
Physiological significance of IRP1
Mice with targeted deletion of Irp1 exhibit extramedullary hematopoiesis, splenomegaly, polycythemia (also known as erythrocytosis)[35–37] and pulmonary hypertension[35]. One of the targets of the IRP/IRE system is HIF2α, also known as endothelial PAS domain protein 1 (EPAS1), which has a 5’-IRE [38]. HIF2α is one of the members of a family of heterodimeric transcriptional factors that mediate the physiological response to low oxygen (hypoxic) conditions, by heterodimerizing with the aryl hydrocarbon receptor nuclear translocator (aka the β subunit) and translocating to the nucleus where it regulates the expression of genes involved in the adaptive response to hypoxia. Under normoxia, HIFα proteins undergo proline hydroxylation by the iron-, oxygen- and α-ketoglutarate- dependent prolyl hydroxylases (PHDs) followed by proteasomal degradation mediated by the VHL (Von Hippel-Lindau) E3 ubiquitin ligase[39–41]. At low oxygen and low iron conditions, stabilized HIF2α transcriptionally activates several target genes, including erythropoietin (EPO)[40]. Binding of IRPs to the HIF2α 5’-IRE inhibits HIF2α translation[18], thereby reducing EPO production. Irp1 is the major IRE binding protein in kidney, liver, heart, brown fat and lungs [33–35]. The Hif2α-IRE binding activity of Irp1 is more than 10-fold higher than Irp2 in kidney lysates [42](Fig. 2). Thus, deletion of Irp1 causes translational de-repression of Hif2α mRNA and subsequent accumulation of Hif2α protein in the kidney, which leads to upregulation of EPO causing increased erythropoiesis and polycythemia[35–37]. Overall, polycythemia has been reported to result from mutations in genes encoding various proteins, including erythropoietin receptor (EPOR), hemoglobins (HBB, HBA), HIF2α (EPAS1), IRP1, and proteins involved in the HIFα regulatory pathways, VHL and PHD-2 (EGLN1)[35–37, 43–48]. There are two types of polycythemia. Primary polycythemia, also known as polycythemia vera, is mainly caused by mutations in JAK2[49] that lead to hypersensitivity to EPO signaling and production of excess red blood cells, whereas secondary polycythemia is caused by increased EPO levels. Secondary polycythemia developed in Irp1−/− mice as early as 4–6 weeks of age[36, 37]. Although two research groups [36, 37] reported polycythemia only in young (4–6 weeks old) Irp1−/− mice, a third group observed polycythemia in adult (6–11 months old) Irp1−/− mice that were fed either a normal or iron deficient diet[35, 50]. When Irp1−/− mice were challenged with an iron deficient diet, Hif2α protein levels increased even further as a result of its stabilization due to impaired Hif2α degradation mediated by PHDs. Hematocrit levels increased up to 65% or more, which caused sluggish blood flow and possibly clots that led to early death of the mice by abdominal hemorrhages[35], which may have been caused by blood vessel occlusion and increased pressure as the blockage impaired circulation and pre-occlusion venous pressure increased, leading to rupture of the relatively delicate venous endothelia. Thus, Irp1−/− mice that were fed a low iron diet developed severe polycythemia, tissue iron deficiency and underwent sudden death, which establishes the essential role of Irp1 in systemic iron homeostasis and erythropoiesis[35]. Mice that inducibly expressed a constitutively active Irp1 developed macrocytic anemia[51], likely because of impaired erythropoiesis caused by translational repression of Hif2α by Irp1, which reduced Epo levels.
Figure 2. Model for the mechanism of preventive action of TEMPOL on VhlR200W mice.
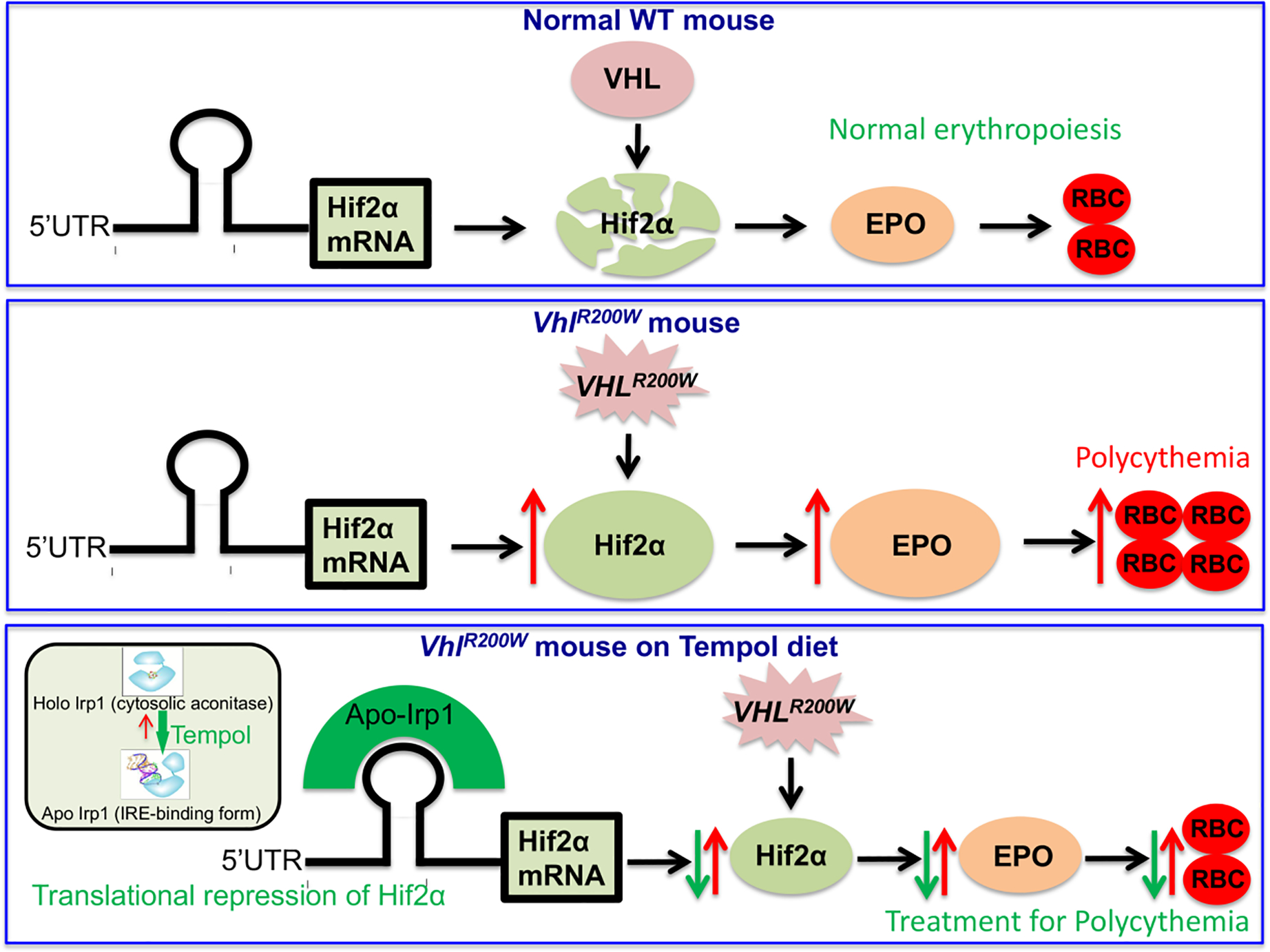
Hif2α expression is regulated at multiple levels. At normal conditions, Vhl degrades Hif2α. In VhlR200W mice, Hifα proteins are not degraded, which increases their levels and activates expressions of their transcriptional targets, including EPO, a Hif2α specific target. Hif2α is also translationally regulated by the IRE-Irp regulatory system (red arrows). TEMPOL treatment increases binding of Irp1 to the Hif2α-IRE, thus reducing Hif2α protein levels, which lead to diminished EPO expression and restoration of normal hematocrit levels (green arrows). (Reproduced from Ghosh et al., [42]).
In addition to polycythemia, Irp1−/− mice developed pulmonary hypertension, and cardiac hypertrophy and fibrosis[35] which emphasizes the crucial role of Irp1 in the physiology of the pulmonary and cardiovascular system. This is not unexpected considering that in lung and heart, Irp1 is the major contributor towards IRE-binding[33], and Hif2α is involved in the pathogenesis of pulmonary hypertension[52]. Right ventricular pressure (RVP), a hallmark of pulmonary hypertension, which is best measured by cardiac catheterization, was about 30% higher in Irp1−/− mice, compared to age-matched WT mice. A strikingly abnormal motion of the interventricular wall during systole was a further indication that pulmonary hypertension was present in Irp1−/− mice[35]. Pulmonary hypertension developed in Irp1−/− mice as early as 3 months of age, and RVP increased only 10% more when mice lived up to 12 months[35]. Endothelin-1, a HIF target, is a potent vasoconstrictor. The mRNA and protein levels of endothelin-1 were 2–3-fold higher in the lungs of Irp1−/− mice compared to WT mice. Strikingly, in cultured primary endothelial cells derived from Irp1−/− mice, endothelin-1 protein levels were more than 40-fold higher and as expected, Hif2α expression was significantly increased[35]. Interestingly, in Irp1−/− mice that were fed a low iron diet, Epo expression and hematocrit levels further increased, whereas endothelin-1 and RVP levels did not change. The reason for this mechanistic difference in the pathophysiology of polycythemia and pulmonary hypertension remains unclear.
Treatment of polycythemia and pulmonary hypertension caused by mutations in genes encoding proteins that regulate Hif2α in mice
The discovery that mice with ablation of Irp1 developed EPO-dependent polycythemia and pulmonary hypertension led us to evaluate activation of the IRE-binding activity of Irp1 as a potential therapy for these diseases in mice [42]. The mouse model chosen for this work carried a homozygous mutation in the VHL tumor-suppressor gene that resulted in a R200W amino acid substitution and it is also known as Chuvash polycythemia mouse model in reference to the patients who carry the same homozygous VHLR200W mutation[53], which is endemic to the Chuvash population of the Russian Federation. VHL targets HIFα proteins for proteasomal degradation. The VHLR200W mutation, both in patients and mice, is known to stabilize HIFα proteins and promote the transcription of multiple HIFα targets including EPO (Fig. 2) due to impaired HIFα protein degradation [54, 55]. The VhlR200W mice were initially treated with a diet supplemented with TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl), a small, stable nitroxide radical that is known to activate the IRE-binding activity of Irp1 by shifting the equilibrium from the holoprotein to the IRE binding apoform[56]. TEMPOL treatment normalized the hematocrit, corrected splenomegaly, lowered Epo production, decreased Hif2α protein levels and increased the lifespans of VhlR200W mice[42]. TEMPOL attenuated the polycythemia of the VhlR200W mice by decreasing Hif2α expression through the increased binding of Irp1 to the Hif2α-IRE[42] (Fig. 2). As expected, TEMPOL did not decrease the elevated hematocrit level of Irp1−/− mice or the even higher hematocrit (as high as 84%) of the VhlR200W;Irp1−/− double mutant mice, confirming that the amelioration of polycythemia by TEMPOL was mediated by Irp1 activation. TEMPOL therapy also attenuated hypoxic polycythemia in mice[42], analogous to human polycythemia, indicating that TEMPOL might potentially be a therapeutic approach for treatment of polycythemia in humans who develop high hematocrits at high-altitude.
Since polycythemia and pulmonary hypertension are caused by impaired degradation of Hif2α in VhlR200W mice, and translational de-repression of Hif2α in Irp1−/− mice, we recently used MK-6482 (previously known as PT2977), a second generation HIF2α inhibitor for the oral treatment of these genetically defined murine models of human diseases[50]. MK-6482 treatment normalized Epo and endothelin-1 levels, reversed polycythemia, decreased RVP, normalized the movement of the cardiac interventricular septum and attenuated pulmonary hypertension in VhlR200W mice, Irp1−/− mice and double mutant VhlR200W;Irp1−/− mice[50]. Moreover, this drug lowered the elevated expression of CxCl-12, which in association with its receptor CXCR-4, promotes fibrocyte influx, potentially causing fibrosis in the hearts and lungs of VhlR200W mice, suggesting that MK-6482, a drug with a favorable safety profile in human[57, 58], could possibly be a therapeutic option for pulmonary and cardiac fibrosis. Pharmacological inhibition of Hif2α by a Hif2α translation inhibitor C76 was found to reduce pulmonary hypertension in PHD2–deficient Egln1Tie2Cre mice and Sugen 5416/hypoxia PAH (pulmonary arterial hypertension) rats[59]. Suppression of Hif2α signaling by another small-molecule, PT2567, diminished pulmonary hypertension in rodents exposed to hypoxia for 4 to 5 weeks[60].
Human patients with IRP1 mutations
Recently, seven novel variants at the ACO1 locus, which encodes IRP1, were found to be associated with altered hemoglobin concentrations in a genome-wide association study from 684,122 individuals from Iceland and the UK[45]. The two most frequent variants were found to be the missense Cys506Ser and the nonsense Lys334Ter mutations. Serine substitution of Cys506 affects one of the three cysteines that directly coordinate the [4Fe-4S] cluster in cytosolic aconitase, leading to disassembly of the cluster and consequently to increased IRE-binding activity of IRP1[45, 61], which, by repressing HIF2α translation, reduces EPO production, leading to anemia. Conversely, the variant Lys334Ter affects an amino acid residue located within domain 2 of IRP1, which together with domains 3 and 4, is involved in interacting with the IRE; the mutation leads to premature truncation of the full-length 889 amino acid protein at position 334, causing loss of its IRE-binding activity, which leads to elevated EPO production and increased risk of polycythemia[45]. Notably, there was no report of pulmonary hypertension for the carriers of Lys334Ter, possibly because the carriers are heterozygous for the mutation. It is also worth noting that loss of only one functional IRP1 allele in the patients was sufficient for the development of subtle polycythemia. In a separate study, a somatic IRP1 mutation, which prematurely truncated the protein, dramatically increased HIF2α and EPO production and induced polycythemia [62]. Patients with increased risk of EPO-dependent polycythemia might potentially be treated with the second generation HIF2α inhibitor, MK-6482, since the drug has been found to attenuate EPO-dependent polycythemia in Irp1−/− mice[50]. Chuvash and other patients with polycythemia and/or pulmonary hypertension caused by upregulation of Hif2α might be treated with MK-6482[50] as well. Patients with only polycythemia caused by mutations in critical genes known to be involved in the HIF2α pathway but without any mutation in Irp1 might be potentially treated with TEMPOL[42].
Physiological significance of IRP2
Mice with targeted deletion of Irp2 develop progressive neurodegeneration, microcytic anemia and erythropoietic protoporphyria[34, 63, 64], and are characterized by significant cytosolic iron accumulations into ferritin heteropolymers throughout the brain and duodenal mucosa and by decreased expression of the iron import protein transferrin receptor 1 [34]. Iron misregulation caused axonal degeneration and a movement disorder characterized by tremor, abnormal gait and motor weakness prominent when the mice were older than 6 months[34]. Complete loss of Irp2 and loss of one allele of Irp1 in mice (Irp+/−Irp2−/−) increased the severity of neurodegeneration, indicating a dosage effect of combining partial Irp1 deficiency with complete Irp2 loss[65]. The decreased TfR1 and increased ferritin expression in Irp2−/− mice caused functional iron deficiency, which led to impaired activities of mitochondrial complexes I and II[34, 66] that rely on iron-sulfur and heme cofactors for function. A second group of researchers working with their Irp2−/− mice also reported decreased TfR1 and increased ferritin expression in brain lysates[67]. Despite the fact that this group observed only a discrete impairment of balance and/or motor coordination, they did not report iron deposition or evidence of neuronal degeneration in the brains of their mice[67]. A third group observed locomotor dysfunction and increased iron deposits in the brain, without cellular degeneration[68]. Genetic ablation of Irp2 also caused microcytic hypochromic anemia in mice[63] and erythropoietic protoporphyria, due to the loss of translational repression of Alas2 mRNA [63]. The functional iron deficiency was reported to impair iron-sulfur cluster assembly in pancreatic β cells compromising biogenesis of the iron-sulfur enzyme Cdkal1, which catalyzes the methylthiolation of the tRNALysUUU, leading to reduced insulin secretion and diabetes in Irp2−/− mice[69]. We refer the readers to a number of excellent reviews in which the phenotypes associated with global and tissue-specific ablation of Irps have been discussed [8, 70, 71].
The neurodegenerative symptoms of Irp2−/− mice were ameliorated by oral treatment with TEMPOL[56]. The axons of TEMPOL-treated Irp2−/− mice were partially spared from degeneration[56, 66]. Moreover, TEMPOL treatment restored complex I activity in Irp2−/− mice, further suggesting that functional iron deficiency was the cause of motor neuronal degeneration[66]. Interestingly, TEMPOL treatment did not prevent anemia in Irp2−/− mice, likely because TEMPOL’s action on converting cytosolic aconitase to the IRE-binding form would suppress translation of erythrocytic Alas, thereby blocking heme biosynthesis[56]. Additionally, in the kidney, TEMPOL reduced EPO expression and accordingly diminished erythropoiesis and promoted development of anemia, by increasing binding of Irp1 to the Hif2α-IRE.
Human patients with IRP2 mutations
The possibility that autosomal recessive IRP2-related disorders would be found in humans had been hypothesized for several years. With the use of exome sequencing, the first patient with bi-allelic loss-of-function variants in IRP2, leading to complete absence of IRP2 protein, was recently identified in a study from our group in collaboration with clinicians at the Toronto Children’s Hospital[72]. The patient, a 16-year-old male, had neurological and hematological features that paralleled those of Irp2−/− mice, including disabling neurodegeneration, a treatment-resistant choreiform movement disorder, microcytic hypochromic anemia unresponsive to iron supplementation and markedly elevated zinc protoporphyrin IX levels. Several months after we reported the first IRP2-deficient patient [72], a second case was described[73]. An Australian patient with progressive neurological symptoms and bi-allelic IRP2 missense variants was identified through trio exome sequencing[73]. Although biochemical studies on patient-derived lymphoblasts were not performed, in silico predictions revealed that the two mutations harbored by the Australian patient, namely substitution of Gly785 into arginine and deletion of the highly conserved Ser444, were likely to be pathogenic and to impair the IRE-binding activity of IRP2[73, 74]. The Australian patient[73] was of particular interest as it expanded the scope of IRP2-related disorders and underscored the possibility that severe clinical symptoms might be present even in the absence of nonsense mutations that would substantially truncate the encoded IRP2 protein[72, 74]. As more IRP2 patients are identified and diagnosed, it may be possible to correlate the spectrum of the disease manifestations with the impact of the mutations on the IRE-binding activity of IRP2, as it is conceivable that less obvious neurological impairments may be attributable to IRP2 variants that have a less deleterious effect on IRE binding.
There is the potential of translating into clinical trials oral treatment with TEMPOL, which was found to mitigate the neurological disorder of Irp2−/− mice[56]. We anticipate that ongoing studies on the molecular mechanisms of cellular iron homeostasis will continue to lead novel clinical and therapeutic discoveries.
In summary, the phenotypes of patients harboring mutations in either IRP1 or IRP2 resemble those of the murine models with genetic ablations of either Irp1 or Irp2 and are consistent with the expression profiles of Irps in different tissues, with Irp1 being the dominant iron regulatory protein in the kidneys whereas IRP2 is relatively more highly expressed in the central nervous system. IRP1 is also highly expressed in adipose tissue. Consistently, IRP1 expression was found to positively correlate with adipogenic markers in subcutaneous and visceral adipose tissue in two human independent cohorts[75]. Because each IRP is equally efficacious at repressing ferritin translation and stabilizing TFR1 mRNA, differences in relative levels of expression of IRPs in differentiated cells and tissues are the most likely cause for differences in the overall phenotypes associated with genetic ablation and or mutations in IRPs [76, 77]. Nevertheless, considering the complexity of the cytosolic environment, including the concentration of IRE-containing mRNAs, the ratios between IRPs and IRE-containing targets, the location of the IRE in the mRNAs and the subcellular distribution of IRPs and IREs, the regulation and interaction of the IRP/IRE system remains a very dynamic and complex process with several critical questions that remain to be elucidated.
The importance of an alternative FPN transcript in erythropoiesis
In addition to the well-known post-translational regulation of FPN levels by hepcidin [12], hepcidin binding to FPN has been reported to directly block FPN iron transport activity [11, 78], by causing a physical obstruction that presumably plays a significant role in regulating iron efflux from mature red blood cells, which have high FPN levels on their membranes, but lack the necessary machinery to facilitate hepcidin-mediated internalization and degradation. Of note, the same direct arrest mechanism of FPN transport activity by hepcidin is likely to play a role in tissues that are less sensitive to the post-translational modulation of FPN levels by hepcidin, such as intestinal epithelial cells [79], where the direct blocking mechanism may help to mitigate fluctuations of blood iron levels between meals. Not surprisingly, mutations that disrupt the hepcidin-FPN interaction cause disorders such as hemochromatosis, iron deficiency and anemia, thus emphasizing the essential interplay between hepcidin and FPN in systemic iron homeostasis [80, 81].
As already mentioned, FPN expression is regulated at the post-transcriptional level by the IRP/IRE system, as the major transcript of FPN mRNA, FPN1a, has an IRE in its 5’UTR, which causes repression of FPN translation and therefore reduces iron export, under low iron conditions [4–6]. However, in intestinal epithelial cells, FPN expression must be able to evade repression by the IRE-IRP system, in order to maintain systemic iron stores, as iron deficiency in intestinal mucosal cells would otherwise repress expression of FPN needed to export iron into the circulation to satisfy overall bodily iron demands. Thus, intestinal epithelial cells generate an alternative FPN mRNA (FPN1b) that does not contain an IRE [82]. The FPN1b mRNA is transcribed from an upstream promoter; however, it still encodes the same protein as the FPN1a transcript. Expression of the mRNA that lacks the 5’-IRE in intestinal epithelial cells ensures that FPN protein synthesis is not repressed by IRPs when intestinal epithelial cells are themselves iron deficient. Thus, the expression of a FPN transcript that lacks a 5’IRE enables the organism to increase systemic iron uptake. Though high expression of FPN might induce cellular iron scarcity in the intestinal epithelium, it enables the organism to transfer iron into the bloodstream before these cells are shed every 4–5 days, which could result in excreting and losing large amounts of needed iron (Fig. 3)[83].
Figure 3. Physiological functions of FPN and its mRNA isoforms in systemic iron homeostasis.
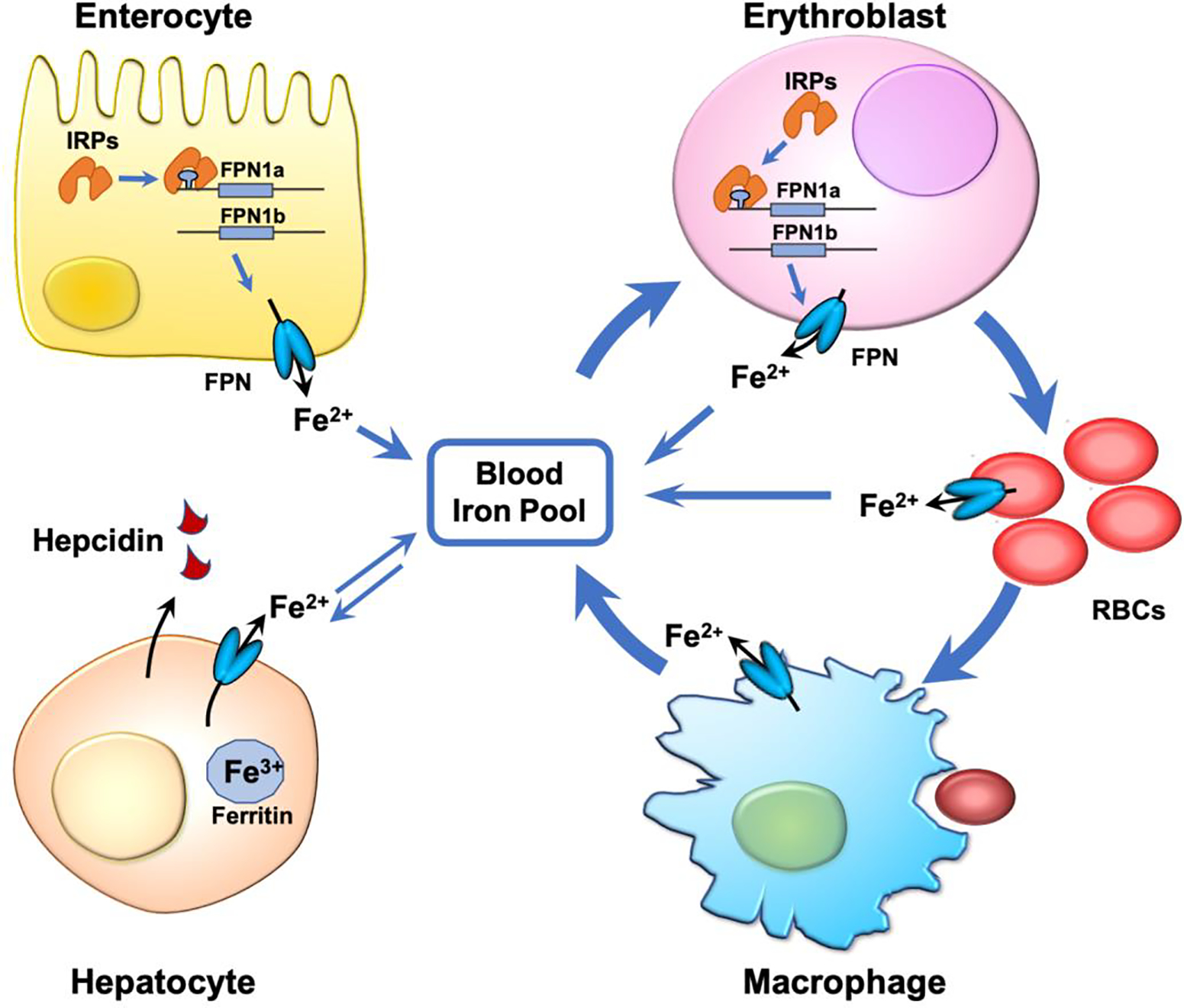
FPN exports iron into the blood circulation, and it is highly expressed in enterocytes, where it enables iron absorption, in erythroblasts and macrophages, where it functions in iron recycling, and in hepatocytes for iron storage. While FPN1a isoform is ubiquitously expressed, FPN1b isoform is selectively expressed in enterocytes and erythroblasts. The existence of these two FPN isoforms enable these two cell types to express FPN protein under iron deficiency, thereby providing iron to the circulating blood where it becomes available to essential cells and tissues. FPN is also abundant in mature RBCs where it exerts a protective effect against iron-induced oxidative stress. See the main text for detail.
FPN1b mRNA is also highly expressed in erythroblasts (Fig. 3). While the FPN1b transcript accounts for ~20% of total FPN mRNA in the small intestine, it accounts for ~40% of FPN mRNA in the bone marrow and ~60% in erythroblasts, suggesting that FPN1b likewise plays an indispensable role in erythropoiesis [82]. During erythroid differentiation and maturation, erythropoietin binds to the EPO receptor of erythroblasts to promote their proliferation and differentiation to proerythroblasts. Proerythroblasts subsequently develop into basophilic, polychromatic, and orthochromatic erythroblasts. Erythroblasts then lose their nuclei and develop into reticulocytes, which lose their mitochondria and mature into RBCs. Throughout this differentiation process, erythroblasts accumulate a large amount of iron via the Tf/TFR1 cycle for hemoglobin synthesis. TFR1 expression increases progressively in erythroblasts and throughout the proerythroblast stages. High levels of TFR1 are maintained through the orthochromatic and reticulocyte stages before its levels drop prior to RBC maturation[84]. In G1E cells, an erythroblast cell line engineered to differentiate from the erythroblastic to the orthochromatic stage upon administration of estrogen, FPN1b mRNA initially accounts for 60% of the total FPN mRNA. However, the proportion of FPN1b decreases to less than 10% by the end of differentiation (equivalent to the orthochromatic stage)[82]. In contrast, FPN1a mRNA increases from less than 40% of the total FPN mRNA at the beginning of differentiation to more than 90%. As differentiation nears completion, thereby largely restoring IRP-dependent regulation of FPN expression. Of note, FPN protein levels gradually decrease during erythroblast differentiation, suggesting that FPN1b mRNA is the major contributor to FPN protein synthesis[10, 11, 82]. The switch from FPN1b to FPN1a mRNA over the course of differentiation suggests that these two mRNAs play distinct roles during erythropoiesis. FPN1b mRNA enables erythroblasts to express FPN regardless of cellular iron status, whereas FPN1a mRNA ensures that sufficient cellular iron is retained in the developing erythroblast to support hemoglobin synthesis. The high expression of FPN1b mRNA in erythroblasts allows these precursors to export iron even when IRPs are highly active. As a result, erythroblasts can retain their iron stores only when hepcidin levels increase due to liver iron sufficiency[85](Fig. 3). Thus, by reducing the amount of iron invested in RBC production, the body can spare iron for other essential cells and organs such as the brain and heart, to survive iron scarcity. Once an iron-replete state is restored, the body can start reactivating erythropoiesis once again. Therefore, the expression of FPN1b mRNA enables the system to finely tune iron distribution among all tissues, ensuring survival of the organism under iron scarcity.
The abundance of FPN in mature RBCs implies that FPN plays a critical role in the mature RBCs[10, 11]. Interestingly, FPN-deficient RBCs have a much shorter lifespan than wild-type and undergo increased hemolysis both in vitro and in vivo due to their inability to export free iron and protect themselves from iron-mediated oxidative damage[10, 11]. The increased hemolysis and shorter lifespan of FPN-deficient RBCs underscore the essential role that FPN plays in the antioxidative system of RBCs[10, 11].
The FPN1b mRNA isoform is conserved among different species and its promoter has a conserved GATA1 transcription factor binding site, consistent with the cell-specific expression in erythroid cells[82, 86]. However, the transcriptional mechanisms that regulate FPN1b and FPN1a expression and their switching of promoter use during erythroid differentiation are not yet understood. Additionally, the FPN1b mRNA is specifically expressed in the epithelial cells of the intestine and in erythroblast precursors. The physiological significance remains to be fully explored in vivo preferably through the use of promoter-specific knockout animal data that would eliminate expression of one of the two alternative transcripts.
The role of ferritin-bound iron availability and ferritinophagy in erythropoiesis
The terminal step of erythroblast maturation relies on the exceptionally high amounts of iron needed to support hemoglobin synthesis. To prevent toxicity due to free cytosolic iron, the majority of the endocytosed iron is stored into ferritin. The recent characterization of two ferritin regulators, poly rC-binding protein 1 (PCBP1) [87] and nuclear receptor coactivator 4 (NCOA4) [88] that facilitate the obligatory flux of iron through ferritin has dissected an important route of iron trafficking in developing erythroid cells, in which PCBP1 was shown to function as an iron chaperone that delivers iron to ferritin and NCOA4 as a selective cargo receptor for the autophagic turnover of ferritin. Consistently, depletion of PCBP1 in murine erythroid cells led to decreased iron incorporation into ferritin, which impaired hemoglobinization[87]. A conditional Pcbp1 knockout (ko) mouse model showed similar phenotypes and developed microcytic anemia regardless of cellular iron availability. Interestingly, prolonged PCBP1 depletion resulted in systemic iron deficiency that was shown to activate the EPO-ERFE (erythroferrone)-hepcidin regulatory axis to increase iron uptake and RBC production[87, 89].
Insights into how iron is delivered to ferritin have been followed by progress in understanding how iron is released from ferritin. Several independent groups have characterized the role of NCOA4 as a receptor for the autophagic degradation of ferritin in the lysosomes by a process called ferritinophagy [87, 88, 90, 91]. Depletion of NCOA4 in cell culture, and in zebrafish and mice resulted in hemoglobinization defects [87, 88, 90]. Similar phenotypes were observed in two different mouse models, the global Ncoa4-ko [92] and the Ncoa4 conditional knockout [93]. In both cases, mice developed microcytic anemia with an overall decrease in RBC count and hemoglobin levels, despite an inappropriate accumulation of tissue iron into ferritin, which was indicative of inefficient iron mobilization from ferritin in the absence of Ncoa4 [92]. Remarkably, the anemia observed in the adult mice was less severe as compared to the mice examined at post-natal stage [93], suggesting the potential activation of an Ncoa4-independent pathway that reversed the progression of acute anemia at adulthood. A separate Ncoa4 ko mouse model, in which Ncoa4 was genetically ablated in the iron-rich Sv129/J mouse strain, provided further insights into the tissue specific physiological role of Ncoa4 [94]. Three- to nine-month-old Ncoa4 ko mice in the iron rich background showed microcytic red cells without anemia. Transferrin saturation, serum iron levels and liver, spleen and kidney non-heme iron content were comparable to those in WT mice, despite the abnormal retention of tissue iron into ferritin, which activated a signal of iron deficiency, with upregulation of TfR1, likely due to a reduced free iron pool secondary to ferritin iron sequestration [94]. Additionally, to assess the erythropoietic capacity of the hematopoietic stem cells, the authors performed a reciprocal bone marrow transplant experiment between WT and Ncoa4 ko mice, in which the lethally irradiated Ncoa4 ko mice were transplanted with the bone marrow derived from the WT mice and viceversa[94]. Two months after the transplant, both genotypes exhibited full recovery with comparable RBC count and hemoglobin levels. The only remaining abnormalities were the low erythrocyte MCV (mean corpuscular volume) and MCH (mean corpuscular hemoglobin) of WT mice transplanted with Ncoa4-ko bone marrow. Overall, these experiments pointed to the loss of Ncoa4 exclusively in bone marrow-derived cells as the leading cause of microcytosis. However, considering not only the normal erythropoiesis reported in the Ncoa4-ko mice but also the ability of ko bone marrow cells to completely reconstitute erythropoiesis in lethally irradiated animals, the authors ruled out that a major defect in erythroid precursors lacking Ncoa4 was likely to occur in vivo[94]. While further investigation on the role of NCOA4 in erythropoiesis is required, it is plausible that NCOA4 plays a crucial role in hemoglobinization and erythropoiesis only during chronic iron deficiency and/or at the post-natal stages and that additional mechanisms, which can bypass the requirement of NCOA4, may be triggered later during the animal’s life. Consistently, a long-term depletion of NCOA4, was found to activate a compensatory mechanism via the HIF-2α-EPO axis to normalize the acute anemia and erythropoiesis defects in the conditional Ncoa4 ko mouse model[93]. Nevertheless, while depletion of Ncoa4 does not seem to have a profound impact on erythroid differentiation [93, 94], ferritinophagy in macrophages has been found to play a significant role in erythropoiesis [94].
NCOA4-mediated ferritinophagy is responsive to cellular iron status and is activated in response to systemic iron deficiency[90]. Conversely, under iron-replete conditions, NCOA4 is targeted for proteasomal degradation[88] by the E3 ubiquitin ligase HERC2 (HECT and RLD domain containing E3 ubiquitin protein ligase 2), thereby promoting the storage of iron safely into the ferritin heteropolymers[88]. Interestingly, the HERC2 binding site on NCOA4 overlaps with the ferritin H-subunit binding site suggesting the possibility that competitive binding may regulate the steady-state levels of NCOA4 in vivo[90]. Given the role of NCOA4 in releasing free iron into the cytosol, an aberrant increase in the activity of NCOA4 may lead to pathologies associated with elevated free iron levels [90, 95]. Therefore, NCOA4 activity must be regulated at multiple levels. In addition to its regulation at the post-translational level by HERC2, the transcriptional regulation of NCOA4 by GATA-1 in erythroid lineages[96], and HIF1α and HIF2α transcriptional factors in hepatocytes has also been reported[97]. NCOA4 was originally discovered in a yeast-two hybrid screen as a coactivator of androgen receptor[98]. Subsequently, it was also reported as a regulator of other nuclear receptors and a repressor of DNA replication[99]. A recent study reported the regulation of NCOA4 by the thyroid hormone, which promoted its binding to chromatin regions and induced a transcriptional program supporting erythropoiesis[100]. Further investigations are required to reconcile the various reported roles of NCOA4 and their potential involvement in cellular iron homeostasis. Taken together, the studies summarized here support a model whereby a balance in PCBP1 and NCOA4 levels regulate iron storage into ferritin and iron mobilization via ferritinophagy during the early to mid-stages of erythroid differentiation.
Iron sulfur cluster cofactors are required at several steps in mammalian heme biosynthesis
As discussed throughout the previous sections of this review, heme biosynthesis and iron-sulfur (Fe-S) cluster biogenesis are the two major pathways that utilize iron in cells [9]. Fe-S clusters (ISCs) are ubiquitous cofactors essential to numerous fundamental cellular processes, including mitochondrial respiration, photosynthesis, metabolism, nitrogen fixation, DNA replication and repair, tRNA modifications, cell growth and proliferation [101, 102]. ISCs are composed of iron and inorganic sulfide and are typically, but not exclusively, ligated by cysteinyl sulfur in proteins [103]. The most common types are the rhombic [2Fe-2S] clusters, which are present in enzymes such as mammalian ferrochelatase, mitochondrial respiratory complexes I and II, ferredoxins, and Rieske proteins, and the tetranuclear [4Fe-4S] clusters [103]. ISCs are by far the most flexible cofactors in facilitating a variety of chemical activities, including electron transfer, as in the mitochondrial respiratory chain complexes, binding of substrates to one of the irons in the cubane clusters of a class of dehydratases that includes mitochondrial and cytosolic aconitases (ACO2 and ACO1), enabling radical reactions, as in the radical S-adenosylmethionine (SAM) family of enzymes [104], and functioning as iron and oxygen sensors [105]. Assembly of ISCs and their insertion into apoproteins involves the function of complex cellular machineries that operate in parallel in the mitochondrial and cytosolic/nuclear compartments of mammalian cells[106–108]. De novo ISC assembly involves an initial critical step catalyzed by a pyridoxal-phosphate (PLP)-dependent homodimeric transaminase, the cysteine desulfurase NFS1, which, in complex with the accessory protein LYRM4, the allosteric effector frataxin, and the acyl-carrier protein, converts cysteine into alanine and donates sulfur for the initial ISC assembly on the main scaffold protein ISCU[101, 107] (Fig. 4A). Transfer of newly assembled ISCs downstream of the main scaffold protein ISCU in mammalian cells relies on the activity of a highly conserved chaperone/cochaperone system analogous to the yeast Ssq1/Jac1 and the bacterial HscA/HscB complexes[109–112]. In mammalian cells, the multifunctional member of the HSP70 family, HSPA9 (also known as GRP75), works with the specialized DnaJ-type III protein, HSC20 (also referred to as DNAJC20 or HSCB), to either directly facilitate ISC transfer to recipient proteins or to secondary carriers, which then target specific recipients [109, 113–116](Fig. 4A). Given the involvement of Fe-S enzymes in several fundamental cellular processes, it is not surprising that defects in the components of the ISC biogenesis pathway cause an increasingly recognized number of rare human diseases[102, 115, 117].
Figure 4. An overview of the two main pathways utilizing iron in mammalian cells.
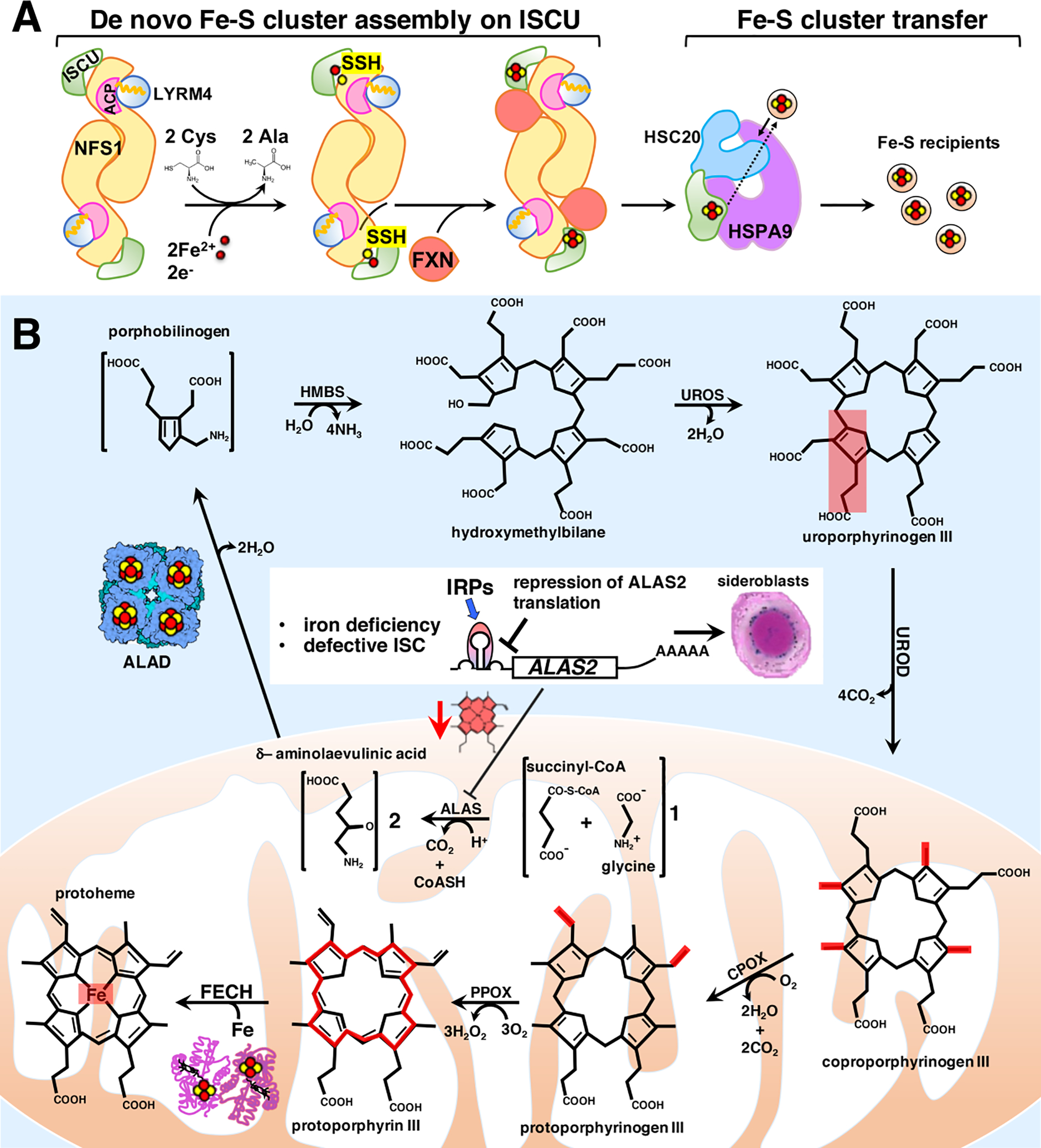
A. Iron-sulfur cluster (ISC) biogenesis in mammalian cells: an overview of the main steps. Nascent ISCs are assembled de novo on the main scaffold protein ISCU. A cysteine desulfurase, NFS1, forms a dimer to which monomers of the primary scaffold ISCU bind at either end. LYRM4 (aka ISD11) and acyl carrier protein (ACP) with its bound acyl chain are structural components of the core complex in eukaryotes. NFS1, aided by its cofactor pyridoxal phosphate (not shown), provides inorganic sulfur, abstracted from cysteine, to the nascent cluster. Transient binding of frataxin (FXN) in a pocket-like region between NFS1 and ISCU promotes sulfur transfer from NFS1 to ISCU. The cluster assembles upon ISCU when iron is provided together with the reducing equivalents needed to generate the final electronic configuration of the cluster. A chaperone-cochaperone complex binds to ISCU and facilitates direct cluster transfer to recipient proteins or to intermediary carriers which then target specific recipients.
B. Overview of the main steps of heme biosynthesis in which the intersections with the ISC biogenesis pathway are highlighted. Heme is synthesized in eight sequential steps that take place in the mitochondrial matrix and in the cytosol of mammalian cells. Four major points of intersection have been characterized between ISC biogenesis and heme biosynthesis in mammalian cells (see the main text for detailed description). Mitochondrial dysfunction due to defects in heme biosynthesis or in ISC biogenesis is the leading cause of a heterogenous group of inherited diseases, known as congenital sideroblastic anemias (see main text for details), characterized by ineffective heme biosynthesis and mitochondrial iron overload in erythroid progenitors, in which iron accumulation in the mitochondria surrounding the nucleus gives erythroblasts their characteristic appearance of ringed sideroblasts.
On the other hand, the most abundant iron cofactor in mammals, heme, consists of the protoporphyrin IX ring complexed with ferrous iron and it is necessary for the function of multiple proteins, including hemoglobin and myoglobin, cytochromes of the electron transport chain, catalase, and nitric oxide synthase. Heme is synthesized in eight sequential enzymatic steps [118](Fig. 4B), the first of which takes place in the mitochondrial matrix, where 5-aminolevulinate synthase (ALAS) catalyzes the condensation of succinyl-CoA with glycine to generate aminolaevulinic acid (ALA) [119]. Vertebrates rely on the activities of two ALAS enzymes, a ubiquitously expressed ALAS1 and the erythroid specific ALAS2 [120, 121]. The mRNA of ALAS2 has an IRE in its 5’-UTR, which permits translational regulation by IRPs. Interestingly, the reaction catalyzed by ALAS2, which is the rate-limiting step of heme biosynthesis, represents one of four major points at which the heme and ISC biosynthetic pathways intersect. Since IRP1 loses its IRE-binding activity when it ligates an ISC, ALAS2 protein levels are indirectly regulated by ISC biogenesis, because in conditions of iron deficiency or defective ISC biogenesis, IRP1 is converted into its apo form, which binds to the IRE present in the 5’UTR of the ALAS2 mRNA and represses its translation (Fig. 4B), thereby preventing accumulation of toxic porphyrin intermediates when heme biosynthesis cannot be sustained due to limited availability of iron. A compelling point of intersection between ISC biogenesis and heme biosynthesis came from studies in zebrafish deficient in the ISC biogenesis protein glutaredoxin 5 (GLRX5, in human)[122]. Genetic experiments demonstrated that in the shiraz zebrafish mutants, characterized by hypochromic anemia, loss of glutaredoxin 5 led to translational repression of ALAS2 mRNA due to activation of IRP1[122]. Subsequent studies in fibroblasts derived from a patient affected by sideroblastic anemia caused by an inactivating mutation in GLRX5 and in cells depleted of GLRX5 by short interfering RNAs (siRNAs) confirmed that hyperactivation of IRP1 discrupted cellular iron homeostasis and repressed ALAS2 translation upon loss of function of GLRX5 [123, 124]. A recent study revealed an additional point of intersection between heme biosynthesis and ISC biogenesis that hinges on the presence of an oxidation-sensitive [2Fe-2S] cluster in the C-terminal domain of the F-box and leucine-rich repeats protein FBXL5[125] that recognizes IRP2 and promotes its iron- and oxygen-dependent degradation. Interestingly, studies for over a decade reported increased IRE-binding activities of IRPs upon loss of components of the ISC biogenesis machinery[109, 123, 126–128]. While loss of the cubane cluster in IRP1 in cells with defective ISC biogenesis accounted for its increased IRE-binding activity, the mechanism that led to stabilization of IRP2 upon loss of ISC biogenesis components had previously remained elusive until the discovery of an ISC in FBXL5[31, 125]. By impairing ISC incorporation into FBXL5, defective Fe-S cluster biogenesis hampers the interaction of FBXL5 with IRP2, leading to stabilization of IRP2 and consequently to the increase in its IRE-binding activity that represses ALAS2 translation (Fig. 4B).
For the sake of completeness, we must say that ALAS1, which catalyzes in non-erythroid cells the same rate-limiting step of heme biosynthesis as ALAS2, lacks a 5’-IRE and is instead subject to a negative feedback regulation by cellular heme content[129, 130].
Once synthesized in mitochondria, ALA is exported to the cytosol, where ALA dehydratase (ALAD) catalyzes the second step of heme biosynthesis by condensing two ALA molecules into porphobilinogen (Fig. 4B). A recent study, contributed by our group in collaboration with colleagues at the Penn State University, shed light on a previously unrecognized connection between heme biosynthesis and ISC biogenesis with the discovery that human ALAD is a Fe-S protein[131]. Our previous investigations had defined the requirement of a three amino acid residue sequence, known as LYR motif, to serve as a binding site for the cochaperone HSC20 dedicated to ISC biogenesis[109, 116]. The LYR motif was experimentally defined as a three amino acid residue sequence that contained an aliphatic residue in the first position, usually leucine, isoleucine, alanine or valine, either tyrosine or phenylalanine in the second position, and arginine or lysine in the third position[116]. We therefore screened heme biosynthetic enzymes for the presence of LYR-like motifs and found that ALAD contained an A306F307R308 motif, that was shown to be required for binding to the cochaperone HSC20 and for acquisition of a [4Fe-4S] cluster by ALAD, yielding an enzyme with a total of 8 cubane clusters in its most active octameric form[131].
After three additional enzymatic reactions in the cytosol and two in mitochondria, the final insertion of ferrous iron into protoporphyrin IX occurs in the mitochondrial matrix and is catalyzed by ferrochelatase (FECH) (Fig. 4B). Ferrochelatase of higher eukaryotes, but not yeast, contains a [2Fe-2S] cluster[132] that was found to contribute to the structural stabilization of the enzyme[133, 134], thereby representing yet another point of convergence between the heme and ISC biosynthetic pathways. Thus, defects that interfere with ISC biogenesis impair heme biosynthesis by repressing ALAS2 synthesis in erythroid cells, and by inactivating the second and the last steps of heme biosynthesis catalyzed by the ISC enzymes ALAD and FECH, respectively (Fig. 4B).
In addition to the already mentioned points of intersection between heme biosynthesis and ISC biogenesis, impaired Fe-S cluster biogenesis has been reported to elicit profound alterations in cellular iron homeostasis[61, 115]. Mutations in several ISC biogenesis factors, including GLRX5[122, 123], HSC20[135] and HSPA9[136] have been reported to cause inherited forms of sideroblastic anemia, a heterogenous group of bone marrow disorders defined by an impaired ability to produce normal red blood cells and by pathological iron accumulation in the mitochondria of erythroid precursors[137, 138] (Fig. 4B). Congenital sideroblastic anemias are inherited diseases of mitochondrial dysfunction due to defects in heme biosynthesis, ISC biogenesis, generalized mitochondrial protein synthesis, or the synthesis of specific mitochondrial proteins involved in oxidative phosphorylation[137]. Interestingly, mitochondrial iron accumulation has also been reported in individuals affected by X-linked sideroblastic anemia and ataxia caused by mutations in the ATP-binding cassette (ABC) transporter of the inner mitochondrial membrane ABCB7[139, 140]. The yeast ortholog of ABCB7, Atm1, has been proposed to export a special sulfur species from the mitochondrial matrix to be utilized in the cytosol as a building block for cytosolic Fe-S cluster biogenesis[141, 142]. However, the phenotype associated with loss-of-function mutations in ABCB7 remains largely unexplained by the proposed role assigned to its yeast ortholog Atm1.
Multiple studies for more than two decades have demonstrated the existence of a de novo cytosolic ISC pathway in mammalian cells, which can supply the elemental components (i.e., iron and sulfur) and the biogenesis proteins required to build ISCs in the cytosol of mammalian cells[102, 108]. Consistent with this model, the pool of NFS1 that localizes to the cytosol[143] is a functional enzyme that mobilizes sulfur from cysteine[144, 145], an observation that obviates the need for the export of a sulfur-containing compound out of mitochondria. Alternative isoforms of the core ISC biogenesis components have been detected in the cytosol of mammalian cells[109, 126, 127, 143, 146, 147], suggesting that ISC biogenesis machineries independently operate in parallel to generate nascent ISCs in multiple subcellular compartments of multicellular eukaryotes.
To interrogate the role of ABCB7 and examine the time-dependent consequences of its loss in mammalian cells, we generated inducible ABCB7-knockdown cell lines [148]. We found that depletion of ABCB7 led to significant loss of mitochondrial Fe-S proteins, which preceded the development of milder defects in cytosolic Fe-S enzymes [148]. Similarly, studies in Atm1-depleted yeast cells reported a profound growth defect caused by mitochondrial dysfunction, which included loss of oxidative phosphorylation and defective heme biosynthesis [149–152]. Consistent with the phenotype observed in patients affected by inactivating mutations in ABCB7 [153], loss of the transporter in erythroid cells altered cellular iron distribution and caused mitochondrial iron overload due to activation of the IRE-binding activities of IRPs in the cytosol and to upregulation of the mitochondrial iron importer, mitoferrin-1 (MFRN1) [148]. Despite the exceptionally large amount of iron imported in mitochondria, erythroid cells lacking ABCB7 showed a profound hemoglobinization defect and underwent apoptosis triggered by oxidative stress [148]. By combining chemical crosslinking, tandem mass spectrometry and mutational analyses, we were able to characterize a complex formed of ferrochelatase, ABCB7 and ABCB10 [148]. We found that a dimeric ferrochelatase physically bridged ABCB7 and ABCB10 homodimers by binding near the nucleotide-binding domains of each ABC transporter. A previous model was proposed based on the identification of a FECH/ABCB10/MFRN1 complex [154], in which the interaction of FECH with ABCB10 and MFRN1 was required to integrate mitochondrial iron import with its utilization for heme synthesis. In the same study [154], a separate interaction of FECH with ABCB7 was also reported. With the identification of an ABCB7-FECH-ABCB10 complex, our studies [148], not only underscored the importance of ABCB7 for the integrity of mitochondrial Fe-S biogenesis and for the maintenance of cellular iron homeostasis, but also provided the biochemical characterization of a multiprotein complex required for heme biosynthesis, suggesting that more definitive experiments employing the coordinated activity of the entire complex may aid identification of its physiological substrates.
Mitochondrial iron accumulation is also a distinctive biochemical feature of cells derived from patients affected by Friedreich ataxia (FRDA) [155–157]. FRDA patients harbor a homozygous GAA repeat expansion within intron 1 of the frataxin (FXN) gene [158], which causes profound loss of the ISC biogenesis protein frataxin (FXN). Several hypotheses have been proposed to explain the intimate connection between Fe–S cluster biogenesis and iron homeostasis. One possibility could be that an ISC protein is directly or indirectly involved in the sensing of iron levels in mitochondria, so that compromised Fe–S biogenesis might be registered as insufficient provision of iron to the mitochondrial compartment. This could result in a feedback regulation and activation of a response that includes the increased delivery of iron to mitochondria and concomitant depletion of the cytosolic iron pool, which finally engages the cell in a vicious cycle in which increased cellular iron uptake further exacerbates mitochondrial iron overload [159, 160]. In support to this idea, transcriptional expression of the mitochondrial iron importer mitoferrin was increased in frataxin-deficient mouse hearts [161], and in muscle biopsies from ISCU myopathy patients [162]. An attractive possibility is also that a molecule exported from mitochondria acts as a signal that drives iron import. Active export of peptides from the mitochondrial matrix to the intermembrane space in S. cerevisiae is accomplished by the ABC transporter Mdl1, which was proposed to be crucial for yet-to-be-determined signal transduction pathways in yeast [163]. Several cellular pathways, such as the stress caused by protein misfolding in the mitochondrial matrix, have been dissected, and they were found to rely on export of signaling molecules, which activated nuclear-encoded mitochondrial genes [164–166]. Uncovering the molecular mechanisms that coordinate functional ISC biogenesis and maintenance of cellular iron homeostasis will likely illuminate the elaborate mechanisms required to manage iron availability and distribution in the cell.
Concluding remarks
As outlined throughout this review, heme biosynthesis and iron-sulfur cluster biogenesis are the two major pathways in mammalian cells that rely on iron utilization and that are regulated by iron availability. Intriguingly, there are several points of intersection between the two pathways. In erythroid cells, the ALAS2 transcript that encodes the first heme biosynthetic enzyme contains a 5’IRE that enables IRPs to repress its translation when cells lack sufficient iron to support heme synthesis. The second enzyme of heme synthesis, ALAD, depends on the insertion of a cubane iron sulfur cluster for function, and the terminal enzyme of the pathway, mammalian ferrochelatase, requires an iron sulfur cluster to catalyze the insertion of iron into protoporphyrin IX.
IRP1 and IRP2 sense cytosolic iron concentrations and post-transcriptionally regulate the expression of iron metabolism genes to optimize iron availability for essential cellular processes. Mutations affecting either IRP1 or IRP2 cause distinctive phenotypes highlighting that, despite their inherently redundant functions, differential expression and relative abundance dictate which IRP dominates regulation of cellular iron homeostasis in a specific cell type and/or tissue. IRP2-deficient patients develop neurodegeneration and anemia, IRP1 loss-of-function mutations cause polycythemia and pulmonary hypertension, and IRP1 gain of function mutations cause anemia.
Erythroid cells express a FPN transcript that cannot be translationally repressed by IRPs, permitting red cells to return iron to the circulation and to other tissues in conditions of systemic iron deficiency. This mechanism protects the organism from rendering other tissues iron deficient due to overwhelming iron consumption by erythropoiesis and may explain why iron-deficiency anemia is one of the first physiological indicators of iron deficiency.
In this review, we have highlighted some of the mechanisms that regulate iron homeostasis and the pathways that utilize iron. Several questions remain unanswered, and further investigations will be required to fully understand mammalian iron homeostasis and the many intersections of iron utilizing pathways at both the cellular and systemic level.
Acknowledgements:
the authors would like to acknowledge support from the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Funding: this work was supported by the Intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
List of Abbreviations (Alphabetical)
- ABC
ATP-binding cassette
- ABCB7
ABC transporter of the inner mitochondrial membrane, subfamily B, member 7
- ACO1
cytosolic aconitase
- ACO2
mitochondrial aconitase
- ACP
acyl carrier protein
- ALA
aminolaevulinic acid
- ALAD
ALA dehydratase
- ALAS
5-aminolevulinate synthase
- CDC14A
cell division cycle 14A
- CLPX
ATP-dependent Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit X
- DcytB
duodenal cytochrome b516
- DMT1
divalent iron transporter 1
- dSDH
drosophila succinate dehydrogenase B
- eALAS/ALAS2
erythroid specific 5-aminolevulinate synthase
- EPO
erythropoietin
- EPOR
erythropoietin receptor
- ERFE
erythroferrone
- FBXL5
F-box and leucine-rich repeat protein 5
- Fe-S
iron-sulfur
- FECH
ferrochelatase
- FPN
ferroportin
- FPN1a
major FPN mRNA transcript with 5’UTR IRE
- FPN1b
alternative FPN mRNA transcript without IRE
- FRDA
Friedreich ataxia
- FXN
frataxin
- GLRX5
glutaredoxin 5
- HBB/HBA
hemoglobin subunit B/A
- HERC2
HECT and RLD domain containing E3 ubiquitin protein ligase 2
- Hif2α/EPASI
hypoxia-inducible factor 2 alpha/endothelial PAS domain protein 1
- IRE
iron-responsive elements
- IRP1/IRP2
iron regulatory protein ½
- ISC(s)
Fe-S cluster(s)
- KO
knockout
- MFRN1
mitoferrin-1
- MRCKa
myotonic dystrophy kinase-related Cdc42-binding kinase α
- NCOA4
nuclear receptor coactivator
- PAH
pulmonary arterial hypertension
- PCBP1
poly rC-binding protein 1
- Pfn2
profilin 2
- PHDs
prolyl hydroxylases
- PLP
pyridoxal-phosphate
- RBC
red blood cell
- RVP
right ventricular pressure
- SAM
S-adenosylmethionine
- siRNAs
short interfering RNAs
- TF
transferrin
- TFR1
transferrin receptor
- UTR
untranslated region
- VHL
Von-Hippel-Lindau
Footnotes
Conflict of interest statement: the authors declare that they have no conflict of interest related to the work discussed in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hentze MW, Muckenthaler MU, Andrews NC, Balancing acts: molecular control of mammalian iron metabolism, Cell 117(3) (2004) 285–97. [DOI] [PubMed] [Google Scholar]
- [2].McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ, An iron-regulated ferric reductase associated with the absorption of dietary iron, Science 291(5509) (2001) 1755–9. [DOI] [PubMed] [Google Scholar]
- [3].Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA, Cloning and characterization of a mammalian proton-coupled metal-ion transporter, Nature 388(6641) (1997) 482–8. [DOI] [PubMed] [Google Scholar]
- [4].Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI, Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter, Nature 403(6771) (2000) 776–81. [DOI] [PubMed] [Google Scholar]
- [5].Abboud S, Haile DJ, A novel mammalian iron-regulated protein involved in intracellular iron metabolism, J Biol Chem 275(26) (2000) 19906–12. [DOI] [PubMed] [Google Scholar]
- [6].McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ, A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation, Mol Cell 5(2) (2000) 299–309. [DOI] [PubMed] [Google Scholar]
- [7].Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JM, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, et al. , Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts, Nature 349(6309) (1991) 541–4. [DOI] [PubMed] [Google Scholar]
- [8].Zhang DL, Ghosh MC, Rouault TA, The physiological functions of iron regulatory proteins in iron homeostasis - an update, Front Pharmacol 5 (2014) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Muckenthaler MU, Rivella S, Hentze MW, Galy B, A Red Carpet for Iron Metabolism, Cell 168(3) (2017) 344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang DL, Ghosh MC, Ollivierre H, Li Y, Rouault TA, Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress, Blood 132(19) (2018) 2078–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang DL, Wu J, Shah BN, Greutelaers KC, Ghosh MC, Ollivierre H, Su XZ, Thuma PE, Bedu-Addo G, Mockenhaupt FP, Gordeuk VR, Rouault TA, Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk, Science 359(6383) (2018) 1520–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J, Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization, Science 306(5704) (2004) 2090–3. [DOI] [PubMed] [Google Scholar]
- [13].Zimmerman C, Klein KC, Kiser PK, Singh AR, Firestein BL, Riba SC, Lingappa JR, Identification of a host protein essential for assembly of immature HIV-1 capsids, Nature 415(6867) (2002) 88–92. [DOI] [PubMed] [Google Scholar]
- [14].Hentze MW, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, Klausner RD, Identification of the iron-responsive element for the translational regulation of human ferritin mRNA, Science 238(4833) (1987) 1570–3. [DOI] [PubMed] [Google Scholar]
- [15].Leibold EA, Munro HN, Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5’ untranslated region of ferritin heavy- and light-subunit mRNAs, Proc Natl Acad Sci U S A 85(7) (1988) 2171–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kim HY, LaVaute T, Iwai K, Klausner RD, Rouault TA, Identification of a conserved and functional iron-responsive element in the 5’-untranslated region of mammalian mitochondrial aconitase, J Biol Chem 271(39) (1996) 24226–30. [DOI] [PubMed] [Google Scholar]
- [17].Dandekar T, Stripecke R, Gray NK, Goossen B, Constable A, Johansson HE, Hentze MW, Identification of a novel iron-responsive element in murine and human erythroid delta-aminolevulinic acid synthase mRNA, EMBO J 10(7) (1991) 1903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sanchez M, Galy B, Muckenthaler MU, Hentze MW, Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency, Nat Struct Mol Biol 14(5) (2007) 420–6. [DOI] [PubMed] [Google Scholar]
- [19].Kohler SA, Henderson BR, Kuhn LC, Succinate dehydrogenase b mRNA of Drosophila melanogaster has a functional iron-responsive element in its 5’-untranslated region, J Biol Chem 270(51) (1995) 30781–6. [DOI] [PubMed] [Google Scholar]
- [20].Koeller DM, Casey JL, Hentze MW, Gerhardt EM, Chan LN, Klausner RD, Harford JB, A cytosolic protein binds to structural elements within the iron regulatory region of the transferrin receptor mRNA, Proc Natl Acad Sci U S A 86(10) (1989) 3574–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sanchez M, Galy B, Dandekar T, Bengert P, Vainshtein Y, Stolte J, Muckenthaler MU, Hentze MW, Iron regulation and the cell cycle: identification of an iron-responsive element in the 3’-untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy, J Biol Chem 281(32) (2006) 22865–74. [DOI] [PubMed] [Google Scholar]
- [22].Cmejla R, Petrak J, Cmejlova J, A novel iron responsive element in the 3’UTR of human MRCKalpha, Biochem Biophys Res Commun 341(1) (2006) 158–66. [DOI] [PubMed] [Google Scholar]
- [23].Luscieti S, Galy B, Gutierrez L, Reinke M, Couso J, Shvartsman M, Di Pascale A, Witke W, Hentze MW, Pilo Boyl P, Sanchez M, The actin-binding protein profilin 2 is a novel regulator of iron homeostasis, Blood 130(17) (2017) 1934–1945. [DOI] [PubMed] [Google Scholar]
- [24].Sanchez M, Galy B, Schwanhaeusser B, Blake J, Bahr-Ivacevic T, Benes V, Selbach M, Muckenthaler MU, Hentze MW, Iron regulatory protein-1 and −2: transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins, Blood 118(22) (2011) e168–79. [DOI] [PubMed] [Google Scholar]
- [25].Beaumont C, Leneuve P, Devaux I, Scoazec JY, Berthier M, Loiseau MN, Grandchamp B, Bonneau D, Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract, Nat Genet 11(4) (1995) 444–6. [DOI] [PubMed] [Google Scholar]
- [26].Girelli D, Corrocher R, Bisceglia L, Olivieri O, De Franceschi L, Zelante L, Gasparini P, Molecular basis for the recently described hereditary hyperferritinemia-cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the “Verona mutation”), Blood 86(11) (1995) 4050–3. [PubMed] [Google Scholar]
- [27].Allerson CR, Cazzola M, Rouault TA, Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia-cataract syndrome, J Biol Chem 274(37) (1999) 26439–47. [DOI] [PubMed] [Google Scholar]
- [28].Ducamp S, Luscieti S, Ferrer-Cortes X, Nicolas G, Manceau H, Peoc’h K, Yien YY, Kannengiesser C, Gouya L, Puy H, Sanchez M, A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria, Haematologica (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, Bruick RK, An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis, Science 326(5953) (2009) 722–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA, Control of iron homeostasis by an iron-regulated ubiquitin ligase, Science 326(5953) (2009) 718–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rouault TA, Maio N, How Oxidation of a Unique Iron-Sulfur Cluster in FBXL5 Regulates IRP2 Levels and Promotes Regulation of Iron Metabolism Proteins, Mol Cell 78(1) (2020) 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Smith SR, Ghosh MC, Ollivierre-Wilson H, Hang Tong W, Rouault TA, Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development, Blood Cells Mol Dis 36(2) (2006) 283–7. [DOI] [PubMed] [Google Scholar]
- [33].Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, Rouault TA, Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis, EMBO J 23(2) (2004) 386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R 3rd, Grinberg A, Love P, Tresser N, Rouault TA, Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice, Nat Genet 27(2) (2001) 209–14. [DOI] [PubMed] [Google Scholar]
- [35].Ghosh MC, Zhang DL, Jeong SY, Kovtunovych G, Ollivierre-Wilson H, Noguchi A, Tu T, Senecal T, Robinson G, Crooks DR, Tong WH, Ramaswamy K, Singh A, Graham BB, Tuder RM, Yu ZX, Eckhaus M, Lee J, Springer DA, Rouault TA, Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2alpha, Cell Metab 17(2) (2013) 271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Anderson SA, Nizzi CP, Chang YI, Deck KM, Schmidt PJ, Galy B, Damnernsawad A, Broman AT, Kendziorski C, Hentze MW, Fleming MD, Zhang J, Eisenstein RS, The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption, Cell Metab 17(2) (2013) 282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wilkinson N, Pantopoulos K, IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation, Blood 122(9) (2013) 1658–68. [DOI] [PubMed] [Google Scholar]
- [38].Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, Iliopoulos O, Small-molecule inhibitors of HIF-2a translation link its 5’UTR iron-responsive element to oxygen sensing, Mol Cell 32(6) (2008) 838–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ, The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis, Nature 399(6733) (1999) 271–5. [DOI] [PubMed] [Google Scholar]
- [40].Semenza GL, Hypoxia-inducible factors in physiology and medicine, Cell 148(3) (2012) 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kaelin WG Jr., Ratcliffe PJ, Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway, Mol Cell 30(4) (2008) 393–402. [DOI] [PubMed] [Google Scholar]
- [42].Ghosh MC, Zhang DL, Ollivierre H, Eckhaus MA, Rouault TA, Translational repression of HIF2alpha expression in mice with Chuvash polycythemia reverses polycythemia, J Clin Invest 128(4) (2018) 1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bento C, Genetic basis of congenital erythrocytosis, Int J Lab Hematol 40 Suppl 1 (2018) 62–67. [DOI] [PubMed] [Google Scholar]
- [44].Oliveira JL, Coon LM, Frederick LA, Hein M, Swanson KC, Savedra ME, Porter TR, Patnaik MM, Tefferi A, Pardanani A, Grebe SK, Viswanatha DS, Hoyer JD, Genotype-Phenotype Correlation of Hereditary Erythrocytosis Mutations, a single center experience, Am J Hematol (2018). [DOI] [PubMed] [Google Scholar]
- [45].Oskarsson GR, Oddsson A, Magnusson MK, Kristjansson RP, Halldorsson GH, Ferkingstad E, Zink F, Helgadottir A, Ivarsdottir EV, Arnadottir GA, Jensson BO, Katrinardottir H, Sveinbjornsson G, Kristinsdottir AM, Lee AL, Saemundsdottir J, Stefansdottir L, Sigurdsson JK, Davidsson OB, Benonisdottir S, Jonasdottir A, Jonasdottir A, Jonsson S, Gudmundsson RL, Asselbergs FW, Tragante V, Gunnarsson B, Masson G, Thorleifsson G, Rafnar T, Holm H, Olafsson I, Onundarson PT, Gudbjartsson DF, Norddahl GL, Thorsteinsdottir U, Sulem P, Stefansson K, Predicted loss and gain of function mutations in ACO1 are associated with erythropoiesis, Commun Biol 3(1) (2020) 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Semenza GL, The Genomics and Genetics of Oxygen Homeostasis, Annu Rev Genomics Hum Genet (2020). [DOI] [PubMed] [Google Scholar]
- [47].Mallik N, Sharma P, Kaur Hira J, Chhabra S, Sreedharanunni S, Kumar N, Naseem S, Sachdeva MUS, Ahluwalia J, Malhotra P, Varma N, Varma S, Das R, Genetic basis of unexplained erythrocytosis in Indian patients, Eur J Haematol 103(2) (2019) 124–130. [DOI] [PubMed] [Google Scholar]
- [48].Zmajkovic J, Lundberg P, Nienhold R, Torgersen ML, Sundan A, Waage A, Skoda RC, A Gain-of-Function Mutation in EPO in Familial Erythrocytosis, N Engl J Med 378(10) (2018) 924–930. [DOI] [PubMed] [Google Scholar]
- [49].James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W, A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera, Nature 434(7037) (2005) 1144–8. [DOI] [PubMed] [Google Scholar]
- [50].Ghosh MC, Zhang DL, Ollivierre WH, Noguchi A, Springer DA, Linehan WM, Rouault TA, Therapeutic inhibition of HIF-2alpha reverses polycythemia and pulmonary hypertension in murine models of human diseases, Blood 137(18) (2021) 2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Casarrubea D, Viatte L, Hallas T, Vasanthakumar A, Eisenstein RS, Schumann K, Hentze MW, Galy B, Abnormal body iron distribution and erythropoiesis in a novel mouse model with inducible gain of iron regulatory protein (IRP)-1 function, J Mol Med (Berl) 91(7) (2013) 871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P, Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia, J Clin Invest 111(10) (2003) 1519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sergeyeva A, Gordeuk VR, Tokarev YN, Sokol L, Prchal JF, Prchal JT, Congenital polycythemia in Chuvashia, Blood 89(6) (1997) 2148–54. [PubMed] [Google Scholar]
- [54].Haase VH, The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease, Kidney international 69(8) (2006) 1302–7. [DOI] [PubMed] [Google Scholar]
- [55].Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH, Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo, J Clin Invest 117(4) (2007) 1068–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ghosh MC, Tong WH, Zhang D, Ollivierre-Wilson H, Singh A, Krishna MC, Mitchell JB, Rouault TA, Tempol-mediated activation of latent iron regulatory protein activity prevents symptoms of neurodegenerative disease in IRP2 knockout mice, Proc Natl Acad Sci U S A 105(33) (2008) 12028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, Wang B, Wehn PM, Yang H, Dixon DD, Czerwinski RM, Du X, Ged EL, Han G, Tan H, Wong T, Xie S, Josey JA, Wallace EM, 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzo nitrile (PT2977), a Hypoxia-Inducible Factor 2alpha (HIF-2alpha) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma, J Med Chem 62(15) (2019) 6876–6893. [DOI] [PubMed] [Google Scholar]
- [58].Choueiri, ER TP; Bauer TM; Merchan JR; Papadopoulos KP; McDermott DF; Michaelson MD; Appleman LJ; Thamake S; Zojwalla NJ; Jonasch E, Phase I/II study of the oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC), J Clin Oncol 38 (2020) abstr 611. [Google Scholar]
- [59].Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, Zhang X, Zhao YY, Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2alpha Inhibitor, Am J Respir Crit Care Med 198(11) (2018) 1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hu CJ, Poth JM, Zhang H, Flockton A, Laux A, Kumar S, McKeon B, Mouradian G, Li M, Riddle S, Pugliese SC, Brown RD, Wallace EM, Graham BB, Frid MG, Stenmark KR, Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension, Eur Respir J 54(6) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rouault TA, Maio N, Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways, J Biol Chem 292(31) (2017) 12744–12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pang Y, Gupta G, Yang C, Wang H, Huynh TT, Abdullaev Z, Pack SD, Percy MJ, Lappin TRJ, Zhuang Z, Pacak K, A novel splicing site IRP1 somatic mutation in a patient with pheochromocytoma and JAK2(V617F) positive polycythemia vera: a case report, BMC Cancer 18(1) (2018) 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cooperman SS, Meyron-Holtz EG, Olivierre-Wilson H, Ghosh MC, McConnell JP, Rouault TA, Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2, Blood 106(3) (2005) 1084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ghosh MC, Ollivierre-Wilson H, Cooperman S, Rouault TA, Reply to “Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse”, Nature Genetics 38 (2006) 969–970. [DOI] [PubMed] [Google Scholar]
- [65].Smith SR, Cooperman S, Lavaute T, Tresser N, Ghosh M, Meyron-Holtz E, Land W, Ollivierre H, Jortner B, Switzer R 3rd, Messing A, Rouault TA, Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies, Annals of the New York Academy of Sciences 1012 (2004) 65–83. [DOI] [PubMed] [Google Scholar]
- [66].Jeong SY, Crooks DR, Wilson-Ollivierre H, Ghosh MC, Sougrat R, Lee J, Cooperman S, Mitchell JB, Beaumont C, Rouault TA, Iron insufficiency compromises motor neurons and their mitochondrial function in Irp2-null mice, PLoS One 6(10) (2011) e25404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Galy B, Holter SM, Klopstock T, Ferring D, Becker L, Kaden S, Wurst W, Grone HJ, Hentze MW, Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse, Nat Genet 38(9) (2006) 967–9; discussion 969–70. [DOI] [PubMed] [Google Scholar]
- [68].Zumbrennen-Bullough KB, Becker L, Garrett L, Holter SM, Calzada-Wack J, Mossbrugger I, Quintanilla-Fend L, Racz I, Rathkolb B, Klopstock T, Wurst W, Zimmer A, Wolf E, Fuchs H, Gailus-Durner V, de Angelis MH, Romney SJ, Leibold EA, Abnormal brain iron metabolism in Irp2 deficient mice is associated with mild neurological and behavioral impairments, PLoS One 9(6) (2014) e98072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Santos M, Anderson CP, Neschen S, Zumbrennen-Bullough KB, Romney SJ, Kahle-Stephan M, Rathkolb B, Gailus-Durner V, Fuchs H, Wolf E, Rozman J, de Angelis MH, Cai WM, Rajan M, Hu J, Dedon PC, Leibold EA, Irp2 regulates insulin production through iron-mediated Cdkal1-catalyzed tRNA modification, Nat Commun 11(1) (2020) 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wilkinson N, Pantopoulos K, The IRP/IRE system in vivo: insights from mouse models, Front Pharmacol 5 (2014) 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ghosh MC, Zhang DL, Rouault TA, Iron misregulation and neurodegenerative disease in mouse models that lack iron regulatory proteins, Neurobiol Dis 81 (2015) 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Costain G, Ghosh MC, Maio N, Carnevale A, Si YC, Rouault TA, Yoon G, Absence of iron-responsive element-binding protein 2 causes a novel neurodegenerative syndrome, Brain 142(5) (2019) 1195–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cooper MS, Stark Z, Lunke S, Zhao T, Amor DJ, IREB2-associated neurodegeneration, Brain 142(8) (2019) e40. [DOI] [PubMed] [Google Scholar]
- [74].Maio N, Ghosh MC, Costain G, Carnevale A, Si Y, Yoon G, Rouault TA, Reply: IREB2-associated neurodegeneration, Brain 142(8) (2019) e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Moreno M, Ortega F, Xifra G, Ricart W, Fernandez-Real JM, Moreno-Navarrete JM, Cytosolic aconitase activity sustains adipogenic capacity of adipose tissue connecting iron metabolism and adipogenesis, FASEB J 29(4) (2015) 1529–39. [DOI] [PubMed] [Google Scholar]
- [76].Kim HY, Klausner RD, Rouault TA, Translational repressor activity is equivalent and is quantitatively predicted by in vitro RNA binding for two iron-responsive element-binding proteins, IRP1 and IRP2, J Biol Chem 270(10) (1995) 4983–6. [DOI] [PubMed] [Google Scholar]
- [77].Meyron-Holtz EG, Ghosh MC, Rouault TA, Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo, Science 306(5704) (2004) 2087–90. [DOI] [PubMed] [Google Scholar]
- [78].Aschemeyer S, Qiao B, Stefanova D, Valore EV, Sek AC, Ruwe TA, Vieth KR, Jung G, Casu C, Rivella S, Jormakka M, Mackenzie B, Ganz T, Nemeth E, Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin, Blood 131(8) (2018) 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Chaston T, Chung B, Mascarenhas M, Marks J, Patel B, Srai SK, Sharp P, Evidence for differential effects of hepcidin in macrophages and intestinal epithelial cells, Gut 57(3) (2008) 374–82. [DOI] [PubMed] [Google Scholar]
- [80].Mayr R, Janecke AR, Schranz M, Griffiths WJ, Vogel W, Pietrangelo A, Zoller H, Ferroportin disease: a systematic meta-analysis of clinical and molecular findings, J Hepatol 53(5) (2010) 941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Pietrangelo A, Ferroportin disease: pathogenesis, diagnosis and treatment, Haematologica 102(12) (2017) 1972–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA, A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression, Cell Metab 9(5) (2009) 461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cheng H, Leblond CP, Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types, Am J Anat 141(4) (1974) 537–61. [DOI] [PubMed] [Google Scholar]
- [84].Nandakumar SK, Ulirsch JC, Sankaran VG, Advances in understanding erythropoiesis: evolving perspectives, Br J Haematol 173(2) (2016) 206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhang DL, Senecal T, Ghosh MC, Ollivierre-Wilson H, Tu T, Rouault TA, Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts, Blood 118(10) (2011) 2868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Cianetti L, Segnalini P, Calzolari A, Morsilli O, Felicetti F, Ramoni C, Gabbianelli M, Testa U, Sposi NM, Expression of alternative transcripts of ferroportin-1 during human erythroid differentiation, Haematologica 90(12) (2005) 1595–606. [PubMed] [Google Scholar]
- [87].Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC, PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis, J Clin Invest 127(5) (2017) 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC, Harper JW, Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Philpott CC, The flux of iron through ferritin in erythrocyte development, Curr Opin Hematol 25(3) (2018) 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Santana-Codina N, Mancias JD, The Role of NCOA4-Mediated Ferritinophagy in Health and Disease, Pharmaceuticals (Basel) 11(4) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, Cantwell J, Luu C, Cornella-Taracido I, Harrington E, Fekkes P, Lei H, Fang Q, Digan ME, Burdick D, Powers AF, Helliwell SB, D’Aquin S, Bastien J, Wang H, Wiederschain D, Kuerth J, Bergman P, Schwalb D, Thomas J, Ugwonali S, Harbinski F, Tallarico J, Wilson CJ, Myer VE, Porter JA, Bussiere DE, Finan PM, Labow MA, Mao X, Hamann LG, Manning BD, Valdez RA, Nicholson T, Schirle M, Knapp MS, Keaney EP, Murphy LO, Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo, Nat Cell Biol 16(11) (2014) 1069–79. [DOI] [PubMed] [Google Scholar]
- [92].Bellelli R, Federico G, Matte A, Colecchia D, Iolascon A, Chiariello M, Santoro M, De Franceschi L, Carlomagno F, NCOA4 Deficiency Impairs Systemic Iron Homeostasis, Cell Rep 14(3) (2016) 411–421. [DOI] [PubMed] [Google Scholar]
- [93].Santana-Codina N, Gableske S, Quiles del Rey M, Malachowska B, Jedrychowski MP, Biancur DE, Schmidt PJ, Fleming MD, Fendler W, Harper JW, Kimmelman AC, Mancias JD, NCOA4 maintains murine erythropoiesis via cell autonomous and non-autonomous mechanisms, Haematologica 104(7) (2019) 1342–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Nai A, Lidonnici MR, Federico G, Pettinato M, Olivari V, Carrillo F, Geninatti Crich S, Ferrari G, Camaschella C, Silvestri L, Carlomagno F, NCOA4-mediated ferritinophagy in macrophages is crucial to sustain erythropoiesis in mice, Haematologica 106(3) (2021) 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ito J, Omiya S, Rusu MC, Ueda H, Murakawa T, Tanada Y, Abe H, Nakahara K, Asahi M, Taneike M, Nishida K, Shah AM, Otsu K, Iron derived from autophagy-mediated ferritin degradation induces cardiomyocyte death and heart failure in mice, Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, Weiss MJ, Global regulation of erythroid gene expression by transcription factor GATA-1, Blood 104(10) (2004) 3136–47. [DOI] [PubMed] [Google Scholar]
- [97].Li X, Lozovatsky L, Sukumaran A, Gonzalez L, Jain A, Liu D, Ayala-Lopez N, Finberg KE, NCOA4 is regulated by HIF and mediates mobilization of murine hepatic iron stores after blood loss, Blood 136(23) (2020) 2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Yeh S, Chang C, Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells, Proc Natl Acad Sci U S A 93(11) (1996) 5517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kollara A, Brown TJ, Expression and function of nuclear receptor co-activator 4: evidence of a potential role independent of co-activator activity, Cell Mol Life Sci 69(23) (2012) 3895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gao X, Lee HY, Li W, Platt RJ, Barrasa MI, Ma Q, Elmes RR, Rosenfeld MG, Lodish HF, Thyroid hormone receptor beta and NCOA4 regulate terminal erythrocyte differentiation, Proc Natl Acad Sci U S A 114(38) (2017) 10107–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Braymer JJ, Lill R, Iron-sulfur cluster biogenesis and trafficking in mitochondria, J Biol Chem 292(31) (2017) 12754–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Maio N, Rouault TA, Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis, Trends Biochem Sci 45(5) (2020) 411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Beinert H, Holm RH, Munck E, Iron-sulfur clusters: nature’s modular, multipurpose structures, Science 277(5326) (1997) 653–9. [DOI] [PubMed] [Google Scholar]
- [104].Landgraf BJ, McCarthy EL, Booker SJ, Radical S-Adenosylmethionine Enzymes in Human Health and Disease, Annu Rev Biochem 85 (2016) 485–514. [DOI] [PubMed] [Google Scholar]
- [105].Mettert EL, Kiley PJ, Reassessing the Structure and Function Relationship of the O2 Sensing Transcription Factor FNR, Antioxid Redox Signal 29(18) (2018) 1830–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kim KS, Maio N, Singh A, Rouault TA, Cytosolic HSC20 integrates de novo iron-sulfur cluster biogenesis with the CIAO1-mediated transfer to recipients, Hum Mol Genet 27(5) (2018) 837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Maio N, Jain A, Rouault TA, Mammalian iron-sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins, Curr Opin Chem Biol 55 (2020) 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rouault TA, The indispensable role of mammalian iron sulfur proteins in function and regulation of multiple diverse metabolic pathways, Biometals 32(3) (2019) 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Maio N, Singh A, Uhrigshardt H, Saxena N, Tong WH, Rouault TA, Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery, Cell Metab 19(3) (2014) 445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Voisine C, Cheng YC, Ohlson M, Schilke B, Hoff K, Beinert H, Marszalek J, Craig EA, Jac1, a mitochondrial J-type chaperone, is involved in the biogenesis of Fe/S clusters in Saccharomyces cerevisiae, Proc Natl Acad Sci U S A 98(4) (2001) 1483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Dutkiewicz R, Schilke B, Cheng S, Knieszner H, Craig EA, Marszalek J, Sequence-specific interaction between mitochondrial Fe-S scaffold protein Isu and Hsp70 Ssq1 is essential for their in vivo function, J Biol Chem 279(28) (2004) 29167–74. [DOI] [PubMed] [Google Scholar]
- [112].Vickery LE, Cupp-Vickery JR, Molecular chaperones HscA/Ssq1 and HscB/Jac1 and their roles in iron-sulfur protein maturation, Crit Rev Biochem Mol Biol 42(2) (2007) 95–111. [DOI] [PubMed] [Google Scholar]
- [113].Maio N, Ghezzi D, Verrigni D, Rizza T, Bertini E, Martinelli D, Zeviani M, Singh A, Carrozzo R, Rouault TA, Disease-Causing SDHAF1 Mutations Impair Transfer of Fe-S Clusters to SDHB, Cell Metab 23(2) (2016) 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Maio N, Kim KS, Singh A, Rouault TA, A Single Adaptable Cochaperone-Scaffold Complex Delivers Nascent Iron-Sulfur Clusters to Mammalian Respiratory Chain Complexes I-III, Cell Metab 25(4) (2017) 945–953 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Maio N, Rouault TA, Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery, Biochim Biophys Acta 1853(6) (2015) 1493–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Maio N, Rouault TA, Mammalian Fe-S proteins: definition of a consensus motif recognized by the co-chaperone HSC20, Metallomics 8(10) (2016) 1032–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Rouault TA, Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease, Dis Model Mech 5(2) (2012) 155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Ponka P, Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells, Blood 89(1) (1997) 1–25. [PubMed] [Google Scholar]
- [119].Ajioka RS, Phillips JD, Kushner JP, Biosynthesis of heme in mammals, Biochim Biophys Acta 1763(7) (2006) 723–36. [DOI] [PubMed] [Google Scholar]
- [120].Kafina MD, Paw BH, Intracellular iron and heme trafficking and metabolism in developing erythroblasts, Metallomics 9(9) (2017) 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Schultz IJ, Chen C, Paw BH, Hamza I, Iron and porphyrin trafficking in heme biogenesis, J Biol Chem 285(35) (2010) 26753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, Weber GJ, Dooley K, Davidson AJ, Schmid B, Paw BH, Shaw GC, Kingsley P, Palis J, Schubert H, Chen O, Kaplan J, Zon LI, Tubingen Screen C, Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis, Nature 436(7053) (2005) 1035–39. [DOI] [PubMed] [Google Scholar]
- [123].Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, Uchida N, Tisdale J, Camaschella C, Rouault TA, Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts, J Clin Invest 120(5) (2010) 1749–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A, The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload, Blood 110(4) (2007) 1353–8. [DOI] [PubMed] [Google Scholar]
- [125].Wang H, Shi H, Rajan M, Canarie ER, Hong S, Simoneschi D, Pagano M, Bush MF, Stoll S, Leibold EA, Zheng N, FBXL5 Regulates IRP2 Stability in Iron Homeostasis via an Oxygen-Responsive [2Fe2S] Cluster, Mol Cell 78(1) (2020) 31–41 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Shi Y, Ghosh MC, Tong WH, Rouault TA, Human ISD11 is essential for both ironsulfur cluster assembly and maintenance of normal cellular iron homeostasis, Hum Mol Genet 18(16) (2009) 3014–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Tong WH, Rouault TA, Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis, Cell Metab 3(3) (2006) 199–210. [DOI] [PubMed] [Google Scholar]
- [128].Crooks DR, Maio N, Lane AN, Jarnik M, Higashi RM, Haller RG, Yang Y, Fan TW, Linehan WM, Rouault TA, Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells, J Biol Chem 293(21) (2018) 8297–8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kubota Y, Nomura K, Katoh Y, Yamashita R, Kaneko K, Furuyama K, Novel Mechanisms for Heme-dependent Degradation of ALAS1 Protein as a Component of Negative Feedback Regulation of Heme Biosynthesis, J Biol Chem 291(39) (2016) 20516–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zheng J, Shan Y, Lambrecht RW, Donohue SE, Bonkovsky HL, Differential regulation of human ALAS1 mRNA and protein levels by heme and cobalt protoporphyrin, Mol Cell Biochem 319(1–2) (2008) 153–61. [DOI] [PubMed] [Google Scholar]
- [131].Liu G, Sil D, Maio N, Tong WH, Bollinger JM Jr., Krebs C, Rouault TA, Heme biosynthesis depends on previously unrecognized acquisition of iron-sulfur cofactors in human amino-levulinic acid dehydratase, Nat Commun 11(1) (2020) 6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Dailey HA, Finnegan MG, Johnson MK, Human ferrochelatase is an iron-sulfur protein, Biochemistry 33(2) (1994) 403–7. [DOI] [PubMed] [Google Scholar]
- [133].Wu CK, Dailey HA, Rose JP, Burden A, Sellers VM, Wang BC, The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis, Nat Struct Biol 8(2) (2001) 156–60. [DOI] [PubMed] [Google Scholar]
- [134].Crooks DR, Ghosh MC, Haller RG, Tong WH, Rouault TA, Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery, Blood 115(4) (2010) 860–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Crispin A, Guo C, Chen C, Campagna DR, Schmidt PJ, Lichtenstein D, Cao C, Sendamarai AK, Hildick-Smith GJ, Huston NC, Boudreaux J, Bottomley SS, Heeney MM, Paw BH, Fleming MD, Ducamp S, Mutations in the iron-sulfur cluster biogenesis protein HSCB cause congenital sideroblastic anemia, J Clin Invest 130(10) (2020) 5245–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Schmitz-Abe K, Ciesielski SJ, Schmidt PJ, Campagna DR, Rahimov F, Schilke BA, Cuijpers M, Rieneck K, Lausen B, Linenberger ML, Sendamarai AK, Guo C, Hofmann I, Newburger PE, Matthews D, Shimamura A, Snijders PJ, Towne MC, Niemeyer CM, Watson HG, Dziegiel MH, Heeney MM, May A, Bottomley SS, Swinkels DW, Markianos K, Craig EA, Fleming MD, Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9, Blood 126(25) (2015) 2734–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Ducamp S, Fleming MD, The molecular genetics of sideroblastic anemia, Blood 133(1) (2019) 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Rouault TA, Mitochondrial iron overload: causes and consequences, Curr Opin Genet Dev 38 (2016) 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM, Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A), Hum Mol Genet 8(5) (1999) 743–9. [DOI] [PubMed] [Google Scholar]
- [140].Pondarre C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD, Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis, Blood 109(8) (2007) 3567–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Lill R, Dutkiewicz R, Freibert SA, Heidenreich T, Mascarenhas J, Netz DJ, Paul VD, Pierik AJ, Richter N, Stumpfig M, Srinivasan V, Stehling O, Muhlenhoff U, The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins, Eur J Cell Biol 94(7–9) (2015) 280–91. [DOI] [PubMed] [Google Scholar]
- [142].Pandey A, Pain J, Dziuba N, Pandey AK, Dancis A, Lindahl PA, Pain D, Mitochondria Export Sulfur Species Required for Cytosolic tRNA Thiolation, Cell Chem Biol 25(6) (2018) 738–748 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Land T, Rouault TA, Targeting of a human iron-sulfur cluster assembly enzyme, nifs, to different subcellular compartments is regulated through alternative AUG utilization, Mol Cell 2(6) (1998) 807–15. [DOI] [PubMed] [Google Scholar]
- [144].Marelja Z, Mullick Chowdhury M, Dosche C, Hille C, Baumann O, Lohmannsroben HG, Leimkuhler S, The L-cysteine desulfurase NFS1 is localized in the cytosol where it provides the sulfur for molybdenum cofactor biosynthesis in humans, PLoS One 8(4) (2013) e60869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Marelja Z, Stocklein W, Nimtz M, Leimkuhler S, A novel role for human Nfs1 in the cytoplasm: Nfs1 acts as a sulfur donor for MOCS3, a protein involved in molybdenum cofactor biosynthesis, J Biol Chem 283(37) (2008) 25178–85. [DOI] [PubMed] [Google Scholar]
- [146].Uhrigshardt H, Singh A, Kovtunovych G, Ghosh M, Rouault TA, Characterization of the human HSC20, an unusual DnaJ type III protein, involved in iron-sulfur cluster biogenesis, Hum Mol Genet 19(19) (2010) 3816–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Li K, Tong WH, Hughes RM, Rouault TA, Roles of the mammalian cytosolic cysteine desulfurase, ISCS, and scaffold protein, ISCU, in iron-sulfur cluster assembly, J Biol Chem 281(18) (2006) 12344–51. [DOI] [PubMed] [Google Scholar]
- [148].Maio N, Kim KS, Holmes-Hampton G, Singh A, Rouault TA, Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis, Haematologica (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kispal G, Csere P, Guiard B, Lill R, The ABC transporter Atm1p is required for mitochondrial iron homeostasis, FEBS Lett 418(3) (1997) 346–50. [DOI] [PubMed] [Google Scholar]
- [150].Leighton J, Schatz G, An ABC transporter in the mitochondrial inner membrane is required for normal growth of yeast, EMBO J 14(1) (1995) 188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Miao R, Kim H, Koppolu UM, Ellis EA, Scott RA, Lindahl PA, Biophysical characterization of the iron in mitochondria from Atm1p-depleted Saccharomyces cerevisiae, Biochemistry 48(40) (2009) 9556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Hausmann A, Samans B, Lill R, Muhlenhoff U, Cellular and mitochondrial remodeling upon defects in iron-sulfur protein biogenesis, J Biol Chem 283(13) (2008) 8318–30. [DOI] [PubMed] [Google Scholar]
- [153].Bottomley SS, Fleming MD, Sideroblastic anemia: diagnosis and management, Hematol Oncol Clin North Am 28(4) (2014) 653–70, v. [DOI] [PubMed] [Google Scholar]
- [154].Chen W, Dailey HA, Paw BH, Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis, Blood 116(4) (2010) 628–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Waldvogel D, van Gelderen P, Hallett M, Increased iron in the dentate nucleus of patients with Friedrich’s ataxia, Ann Neurol 46(1) (1999) 123–5. [DOI] [PubMed] [Google Scholar]
- [156].Foury F, Roganti T, Deletion of the mitochondrial carrier genes MRS3 and MRS4 suppresses mitochondrial iron accumulation in a yeast frataxin-deficient strain, J Biol Chem 277(27) (2002) 24475–83. [DOI] [PubMed] [Google Scholar]
- [157].Martelli A, Puccio H, Dysregulation of cellular iron metabolism in Friedreich ataxia: from primary iron-sulfur cluster deficit to mitochondrial iron accumulation, Front Pharmacol 5 (2014) 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M, Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion, Science 271(5254) (1996) 1423–7. [DOI] [PubMed] [Google Scholar]
- [159].Li J, Kogan M, Knight SA, Pain D, Dancis A, Yeast mitochondrial protein, Nfs1p, coordinately regulates iron-sulfur cluster proteins, cellular iron uptake, and iron distribution, J Biol Chem 274(46) (1999) 33025–34. [DOI] [PubMed] [Google Scholar]
- [160].Foury F, Talibi D, Mitochondrial control of iron homeostasis. A genome wide analysis of gene expression in a yeast frataxin-deficient strain, J Biol Chem 276(11) (2001) 7762–8. [DOI] [PubMed] [Google Scholar]
- [161].Huang ML, Becker EM, Whitnall M, Suryo Rahmanto Y, Ponka P, Richardson DR, Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant, Proc Natl Acad Sci U S A 106(38) (2009) 16381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Crooks DR, Natarajan TG, Jeong SY, Chen C, Park SY, Huang H, Ghosh MC, Tong WH, Haller RG, Wu C, Rouault TA, Elevated FGF21 secretion, PGC-1alpha and ketogenic enzyme expression are hallmarks of iron-sulfur cluster depletion in human skeletal muscle, Hum Mol Genet 23(1) (2014) 24–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Young L, Leonhard K, Tatsuta T, Trowsdale J, Langer T, Role of the ABC transporter Mdl1 in peptide export from mitochondria, Science 291(5511) (2001) 2135–8. [DOI] [PubMed] [Google Scholar]
- [164].Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D, The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans, Mol Cell 37(4) (2010) 529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ, A mitochondrial specific stress response in mammalian cells, EMBO J 21(17) (2002) 4411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D, Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones, J Cell Sci 117(Pt 18) (2004) 4055–66. [DOI] [PubMed] [Google Scholar]
- [167].Rouault TA, Biochemistry. If the RNA fits, use it, Science 314(5807) (2006) 1886–7. [DOI] [PubMed] [Google Scholar]