

Abstract
The remarkable power and specificity of enzyme catalysis relies on the dynamic alignment of enzyme, substrates and cofactors, yet the role of dynamics has usually been approached from the perspective of the protein. We have been using an underappreciated NMR technique, subtesla high resolution field cycling 31P NMR relaxometry, to investigate the dynamics of enzyme-bound substrates and cofactor on guanosine-5’-monophosphate reductase (GMPR). GMPR forms two dead end, yet catalytically competent, complexes that mimic distinct steps in the catalytic cycle: E•IMP•NADP+ undergoes a partial hydride transfer reaction while E•GMP•NADP+ undergoes a partial deamination reaction. A different cofactor conformation is required for each partial reaction. Here we report the effects of mutations designed to perturb cofactor conformation and ammonia binding with the goal of identifying the structural features that contribute to the distinct dynamic signatures of the hydride transfer and deamination complexes. These experiments suggest that Asp129 is a central cog in a dynamic network required for both hydride transfer and deamination. In contrast, Lys77 modulates the conformation and mobility of substrates and cofactors in a reaction specific manner. Thr105 and Tyr318 are part of a deamination-specific dynamic network that includes the 2′-OH of GMP. These residues have comparatively little effect on the dynamic properties of the hydride transfer complex. These results further illustrate the potential of high resolution field cycling NMR relaxometry for the investigation of ligand dynamics. In addition, exchange experiments indicate that NH3/NH4+ has high affinity for the deamination complex but low affinity for the hydride transfer complex, suggesting that the movement of ammonia may gate the cofactor conformational change. Collectively these experiments reinforce the view that enzyme, substrates and cofactor are linked in intricate, reaction-specific, dynamic networks, and demonstrate that distal portions of the substrates/cofactors are critical features in these networks.
Keywords: oxidoreductases, enzyme catalysis, NMR spectroscopy, cofactor dynamics, dynamic networks
Graphical Abstract
INTRODUCTION
The extraordinary catalytic power of enzymes relies on the dynamic alignment of enzyme, substrates and cofactors. Many laboratories have experimentally investigated the role of protein dynamics in enzyme catalysis 1-7. However, far fewer investigations have probed the dynamics of enzyme-bound ligands, as seen in the prior work of Peng (2003) 8. Most of these studies have focused on the site of chemical transformation 9-11. Despite the widespread occurrence of phosphorylated substrates in metabolism, even fewer studies have addressed the dynamics of enzyme-bound phosphates 12, 13.
While elucidating dynamics at the site of chemical transformation is clearly critical for understanding the origins of catalysis, enzymes also accelerate reactions by harvesting binding energy associated with distal portions of substrates and cofactors. Several mechanisms underlie this phenomenon: (i) in some cases, remote portions appear to simply tether the substrate to the active site, enforcing productive binding modes, as observed in ketosteroid isomerase 14; (ii) remote interactions can stabilize mobile loops, thereby positioning key catalytic residues 15; (iii) interactions with remote portions of the substrate can become optimal during the transition state, as observed in tyrosyl-tRNA synthetase 16; (iv) the electrostatic potential of the active site can be modulated by remote substrate moieties; and (v) lastly, remote portions of the substrate can couple motions across the active site 12, 13.
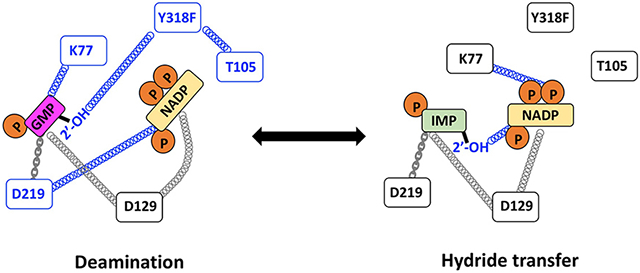
Guanosine-5′-monophosphate reductase (GMPR; UniProt accession ID P60560) is an excellent system to address questions of substrate/cofactor dynamics. GMPR catalyzes a two-step reaction involving deamination of GMP to form a covalent enzyme intermediate E-XMP* and the subsequent reduction of E-XMP* with NADPH to produce IMP and NADP+ (Figure 1A; 17). The cofactor is present throughout the catalytic cycle and participates in the deamination reaction, helping to activate the ammonia leaving group with the assistance of the Thr188-Glu289 dyad 17. Both ammonia release and hydride transfer occur on the same face of E-XMP*, so each step must utilize a distinct cofactor conformation. Two cofactor conformations, termed IN and OUT, are observed in the x-ray crystal structure of the inactive E•IMP•NADPH complex (Figure 1B,C) 17. In the IN conformation, the nicotinamide C4 is in contact with C2 of IMP, as would be required for hydride transfer. In contrast, the nicotinamide C4 is 6.6 Å from C2 of IMP in the OUT conformation, as might be expected for deamination.
Figure 1. The structure and reactions of GMPR.

(A) The GMPR reaction. (B) The IN conformation as observed in subunit B of 2C6Q 17. (C) The OUT conformation as observed in subunit D of 2C6Q. Protein is shown in purple, IMP is gray, NADPH is green and hydrogen bonds are cyan. The side chain of Met269 has been removed for clarity. E. coli GMPR numbering is used for consistency with experiments. Panels B and C rendered with UCSF Chimera 18. (D) Partial reactions catalyzed by GMPR. Panel adapted from reference 12.
Most importantly, GMPR can form two dead end, yet catalytically competent, complexes that mimic distinct steps in the catalytic cycle (Figure 1D)12, 13, 17. The E•IMP•NADP+ complex (hereafter the hydride transfer complex) undergoes a partial hydride transfer reaction to produce E-XMP*•NADH. In the absence of ammonia, this complex is stable for days. The E•GMP•NADP+ complex (hereafter the deamination complex) undergoes a partial deamination reaction, producing E-XMP*•NADP+•NH3, which cannot complete the catalytic cycle in absence of NADPH. Intriguingly, only ~20% of the enzyme is converted into E-XMP*, which indicates that ammonia is tightly bound. As detailed below, our previous NMR relaxation experiments indicate that the phosphate of GMP has a different binding mode than that of IMP in these complexes, which makes the OUT conformation observed in the crystal structure of E•IMP•NADPH an uncertain model for the structure of the catalytically competent deamination complex.
We have been utilizing subtesla high-resolution field-cycling 31P NMR relaxometry to interrogate the dynamics of enzyme-bound substrates and cofactor in GMPR 12, 13. This variant of field-cycling 31P NMR relaxometry enables the measurement of the spin-lattice/longitudinal relaxation rates (R1) over a wide range of magnetic fields (11.7 to 0.003 T). Samples are excited in the probe at high magnetic field, shuttled up the bore of the magnet to a lower field for relaxation, then moved back to the high field for detection of residual magnetization 19, 20. This shuttling preserves the chemical shift differences of nuclei, allowing the measurement of R1 for different phosphorylated ligands. The ability to observe relaxation at very low and varied magnetic fields allows the measurement of high dipolar R1 values that can provide information about the orientation and relative mobility of enzyme-bound ligands 12, 13, 21, 22. R1 also depends on the correlation time for the phosphorylated ligand. If the ligand is relatively rigid when bound, the correlation time will be comparable to the overall rotation of the protein complex in solution. Note that these measurements require that the ligands are in fast exchange, which means that the experiments probe the Michaelis complexes.
Our previous work revealed that the hydride transfer and deamination complexes of GMPR display distinct dynamic properties reflected in the molecular dipolar correlation time, τD, and the maximum dipolar relaxation rate, RD0, and the ratio τD/RD0 (Table 1; 12, 13). The value of τD/RD0 is related to the averaged effective distance (reff) between the 31P nuclei and the 1H relaxers, both intramolecular and intermolecular, and thus describes the conformation of the ligand and the structure of the phosphate binding site. Previous experiments indicated that the cofactor molecule is more rigid than the substrate IMP in the hydride transfer complex while GMP is more restrained than the cofactor in the deamination complex. The substrate phosphate binding sites are similar in the hydride transfer complex and the binary E•IMP and E•GMP complexes. These observations are consistent with x-ray crystal structures 12, 13. However, the value of τD/RD0 is much higher for GMP in the deamination complex (24 x10−8 s2 and 3.3 x 10−8 s2 for GMP and IMP, respectively), indicating that the substrate phosphate binding site is different from that in the hydride transfer complex. Thus although the OUT conformation of the cofactor in the inactive E•IMP•NADPH crystal structure is consistent with expectations for the deamination reaction, the substrate binding site is not. Therefore the crystal structure is an uncertain model for the deamination complex. We have proposed that the distinct dynamic signatures of the hydride transfer and deamination complexes reflect the behavior of catalytically productive substrate/cofactor binding modes, and therefore provide valuable insight into the dynamics of substrate/cofactor in Michaelis complexes 12, 13.
Table 1.
Characteristics of 31P field cycling NMR parameters associated with WT GMPR in the hydride transfer (E•IMP•NADP+, HT) and deamination (E•GMP•NADP+, DA) complexes.
| 31P | Comparison | P a | Implication |
|---|---|---|---|
| Substrate mono-P | τD (HT) ~ τD (DA) | 0.13 | Bound substrate 31P mobility is the same for both IMP and GMP; τD smaller than that of overall GMPR rotation, suggesting that the substrate is mobile in the active site |
| RD0 (HT) < RD0 (DA) | 0.015 | GMP interacts with fewer 1H in DA than IMP in HT | |
| τD/RD0 (HT) < τD/RD0 (DA) | 0.005 | Effective rPH is longer for bound GMP (1H fewer or farther away) | |
| NADP+ mono-P | τD (HT) > τD (DA) b | 0.004 | Bound NADP+ is more mobile in DA |
| RD0 (HT) > RD0 (DA) | 0.009 | 1H dipolar relaxation is significantly higher in HT | |
| τD/RD0 (HT) < τD/RD0 (DA) | 0.014 | rPH is shorter for the bound cofactor mono-P in HT | |
| NADP+ di-P b | τD (HT) > τD (DA) b | 0.049 | Bound NADP+ is more mobile in DA |
| RD0 (HT) > RD0 (DA) | 0.029 | 1H dipolar relaxation is significantly higher in HT | |
| τD/RD0 (HT) < τD/RD0 (DA) | 0.041 | Effective rPH is smaller for HT | |
| IMP vs NADP+ b | τD (IMP) < τD (NADP+) | 0.003 | The cofactor is more rigid than substrate in HT; cofactor τD approximates that of overall GMPR rotation |
| GMP vs NADP+ di-P b | τD (GMP) > τD (NADP+) | 0.024 | The cofactor has more mobility than substrate in DA |
The comparisons were done with an unpaired student t-test and significance indicated by P<0.05.
Statistics are given for only one of the diphosphate nuclei because the values are indistinguishable.
Here we report the effects of mutations designed to perturb cofactor conformation and ammonia binding with the goal of identifying the structural features that contribute to the distinct dynamic signatures of the hydride transfer and deamination complexes of GMPR. These experiments suggest that Asp129 is a central cog in a dynamic network required for both hydride transfer and deamination. In contrast, Lys77 modulates the conformation and mobility of substrates and cofactors in a reaction specific manner. Thr105 and Tyr318 are part of a deamination-specific dynamic network that includes the 2′-OH of GMP. These residues have comparatively little effect on the dynamic properties of the hydride transfer complex. Together these results provide a preliminary map of the dynamic networks that govern GMPR catalysis, and further illustrate the potential of high resolution field cycling NMR relaxometry for the investigation of ligand dynamics.
MATERIALS AND METHODS
Materials.
NADP+ was obtained from Roche Diagnostics. IMP was purchased from MP Biomedicals. GMP and NADPH were purchased from Sigma. D2O (99.9%), deuterated Tris-d11 (98%), 15N-ammonia and uniformly labeled 15N-GMP were obtained from Cambridge Isotopes.
Expression and purification of E. coli GMPR.
All proteins were His-tagged and expressed in BL21(DE3) ΔguaC cells, which lack endogenous GMPR, as previously described 17. GMPR mutant Thr188A was reported previously 17.
Enzyme kinetics.
Standard GMPR assays were conducted at 25°C in 75 mM Tris•HCl, pH 7.8, 100 mM KCl, 1 mM EDTA and 1 mM DTT (assay buffer). Enzyme activity was monitored by observing the changes in NADPH concentration spectrophotometrically at 340 nm. Initial velocity data for the forward reaction were obtained by varying both GMP and NADPH. Data were typically fit to the two substrate random bi-bi (sequential) equation using SigmaPlot, where v is the initial velocity, Vm is the maximal velocity, A is GMP, B is NADPH, α is the dissociation constant, Ka is the Michaelis constant for substrate A, Kb is the Michaelis constant for substrate B:
| [1] |
For mutant enzymes with low activity, data were fit to an ordered sequential mechanism with GMP binding first using Dynafit (BioKin Ltd., Watertown MA) 23.
Initial velocities for the backward reaction were obtained at fixed ammonium (20 mM), varying either NADP+ (20 to 640 μM) at fixed saturating (for wild type) IMP (1 mM) or varying IMP (20 to 640 μM) at fixed saturating (for wild type) NADP+ (1 mM). Data were fit to the Michaelis-Menten equation (Equation 2):
| [2] |
Proton uptake experiments.
Changes in proton concentration were measured by monitoring the change in absorbance at 560 and 572 nm of phenol red (30 μM). The following enzyme concentrations were used: WT = 1 μM, K77A = 5 μM, D129A = 5 μM, Y318F = 10 μM, [GMP] = 500 μM, [NADPH] = 500 μM; for backward reactions (both partial and whole reaction), WT = 10 μM, [IMP] = 500 μM, [NADP+] = 500 μM, [NH4+] = 20 mM. The assay buffer contained 0.5 mM Tris•HCl, pH 7.8, 150 mM KCl, 1 mM DTT.
Stopped flow experiments.
A stopped-flow spectrophotometer (Applied Photophysics SX.17MV) was used to conduct pre-steady-state experiments. The changing color of indicator (phenol red) in proton uptake assay was monitored by absorbance at 560 nm at 25°C; the production of NADPH in partial backward reaction or whole backward reaction was monitored by absorbance at 340 nm at 25°C, with a 420 nm cut-off filter. The following enzyme concentrations were used in the proton uptake assay and backward reaction (both partial and whole reaction): 10 μM WT; 30 μM T188A; 30 μM Y318A; and 10 μM Y318F. The proton uptake assay used 500 μM GMP, 500 μM NADPH. Backward reactions (both partial and whole reaction) used 500 μM IMP, 500 μM NADP+, and 20 mM NH4+. For the partial hydride transfer reaction, data were fit to a single exponential (Equation 3):
| [3] |
High resolution field cycling NMR measurements.
Instrumentation and experimental details for the GMPR/ligand field cycling have been reported earlier (4,5). Samples contained 400 μM GMPR and 1.6 mM ligands in 75 mM Tris-HCl, pH 7.8, with 100 mM KCl, 1 mM DTT, and 0.5 mM EDTA in 24% D2O. The 31P field-cycling spin–lattice relaxation rate (R1) measurements were obtained at 25°C on a Varian INOVA 500 spectrometer using a standard 10 mm Varian probe in a device that was built to move the sample from the conventional sample probe location at 11.74 T to a higher position where the magnetic field can be as low as 0.04 T 19. Magnetic fields between 0.003 and 0.04 T were accessed with a permanent magnet mounted at the top of the dewar 20.
Measurements of R1 and data analysis have been described in detail 12, 13. The R1 at any field is equal to RD +RCSA + kD, where the constant kD is attributed to fast dipolar relaxation of the 31P by the protons to which it is coupled where we see only the maximum relaxation rate, and RCSA is equal to kCSA ωP2 in the field range we have available (kCSA is a constant). Equation 4 describes the dependence of the R1 on the angular frequency of the nucleus, ωP, which for 31P is γP Brelax, where γP is the magnetogyric ratio of the nucleus and Brelax is the magnetic field 21.
| [4] |
The spectral density J(ω) is equal to 2τ (1+ω2τ2) The key parameters extracted from Eqn. 4 include a correlation time for the dipolar interaction (τD) and a dipolar relaxation rate extrapolated to zero field, RD0. An averaged effective distance between the 31P nuclei and the relaxer 1H that are nearby is proportional to the sixth root of τD/RD0 (Equation [5]).
| [5] |
The effective distance reff is not a true distance because the number and geometry of the 1H relaxers are not known (and the equation as shown assumes a single relaxer). Nonetheless, τD/RD0 provides a very useful probe of protein/ligand interactions. Since the GMPR-bound and free ligands must be in fast exchange to detect the protein-induced relaxation behavior, τD and RD0 characterize the dynamics of substrate and cofactor binding in the Michaelis complex, and presumably describe catalytically competent binding modes.
15N NMR measurements.
Samples for NMR experiments were prepared in 25 mM Tris·HCl, pH 7.8, 0.5 mM TCEP, and 10% of D2O. To assess GMP binding to protein as well as look for bound ammonia, 8 mM uniformly 15N- labeled GMP was mixed with 8 mM NADP+ in the absence or presence of 0.2 μM GMPR. To monitor 15NH4+ exchange into GMP, 0.02 μM GMPR was incubated for one hour with 50 mM 15NH4Cl and 1.3 and 2 mM of IMP and NADP+. The protein was subsequently removed by filtration and the rest of the sample was subjected to NMR analysis. NMR data were acquired at 25°C on a Bruker Avance spectrometer (Brandeis University Landsman Research Facility) operating at 800.13 and 81.086 MHz for 1H and 15N, respectively. Standard TROSY-based pulse sequences were used to acquire two-dimensional (2D) 1H–15N HSQC with 2048 (1H) × 256 (15N) complex points and 128-256 scans per t1 increment. Data processing was performed using Bruker Topspin version 4.0.6. and internal Bruker referencing, where the 15N chemical shifts are calculated relative to liquid NH3 at 25 °C.
Cluster analysis.
Field cycling parameters were expressed as the ratio of mutant to wild-type hydride transfer complex and converted to log2. Enzyme complexes were clustered using Euclidean distance algorithm with centroid linkages in Cluster 3.0 24 and visualized using TreeView 25.
RESULTS
Identification of candidate residues.
We inspected the x-ray crystal structure of the inactive E•IMP•NADPH complex of human GMPR type 2 (hGMPR2; PDB code 2C6Q) to identify residues that appear to make conformation-specific contacts with the cofactor, hypothesizing that such interactions might underlie the distinct dynamic signatures of the hydride transfer and deamination complexes of GMPR. Three candidate residues were identified: Lys77, Thr105 and Asp129 (Figure 1B,C; reference 17). The side chain of Lys77 forms a hydrogen bond with one of the di-P oxygens in the OUT conformation. In the IN conformation, Lys77 forms water mediated hydrogen bonds with the 3’-OH of the nicotinamide ribose and an axial O of the adenosine phosphate (Figure 1C). Thr105 forms two hydrogen bonds with the cofactor in the OUT conformation, one between the side chain OH and the adenosine 5’-O and another between the backbone nitrogen and an axial O of the adenosine diphosphate. These interactions are absent in the IN conformation. Lastly, the Asp129 carboxylate forms a hydrogen bond with the 2’-OH of the nicotinamide nucleoside in the IN conformation that is absent in the OUT conformation. We substituted these residues with Ala, reasoning that if two residues are part of the same dynamic network, these substitutions are likely to have similar effects on the field cycling parameters.
Kinetic characterization of mutant enzyme activity.
We characterized the effects of the mutations on the forward reaction (E+GMP+NADPH), the reverse reaction (E+IMP+NADP++NH3) and the partial hydride transfer reaction (E+IMP+NADP+) as summarized in Table 2. Note that while we can measure the partial hydride transfer reaction, we do not have a direct measure of the partial deamination reaction. Hydride transfer is rate-limiting in the forward reaction 17, so the effects of a mutation on the deamination reaction are masked. In contrast, the reverse reaction is clearly limited by ammonia since concentrations are not saturating. Therefore, the reverse reaction is the best probe of the deamination step, although the rates of ammonia binding and amination cannot be deconvoluted. Based on the inspection of the crystal structure described above, we expected the K77A mutation to affect both hydride transfer and deamination, T105A to selectively perturb deamination and D129A to selectively perturb hydride transfer. Kinetic analysis was largely consistent with these expectations in the cases of K77A and T105A, but D129A disrupted both steps.
Table 2: Kinetic Parameters of wild-type and mutant GMPRs.
All reactions were conducted at 25°C in 75 mM Tris, pH 7.8, 100 mM, KCl, 1 mM EDTA and 1 mM DTT. Amp = [NADPH]/[E].
| Enzyme | E + GMP + NADPH a | E + IMP + NADP+ + NH4+ b | E + IMP + NADP+ | |||||
|---|---|---|---|---|---|---|---|---|
|
kcat (s−1) |
Km GMP (μM) |
Km NADPH (μM) |
Vmax (s−1) |
Km IMP (μM) |
Km NADP+ (μM) |
kobs (s−1) |
Amp | |
| WT c | 0.35±0.01 | 3.2±0.5 | 10.1±0.8 | 0.018±0.009 | 23±6 | 50±16 | 0.49±0.01 | 0.16 |
| K77A | 0.05±0.01 d | 12±4 | 1050±560 d | (1.6±0.6)x10−4 | 74 ± 24 | 750±70 | 0.016±0.008 | 0.07 |
| T105A | 0.45±0.2 | 5.4±1.0 | 64±8 | 0.005±0.001 | 40±6 | 130±40 | 0.29±0.06 | 0.15 |
| D129A | 0.009±0.001 | 4.1±0.5 | 15±3 | (1.7±0.3)x10−4 | 120±50 | 167±45 | 0.0047±0.0002 | 0.08 |
| T188A | 0.002±0.0003 e | 100±30 | 100±40 | b.d. f | b.d. f | b.d. f | 0.044±0.001 | 0.03 |
| Y318A | 0.0023±0.0005 g | 40±11 g | 190±70 g | b.d. f | b.d. f | b.d. f | 0.20±0.01 | 0.10 |
| Y318F | 0.5±0.1 | 29±7 | 260±98 | (4.5±0.5)x10−4 | 32±8 | 90±13 | 0.33±0.01 | 0.28 |
| WT(dG/IMP) | 0.023 ± 0.002 | 15 ±3 | 16 ± 4 | 0.002 ± 0.001 | 140 ± 20 | 109 ± 28 | 0.49±0.01 | 0.16 |
| D219A | 0.013 ± 0.003 | 380 ± 130 | 120 ± 20 | (4.0 ± 0.3) x 10−4 | 1100 ± 240 | 420 ± 220 | 0.064 ± 0.001 | 0.10 |
GMP concentration was varied from 10 to 160 μM, NADPH was varied from 10 to 160 μM.
IMP concentration was varied from 20 to 640 μM at fixed 1 mM NADP+ and 20 mM NH4+, while NADP+ concentration was varied from 20 to 640 μM at fixed 1 mM IMP and 20 mM NH4+.
Values from 17.
Experiments used cuvettes with pathlengths of 0.1 and 0.2 cm.
Previously reported value of 0.00088 s−1 was acquired at 1 mM GMP and 1 mM NADPH 17.
‘b.d.’ means below detection (< 0.5% WT activity).
Data were fit to a sequential ordered mechanism using Dynafit (BioKin Ltd) 23.
Data from 13
The K77A mutation disrupted both the deamination and hydride steps. In the forward reaction, the value of kcat decreased by a factor of 7, and the values of Km for GMP and NADPH increased by 4- and 100-fold, respectively (Table 2). The partial hydride transfer reaction was impaired by a factor of 30. Moreover, the value of kobs for the partial hydride transfer reaction was similar to the value of kcat for the reduction of GMP, suggesting that hydride transfer remains rate-limiting. Likewise, the value of Vmax for the reverse reaction was decreased by a factor of 100. Thus Lys77 is important for both steps of the GMPR reaction.
In contrast, the T105A mutation had little effect on catalytic activity. In the forward reaction, no effect was observed on the values of kcat and Km(GMP), although the value of Km(NADPH) increased by 6-fold (Table 2). In the reverse reaction, the values of Km of IMP and NADP+ increased by 2-2.5-fold and Vmax decreased by a factor of 3.5. The rate constant for the partial hydride transfer reaction decreased by less than a factor of 2 (Table 2). These observations suggest that ammonia binding and/or deamination are slightly more affected by the mutation than hydride transfer, although overall the effects are modest.
Like K77A, the D129A mutation disrupted both steps in the GMPR reaction. In the forward reaction, the value of kcat decreased by a factor of 35 although the values of Km were unchanged (Table 2). The value of Vmax in the reverse reaction decreased by a factor of 100 while the values of Km(IMP) and Km(NAD+) increased by 6- and 3-fold, respectively. The value of kobs for the partial hydride transfer decreased by a factor of 100. Thus like Lys77, Asp129 is important for both steps of the GMPR reaction.
Characterization of 31P relaxation.
Field cycling NMR probes the mobility of protein-bound phosphates and structure of the binding mode. An increase in the value of τD indicates loss of mobility; a decrease indicates increased mobility. If τD is not perturbed, a change in RD0 indicates a change in the number or proximity of 1H relaxers. The value of τD/RD0 is related to the averaged effective distance (reff) between the 31P nucleus and the 1H relaxers, with smaller values of τD/RD0 meaning closer relaxers. If the mobility of the 31P nucleus changes, but the structure of the binding mode is unchanged, then values of τD and RD0 will change proportionally so that τD/RD0 remains constant.
Figure 2 summarizes the field cycling parameters for the GMPR mutants. The results generally match expectations based on the kinetic behavior. The dynamic behavior of both the hydride transfer and deamination complexes are perturbed by the K77A and D129A mutations while the Thr105A has comparatively subtle effects on the deamination complex alone.
Figure 2. 31P NMR field cycling parameters extracted for wild type and mutant GMPRs.
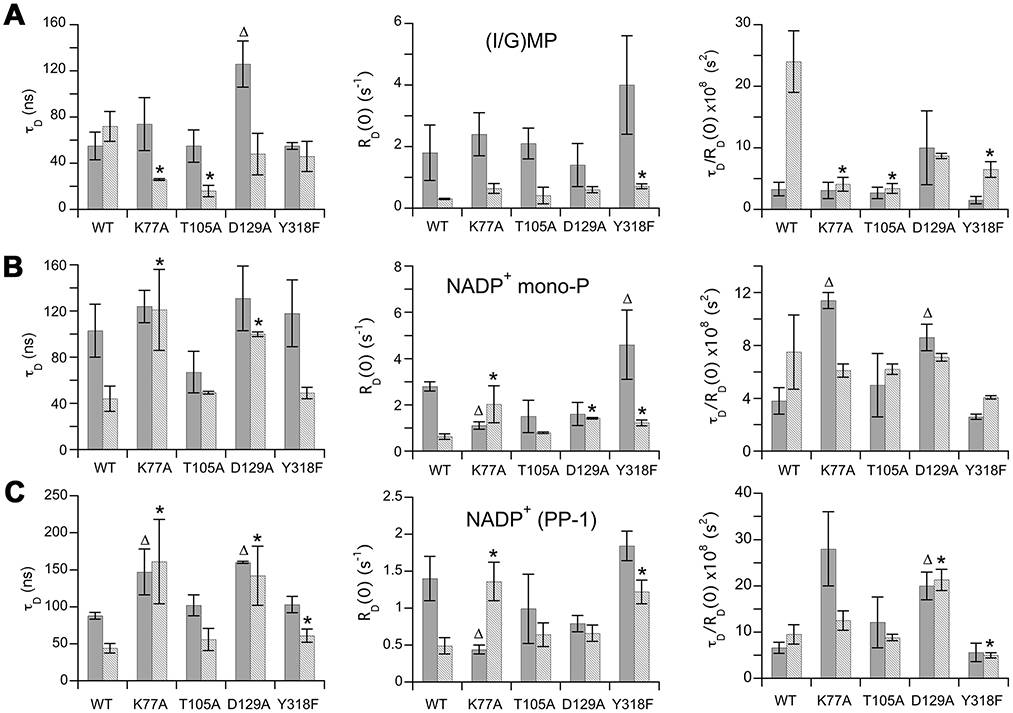
The values for the partial hydride transfer complex (E•IMP•NADP+) are shown in solid gray and the values for the partial deamination complex (E•GMP•NADP+) are shown in a lighter gray. Wild type data are from 12. The values are listed in Table S1. The parameters of the two cofactor pyrophosphate 31P nuclei are indistinguishable, so only PP-1 is shown here. Triangles denote hydride transfer complexes that differ from wild-type; asterisks denote deamination complexes that differ from wild-type (P<0.05 in an unpaired t-test, values in Figure S1 and Table S2).
Lys77 coordinates cofactor conformation and dynamics with substrate.
The K77A mutation has no effect on the substrate in the hydride transfer complex (Figure 2, Figure S2 and Table S1). Cofactor relaxation is perturbed: the NADP+ mono-P exhibits a decreased RD0, and increased τD/RD0, which implies that the proximity or number of 1H relaxers has decreased. A similar decreased RD0 and increased τD/RD0 is observed for the cofactor di-P. Note that cofactor binding was not saturating in the field cycling experiments, so the value of RD0 is probably underestimated. Adjusting for the fraction bound (0.67 based on Km) increases RD0 values by a factor of 1.5 with corresponding decreases in τD/RD0 values by a factor of 0.67. These adjustments do not change the findings. Thus Lys77 modulates cofactor conformation in the hydride transfer complex, but does not influence the mobility or binding mode of IMP.
In contrast, K77A clearly perturbs substrate relaxation in the deamination complex. The value of τD is roughly 2-fold lower, indicating that GMP is more mobile within the active site than in the wild-type enzyme. The value of RD0 appeared to increase slightly (but not quite to statistical significance), and the value of τD/RD0 decreased substantially, indicating that now there are more 1H dipoles relaxing the bound GMP. Thus the binding mode of GMP in the K77A deamination complex resembles that of IMP in the wild-type hydride transfer complex. The K77A mutation also changed cofactor relaxation. The values of τD increased for both the cofactor mono- and di-Ps and there was no longer a difference in the τD for the hydride transfer versus deamination complex. The values of RD0 also changed compared to WT. RD0 was higher for the deamination complex while it was lower for the hydride transfer complex. The value of τD/RD0 was much higher in the hydride transfer complex, indicating a change in the 1H dipoles contributing to cofactor relaxation. The τD/RD0 for the deamination complex now resembles that for the wild-type hydride transfer complex. As in the case of the hydride transfer complex, cofactor binding was not saturating in the field cycling experiments, and again adjusting for the fraction bound (0.62) does not change the findings. These observations indicate that Lys77 participates in distinct dynamic networks in each complex.
Substitution of Thr105 with Ala has little effect on the relaxation of enzyme-bound substrates and cofactor.
The T105A mutation had little effect on the relaxation of either cofactor or substrate in the hydride transfer, as expected given the very small changes in the kinetics of this reaction (Figure 2, Figure S3 and Table S1). The relaxation behavior of cofactor in the deamination complex is also very similar to that of wild-type. However, the values of both τD and τD/RD0 for GMP are much smaller than those observed in the wild-type deamination complex. Instead, these values resemble those of IMP in the wild-type hydride transfer complex. It is notable that although Thr105 directly interacts with the cofactor in the OUT conformation in the crystal structure, the T105A mutation has the biggest effects on the binding mode and mobility of GMP.
Asp129 is the linchpin that controls the distinct dynamic signatures of the hydride transfer and deamination complexes.
Unfortunately, we were only able to obtain one dataset for the D129A deamination complex with adequate R1 values at low field to obtain NMR parameters for GMP (NADP+ 31P were monitored in both samples). Remarkably the relaxation behavior of the cofactor was virtually identical in both the hydride transfer and deamination complexes of D129A (Figure 2, Figure S4 and Table S1). The values of τD/RD0 for the di-P were larger than those observed in either complex of the wild-type enzyme suggesting a new cofactor binding mode. The relaxation behavior of the substrates in D129A was also affected. The mobility of IMP decreased as evidenced by the high value of τD. The values of τD/RD0 were similar for IMP and GMP in the D129A complexes, and different from the values observed in either the wild-type hydride transfer or deamination complexes. Interestingly, similar values of τD/RD0 were observed for the wild-type hydride transfer and deamination complexes of the 2’-deoxy substrates (10x10−8 and 11x10−8 s2, for dIMP and dGMP, respectively 13).
Ammonia release.
The distinct dynamic properties of the hydride transfer and deamination complexes derive from the presence/absence of a single heavy atom, the amine of GMP. We previously suggested that ammonia might gate the transition between the two dynamic states of the cofactor 12. The formation of ammonia requires the uptake of one proton, likely from the Thr188-Glu289 dyad. The subsequent formation of ammonium requires a second proton, presumably from solvent (Figure 1A). We used phenol red to monitor proton uptake as a proxy for NH3/NH4+ formation. The rate constant for proton uptake (kH+ = 1.9±0.4 s−1; note 2H+ consumed/NADPH consumed) was in good agreement with the rate determined by monitoring loss of NADPH (0.8 s−1), validating the assay. Previous experiments found that the partial deamination reaction (E+GMP+NADP+) produced approximately 20% conversion to E-XMP* as measured by the incorporation of radioactivity into protein from [14C]-GMP. The sub-stoichiometric accumulation of E-XMP* suggests that NH3/NH4+ remains largely bound to the enzyme. The addition of ligands has been shown to trap NH4+ on proteins 26, providing precedence of our hypothesis that NH3/NH4+ does not dissociate from the deamination complex of GMPR. To further interrogate the release of NH3/NH4+, we measured proton uptake during the partial deamination reaction with 10 μM GMPR. Approximately 4 μM H+ would be produced with 20% conversion to E-XMP* if NH3/NH4+ was released from the enzyme, which would generate an absorbance change of approximately 0.004 (Figure S5). An NADP+ associated increase in absorbance was observed in both the enzyme/NADP+ and GMP/NADP+ control samples (Figure 3). The origin of this increase is likely due to small differences in the pH of the NADP+ containing samples. A small increase in absorbance of approximately 0.001 above this background was observed when enzyme was mixed with GMP and NADP+. This suggests that no more than 25% of the NH3/NH4+ produced in the partial deamination reaction has been released. Similar results were obtained when the reaction contained 500 μM GMPR. These observations further suggest that little NH3/NH4+ is released from the deamination complex, which implies that the value of Kd for NH3/NH4+ binding to E-XMP*•NADP+ is no higher than 0.2 μM.
Figure 3. Proton uptake during the partial deamination reaction.

When [E] = 10 μM, 2 μM ammonia should be produced, which, if released, will result in the uptake of 4 μM protons, and a change in absorbance of 0.004. However, the GMP-dependent change in absorbance is no more than 0.001. Proton uptake was monitored by following the change in absorbance of phenol red (30 μM) at 560 nm when GMP (500 μM) and NADP+ (500 μM) were added to GMPR (10 μM) (red trace; final concentrations; phenol red was present in both syringes). Conditions: 0.5 mM Tris-HCl, pH 7.8, 150 mM KCl, 1 mM DTT and 0.5 mM EDTA. Controls: proton uptake for GMPR and GMP (blue); GMPR and NADP+ (black); GMP and NADP+ in absence of enzyme (green).
We attempted to detect bound NH3/NH4+ using 15N-edited NMR methods. 15NH3/15NH4+ in solution is not observed in 1H,15N-HSQC spectra. However, 15NH3/15NH4+ can be observed when bound to a protein if the exchange of ammonium protons with bulk solvent is sufficiently slow 26. In this event, the bound species will have a different chemical shift than that of ammonium ion in bulk solution. Therefore we obtained HSQC spectra with uniformly labeled 15N-GMP in the absence/presence of GMPR and NADP+ (Figure 4A). The partial deamination reaction should generate E-XMP•NADP•NH3. If the time-scale for the exchange of [15N]-GMP and enzyme-bound NH3/NH4+ is in the slow exchange regime, a new 15N peak should be observed reflecting bound 15NH3/15NH4+. Slow exchange is likely given the large chemical shift difference between the two species (δH for the 15NH2 group is 6.5 ppm, while 15N-1 is 8.1 ppm 27 with δN for the two species even further apart). However, no new 15N peak was observed. Possible explanations for the lack of a detectable signal include: (1) the binding site is solvent exposed and fails to shield bound 15NH3/NH4+ protons from exchanging with solvent, or (2) the linewidth of the enzyme-bound 15NH3/NH4+ is too broad to observe (although with other enzymes, protein-bound ammonia resonances are considerably narrower than expected 26).
Figure 4. Ammonia release from GMPR during the partial deamination reaction.

(A) HSQC spectra of uniformly [15N]-labeled GMP. Samples contained GMP (8 mM) and NADP+ (8 mM) in the absence (blue) and presence (red) of GMPR (200 μM). (B-D) Ammonia exchange into GMP. (B) [15NH2]-GMP produced by incubating IMP (1.3 mM), NADP+ (1.3 mM) and 15N-ammonia (50 mM) with GMPR (200 μM). Enzyme was removed by filtration prior to obtaining the HSQC spectrum. The exchange of [15N]-ammonia into GMP catalyzed by wild-type (C) and Y318F GMPR (D). Reactions contained enzyme (500 μM), [15N]-ammonia (50 mM), GMP (1.3 mM) and NADP+ (1.3 mM). The cumulative time of data acquisition is shown.
[15N]-GMP is in fast change when it binds to the enzyme, and changes in the linewidth (Δν1/2) and chemical shift (δ) provide information on the E•GMP•NADP+ complex (Figure 4A). For these experiments the ratio of ligand to protein was 16:1. The 15N Δν1/2 roughly doubled in the presence of enzyme for both N-1 and the -NH2. The 15N linewidths are quite broad so that δH provides a better measure of protein binding. The δH for the GMP 15NH2 showed little change in the presence of enzyme. However, δH for the N-1 group shifted upfield by about 0.05 ppm, leading to ΔδH ~ 0.8 ppm for the bound GMP 15N in E•GMP•NADP+. The δH and δN of nucleobases are sensitive to hydrogen bonding 28. The magnitude of the upfield chemical shift is consistent of a strong interaction of N-1 with the protein. Inspection of the crystal structure of E•IMP•NADPH reveals that the N-1 of IMP forms hydrogen bonds with the main chain carbonyl of Glu289 in both the IN and OUT conformations. An analogous interaction is likely to be formed between enzyme and GMP.
The failure to observe NH3/NH4+ release might also be explained if the partial deamination reaction only proceeded to the tetrahedral intermediate E-GMP† (Figure 1A). The subsequent isolation of the protein by acid precipitation would likely convert E-GMP† to E-XMP*, explaining previous results. Therefore, we measured the exchange of [15N]-ammonium into unlabeled GMP to determine if E-XMP* was indeed formed and NH3/NH4+ was released. [15N]-GMP was easily observed within 2 h (Figure 4B,C), confirming the formation of E-XMP* and the exchange of enzyme-bound 14NH3 with a large reservoir of free [15N]-NH3/NH4+.
Lastly, we examined the effect of a large excess of NH3/NH4+ on the partial hydride transfer reaction. The addition of 20 mM NH4+ had no effect on the reaction of IMP and NADP+ as indicated by the progress curve for production of NADPH (kobs = 0.64 ± 0.01 s−1, [NADPH]/[E] = 0.48). This observation is not surprising given that the Km for ammonium is greater than 20 mM in the reverse reaction 17. These observations suggest that the ammonium ion or free ammonia has low affinity for the hydride transfer complex.
Identification of potential ammonia binding sites.
Inspection of the x-ray crystal structure of the inactive E•IMP•NADPH complex revealed two potential NH3/NH4+ binding sites (Figure 5). As noted previously 17, a density modeled as water is found 3.0 Å from C2 of E-XMP* in the OUT conformation, within hydrogen bonding distance to both the OH of Thr188 and the nicotinamide carbonyl, as might be expected for the NH3 immediately after leaving GMP (or for NH3 poised to attack E-XMP* to form GMP). We suggest that this is the “reactive NH3 binding site”. In the IN complex, the nicotinamide now occupies this site. Another putative water molecule is observed 5.8 Å from C2 of E-XMP*, where it forms hydrogen bonds to the backbone carbonyl of Ala131’ and the OH of Tyr318’ (Figure 5; the prime denotes a residue from the adjacent subunit). These residues are conserved in EcGMPR and other GMPRs. This second water molecule is also within 2.9 Å of the nicotinamide carbonyl, which has rotated 180 degrees from its position in the OUT conformation. We suggest that this water molecule identifies an “ammonia holding site”. This site is shielded from bulk water, which would prevent the release of ammonia into solvent, which might explain the failure to observe bound NH3/NH4+ in the 15N experiment (there is very little NH3/NH4+ and the peak is likely to be broad).
Figure 5. Proposed ammonia binding sites.

(A) The “reactive” NH3 binding site, the proposed site of NH3 immediately after deamination. E•IMP•NADPH is from 2C6Q subunit D. HOH2302.D is colored blue to represent the NH3. (B) The NH3 “holding” site, where NH3/NH4+ resides during the hydride transfer reaction. HOH2132.B is colored blue to represent the NH3. E•IMP•NADPH is from 2C6Q subunit B. Hydrogen bonds are shown in cyan, numbers are distance in Å between the atoms connected by the dotted lines. Figure rendered with UCSF Chimera 18.
Substitution of Thr188 and Tyr318 selectively perturb deamination.
We previously reported that the substitution of Ala at Thr188 decreased the value of kcat for the forward reaction with GMP by a factor of 400, but had a much smaller effect on the reaction with 2-Cl-IMP (kcat decreased by a factor of 15), as expected if Thr188 was involved in activating the ammonia leaving group 17). Here we report further characterization of the T188A reaction. The reverse reaction could not be detected (the value of Vmax at 20 mM NH4+ is less than 0.5% of that for the wild-type GMPR). The partial hydride transfer reaction could be observed, although the value of kobs was decreased by a factor of 10 (Table 2). Nonetheless, kobs is 10-fold greater than the value of kcat for the reduction of GMP, suggesting that the deamination step is rate-limiting. These observations further demonstrate that Thr188 plays a crucial role in the deamination step.
We substituted Tyr318 with Ala and Phe with the expectation that loss of the Tyr OH would disrupt the putative ammonia holding site. The Ala substitution profoundly decreased enzymatic activity. In the forward reaction, the value of kcat decreased by a factor of 150 while the values of Km(GMP) and Km(NADPH) increased by 13- and 19-fold, respectively. The reverse reaction could not be detected for GMPR/Y318A (< 0.5% of the activity of wild-type GMPR). In contrast, the partial hydride transfer reaction was relatively unperturbed by the mutation (Table 2), decreasing by only 2.5-fold. These observations suggest that deamination has become rate-limiting in the reaction of Y318A.
Y318F remained a proficient catalyst.
The Phe substitution increased the values of Km(GMP) and Km(NADPH) by 9-fold and 26-fold, respectively (Table 2). Nonetheless, the value of kcat was similar to that of the wild-type enzyme. Likewise, the value of kobs for the partial hydride transfer reaction was also similar to that of the wild-type enzyme, indicating that the mutation had little effect on hydride transfer, and that hydride transfer remains rate-limiting in the forward direction. Unlike Y318A, the reverse reaction could be observed, but the value of Vmax at 20 mM NH4+ was decreased by a factor of 40. Since the forward and partial hydride transfer reactions are unaffected, the mutation must selectively disrupt the deamination step.
We measured the exchange of [15N]-NH4+ into GMP to further characterize the role of Tyr318 in the deamination reaction. In contrast to the wild-type enzyme (Figure 4C), [15N]-GMP was barely visible after 1.6 h with Y318F (Figure 4D). The signal is so low that a high vertical gain is required for observation, which also reveals additional 15N resonances likely related to natural abundance 15N signals from a minor amount of guanine derivative present in the sample. These signals become insignificant as more [15N]-GMP is produced. Thus the Y318F mutation perturbs the ammonia exchange reaction, confirming that the hydroxyl of Tyr318 plays an important role in the deamination reaction. These observations are consistent with the hypothesis that Tyr318 is part of the ammonia holding site.
Tyr318 is critical for the distinct dynamic states associated with deamination.
The relaxation behavior of IMP in the hydride transfer complex of GMPR/Y318F was essentially indistinguishable from that of wild-type enzyme, with the exception of a small change in the value of RD0 for the cofactor mono-P (Figure 2, Figure S6 and Table S1). The value of RD0 and τD for the cofactor are larger than those of IMP, as observed in the wild-type enzyme complex, demonstrating that the mutant retains the characteristic dynamic state of the hydride transfer complex, as expected given the modest effects on the kinetics of the partial hydride transfer reaction. However, significant differences are observed in the relaxation of the deamination complex. Most notably, the values of τD/RD0 for the substrate mono-P and the cofactor di-Ps are decreased compared to those of the wild-type deamination complex. These observations suggest that the hydroxyl of Tyr318 plays a critical role in the dynamics of the deamination reaction, as expected if the movement of ammonia gates a cofactor conformational change.
DISCUSSION
Cluster analysis reveals distinct dynamic networks associated with hydride transfer and deamination complexes.
We hypothesized that if two residues are part of the same dynamic network, Ala substitutions of these residues are likely to have the similar effects on the field cycling parameters. Therefore we used clustering analysis to identify complexes with similar relaxation behavior, including the D219A, dIMP and dGMP complexes reported previously (please note the unfortunate similarity in the names of Asp219 characterized previously and Asp129 reported herein) 13. Since the field cycling parameters are similar for substrates in wild-type E•IMP, E•GMP and E•IMP•NADP+ complexes, we compared all complexes to the wild-type hydride transfer complex by expressing the field cycling parameters as log2 ratios to better highlight changes. Three clusters emerged as shown in Figure 6 (Figure S7 shows the associated heat map and Table S3 presents the field cycling parameters in the order of the clusters).
Figure 6. Cluster analysis of dynamic behavior of GMPR complexes.

A. The E•IMP•NADP+ and E•GMP•NADP+ complexes are dark green are dark pink, respectively. The analogous complexes with deoxy substrates are light green and light pink. The relative rate constants for the partial hydride transfer reaction are shown to the right of E•(d)IMP•NADP+ complexes. The relative rate constants for the reverse reaction are shown to the right of the E•(d)GMP•NADP+ complexes. B. Cartoon of proposed dynamic networks. Dynamic interactions are denoted by springs, blue denotes reaction specific interactions. Chains denote direct interactions that have modest effects on the dynamic networks. Cyan lines represent direct interactions as observed in the crystal structure of the inactive E•IMP•NADPH complex (Figure 1)17.
Cluster 1 contains the wild type and three mutant hydride transfer complexes (T105A, Y318F and D219A) plus the K77A deamination complex. The aberrant relaxation behavior of the K77A deamination complex readily explains its low activity. It is clear that Lys77 plays a critical role in a deamination specific dynamic network centering on the substrate. As noted above, the catalytic activities of the other three mutant complexes are modestly compromised compared to that of the wild-type hydride transfer reaction (the relative rate constants range from 0.11 to 0.67). Thus both activity and the dynamic network are largely preserved despite these substitutions, indicating that Thr105, Asp219 and Tyr318 do not play a substantial role in the networks required for hydride transfer. The D219A mutation is perhaps the most notable: Asp219 forms hydrogen bonds with both ribose hydroxyls of IMP, increasing the value of Km by 50-fold, yet the complex maintains relaxation behavior very similar that of the wild-type. We suggest that Asp219 may mainly serve to tether IMP in the hydride transfer complex.
Cluster 2 contains the wild-type and three mutant deamination complexes (T105A, Y318F and dGMP) plus the wild-type dIMP hydride transfer complex. Curiously, the dIMP hydride transfer complex is the most similar to the wild-type deamination complex, explaining why dIMP is a poor substrate. Thus the substrate 2′-OH is a critical component of a hydride transfer specific dynamic network. The three other deamination complexes display severely compromised activity (the relative rate constants range from 0.025 to 0.11). All three complexes display very similar relaxation parameters: the substrate 31Ps are closer to relaxers and the values of τD are smaller than in the wild-type deamination complex, suggesting that the substrate is more mobile. In contrast, these substitutions have comparatively little effect on the cofactor parameters. The absence of an effect on the cofactor in the T105A deamination complex is especially curious. Thr105 interacts directly with the cofactor in the OUT conformation of E•IMP•NADPH (Figure 1B,C), yet the biggest changes in relaxation are observed in the substrate (Figure 2). Collectively, these observations suggest that Thr105, Tyr318 and the substrate 2'-OH are part of a deamination specific dynamic network.
The last cluster contains both D129A complexes, the K77A hydride transfer complex and the D219A deamination complex. As noted earlier, the hydride transfer and deamination complexes of D129A are indistinguishable. Both the substrate and cofactor 31P nuclei are constrained and farther from relaxers than in the wild-type hydride transfer complex. Asp129 interacts directly with the cofactor and indirectly with the substrate via a water network in the IN conformation (Figure 1B,C). Although we do not have a crystal structure for E•GMP•NADP+, the OUT conformation of the E•IMP•NADPH complex suggests that Asp129 may interact indirectly via hydrogen bonding networks with both substrate and cofactor (Figure B,C). The Ala substitution eliminates these interactions, yet the substrates and cofactors become more constrained in the active site. Collectively these observations suggest that Asp129 is a central cog in a dynamic network required for both hydride transfer and deamination. We propose that Asp129 couples motions of the substrate, cofactor and enzyme. Networks of hydrogen bonds, involving Asp219, Lys177 and water molecules, connect Asp129 to the substrate 2’-OH in both the IN and OUT conformations (Figure 1B,C). Perhaps the coupled motions of these residues, together with the association/dissociation of water molecules, modulate the conformation and mobility of substrates and cofactors.
Like the D129A complexes, the D219A deamination and K77A hydride transfer complexes display high values of τD that indicate both substrate and cofactors are constrained. The cofactor 31Ps are also farther from relaxers than in the wild-type hydride transfer complex. Thus Asp219 and Lys77 determine the cofactor binding mode and play critical roles in reaction specific dynamic networks. Asp129 also influences substrate binding mode in the deamination complex, while Lys77 has no effect on substrate binding mode in the hydride transfer complex.
The movement of ammonia may gate the two dynamic states.
The experiments described above indicate that NH3/NH4+ has high affinity for the deamination complex but low affinity for the hydride transfer complex. We suggest two potential explanations for this apparent conundrum. First, it is possible that NH3/NH4+ has different affinities in the presence of reduced and oxidized cofactor, i.e., low affinity for E-XMP*•NADPH but high affinity for E-XMP*•NADP+. In this scenario, the high affinity of NH3/NH4+ for E-XMP*•NADP+ prevents inadvertent inactivation if E•GMP•NADP+ forms in a cell. While we cannot rule out this scenario, we favor a model where the NH3/NH4+ affinity of E-XMP*•NADPH is similar to that of E-XMP*•NADP+, such that NH3/NH4+ is not readily released from either E-XMP* complex. Instead NH3/NH4+ is released from a final product complex E•IMP•NADP+•NH3. We further suggest that the movement of NH3/NH4+ from the “reactive” site to the “holding” site changes the dynamic properties of the E-XMP*•NADPH complex from those conducive to deamination to those that favor hydride transfer. Such dynamic conformational transitions have been well documented from the perspective of the protein. 1-7
CONCLUSIONS
The experiments reported herein suggest that GMPR catalysis involves both general and reaction-specific dynamic networks connecting enzyme, substrates and cofactor (summarized in Figure 6B). The operation these networks is difficult to rationalize with the currently available crystal structures. Counterintuitively, loss of an interaction often decreases the mobility of a bound ligand, while in other cases the largest effects on relaxation occur far from the structural perturbation. Computational approaches will hopefully resolve these issues.
We also suggest that the catalytic cycle involves the movement of ammonia to a holding site where it interacts with the hydroxyl of Tyr318. The Tyr318 hydroxyl differentiates the distinct dynamic states associated with deamination and hydride transfer, suggesting that the movement of ammonia may gate the cofactor conformational. These findings reinforce the view that a subtle balance of substrate and cofactor dynamics is required for efficient catalysis, and further demonstrate that distal portions of the substrates/cofactors are critical features in the dynamic networks that modulate catalysis.
Supplementary Material
Acknowledgements:
This work was supported by the National Institutes of Health (GM054403 to LH). Molecular graphics images were produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIGMS P41-GM103311).
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
Supporting Information. Field cycling data including fits and parameter tables with statistical analysis, standard curve for proton uptake experiments, heat map of cluster analysis and field cycling parameters arranged by cluster.
REFERENCES
- [1].Boehr DD, Dyson HJ, and Wright PE (2006) An NMR perspective on enzyme dynamics, Chem Rev 106, 3055–3079. [DOI] [PubMed] [Google Scholar]
- [2].Pasat G, Zintsmaster JS, and Peng JW (2008) Direct 13C-detection for carbonyl relaxation studies of protein dynamics, Journal of magnetic resonance 193, 226–232. [DOI] [PubMed] [Google Scholar]
- [3].Ma B, and Nussinov R (2010) Enzyme dynamics point to stepwise conformational selection in catalysis, Curr Opin Chem Biol 14, 652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Klinman JP (2015) Dynamically achieved active site precision in enzyme catalysis, Acc Chem Res 48, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Osuna S, Jimenez-Oses G, Noey EL, and Houk KN (2015) Molecular dynamics explorations of active site structure in designed and evolved enzymes, Acc Chem Res 48, 1080–1089. [DOI] [PubMed] [Google Scholar]
- [6].Callender R, and Dyer RB (2015) The dynamical nature of enzymatic catalysis, Acc Chem Res 48, 407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Palmer AG 3rd. (2015) Enzyme dynamics from NMR spectroscopy, Acc Chem Res 48, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Peng JW (2003) New probes of ligand flexibility in drug design: transferred (13)C CSA-dipolar cross-correlated relaxation at natural abundance, J Am Chem Soc 125, 11116–11130. [DOI] [PubMed] [Google Scholar]
- [9].Deng H, Vu DV, Clinch K, Desamero R, Dyer RB, and Callender R (2011) Conformational heterogeneity within the Michaelis complex of lactate dehydrogenase, J Phys Chem B 115, 7670–7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deng H, Vedad J, Desamero RZB, and Callender R (2017) Difference FTIR Studies of Substrate Distribution in Triosephosphate Isomerase, J Phys Chem B 121, 10036–10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vaughn MB, Zhang J, Spiro TG, Dyer RB, and Klinman JP (2018) Activity-Related Microsecond Dynamics Revealed by Temperature-Jump Forster Resonance Energy Transfer Measurements on Thermophilic Alcohol Dehydrogenase, J Am Chem Soc 140, 900–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rosenberg MM, Redfield AG, Roberts MF, and Hedstrom L (2016) Substrate and Cofactor Dynamics on Guanosine Monophosphate Reductase Probed by High Resolution Field Cycling 31P NMR Relaxometry, J Biol Chem 291, 22988–22998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rosenberg MM, Redfield AG, Roberts MF, and Hedstrom L (2018) Dynamic Characteristics of Guanosine-5′-monophosphate Reductase Complexes Revealed by High-Resolution (31)P Field-Cycling NMR Relaxometry, Biochemistry 57, 3146–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schwans JP, Kraut DA, and Herschlag D (2009) Determining the catalytic role of remote substrate binding interactions in ketosteroid isomerase, Proc Natl Acad Sci U S A 106, 14271–14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Richard JP, Amyes TL, Goryanova B, and Zhai X (2014) Enzyme architecture: on the importance of being in a protein cage, Curr Opin Chem Biol 21, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Leatherbarrow RJ, and Fersht AR (1987) Investigation of transition-state stabilization by residues histidine-45 and threonine-40 in the tyrosyl-tRNA synthetase, Biochemistry 26, 8524–8528. [DOI] [PubMed] [Google Scholar]
- [17].Patton GC, Stenmark P, Gollapalli DR, Sevastik R, Kursula P, Flodin S, Schuler H, Swales CT, Eklund H, Himo F, Nordlund P, and Hedstrom L (2011) Cofactor mobility determines reaction outcome in the IMPDH and GMPR (beta-alpha)8 barrel enzymes, Nat Chem Biol 7, 950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera- a visualization system for exploratory research and analysis, J. Comp. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- [19].Redfield AG (2012) High-resolution NMR field-cycling device for full-range relaxation and structural studies of biopolymers on a shared commercial instrument, Journal of biomolecular NMR 52, 159–177. [DOI] [PubMed] [Google Scholar]
- [20].Roberts MF, and Redfield AG (2004) High-resolution 31p field cycling NMR as a probe of phospholipid dynamics, J Am Chem Soc 126, 13765–13777. [DOI] [PubMed] [Google Scholar]
- [21].Roberts MF (2018) High-resolution applications of shuttle field-cycling NMR, In Field Cycling NMR Relaxometry: Instrumentation, Model Theories and Applications (Kimich R, Ed.), pp 385 – 404 Royal Society Press. [Google Scholar]
- [22].Pu M, Feng J, Redfield AG, and Roberts MF (2009) Enzymology with a spin-labeled phospholipase C: soluble substrate binding by 31P NMR from 0.005 to 11.7 T, Biochemistry 48, 8282–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kuzmic P (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase, Anal. Biochem 237, 260–273. [DOI] [PubMed] [Google Scholar]
- [24].de Hoon MJ, Imoto S, Nolan J, and Miyano S (2004) Open source clustering software, Bioinformatics 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- [25].Saldanha AJ (2004) Java Treeview--extensible visualization of microarray data, Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- [26].Werbeck ND, Kirkpatrick J, Reinstein J, and Hansen DF (2014) Using (1)(5)N-ammonium to characterise and map potassium binding sites in proteins by NMR spectroscopy, Chembiochem 15, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shallop AJ, Gaffney BL, and Jones RA (2003) Use of 13C as an indirect tag in 15N specifically labeled nucleosides. Syntheses of [8-13C-1,7,NH2-15N3]adenosine, -guanosine, and their deoxy analogues, J Org Chem 68, 8657–8661. [DOI] [PubMed] [Google Scholar]
- [28].Hahn U, Desai-Hahn R, and Ruterjans H (1985) 1H and 15N NMR investigation of the interaction of pyrimidine nucleotides with ribonuclease A, Eur J Biochem 146, 705–712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.