

Abstract
Purpose
Typical hydrophobic amino acids (HAAs) are important motifs for self-assembling peptides (SAPs), but they lead to low water-solubility or compact packing of peptides, limiting their capacity for encapsulating hydrophobic drugs. As an alternative, we designed a peptide GQY based on atypical HAAs, which could encapsulate hydrophobic drugs more efficiently. Although hydrophobic general anesthetics (GAs) have been formulated as lipid emulsions, their lipid-free formulations have been pursued because of some side effects inherent to lipids. Using GAs as targets, potential application of GQY as a carrier for hydrophobic drugs was evaluated.
Methods
Thioflavin-T (ThT) binding test, dynamic light scattering (DLS) and transmission electron microscopy (TEM) were used to examine the self-assembling ability of GQY. Pyrene and 8-Anilino-1-naphthalenesulfonic acid (ANS) were used to confirm formation of hydrophobic domain in GQY nanoparticles. Using pyrene as a model, GQY’s capacity to encapsulate hydrophobic drugs was evaluated. GAs including propofol, etomidate and ET26 were encapsulated by GQY. Loss of righting reflex (LORR) test was conducted to assess the anesthetic efficacy of these lipid-free formulations. Paw-licking test was used to evaluate pain-on-injection of propofol-GQY (PROP-GQY) formulation. Hemolytic and cytotoxicity assay were used to evaluate biocompatibility of GQY.
Results
Stable nanoparticles containing plenty of hydrophobic cavities could be formed by GQY, which could encapsulate hydrophobic drugs at very high concentration and form stable suspensions. Propofol, etomidate and ET26 formulated by GQY showed anesthetic efficacy comparable to their currently available formulations. Unlike clinic lipid emulsion, PROP-GQY formulation did not cause pain-on-injection in rats. Neither obvious cytotoxicity nor hemolytic activity of GQY was observed.
Conclusion
GQY could encapsulate GAs to obtain stable and effective formulations. As a lipid-free carrier, GQY exhibited considerable biocompatibility and other side benefits such as reducing pain-on-injection. More SAPs based on atypical HAAs could be designed as promising carriers for hydrophobic drugs.
Keywords: self-assembling peptides, nanoparticles, hydrophobic drugs, lipid-free formulations, general anesthetics
Introduction
In the past two decades, various self-assembling peptides (SAPs) composed of natural amino acids have been developed as novel drug carriers, which have shown intriguing advantages including controllability in synthesis, biocompatibility and biodegradability in application, as well as modifiability for different purposes.1–3 However, despite the fact that many drugs in clinic or under investigation are hydrophobic molecules with poor water-solubility, the application of SAPs as carriers for hydrophobic drugs has not been well exploited yet. Although surfactant-like peptides (SLPs) forming nanovesicles or nanomicelles have been thought to be promising carriers for hydrophobic compounds, their drug-loading capacity was limited by a relatively small inner hydrophobic cavity.4,5 In this regard microfluidic technologies were usually needed to manipulate the size of SLPs vesicles to improve their drug-loading capacity.6 Mainly composed of highly hydrophobic amino acids (HAAs), SLPs also exhibited low water-solubility on their own, which may further limit the concentration of hydrophobic drugs they could reach. On the other hand, ionic self-complementary peptides such as RADA16 have also been used to encapsulate hydrophobic compounds.7,8 Although RADA16 showed much higher water-solubility, it is well-known to form nanofibers with their hydrophobic domain tightly packed inside, making their drug-loading behavior complicated and inefficient.
In many SAP systems such as the SLPs and ionic self-complementary peptides mentioned above, typical HAAs such as alanine, valine, leucine, isoleucine, and phenylalanine have been believed to be very important motifs. These typical HAAs provide strong hydrophobic interaction to support the self-assembling process, but also lead to compact packing between peptide monomers, leaving limited hydrophobic cavity for drug loading. In order to disperse and stabilize hydrophobic drugs at high concentration, the balance between the overall hydrophobicity of SAPs and their drug-loading capacity should be pursued.
As well as typical HAAs, glycine and glutamine also commonly existed in many SAPs.9–11 In our previous study, we found that glutamine could provide considerable hydrophobic interaction by dimethanediyl group in its side chain, although it has been usually categorized as a hydrophilic amino acid in classic textbooks.11 Similarly, although tyrosine has also been categorized as a hydrophilic amino acid, a hydrophobic benzene ring in its side chain also endows it with considerable hydrophobicity, making it an important component in some drug-loading systems.12–14 Based on these findings, we supposed glycine, glutamine and tyrosine could be regarded as atypical HAAs, which could be promising candidates for designing SAPs with well-balanced hydrophobicity and drug-loading capacity.
The intravenous general anesthetics (GAs) propofol and etomidate are highly hydrophobic drugs with very poor water-solubility, while they need to be administrated at relatively high concentration to induce and maintain anesthesia. Currently available clinical formulations of propofol and etomidate are emulsions using natural lipids as carriers. However, lipid emulsion has some innate drawbacks including microbial contamination,15,16 lipid metabolism abnormality,17,18 and pain-on-injection.19 Propofol infusion syndrome, a rare but serious side effect that more commonly occurs in patients with long-term infusion of high-dose propofol and is characterized by unexplained metabolic acidosis, rhabdomyolysis, cardiac failure and renal failure, has also been believed to be related with lipid formulations.20 For these reasons, lipid-free formulations of these drugs have been widely exploited based on various strategies including prodrugs21,22 and alternative carrier materials.23–25 Furthermore, our group has synthesized ET26, an analog of etomidate to minimize its suppression of adrenal cortical function,26 and it is also a hydrophobic molecule in need of suitable carrier materials to develop its effective formulation.
In this study, we explored the hydrophobic drug-loading capacity of GQQQQQY (abbreviated as GQY), a SAP composed of atypical HAAs. As shown in Figure 1, GQY contains five glutamine residues with dimethanediyl group and one tyrosine residue with benzene ring. Hydrophobic interaction among these groups could drive GQY to self-assemble into nanoparticles, which contain plenty of hydrophobic cavities for loading hydrophobic compounds. Using pyrene as a model molecule, the capacity of GQY to encapsulate hydrophobic drugs was evaluated. Using propofol, etomidate and ET26 as target drugs, their lipid-free formulations were prepared. Anesthetic effect and side benefits of these formulations were compared with current available formulations of these drugs. As a potential hydrophobic-drug carrier for clinical application, GQY’s biocompatibility was also evaluated.
Figure 1.

Self-assembling and drug-loading model of GQY. (A) Chemical structure of GQY. (B) Chemical structure of pyrene and GAs including propofol, etomidate and ET26. (C) Schematic illustration of drug loading by GQY. Hydrophobic groups in the side chain of glutamine and tyrosine are shown in purple.
Materials and Methods
Materials
Peptide GQY with purity over 98% was purchased from Shanghai Bootech BioScience & Technology Co. Ltd (Shanghai, China) as lyophilized powder. Pyrene crystal was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Etomidate active pharmaceutical ingredient (API) was purchased from Dalian Meilun Biotechnology Co., Ltd (Dalian, China). Etomidate lipid emulsion (FORRY) was purchased from Nhwa Pharma. Corporation (Xuzhou, China). Propofol (purity >99%) was provided by Zhongke Taidou Chemical Co., Ltd (Shandong, China). Propofol lipid emulsion (DIPRIVAN) was purchased from AstraZeneca UK (London, UK). ET26 (purity >99%) and its hydrochloride (ET26-HCl, purity >99%) were synthesized in our lab as previously reported.27
Formulation Preparation
GQY powder was dissolved in Milli-Q water to get 2.5 or 5 mM GQY solution. The GQY solution was treated with ultrasound bath for 20 min after being rotated up and down, and incubated at room temperature (RT) for 24 h before use. Pyrene crystal was added to 2.5 mM GQY solution, and etomidate or ET26 were added to 5 mM GQY solution and treated with ultrasound bath for 30 min to reach the final drug concentration of 5 mM for pyrene-GQY, 4 mg/mL for etomidate-GQY (ET-GQY for short) and 10 mg/mL for ET26-GQY. To fully disperse pyrene and etomidate in GQY solution, pyrene-GQY and ET-GQY was magnetically stirred for 40 min after ultrasound.
Alternatively, 5 mM GQY solution in 5% glucose injection (GI) was used to prepare propofol-GQY (PROP-GQY for short) formulation with propofol concentration of 10 mg/mL. The procedure was the same as that for ET26-GQY.
Thioflavine (ThT)-Binding Test
ThT-binding fluorescence was used to monitor the self-assembling behavior and thermal stability of GQY. Briefly, GQY was dissolved in Milli-Q water to get 5 mM GQY solution, which was incubated at 25°C, 40°C, 60°C or 80°C for 5 min before measurement. ThT stock solution with concentration of 1 mM was added into GQY solution at a volume ratio of 1:100. After incubation at RT for 2–5 min, the mixture was pipetted in Quartz cuvette to be examined. The excitation wavelength was set to 450 nm and emission spectrum between 460 and 600 nm was recorded by spectrofluorophotometer (Horiba iHR320).
Transmission Electron Microscopy (TEM) Observation
TEM (Tecnai G2 F20, FEI, USA) was applied to observe the nanostructure of GQY and drug-GQY complexes. Briefly, 10 μL of properly diluted sample was set on the surface of a copper grid for about 3 min, after which excess solution was blotted with filter paper. Then the sample was negatively stained for 2 min using 10 μL of 2% phosphotungstic acid, after which the staining solution was blotted with filter paper and the grid was air-dried for TEM observation.
Nanoparticle Size and Zeta Potential Measurement
The size distribution and zeta potential of different samples including GQY solution and drug-GQY complexes were measured by dynamic light scattering (DLS) using Zetasizer Nano ZS90 (Malvern Panalytical, Malvern, UK). Briefly, each sample was diluted 10–50 times to reach a proper count rate for DLS measurement. One milliliter of each diluted sample was pipetted into a disposable cuvette or a potential cell. For each sample, the size distribution plot and zeta potential plot were collected three times and averaged values were calculated.
Detection of Hydrophobic Region in GQY
Pyrene fluorescence in GQY solution or in Milli-Q water was compared to detect the formation of hydrophobic region in GQY. Pyrene crystal dissolved in dimethyl sulphoxide was prepared as a stock solution with concentration of 2 mM. In each 499 μL of GQY solution (5 mM in Milli-Q water) or Milli-Q water, 1 μL of stock solution was added (the final concentration of pyrene was 4 μM). The mixture was incubated at RT for 2–5 min before measurement. Fluorescence spectra ranging from 360–440 nm were measured with excitation wavelength of 336 nm. Obtained spectra were normalized to get a same value of the first peak (I1), and the value of the third peak (I3) was compared.
In addition, 8-Anilino-1-naphthalenesulfonic acid (ANS) fluorescence was measured to confirm the formation of hydrophobic region in GQY. ANS dissolved in PBS (pH 7.4) with the concentration of 2 mM was added to 5 mM GQY or control (Milli-Q water) at a volume ratio of 1:100. After incubation at RT for 2–5 min, the fluorescence spectra ranged from 400–600 nm was measured by spectrofluorophotometer with excitation wavelength of 350 nm.
Pyrene-GQY Stability
The pyrene-GQY solution was kept in RT for a month to examine its stability and the first day of termination of stirring was regarded as day 0. At each time interval, an aliquot of pyrene-GQY solution was taken out and diluted 50 times with Milli-Q water, and pyrene-fluorescence was measured with a spectrofluorophotometer (Horiba iHR320). We tested pyrene-GQY stability through comparing intensity of fluorescence of pyrene-GQY between 360–600 nm with excitation wavelength of 336 nm. The particle size, zeta potential and TEM images of pyrene-GQY was also continuously collected for 30 days.
Drug-Loading Capacity and Encapsulation Efficiency
Centrifugation was performed to examine the drug-loading capacity and encapsulation efficiency for the three GQY-based GAs formulations. Briefly, 600 μL of PROP-GQY, ET-GQY or ET26-GQY was pipetted into an EP tube and centrifuged for 10 min at 20,000 rpm, 4°C. High performance liquid chromatography (Shimadzu LC-20AD) was used to measure the concentration of free drugs in the supernatant with a detection wavelength at 254 nm. Swell ChromPlus C18 column (4.6 mm × 150 mm, 5 μm) was applied to separate analytes and the column temperature was set at 40°C. The mobile phase A and B was 0.1% trifluoroacetic acid aqueous solution and acetonitrile respectively and the ratio of A to B for etomidate and ET26 was 70: 30, and 30: 70 for propofol. The flow rate was set at 1.0 mL/min and the injection volume was 5 μL. The percentage of drug-loading capacity and encapsulation efficiency of each formulation was calculated according to the following equations:
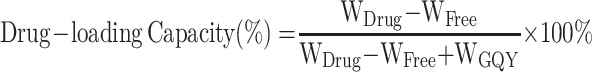 |
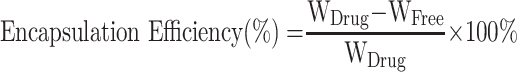 |
In the equations, WDrug was the total weight of GAs added into the formulation, WFree was the weight of free drug in the supernatant, and WGQY was the weight of GQY nanoparticles.
Animals
All animal experiment procedures were approved by the Animal Ethical Committee of West China Hospital, Sichuan University (Ethical approval number, 2020018A) and conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals by the United States National Institutes of Health.28 Young male (8 weeks old) Sprague Dawley rats weighing 230–340 g and 18 male weaning rats weighing 101–114 g (2 weeks old) were purchased from Dossy Biological Technology Co. Ltd (Chengdu, China). All rats were housed at 25 ± 1°C and 60% humidity in the Animal Experimental Center of Sichuan University (Chengdu, China) with a 12-hour light/dark cycle (lights on at 7:00 a.m.) and had access to water and food ad libitum. The rats were habituated to the experimental environment 1 week before testing.
Pharmacodynamic Study
Each rat was placed in a holder to expose its tail. Venipuncture of the lateral caudal vein was performed in the posterior third of the tail after it was sterilized with 75% ethanol alcohol. Test drugs (PROP-GQY, DIPRIVAN, ET-GQY, FORRY, ET26-GQY, ET26-HCl, and GQY) were diluted with 0.9% normal saline (NS) to a fixed volume of 0.6 mL and singly injected into the rat at a rate of 0.1 mL/s. Each rat kept in the cage individually was observed carefully and assessed whether it was sedated according to the modified sedation score (Table S1).27 The rat was placed supine when it had ataxia. If the rats failed to right themselves, they had loss of righting reflex (LORR). The LORR duration no less than 30 s was defined as positive.29,30 The recording from positive to negative or negative to positive was defined as a cross. Once 5 crosses in the same direction were recorded, the test was terminated.27 The Dixon and Mood method31 was applied to calculate the median effective doses (ED50) values with 95% confidence intervals (CIs).
After establishing the ED50 of each formulation, pharmacodynamic study was carried out by giving each formulation at a dose of 2 ED50 to rats through lateral caudal vein. The onset time, LORR duration, recovery time and adverse effects were carefully observed and recorded.
Paw-Licking Test
Paw-licking test32,33 was performed to compare pain-on-injection between two propofol formulations. Eighteen weaning rats were randomly divided into 3 groups. Each rat was kept in the steel holder to expose its right hind limb. The footpad of right hind paw pre-cleaned with 75% alcohol was singly injected with 0.9% NS, DIPRIVAN or PROP-GQY. Licking or lifting frequently of the right hind paw was defined as occurrence of pain after injection. The onset and duration of paw-licking and times of lifting the right hind paw within 10 min after treatment was observed and recorded carefully.
Serum Corticosterone Measurement
Adult male SD rats were randomly divided into 3 groups including ET-GQY, ET26-GQY and GQY groups (n = 9). Lateral tail vein was cannulated with 24 G intravenous catheter as the pathway to give drugs and collect blood samples. To inhibit endogenous adrenocorticotropic hormone (ACTH) production and baseline corticosterone production, dexamethasone (DXM) at dose of 0.5 mg/kg was given to each rat after the blank blood was collected. Two hours after DXM administration, blood was collected as the baseline of corticosterone. Then, 2 ED50 dose of test drugs was singly injected following 0.2 mg/kg DXM administration. Then 15 min later, ACTH1–24 (Sigma-Aldrich Co., St Louis, MO) was given at dose of 25 μg/kg to stimulate corticosterone production.34 Blood was collected at 15, 30, 60 and 90 min after ACTH treatment. Approximately 0.3 mL of blood was drawn for each blood collection and isovolumic 0.9% NS was given. Heparin (10 U/mL) was given to avoid blood clotting after drug or NS administration. All drugs were given at speed of 0.1 mL/s.
Blood samples were kept at RT for up to 60 min to clot. After that, blood samples were centrifuged at 3500 g for 5 min. The supernatant was transferred to a clean EP tube and centrifuged at 16,000 g for 5 min to further separate any cell or particles. The supernatant was transferred to a clean EP tube and stored at −80°C. Liquid chromatography-tandem mass spectrometry was employed to measure corticosterone concentration. After being thawed at RT, 50 μL of sample was added into a 1.5 mL EP tube with 150 μL of acetonitrile which contained corticosterone-d8 as internal standard. The mixture was fully vortexed and centrifuged at 20,000 rpm at 4°C for 10 min, and the supernatant was added into the vials to be measured.
Hemolytic Assay
Approximately 1.5 mL of whole blood was collected from a healthy adult volunteer who provided written informed consent to participate. Collection and use of the blood for the study were approved by the Clinical Ethical Committee of West China Hospital. About 200 μL of PBS was added to the whole blood to wash the erythrocytes. The tube filled with blood was inverted gently and then centrifuged at 1000 rpm, 4°C for 10 min. The supernatant was removed with a pipette. The above steps were repeated 3–4 times until the supernatant was clear without red color. Finally, 1 mL of red blood cells was obtained and added into 24 mL of PBS to get red blood cell suspension, which was kept on ice before use. The erythrocytes suspension was incubated with GQY at different concentrations at 37°C for 1 h. After that, all samples were placed on ice to stop the interaction and then centrifuged at 4°C, 1000 g for 10 min. The supernatant was transferred into a 96-well plate and examined at 541 nm using Eon microplate reader (Biotek Inc., USA). PBS and 1% Triton X-100 were used as the negative and positive control, respectively. The hemolytic percent ratio was calculated using the following equation:
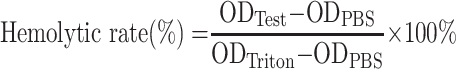 |
Cytotoxicity
Rat kidney fibroblast cell NRK-49F and mouse fibroblast cell L929 used for evaluating the biocompatibility of GQY were obtained from the Metabolomics Laboratory of West China Hospital. The use of these cell lines was approved by the Animal Ethical Committee of West China Hospital, Sichuan University. L929 cells were maintained in RPMI 1640 medium and NRK-49F cells were maintained in Dulbecco’s modified Eagle’s medium, both of which were supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin solution. A cell count kit (CCK-8, Saint-Bio, China) was used to quantitatively evaluate the cell viability, which indicated the cytotoxicity of GQY to each cell. GQY stock solutions with concentration of 10 mM were prepared in Milli-Q water or GI. Cell culture medium containing GQY with different concentrations ranging from 0.01 to 2 mM was prepared by mixing GQY stock solutions (in Milli-Q water or in GI) with full medium properly. L929 and NRK-49F cells were seeded in 96-well plates at 7×103 and 4×103 per well, respectively. After overnight incubation, the original medium was replaced by fresh complete medium or medium containing 0.01 mM, 0.1 mM, 0.5 mM, 1.0 mM, or 2.0 mM of GQY. Each sample was set in quadruplicate. After 24 hours of incubation, the original medium was removed and serum-free medium with 10% CCK-8 reagent was added to each well. The plate was then incubated at 37 °C in 5% CO2 for 1 to 1.5 h. Non-treated cell was used as negative control, and medium without cell was used as blank control. The OD value at 490 nm was quantified using Eon microplate reader. The relative cell viability was calculated according to the following equation:
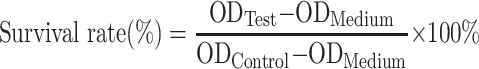 |
Statistical Analysis
SPSS version 24.0 (IBM, Chicago, IL, USA) was applied to analyze all data. All measurement data were reported as mean ± standard deviation or median with interquartile range. LORR and sedation duration in pharmacodynamic study between two formulations were compared using student independent sample t-test or Kruskal–Wallis test. Continuous variable in corticosterone assay was assessed by repeated measures analysis of variance. All P values were two-tailed in this study and P < 0.05 was considered statistically significant.
Results and Discussion
Formation of Stable GQY Nanoparticles with Hydrophobic Domain
As shown in Figure 2A, GQY with concentration of 5 mM can be well dispersed in water and forms a translucent solution with pH of 4.0. TEM image showed that GQY formed nanoparticles with a relatively uniform diameter of less than 100 nm. As shown in Figure 2B and C, adjusting the pH of water solution to 7.4 or dissolving the peptide in GI leads to the formation of nanoparticles with similar morphology and slightly smaller size, suggesting that the nanoparticles formed by GQY were relatively stable under physiological pH and compatible with different clinical solvents. DLS showed that the particle size of GQY in pure water with pH 4.0, pH 7.4 or in GI was (249.57 ± 3.40), (165.60 ± 11.93) and (254.30 ± 3.30) nm (n = 3), respectively, with their presentative plots shown in Figure 2D. The zeta potential of the above three GQY solutions was (18.87 ± 0.42), (−29.83 ± 1.02) and (19.60 ± 0.36) mV, respectively, with their representative plots shown in Figure 2E. The size of GQY particles measured by DLS was larger than that by TEM, which might be attributed to the aggregation of GQY particles. This is not surprising since zeta potential data showed all samples exhibit weak surface charge. Nevertheless, the sample with pH 7.4 showed relatively higher net surface charge (−29.83 ± 1.02 mV), which led to the formation of smaller particles. These results suggested that pH change only slightly changed the size of GQY aggregates by changing their surface charge, while their morphology of nanoparticles was relatively stable. Furthermore, the self-assembling behavior of GQY also showed considerable thermostability. As shown in Figure 2F, compared with GQY solution at RT, ThT-binding fluorescence of the peptide only slightly decreased with the increase of temperature, and the peptide still retained its self-assembling ability when the temperature was increased up to 80°C. These results indicated that GQY could form stable nanoparticles with considerable resistance to the change of solvent parameter and temperature, ensuring its application in physiological conditions and stability in storage.
Figure 2.

Formation of stable nanoparticles by GQY. (A–C) TEM images of GQY nanoparticles formed in H2O with pH 4.0 (A), in H2O with pH 7.4 (B) and in GI (C). Insertions show photographs of corresponding GQY solutions. (D) Representative particle size distribution of GQY in H2O with pH 4.0 and 7.4, and GQY in GI. (E) Representative Zeta potential plot of GQY in H2O with pH 4.0 and 7.4, and GQY in GI. (F) ThT-binding fluorescence spectra of GQY in H2O at different temperatures.
Using pyrene and ANS as fluorescent probes, we further confirmed the formation of hydrophobic domain in the GQY nanoparticles. We know the fluorescent spectrum of pyrene monomers shows five peaks in the range of 360–440 nm. Compared with a hydrophilic environment, the ratio between fluorescence intensity of the 1st peak (I1) and the 3rd peak (I3) would decrease when pyrene was in a hydrophobic environment. As shown in Figure 3A, when the I1 peaks were normalized, I3 of pyrene in GQY was significantly higher than that in water (Figure 3A), which suggested that pyrene monomers were embedded in hydrophobic domains in the GQY nanoparticles. In addition, ANS-binding fluorescent spectrum of GQY showed significant blue-shift and enhancement compared with ANS dissolved in water, which was also a characteristic of the existence of hydrophobic domain (Figure 3B). These results further confirmed that the self-assembly of GQY mainly relied on hydrophobic interaction, which also leads to the formation of plenty of hydrophobic domains for encapsulating hydrophobic drugs.
Figure 3.

Formation of hydrophobic domain. (A) Fluorescence spectra of pyrene monomer in 5 mM GQY solution or H2O. (B) ANS-binding fluorescence spectra of 5 mM GQY or H2O.
Drug Loading Capacity of GQY
Pyrene composed of four benzene rings is a highly hydrophobic molecule with extremely low solubility in water (about 7×10−4 mM). Because of its special fluorescence, pyrene was usually used as a model molecule to evaluate the capacity of carrier materials to encapsulate hydrophobic drugs. In our previous study, we have used a SLP to encapsulate and disperse pyrene, which increased its concentration in aqueous solution to about 0.655 mM. However, due to the limited drug-loading capacity of SLP, the suspension obtained in that study contained many amorphous micro-scale particles and was very unstable.5 In the current study, 5 mM pyrene could be well dispersed in 2.5 mM GQY solution and formed a thick-milky suspension (Figure 4A). As shown in the TEM images in Figure 4B, pyrene-GQY formed homogeneous nanoparticles with diameter of less than 100 nm, and the morphology and size of the nanoparticles did not show obvious change within 30 days. As shown in Figure 4C, DLS revealed that the average size of pyrene-GQY nanoparticles did not change significantly within the first 14 days, but slightly increased from (370.91 ± 13.04) nm to (488.77 ± 19.38) nm after 30 days. At the same time, zeta potential of the pyrene-GQY nanoparticles also slightly decreased from (26.51 ± 1.53) mV to (22.10 ± 0.53) mV. These results indicated that the individual morphology of pyrene-GQY nanoparticles kept very stable and their overall size was also stable for at least 14 days. After storage for 30 days, they tended to undergo mild aggregation as their surface charge decreased. This situation might be caused by the natural degradation of GQY peptide, which could be improved by modifying the peptide with protecting groups such as N-terminal acetyl and C-terminal amide.
Figure 4.

Pyrene-GQY nanoparticles and stability. (A) Photograph of pyrene-GQY suspension with 5 mM pyrene dispersed in 2.5 mM GQY solution. (B) TEM images of pyrene-GQY nanoparticles collected at different time from 0–30 days. (C) Size distribution and zeta potential of pyrene-GQY nanoparticles within 30 days. (D) Fluorescence spectra of pyrene-GQY within 30 days.
Even at its current version, this mild aggregation tendency did not affect the overall stability of pyrene-GQY formulation. As shown in Figure 4D, pyrene-GQY suspension exhibited a fluorescence peak between 440 and 500 nm, which was defined as the excimer peak representing the existence of pyrene nanoparticles in solution. The pyrene excimer peak did not change significantly within 30 days, suggesting a homogeneous and stable distribution of encapsulated pyrene nanoparticles in the suspension. These results indicated that GQY has a very high drug-loading ability, and the suspension containing pyrene encapsulated in GQY was relatively stable for at least a month, implicating GQY’s potential as an efficient carrier for hydrophobic drugs.
Characterization of GQY-Based GAs Formulations
Propofol and etomidate are hydrophobic GAs commonly used in clinic. Currently, the two drugs are formulated with natural lipids to reach a high drug concentration in the emulsions, i.e., 10 mg/mL for propofol and 2 mg/mL for etomidate. Due to potential side effects caused by natural lipids, lipid-free carrier systems for these GAs have been pursued for many years. On the other hand, our group has previously synthesized ET26 (an analog of etomidate) as a potential GA, which is also in need of hydrophobic drug carriers. In this study, GQY was used to encapsulate propofol, etomidate and ET26. As shown in Figure 5A–C, 10 mg/mL propofol, 4 mg/mL etomidate and 10 mg/mL ET26 were well dispersed in 5 mM GQY solution, forming milky suspensions. TEM images showed that PROP-GQY, ET-GQY and ET26-GQY formed nanoparticles with diameter of about 100 nm, which was slightly bigger than empty nanoparticles formed by GQY. As shown in Figure 5D, DLS revealed that the particle size of PROP-GQY, ET-GQY and ET26-GQY was (293.47 ± 23.60), (306.87 ± 22.25) and (367.73 ± 23.60) nm, respectively (n = 3). Like pure GQY solution, the size measured by DLS was larger than that under TEM, which could be explained as the aggregation of nanoparticles. As shown in Figure 5D, zeta potential of the above drug-GQY complex was (22.67 ± 0.74) mV for PROP-GQY, (−8.22 ± 0.35) mV for ET-GQY and (−13.30 ± 0.20) mV for ET26-GQY, respectively (n = 3). The weak surface charge also suggested that nanoparticles of the system tend to aggregate.
Figure 5.

Formation of drug-GQY nanoparticles. (A–C) TEM images of nanoparticles in PROP-GQY (A), ET-GQY (B) and ET26-GQY (C) formulations. Insertions show photographs of corresponding formulation. (D) Particle size and zeta potential of different formulations.
Although GQY nanoparticles with similar original pH value and zeta potential were used to encapsulate different GAs, the obtained formulations showed different zeta potential as shown in Figure 5D. This difference was caused by the different chemical property of drugs encapsulated, which led to different pH of the formulations as shown in Table 1. When etomidate or ET26 was added in the GQY solution, their imidazole groups could absorb free protons disassociated from the -COOH group of the peptide. On one hand, this will decrease the concentration of free protons in the solution, so that the pH value would increase. On the other hand, this would promote the disassociation of -COOH groups on peptides, exposing more -COO− groups, so that the nanoparticles would become negatively charged. But this reaction did not occur in the PROP-GQY system since there are no proton-absorbing groups in propofol. Thus, PROP-GQY showed positive surface charge similar to the empty GQY nanoparticles while ET-GQY and ET26-GQY showed negative charges.
Table 1.
Drug-Loading Capacity and Encapsulation Efficiency of GQY for GAs
| Variables | PROP-GQY | ET-GQY | ET26-GQY |
|---|---|---|---|
| pH | 2.92 | 4.23 | 6.01 |
| Drug-loading capacity (%) | 68.73 ± 0.19 | 41.76 ± 0.08 | 57.87 ± 0.10 |
| Encapsulation efficiency (%) | 96.59 ± 0.87 | 78.78 ± 0.28 | 60.35 ± 0.25 |
The drug-loading capacity and encapsulation efficiency of the three GQY-based GAs is shown in Table 1. It should be noted that PROP-GQY showed much higher drug-loading capacity and encapsulation efficacy than ET-GQY and ET26-GQY. It is likely that protonated imidazole groups could make a portion of etomidate or ET26 become soluble, so that the amount of their insoluble form encapsulated in GQY nanoparticles was decreased. However, compared with previously reported nanocarriers for propofol or etomidate, GQY showed an excellent drug-loading capacity.25,35 These results indicated that GQY could be an efficient carrier for loading these GAs to a concentration as high as, or even higher than their currently available formulations, which would ensure the dosages needed for general anesthesia.
Pharmaceutic Efficacy of GQY-Based GA Formulations
As shown in Table 2, PROP-GQY and ET-GQY formulations exhibited anesthetic efficacy similar to their clinical formulations, i.e., DIPRIVAN and FORRY, respectively. The ED50 with 95% CI of PROP-GQY was 4.90 (4.72–5.08), which was the same as that of DIPRIVAN. Similarly, the ED50 of ET-GQY was close to FORRY ([0.70 (0.66–0.75)] vs [0.65 (0.61–0.69)]). Both PROP-GQY and ET-GQY took effect immediately after intravenously administering the drugs at a dose of 2 ED50, although the onset time of PROP-GQY was slightly slower than that of DIPRIVAN. Considering the duration of LORR and sedation under a dose of 2 ED50, both PROP-GQY and ET-GQY showed efficacy similar to DIPRIVAN and FORRY, respectively. Additionally, no obvious adverse reaction was observed in rats that received two propofol formulations. Two out of eight rats in etomidate group and four in ET-GQY group developed myoclonus, which might be induced by etomidate. These results demonstrated that GQY was not only as efficient as lipids in drug loading, but also achieved anesthetic efficacy equivalent to lipid-based formulations. Furthermore, lyophilized ET-GQY formulation also showed considerable anesthetic efficacy after resuspension (Table S2), indicating a promising way to prepare powder-form GA formulations which might be more stable, easy to store and transport.
Table 2.
Pharmacodynamics Data of Different Formulations
| Drugs | ED50 (mg/kg) | Onset Time (min) | P | LORR Duration (min) | P | Sedation Duration (min) | P |
|---|---|---|---|---|---|---|---|
| Propofol | |||||||
| PROP-GQY | 4.90 (4.72–5.08) | 0.20 ± 0.09 | 0.001 | 8.33 ± 1.86 | 0.51 | 13.60 ± 2.34 | 0.30 |
| DIPRIVAN | 4.90 (4.72–5.08) | 0.14 ± 0.01 | 9.16 ± 1.20 | 12.60 ± 1.14 | |||
| Etomidate | |||||||
| ET-GQY | 0.70 (0.66–0.75) | 0.11 ± 0.02 | 0.57 | 6.50 ± 1.33 | 0.82 | 12.36 ± 2.63 | 0.60 |
| FORRY | 0.65 (0.61–0.69) | 0.12 ± 0.01 | 6.21 ± 1.49 | 13.15 ± 3.28 | |||
| ET26 | |||||||
| ET26-GQY | 2.43 (2.26–2.61) | 0.11 ± 0.02 | 0.001 | 3.10 ± 0.71 | 0.20 | 8.98 ± 0.74 | 0.001 |
| ET26-HCl | 1.87 (1.74–2.00) | 0.18 ± 0.01 | 3.92 ± 1.41 | 11.66 ± 1.67 |
ET26 as an analog of etomidate was also a hydrophobic drug. Previously, 35% propylene glycol was used as a solvent for ET26-HCl in our lab. However, propylene glycol as an organic solvent had some drawbacks such as hemolysis and pain-on-injection.36,37 In this study, GQY was also tested as a potential carrier for ET26. As shown in Table 2, the ED50 of ET26-GQY was comparable with that of ET26-HCl ([2.43 (2.26–2.61) mg/kg] vs [1.87 (1.74–2.00) mg/kg]). Pharmacodynamic study showed that at a dose of 2 ED50, the LORR duration of ET26-GQY (3.10 ± 0.71) was similar to that of ET26-HCl (3.92 ± 1.41, P = 0.20). The duration of sedation in ET26-GQY group (8.98 ± 0.74) was slightly shorter than that in ET26-HCl group (11.66 ± 1.67, P = 0.001), which suggested that ET26-GQY may show an advantage in rapid recovery. Furthermore, it should be noted that severe hematuria was observed in all rats injected with 2 ED50 dose of ET26-HCl, which was mainly caused by 35% propylene glycol as solvent in that group.36,37 On the contrary, no hematuria was observed in the ET26-GQY group. Additionally, myoclonus occurred in four rats in both two groups but no another serious events such as respiratory depression were observed.
Corticosterone Level After Administrating ET26-GQY
We further studied the adrenocortical function of rats injected with GQY, ET-GQY or ET26-GQY. After ACTH administration, the serum corticosterone concentrations of rats in GQY and ET26-GQY groups were significantly higher than that in the ET-GQY group, suggesting that adrenocortical function was suppressed by etomidate but not by ET26 or GQY as carrier (Figure 6). This tendency was similar to the results in our previous study,27 indicating that GQY as delivery carrier did not affect the advantage of ET26 in maintaining the adrenocortical function during general anesthesia.
Figure 6.

Serum corticosterone level. (A) Flow chart of blood collection for serum corticosterone determination, a–f: time for blood collection, which was counterpart of the x axis labels in Figure 6B. (B) The serum corticosterone level after ACTH stimulation and intravenous injection of 2 ED50 GQY, ET-GQY or ET26-GQY (n = 9). #P < 0.05, ET-GQY versus GQY. *P < 0.05, ET-GQY versus ET26-GQY. “1st DXM 2h” defined the timepoint that was 2 hours after 1st DXM was given, “ACTH 15min/30min/60min/90min” are the timepoints that were 15, 30, 60 or 90 minutes after ACTH was administered.
Relieved Pain-on-Injection by PROP-GQY Formulations
Pain-on-injection as a commonly reported side effect has long been a flaw of DIPRIVAN, a lipid emulsion of propofol currently used in clinic. Although the mechanism of pain-on-injection induced by propofol is still unclear, a previous study has proposed that lipid might contribute to this symptom by activating the kallikrein-kinin system and subsequently increasing the bradykinin production, which increased the contact between the aqueous free propofol and free nerve endings at the injected vein and finally caused pain.38 Currently, administering propofol mixed with lidocaine were commonly used in clinic to reduce pain-on-injection,39,40 but it has been found that the particle size of formulation would increase after lidocaine was added into DIPRIVAN,41 which made the formulation unstable and might lead to some adverse events. For these reasons, lipid-free formulation of propofol may have innate advantage in reducing pain-on-injection.
In this study, paw-licking test was used to compare pain-on-injection between different propofol formulations based on lipid or GQY. As shown in Table 3, none of the rats that received NS or PROP-GQY displayed paw-licking behavior, while 4 of 6 rats in DIPRIVAN group showed paw-licking behavior with a median duration of 5.99 (0, 47.41) seconds. Except for paw-licking, the behavior of paw-lifting also demonstrated pain induced by injection of formulations. As shown in Table 3, all rats treated with DIPRIVAN showed paw-lifting behavior with a median frequency of 15.5 (8.5, 23.5) times, while only 2 of 6 rats injected with PROP-GQY displayed paw-lifting behavior with median frequency of 0 (0, 3) times. These data indicated that unlike lipid emulsion of propofol, the GQY formulation showed little irritating effect on injection, suggesting its potential application as a pain-free propofol formulation. Furthermore, GQY formulation did not support the growth of Staphylococcus aureus compared with the formulation based on lipid emulsion (Figure S1).
Table 3.
Pain-on-Injection of Different Formulations
| Events | 0.9% NS | DIPRIVAN | PROP-GQY | P |
|---|---|---|---|---|
| Paw-licking | ||||
| Number of animals | 0/6 | 4/6 | 0/6 | 0.015 |
| Median duration† (seconds) | 0 | 5.99 (0, 47.41) | 0 | 0.009 |
| Paw-lifting | ||||
| Number of animals | 0 | 6/6 | 2/6 | 0.002 |
| Median frequency† (times) | 0 | 15.5 (8.5, 23.5) | 0 (0, 3) | 0.001 |
Note:†Values are presented as median (interquartile range).
Biocompatibility and Safety of GQY
NRK-49F and L929 cells were used to assess potential cytotoxicity of GQY. As shown in Figure 7A, at concentration no higher than 1 mM, GQY in Milli-Q or in GI showed little effect on the viability of rat NRK-49F cells. Although the medium containing 2 mM GQY slightly decreased the cell viability, this effect might be caused by the fact that the medium contained only 80% full medium so that less nutrient was actually provided. Similarly, GQY in Milli-Q or in GI did not show obvious cytotoxicity against mouse L929 cells at concentrations up to 2 mM (Figure 7B), indicating biocompatibility of the peptide.
Figure 7.

Biocompatibility of GQY. (A) Cytotoxicity of GQY in NRK-49F cells. (B) Cytotoxicity of GQY in L929 cells. (C) Hemolytic activity of GQY.
In addition, in vitro hemolytic activity of GQY was assayed to evaluate its safety for intravenous injection. As shown in Figure 7C, as the concentration of GQY rose, the hemolysis percent ratio tended to increase slightly. However, even when the concentration of GQY increased to 1 mM, the hemolysis ratio kept below 6%, which was within an acceptable range.42 It should also be noted that at this concentration, GQY in Milli-Q water showed higher hemolytic ratio than that in GI, suggesting that the hemolytic activity might be caused by osmotic pressure rather than the peptide itself. Furthermore, since all GQY-based formulations will be diluted before intravenous injection, the low hemolytic activity observed in this study was actually negligible. As shown by the results of hemolytic assay and cytotoxicity assay, GQY showed excellent biocompatibility similar to other peptide-based biomaterials.43 Combining the well-evaluated safety of propofol, etomidate and ET26, the GQY-based formulations investigated in our current study are promising candidates for clinical application.
Conclusions
In conclusion, in this study we evaluated GQY for its potential application as a hydrophobic-drug carrier. GQY in aqueous solution could self-assemble to form nanoparticles, which could efficiently encapsulate a high dose of hydrophobic GAs including propofol, etomidate and ET26. All GQY-based formulations exhibited anesthetic efficacy comparable with currently available formulations based on lipid emulsion or propylene glycol. Additionally, PROP-GQY formulation did not cause obvious pain-on-injection as lipid emulsion of propofol did. With the absence of lipid that could boost the growth of bacteria, GQY-based formulations were also more resistant to microbial contamination. Furthermore, GQY as a peptide composed of natural amino acids also showed excellent biocompatibility and safety. These results suggested that GQY could be used as a high-loading carrier material, which would have great potential in exploiting stable lipid-free formulations of hydrophobic drugs.
On the other hand, as we know GQY is the first SAP carrier material completely composed of atypical HAAs. Our study suggested that using atypical HAAs to design SAP might be a promising strategy to obtain carrier materials with well-balanced hydrophobicity and drug-loading capacity. Using GQY as a blueprint, more SAPs based on atypical HAAs could be designed and their potential for encapsulating hydrophobic drugs could be further exploited.
Acknowledgment
This work was funded by the National Natural Science Foundation of China (No. 81973274). The authors are thankful to the Metabolomics Laboratory of West China Hospital for providing NRK-49F cells and L929 cells.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Gong ZY, Liu XY, Dong JH, et al. Transition from vesicles to nanofibres in the enzymatic self-assemblies of an amphiphilic peptide as an antitumour drug carrier. Nanoscale. 2019;11(33):15479–15486. doi: 10.1039/C9NR02874A [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Jiang N, Fu B, Huang F, Liu JF. Self-assembling peptide-based nanodrug delivery systems. Biomater Sci. 2019;7(12):4888–4911. doi: 10.1039/C9BM01212E [DOI] [PubMed] [Google Scholar]
- 3.Peng F, Zhang WS, Qiu F. Self-assembling peptides in current nanomedicine: versatile nanomaterials for drug delivery. Curr Med Chem. 2020;27(29):4855–4881. doi: 10.2174/0929867326666190712154021 [DOI] [PubMed] [Google Scholar]
- 4.Fatouros DG, Lamprou DA, Urquhart AJ, et al. Lipid-like self-assembling peptide nanovesicles for drug delivery. ACS Appl Mater Interfaces. 2014;6(11):8184–8189. doi: 10.1021/am501673x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen YZ, Tang CK, Zhang J, Gong M, Su B, Qiu F. Self-assembling surfactant-like peptide A6K as potential delivery system for hydrophobic drugs. Int J Nanomedicine. 2015;10:847–858. doi: 10.2147/IJN.S71696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan KH, Tay JJJ. Advancement of peptide nanobiotechnology via emerging microfluidic technology. Micromachines (Basel). 2019;10:627. doi: 10.3390/mi10100627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng C, Wei WP, Wang YH, et al. Study of the interaction between self-assembling peptide and mangiferin and in vitro release of mangiferin from in situ hydrogel. Int J Nanomedicine. 2019;14:7447–7460. doi: 10.2147/IJN.S208267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Briuglia ML, Urquhart AJ, Lamprou DA. Sustained and controlled release of lipophilic drugs from a self-assembling amphiphilic peptide hydrogel. Int J Pharm. 2014;474(1–2):103–111. doi: 10.1016/j.ijpharm.2014.08.025 [DOI] [PubMed] [Google Scholar]
- 9.Ishida A, Watanabe G, Oshikawa M, Ajioka I, Muraoka T. Glycine substitution effects on the supramolecular morphology and rigidity of cell-adhesive amphiphilic peptides. Chemistry. 2019;25(59):13523–13530. doi: 10.1002/chem.201902083 [DOI] [PubMed] [Google Scholar]
- 10.Paul S, Kumari K, Paul S. Molecular insight into the effects of enhanced hydrophobicity on amyloid-like aggregation. J Phys Chem B. 2020;124(45):10048–10061. doi: 10.1021/acs.jpcb.0c06000 [DOI] [PubMed] [Google Scholar]
- 11.Chen YZ, Xing ZH, Liao DQ, Qiu F. Neglected hydrophobicity of dimethanediyl group in peptide self-assembly: a hint from amyloid-like peptide GNNQQNY and its derivatives. J Phys Chem B. 2018;122(46):10470–10477. doi: 10.1021/acs.jpcb.8b09220 [DOI] [PubMed] [Google Scholar]
- 12.Gu XL, Qiu M, Sun HL, et al. Polytyrosine nanoparticles enable ultra-high loading of doxorubicin and rapid enzyme-responsive drug release. Biomater Sci. 2018;6(6):1526–1534. doi: 10.1039/C8BM00243F [DOI] [PubMed] [Google Scholar]
- 13.Costache AD, Sheihet L, Zaveri K, Knight DD, Kohn J. Polymer-drug interactions in tyrosine-derived triblock copolymer nanospheres: a computational modeling approach. Mol Pharm. 2009;6(5):1620–1627. doi: 10.1021/mp900114w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheihet L, Piotrowska K, Dubin RA, Kohn J, Devore D. Effect of tyrosine-derived triblock copolymer compositions on nanosphere self-assembly and drug delivery. Biomacromolecules. 2007;8(3):998–1003. doi: 10.1021/bm060860t [DOI] [PubMed] [Google Scholar]
- 15.Ravenelle F, Gori S, Le Garrec D, et al. Novel lipid and preservative-free propofol formulation: properties and pharmacodynamics. Pharm Res. 2008;25(2):313–319. doi: 10.1007/s11095-007-9471-5 [DOI] [PubMed] [Google Scholar]
- 16.Noble RMN, Salim SY, Walker B, et al. Survival of staphylococcus epidermidis in propofol and intralipid in the dead space of intravenous injection ports. Anesth Analg. 2019;129(1):e20–e22. doi: 10.1213/ANE.0000000000002654 [DOI] [PubMed] [Google Scholar]
- 17.Devlin JW, Lau AK, Tanios MA. Propofol-associated hypertriglyceridemia and pancreatitis in the intensive care unit: an analysis of frequency and risk factors. Pharmacotherapy. 2005;25(10):1348–1352. doi: 10.1592/phco.2005.25.10.1348 [DOI] [PubMed] [Google Scholar]
- 18.Devaud JC, Berger MM, Pannatier A, et al. Hypertriglyceridemia: a potential side effect of propofol sedation in critical illness. Intensive Care Med. 2012;38(12):1990–1998. doi: 10.1007/s00134-012-2688-8 [DOI] [PubMed] [Google Scholar]
- 19.Rodrigues TA, Alexandrino RA, Kanczuk ME, Gozzani JL, Mathias LA. A comparative study of non-lipid nanoemulsion of propofol with solutol and propofol emulsion with lecithin. Rev Bras Anestesiol. 2012;62(3):325–334. doi: 10.1016/S0034-7094(12)70133-2 [DOI] [PubMed] [Google Scholar]
- 20.Krajčová A, Waldauf P, Anděl M, Duška F. Propofol infusion syndrome: a structured review of experimental studies and 153 published case reports. Crit Care. 2015;19(1):398. doi: 10.1186/s13054-015-1112-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y, Yang J, Liu J, Wang Y, Zhang WS. Efficacy comparison of the novel water-soluble propofol prodrug HX0969w and fospropofol in mice and rats. Br J Anaesth. 2013;111(5):825–832. doi: 10.1093/bja/aet218 [DOI] [PubMed] [Google Scholar]
- 22.Deng T, Mao XL, Li Y, Bo SW, Yang ZG, Jiang ZX. Monodisperse oligoethylene glycols modified Propofol prodrugs. Bioorg Med Chem Lett. 2018;28(22):3502–3505. doi: 10.1016/j.bmcl.2018.10.009 [DOI] [PubMed] [Google Scholar]
- 23.Ravenelle F, Vachon P, Rigby-Jones AE, et al. Anaesthetic effects of propofol polymeric micelle: a novel water soluble propofol formulation. Br J Anaesth. 2008;101(2):186–193. doi: 10.1093/bja/aen147 [DOI] [PubMed] [Google Scholar]
- 24.Boscan P, Rezende ML, Grimsrud K, Stanley SD, Mama KR, Steffey EP. Pharmacokinetic profile in relation to anaesthesia characteristics after a 5% micellar microemulsion of propofol in the horse. Br J Anaesth. 2010;104(3):330–337. doi: 10.1093/bja/aep377 [DOI] [PubMed] [Google Scholar]
- 25.Wu AL, Wang YY, Min S, Liu H, Xie F. Etomidate-loaded micelles for short-acting general anesthesia: preparation, characterizations, and in vivo studies. J Drug Deliv Sci Technol. 2018;46:156–161. doi: 10.1016/j.jddst.2018.05.013 [DOI] [Google Scholar]
- 26.Wang B, Chen SM, Yang J, Yang LH, Liu J, Zhang WS. ET-26 hydrochloride (ET-26 HCl) has similar hemodynamic stability to that of etomidate in normal and uncontrolled hemorrhagic shock (UHS) rats. PLoS One. 2017;12(8):e0183439. doi: 10.1371/journal.pone.0183439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang B, Yang J, Chen J, et al. An etomidate analogue with less adrenocortical suppression, stable hemodynamics, and improved behavioral recovery in rats. Anesth Analg. 2017;125(2):442–450. doi: 10.1213/ANE.0000000000002063 [DOI] [PubMed] [Google Scholar]
- 28.National Research Council Committee for the Update of the Guide for the C, Use of Laboratory A. The National Academies Collection: Reports Funded by National Institutes of Health. Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academies Press (US); 2011. Copyright © 2011, National Academy of Sciences. doi: 10.17226/12910 [DOI] [Google Scholar]
- 29.Dickinson R, White I, Lieb WR, Franks NP. Stereoselective loss of righting reflex in rats by isoflurane. Anesthesiology. 2000;93(3):837–843. doi: 10.1097/00000542-200009000-00035 [DOI] [PubMed] [Google Scholar]
- 30.Kilpatrick GJ, McIntyre MS, Cox RF, et al. CNS 7056: a novel ultra-short-acting benzodiazepine. Anesthesiology. 2007;107(1):60–66. doi: 10.1097/01.anes.0000267503.85085.c0 [DOI] [PubMed] [Google Scholar]
- 31.Dixon WJ. Staircase bioassay: the up-and-down method. Neurosci Biobehav Rev. 1991;15(1):47–50. doi: 10.1016/S0149-7634(05)80090-9 [DOI] [PubMed] [Google Scholar]
- 32.Lee JM, Park KM, Lim SJ, Lee MK, Kim CK. Microemulsion formulation of clonixic acid: solubility enhancement and pain reduction. J Pharm Pharmacol. 2002;54(1):43–49. doi: 10.1211/0022357021771904 [DOI] [PubMed] [Google Scholar]
- 33.Date AA, Nagarsenker MS. Design and evaluation of microemulsions for improved parenteral delivery of propofol. AAPS PharmSciTech. 2008;9(1):138–145. doi: 10.1208/s12249-007-9023-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cotten JF, Husain SS, Forman SA, et al. Methoxycarbonyl-etomidate: a novel rapidly metabolized and ultra-short-acting etomidate analogue that does not produce prolonged adrenocortical suppression. Anesthesiology. 2009;111(2):240–249. doi: 10.1097/ALN.0b013e3181ae63d1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Zhang Y, Fan Y, et al. Preparation and evaluation of novel mixed micelles as nanocarriers for intravenous delivery of propofol. Nanoscale Res Lett. 2011;6(1):275. doi: 10.1186/1556-276X-6-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doenicke A, Roizen MF, Hoernecke R, Mayer M, Ostwald P, Foss J. Haemolysis after etomidate: comparison of propylene glycol and lipid formulations. Br J Anaesth. 1997;79(3):386–388. doi: 10.1093/bja/79.3.386 [DOI] [PubMed] [Google Scholar]
- 37.Doenicke AW, Roizen MF, Hoernecke R, Lorenz W, Ostwald P. Solvent for etomidate may cause pain and adverse effects. Br J Anaesth. 1999;83(3):464–466. doi: 10.1093/bja/83.3.464 [DOI] [PubMed] [Google Scholar]
- 38.Nakane M, Iwama H. A potential mechanism of propofol-induced pain on injection based on studies using nafamostat mesilate. Br J Anaesth. 1999;83(3):397–404. doi: 10.1093/bja/83.3.397 [DOI] [PubMed] [Google Scholar]
- 39.Gehan G, Karoubi P, Quinet F, Leroy A, Rathat C, Pourriat JL. Optimal dose of lignocaine for preventing pain on injection of propofol. Br J Anaesth. 1991;66(3):324–326. doi: 10.1093/bja/66.3.324 [DOI] [PubMed] [Google Scholar]
- 40.Shimizu T, Inomata S, Kihara S, Toyooka H, Brimacombe JR. Rapid injection reduces pain on injection with propofol. Eur J Anaesthesiol. 2005;22(5):394–396. doi: 10.1017/S0265021505230673 [DOI] [PubMed] [Google Scholar]
- 41.Park JW, Park ES, Chi SC, Kil HY, Lee KH. The effect of lidocaine on the globule size distribution of propofol emulsions. Anesth Analg. 2003;97(3):769–771. doi: 10.1213/01.ANE.0000074797.70349.CA [DOI] [PubMed] [Google Scholar]
- 42.Li GL, Fan YT, Li XR, et al. In vitro and in vivo evaluation of a simple microemulsion formulation for propofol. Int J Pharm. 2012;425(1–2):53–61. doi: 10.1016/j.ijpharm.2012.01.011 [DOI] [PubMed] [Google Scholar]
- 43.Chan KH, Lee WH, Ni M, Loo Y, Hauser CAE. C-terminal residue of ultrashort peptides impacts on molecular self-assembly, hydrogelation, and interaction with small-molecule drugs. Sci Rep. 2018;8(1):17127. doi: 10.1038/s41598-018-35431-2 [DOI] [PMC free article] [PubMed] [Google Scholar]