

Abstract
Purpose:
Mutations in KRAS/NRAS (RAS) predict lack of anti-EGFR efficacy in metastatic colorectal cancer (mCRC). However, it is unclear if all RAS mutations have similar impact and atypical mutations beyond those in standard guidelines exist.
Experimental Design:
We reviewed 7 tissue and 1 cfDNA cohorts of 9485 patients to characterize atypical RAS variants. Using an in-vitro cell-based assay (FACT), Ba/F3 transformation and in-vivo xenograft models of transduced isogenic clones, we assessed signaling changes across mutations.
Results:
KRAS exon 2, extended RAS, and atypical RAS mutations were noted in 37.8%, 9.5%, and 1.2% of patients, respectively. Among atypical variants, KRAS L19F, Q22K and D33E occurred at prevalence ≥0.1%, while no NRAS codon 117/146 and only one NRAS codon 59 mutation was noted. Atypical RAS mutations had worse overall survival than RAS/BRAF wild-type mCRC (HR 2.90, 95% CI 1.24–6.80, P=0.014). We functionally characterized 114 variants with the FACT assay. All KRAS exon 2 and extended RAS mutations appeared activating. Of 57 atypical RAS variants characterized, 18 (31.6%) had signaling below wild-type, 23 (40.4%) had signaling between wild-type and activating control, and 16 (28.1%) were hyperactive beyond the activating control. Ba/F3 transformation (17/18 variants) and xenograft model (7/8 variants) validation was highly concordant with FACT results and activating atypical variants were those that occurred at highest prevalence in clinical cohorts.
Conclusion:
We provide best available evidence to guide treatment when atypical RAS variants are identified. KRAS L19F, Q22K, D33E and T50I are more prevalent than many guideline included RAS variants and functionally relevant.
Keywords: colon, rectal, malignancy, signaling, RAS
Introduction:
Mutations in KRAS/NRAS (RAS) are important biomarkers in metastatic colorectal cancer (mCRC) that predict lack of benefit from anti-EGFR antibodies and occur in ~60% of mCRCs(1–3). Identification of these alterations is important not only to avoid potential toxicity of ineffective therapy, but RAS mutations may also predict worse outcome following anti-EGFR treatment. In the PRIME study, patients with RAS mutant mCRC treated with panitumumab + FOLFOX4 had a worse median progression free survival (mPFS) than patients who received FOLFOX4 alone (HR 1.31, 95% CI 1.07–1.60, P=0.008)(4,5). Due to the strength of RAS mutations as a predictive biomarker, testing for these alterations has become essential prior to treatment with cetuximab or panitumumab(6).
While the evidence supporting KRAS exon 2 mutations as predictive is robust, with a positive interaction test in placebo-controlled trials, the predictive nature of many of the less common variants remains less clear(2–4,7). For example, only 7 patients with codon 59 mutations were identified in the PRIME trial that demonstrated extended RAS variants had clinical relevance(4). These mutations were not part of the extended RAS mutation analysis, but rather were assessed in a post-hoc analysis that showed removing them from the wild-type population resulted in decreasing the hazard ratio in favor of adding anti-EGFR therapy. Even among guideline cited variants, significant work went into evaluating whether KRAS G13D mutations may still benefit from anti-EGFR therapy based on retrospective evidence(8,9). This led to the prospective ICECREAM trial which showed a 0% response rate (RR) following single agent cetuximab among KRAS G13D mutant mCRC(10).
Beyond extended RAS mutations, other “atypical” variants in RAS have been noted and their clinical relevance remains unclear. As we move beyond hot-spot to full gene coverage, these mutations are increasingly observed. Despite G13D being one of the most common RAS mutations, the ICECREAM study took over 2 years to recruit 53 patients and demonstrated the difficulty in studying rare variants prospectively. If a similar prospective strategy is utilized to validate other less common atypical variants, it is unlikely to succeed and alternative strategies are required. With this in mind, we aimed to describe the prevalence of atypical RAS variants across a pooled cohort of 9485 patients and explore their functional and clinical significance using in-vitro functional data with cross platform validation, in-vivo mouse experiments, and retrospective clinical data. We hope this data will provide guidance to clinicians treating patients with uncommon alterations and although this work does not confirm the predictive nature of each individual variant, it provides best available evidence to guide patient care.
Methods:
This study was completed after receiving institutional review board approval and performed according to the Declaration of Helsinki and following institutional guidance for the care of animals. A waiver of consent was obtained for the retrospective review of patient records.
Definitions
KRAS exon 2 mutations were defined as KRAS codon 12 & 13 mutations, extended RAS mutations were defined as KRAS codon 59, 61, 117, 146 and NRAS codon 12, 13, 59, 61, 117, 146 mutations, and atypical RAS mutations were defined as all other mutations not currently included in standard guidelines in KRAS/NRAS(6). For patients with more than one mutation in KRAS/NRAS, variants were considered independently for calculating the specific mutations’ prevalence. These patients were categorized into KRAS exon 2, extended, or atypical with preference given in descending order from KRAS exon 2 to extended to atypical for subsequent analysis.
Patient Population
The 8 cohorts studied are summarized in Table 1 and consisted of 7 tissue-based and 1 cell-free DNA (cfDNA) cohort (Guardant Health, Redwood City, CA). A total of 9485 patients were assessed. The MD Anderson (MDA) CMS 46, MDA T200, a portion (377/1078 patients) of the Mayo cohort, and a portion of the Caris Life Sciences cohort (62/2200 patients) had clinical annotation available for comparison of baseline characteristics and clinical outcomes. Mayo patients with clinical outcomes included all RAS mutant cases treated at the Mayo Clinic, Rochester MN. Caris Life Sciences patients with clinical outcomes included patients with extended and atypical RAS mutations treated at Georgetown University Medical Center, the West Cancer Center, and Karmanos Cancer Institute.
Table 1.
Cohorts utilized to characterize relative prevalence of RAS mutations in colorectal cancer and their characteristics.
| Cohort | MDA CMS 46 (32) | Mayo | MDA T200 (33) | CARIS | Project Genie (34) | TCGA (35) | NHS & HPFS (36) | cfDNA (37) |
|---|---|---|---|---|---|---|---|---|
| Patients | 1877 | 1078 | 207 | 2200 | 2081 | 228 | 619 | 1397 |
| RAS Coverage | Hot spot | Hot spot | All exons | All exons | Mixed | All exons | All exons | All exons |
| Assay Type | 46 Gene Multiplex | 46 Gene Multiplex | 201 Gene; Capture Based | 592 Gene; Hybrid Capture Based | Mixed | Exome | Exome | 54 to 73 Gene cfDNA |
| Assay Depth | ≥250X | ≥250X | Median 906X (tumor) | >750X | Varied by Platform | >20X for 80% of exons | Median 88X (tumor) | 8000X |
| Tumor Cellularity | >20% | >20% | >20% | >20% | >10% | ≥60% | Average 45% | n/a |
| Stage of Patients | Stage IV | Stage IV | Stage IV | Mostly Stage IV | Mostly Stage IV | Stage I-IV | Stage I-IV | Mostly Stage IV |
| Publicly Available | No | No | No | No | Yes | Yes | Yes | No |
MSI Testing
Microsatellite instability (MSI) status was retrospectively reviewed from patient’s charts where access to charts was available and only evaluated in patients with testing performed as part of their standard care. Testing consisted of a mixture of immunohistochemical (IHC) staining for mismatch repair protein deficiency (MLH1, MSH2, PM2, MSH6) and polymerase chain reaction microsatellite assessment.
Statistical Methods for Clinical Cohorts
Categorical characteristics were compared using χ2 test or Fisher’s exact tests as appropriate, while continuous variables were compared with the Mann-Whitney or Kruskal-Wallis tests when a median is reported and the Student’s t-test or ANOVA when averages are shown. P<0.05 was considered significant for all analyses. Right sided tumors were defined based on pathology and surgical reports as those occurring from the cecum up to but not including the splenic flexure. Left sided tumors were defined as those occurring from the splenic flexure to the rectum. Relative variant allele frequency (rVAF) was defined by dividing the allele frequency of a mutation by the maximum allele frequency of any somatic mutation detected in the same sample. OS was defined as the time from diagnosis with stage IV CRC until death or last follow up. Patients alive at the time of last follow up were censored. OS was summarized using Kaplan-Meier curves and compared using the log-rank test and Cox-regression. Where multivariate models were performed, a forward likelihood ratio selection was used. Variables with P<0.05 were included and P>0.1 were excluded during stepwise assessment. All variables met the proportional hazards assumption and were chosen based on differences in baseline characteristics between groups or known prognostic features in CRC. Age was entered as a dichotomy (<60 vs ≥60). Analysis was performed using Graph Pad Prism software version 5.0 (La Jolla, California), SPSS version 22.0 (Armonk, New York) and R studio version 3.30 (Boston, MA).
Functional Validation of Variants
Functional significance was assessed for all RAS variants (a) detected at MDA among patients who received a CMS 46 NGS assay for any malignancy, (b) present in a patient with CRC in the CARIS Life Sciences Molecular Diagnostics database, or (C) noted to be of clinical significance or with prior functional annotation in PubMed or COSMIC(11). For example, KRAS P34R is associated with cardiofaciocutaneous syndrome (CFC) and KRAS T58I is associated with Noonan syndrome. They have both been well characterized to increase cellular proliferation, decrease KRAS GTPase activity, and stimulate down-stream phosphorylation of MEK, serving as reasonable controls for activating atypical alterations(12,13)
Novellus Functional Annotation for Cancer Treatment (FACT) Assay
Variants were functionally characterized using an in-vitro cell‐based assay (FACT) designed to analyze oncogenic activity based on signalling activation (Novellus, Jerusalem, Israel). Variants were generated on a wild‐type expression vector then transfected into a live-cell assay with fluorescently tagged ERK2 that is part of the MAPK/ERK pathway and which shuttles from the cytoplasm to the nucleus upon pathway activation(14). Cells were then fixed and scanned by a fluorescent microscope to detect reporter localization that generated nuclear‐to‐cytoplasmic ratios (NCR) to provide comparisons between signaling activity for each variant. Further details are available in the supplementary methods.
NCR values were normalized and scored according to the activation levels of wild-type and KRAS G13D mutations, so that 0% represents wild-type activity and 100% is the activity of the KRAS G13D. This was achieved using standard rescaling methods: score = (MT – KRAS wild-type)/(KRAS G13D- KRAS wild-type), where MT is the reported NCR of the studied mutation and wild-type is the reported NCR of the wild‐type condition (15,16). Graphical presentation represents mean and 95% confidence interval.
Analysis comparing OS based on functional activity as defined by the FACT assay was performed using the clinical cohorts and by defining a cut point using histograms demonstrating the distribution of patients with mutations at each activity level (Supplemental Figure 1). This approach was used as there were very few patients with non-activating mutations. As such, the cut points still represent variants that have functional activity significantly above wild-type. Cut point selection and sensitivity analysis are outlined in the results section.
Ba/F3 Transformation Assay
In order to validate findings of the Novellus FACT assay, we utilized a Ba/F3 Transformation Assay. A selection of 17 RAS mutations representing KRAS exon 2, extended, atypical, activating, and non-activating (per the FACT assay) mutations were assessed using the previously described Ba/F3 transformation assay(17–19). Wild-type cell viability was assessed with 4 technical repeats and each mutation was assessed with 2 technical repeats. Cell viability was compared to wild-type construct using an unpaired t-test. Full details of the methodology for the Ba/F3 Transformation assay are available in the supplemental methods.
In-vivo Validation of Functional Significance and Response to Cetuximab
We created stable RAS mutant SW48 cell lines that were used in mouse xenograft models to assess whether functional annotation was concordant with response to cetuximab in animal models. Full details of cell line generation are available in supplemental methods. BALB/c nu/nu 6–8-week old athymic nude mice were maintained in the MDA animal facilities under Institutional Animal Care and Use Committee approved protocols. Wild-type SW48 and mutant KRAS/NRAS transduced isogenic clones were used to establish xenografts. Approximately 3×106 cells/mouse were injected subcutaneously into right posterior flanks of 5 mice per group. Tumor establishment was monitored twice/week and when tumor volume reached 100–200 mm3 mice were randomized into treatment groups. The treatment groups received either 250µL saline or cetuximab (0.5 mg/mouse) twice a week via intraperitoneal injection for 21 days at which point mice were euthanized for tumor collection.
Results:
Prevalence of RAS Mutation Classes
The prevalence of missense, nonsense, and indel mutations in RAS across the 8 cohorts is demonstrated in Figure 1. RAS mutations were noted in 4596/9485 patients (48.5%) and varied between cohorts (P<0.0001) with a range in prevalence from 32.1% to 53.3%. Data from the Project Genie collaboration includes patients with a variety of next generation sequencing (NGS) platforms. Given the heterogeneity in techniques used, data is presented for the entire available version 1 Genie cohort (N=1879) and for only those patients who utilized assays that would cover all exons of KRAS/NRAS (depicted with a *, N=1149). There was no statistically significant difference in RAS mutation frequency (P=0.65) or distribution of RAS mutation class (P=0.93) between versions of the cohort.
Figure 1.
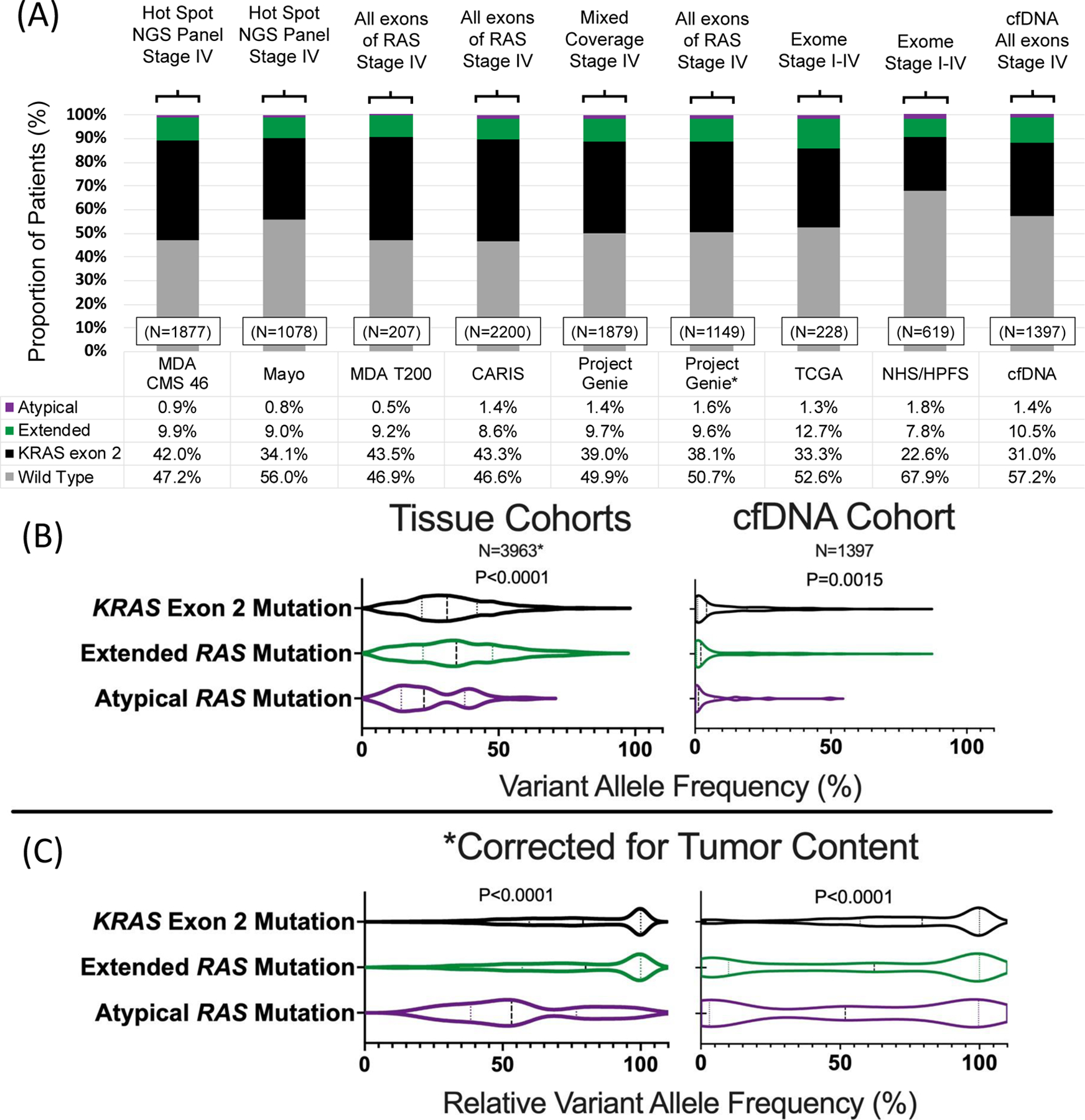
(A) Prevalence of RAS mutations in colorectal cancer across seven tissue and one cfDNA cohort, (B) variant allele frequency of RAS mutation by class and (C) relative variant allele frequency of RAS variants by class.
*Includes patients with high depth sequencing and known variant allele frequencies from MDA CMS 46, MDA T200 and Project Genie cohorts
A total of 3582/9485 (37.8%) patients had KRAS exon 2 mutations, 898/9485 (9.5%) had extended RAS mutations and 116/9485 (1.2%) had atypical RAS mutations. 13/129 atypical variants (10.1%) occurred in patients who had a co-occurring typical or extended mutation and they were categorized according to their more common variant while only 37/4518 (0.8%) extended or exon 2 variants occurred in patients with co-occurring RAS alterations (P<0.0001). Of atypical variants detected, 90 occurred in KRAS and 39 in NRAS. Prevalence of individual variants is summarized in Supplemental Table 1 and 2, and although most atypical variants occur at low frequencies, certain variants are more common than guideline cited variants. For example, KRAS Q22K occurred in 16/9485 (0.2%) patients, and yet not a single NRAS codon 117 or 146 variant was detected and only 1 NRAS codon 59 variant was detected. Other atypical variants occurring at frequencies ≥0.1% include KRAS L19F (7/9485) and KRAS D33E (7/9485).
Clonality of RAS Mutations Based on Category of Mutation and Co-Mutations
Data on variant allele frequency was available in the MDA CM 46, MDA T200, Project Genie, and cfDNA cohorts (N=5360). Tissue and cfDNA results are provided separately due to inherent differences in allele frequency distribution (Figure 1B & 1C). In both tissue and cfDNA, we noted that KRAS exon 2 and extended RAS mutations had significantly higher VAFs than atypical RAS mutations (P<0.0001 & P=0.0015, respectively), even after correcting for tumor content using the rVAF (both P<0.0001). Additionally, atypical mutations demonstrated a more diffuse distribution of clonality, while KRAS exon 2 and extended RAS mutations appeared highly clonal.
Among these same cohorts, we assessed prevalence and clonality of concurrent RAS/BRAF V600 mutations. We defined mutations as subclonal if they occurred at rVAF<10%. In tissue, there were 8 concurrent RAS/BRAF V600 mutations, all of which were clonal, and 3 (37.5%) of which were atypical mutations, a significantly higher prevalence of atypical mutation than in BRAF V600 wild-type CRC (P<0.0001). In cfDNA, there were 10 concurrent RAS/BRAF V600 mutations, of which 7 were subclonal for both the RAS and BRAF partner. Atypical mutations accounted for 2/10 cases (20%), again significantly more prevalent than in BRAF V600 wild type CRC (P=0.037).
Clinical Characteristics and Outcomes of Patients with RAS and BRAF V600 Mutations
We reviewed baseline characteristics and clinical outcomes among 2581 patients with available clinical data. As seen in Table 2, BRAF V600 mutations were associated with older age than all other groups, while KRAS, NRAS, and wild-type cancers did not differ in age distribution (all pairwise P>0.05). KRAS, NRAS, and BRAF V600 mutations more commonly associated with female gender (all pairwise P<0.002) and right sided tumors (all pairwise P<0.004), while BRAF V600 mutations were more commonly associated with MSI-H status (P<0.0001) but RAS mutant tumors did not differ from wild-type tumors regarding MSI status (all pairwise P>0.15). NRAS mutations more commonly co-occurred with additional RAS alterations than KRAS or BRAF V600 alterations (P<0.0001).
Table 2.
Clinical characteristics associated with class of RAS mutation.
|
RAS/BRAF Wild-type N=1069 |
KRAS Mutant N=1226 |
NRAS Mutant N=127 |
BRAF V600 Mutant N=168 |
P |
RAS/BRAF Wild-type N=1069 |
Exon 2 KRAS Mutation N=1033 |
Extended RAS Mutation N=292 |
Atypical RAS Mutation N=21 |
P | |
|---|---|---|---|---|---|---|---|---|---|---|
| Characteristic | ||||||||||
| Median Age (IQR) | 54 (45–62) | 55 (45–63) | 57 (47–65) | 60 (54–67) | <0.0001 | 54 (45–62) | 55 (45–63) | 57 (47–65) | 55 (45–67) | 0.018 |
| Sex | ||||||||||
| Female | 394 (37%) | 593 (48%) | 65 (51%) | 93 (55%) | <0.0001 | 394 (37%) | 505 (49%) | 137 (47%) | 11 (52%) | <0.0001 |
| Male | 675 (63%) | 633 (52%) | 62 (49%) | 75 (45%) | 675 (63%) | 528 (51%) | 155 (53%) | 10 (48%) | ||
| Location* | ||||||||||
| Right | 216 (20%) | 477 (39%) | 40 (31%) | 116 (70%) | <0.0001 | 216 (20%) | 399 (39%) | 104 (36%) | 10 (48%) | <0.0001 |
| Left | 849 (80%) | 742 (61%) | 87 (69%) | 50 (30%) | 849 (80%) | 631 (61%) | 184 (64%) | 11 (52%) | ||
| MSI Status* | ||||||||||
| MSS | 766 (96%) | 870 (96%) | 99 (99%) | 99 (83%) | <0.0001 | 766 (96%) | 735 (97%) | 218 (96%) | 13 (87%) | 0.25 |
| MSI-H | 30 (4%) | 35 (4%) | 1 (1%) | 20 (17%) | 30 (4%) | 26 (3%) | 8 (4%) | 2 (13%) | ||
| Unknown | 273 | 321 | 27 | 49 | 273 | 272 | 66 | 6 |
Percent represent the proportion of patients with a known result for that variable.
Total n=2581 patients, however patients with multiple variants may be included in multiple categories for comparisons between KRAS, NRAS, and BRAF V600 but not RAS mutation class. All RAS mutation categories (atypical, extended, and KRAS exon 2) are grouped together by gene on the left half of the table.
IQR=interquartile range
Patients with RAS/BRAF V600 wild-type CRC had better overall survival (OS) than patients with KRAS (P<0.0001), NRAS (P<0.0001), or BRAF V600 mutant tumors (P<0.0001) (Figure 2A). KRAS mutations were associated with a better prognosis than NRAS (Hazard Ratio (HR) 0.75, 95% Confidence Interval (CI) 0.58–0.97, P=0.012) or BRAF V600 mutations (HR 0.55, 95% CI 0.43–0.70, P<0.0001). NRAS mutations were also associated with better prognosis than BRAF V600 mutations (HR 0.75, 95% CI 0.57–1.00, P=0.047). In a multivariate model controlling for age, gender, MSI status and primary tumor location, KRAS (HR 1.41, 95% CI 1.23–1.62), NRAS (HR 1.83, 95% CI 1.40–2.40, P<0.0001), and BRAF V600 (HR 2.26, 95% CI 1.75–2.91, P<0.0001) had worse prognosis than wild-type tumors with only right sided location remaining significant in the model for controlled co-variates (HR 1.34, 95% CI 1.17–1.53, P<0.0001). When directly comparing KRAS to NRAS, there was no statistical difference between genes after controlling for co-variates.
Figure 2.
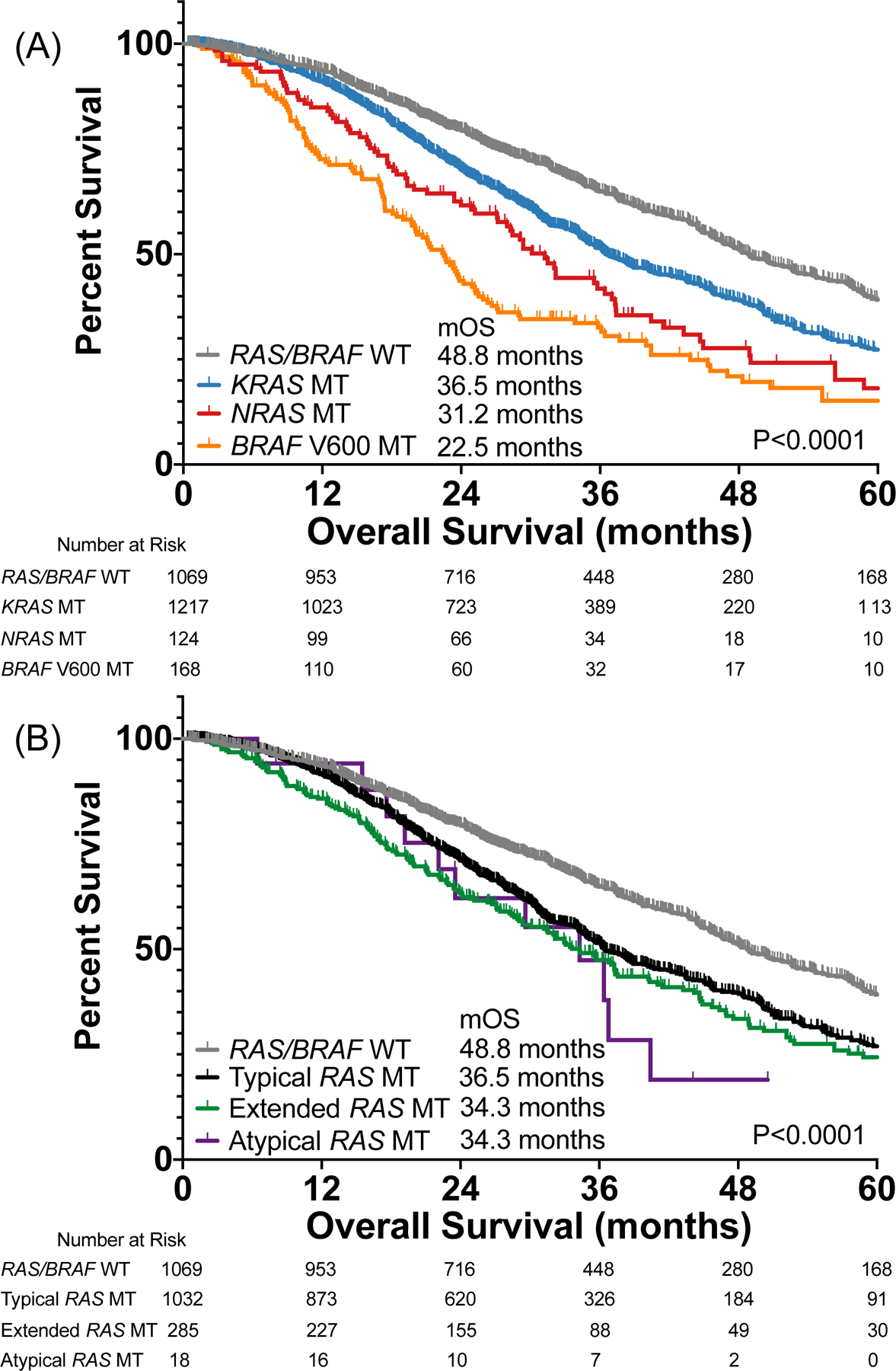
Impact of (A) RAS/BRAF mutations and (B) RAS mutation class on overall survival among patients with metastatic colorectal cancer.
Clinical Characteristics and Outcomes Based on RAS Mutation Class
Extended RAS mutations were associated with older age than wild-type tumors (P=0.010), but other RAS mutation classes did not differ (P>0.14). KRAS exon 2 (P<0.0001) and extended (P=0.0018) RAS mutations were more common in women, while atypical mutations did not have different sex distribution from wild-type tumors. KRAS exon 2 (P<0.0001), extended (P<0.0001), and atypical (P=0.0022) RAS mutations were more likely to be right sided than wild-type tumors. There was no difference in MSI status based on RAS mutation class (P=0.25).
KRAS exon 2 (HR 1.48, 95% CI 1.32–1.67, P<0.0001), extended (HR 1.59, 95% CI 1.31–1.93, P<0.0001) and atypical (HR 2.07, 95% CI 0.89–4.85, P=0.014) RAS mutations had worse OS than RAS/BRAF V600 wild-type tumors (Figure 2B). There was no difference in survival among RAS mutation classes (all pairwise P>0.28). In a multivariate model controlling for age, sex, MSI status, and primary tumor location we found that KRAS exon 2 (HR 1.44, 95% CI 1.24–1.66, P<0.0001), extended (HR 1.46, 95% CI 1.19–1.78, P<0.0001), and atypical mutations (HR 2.27, 95% CI 1.21–4.27, P<0.0001) all had worse prognosis than wild-type tumors but there was no statistically significant difference between class when assessing prognosis among only patients with RAS mutations. In fact, if we included class of mutation and whether the mutation occurred in KRAS or NRAS in the model as two separate co-variates, we found that class of mutation was not retained in the model and only the presence of the mutation in KRAS (HR 1.41, 95% CI 1.23–1.62, P<0.0001) or NRAS (HR 1.84, 95% CI 1.40–2.41, P<0.0001) remained in the model. This suggests that differences in prognosis based on mutation class may be defined more by distribution of these variants across the two genes rather than class, however a test of interaction was not significant.
Functional Characterization of RAS Variants Using Novellus FACT Assay
We characterized 62 KRAS and 52 NRAS variants using the Novellus FACT assay. As seen in Figure 3, KRAS and NRAS showed a large dynamic range of functional activity. Values represent relative signaling compared to wild-type (0%) and a known activating control mutation for KRAS (G13D = 100%) and NRAS (Q61R = 100%). Mean activity on this relative scale ranged from 26% below wild-type signaling for a KRAS E76G mutation to 313% for a KRAS Q22K mutation. All exon 2 and extended mutations were more active than wild-type RAS, indicating the utility of the assay (Figure 3). KRAS alterations had a higher median score relative to NRAS variants (P=0.0002). Of 57 atypical RAS variants, 18 resulted in signaling below wild-type, 23 had signaling between wild-type and activating control, and 16 were more active than the activating control. KRAS atypical variants (23/31) and NRAS atypical variants (16/26) showed similar ratios of variants that were more activating than wild-type (OR 1.80, 95% CI 0.57–5.07, P=0.39) but atypical KRAS variants were more likely to be hyper-activating with signaling above the KRAS activating control (15/31) than NRAS variants (1/26) (OR 23.44, 95% CI 3.45–255.80, P=0.0002). There was no difference between KRAS and NRAS in terms of the number of atypical variants with signaling below wild-type (P=0.31).
Figure 3.
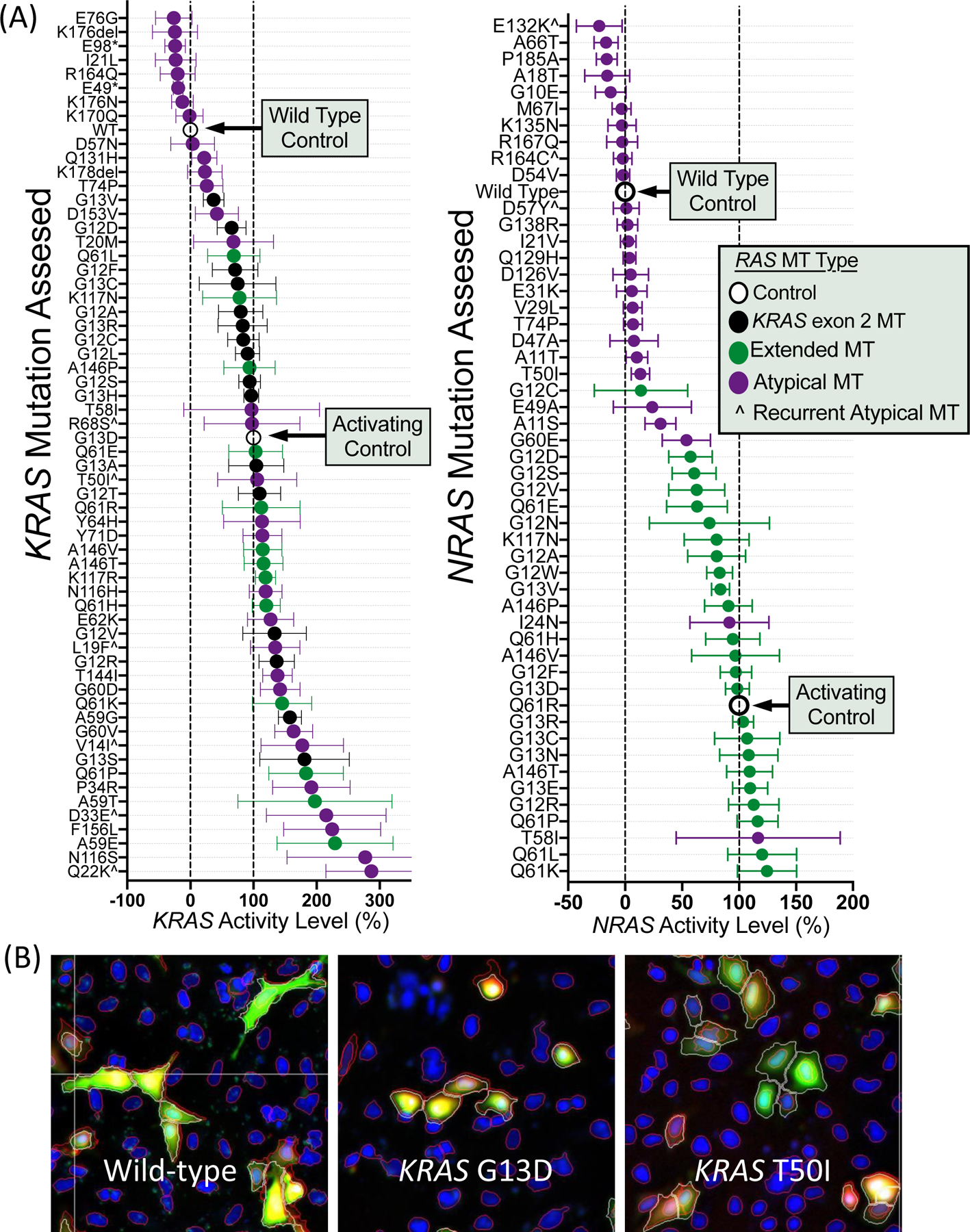
Functional characterization of MAPK signaling for (A) 114 RAS variants assessed using the Novellus FACT assay with (B) representative fluorescent microscopy images from the FACT assay. Values in (A) represent mean +/− 95% confidence interval.
Orthogonal Validation of Functional Status
Using a Ba/F3 transformation assay, we selected a subset of variants for validation from the FACT assay results(19). Variants chosen for validation focused on atypical mutations that were highly prevalent (KRAS L19F, Q22K, and D33E) and a sampling of exon 2, extended, atypical, activating, and inactivating mutations across KRAS and NRAS as comparators. As seen in Figure 4, 17/18 tested variants were concordant between the FACT and Ba/F3 assay. Only KRAS L19F was discordant, lacking transformation activity in the Ba/F3 assay but showing increased signaling in the FACT assay. Importantly, concordance was shown for both the activating alterations in the FACT assay (15/16) and the non-activating alterations (2/2). Using a large library of previously classified BRAF variants, we also compared protein expression levels using reverse phase protein arrays (RPPA) and the Ba/F3 viability and demonstrate that there is no correlation between relative protein level and cell viability (r=0.054, P=0.68), suggesting transformation is due to functional alterations rather than potential differences in protein expression (Supplemental Figure 2).
Figure 4.
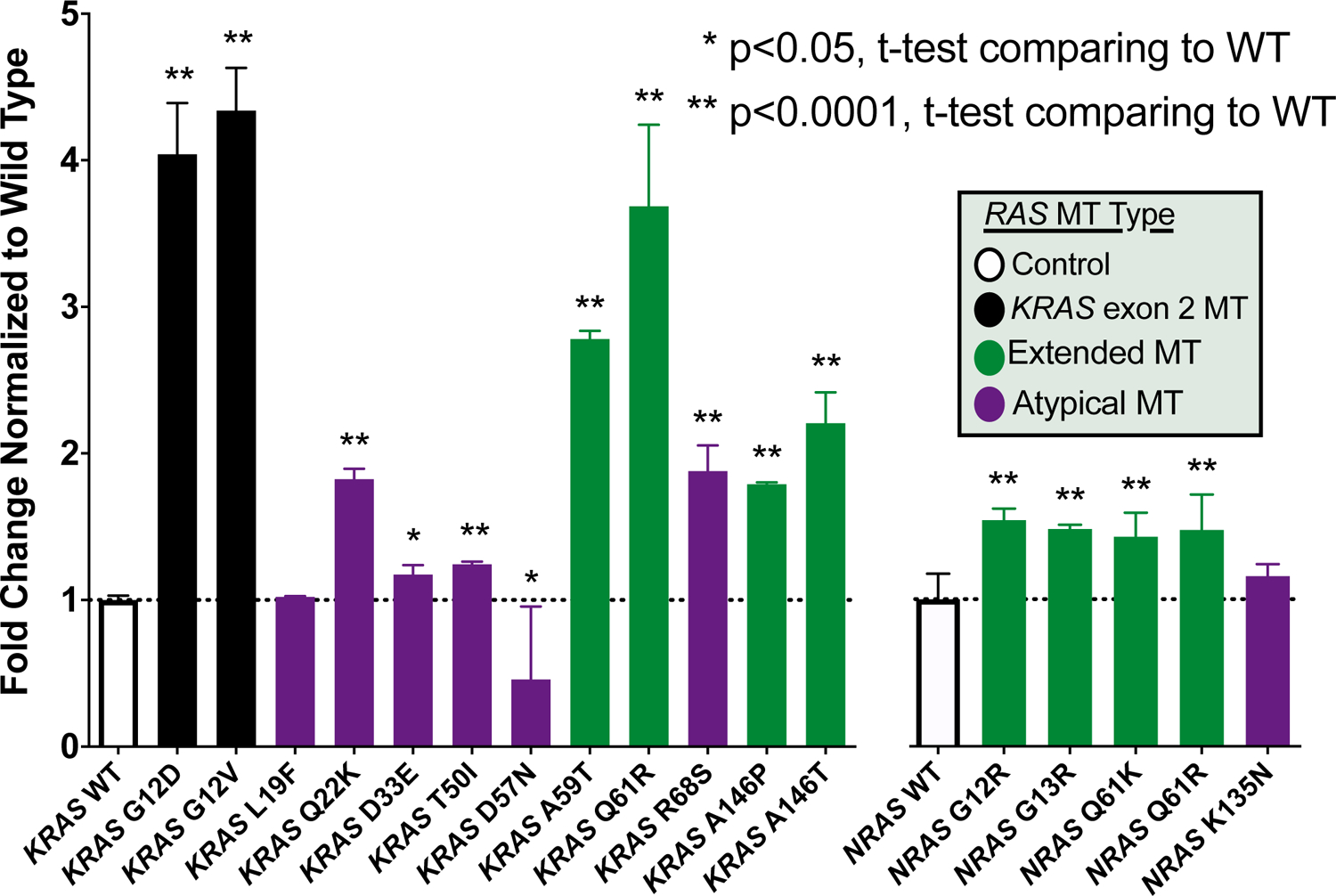
Functional annotation of RAS mutation activation status using the Ba/F3 transformation assay.
Next, we further evaluated the well characterized KRAS G12D and NRAS Q61K mutations and atypical variants with prevalence of ≥0.1% in Supplemental Table 1 and 2 using in-vivo xenograft models derived from transduced isogenic clones of the SW48 cell line. As none of the atypical variants for NRAS were present at this threshold, we randomly selected one atypical alteration (K135N) that was non-activating for characterization. As shown in Figure 5, the SW48 parental and wild-type transduced controls show tumor suppression following cetuximab in xenograft models. Similarly, the well described KRAS G12D and NRAS Q61K mutant xenograft models show reduced growth suppression with cetuximab. The KRAS and NRAS mutations fell into three classes when mice were treated with cetuximab, those that completely blocked the effects of cetuximab (KRAS G12D, KRAS T50I, and NRAS Q61K), those that were associated with decreased activity of cetuximab (KRAS Q22K and KRAS D33E) and those that resulted in response to cetuximab that was similar to wild-type xenografts (KRAS L19F, KRAS D57N, and NRAS K135N). Among atypical variants assessed, only KRAS T50I showed a lack of tumor growth suppression with cetuximab while KRAS Q22K and D33E showed proliferation that was intermediate between wild-type and known activating mutations. Of tested variants, 7/8 were concordant with the FACT assay if the intermediate category was considered similar to activating in the FACT assay and 8/8 were concordant with the Ba/F3 assay. KRAS L19F was inactivating by the Ba/F3 assay and xenograft models but was considered activating with the FACT assay.
Figure 5.
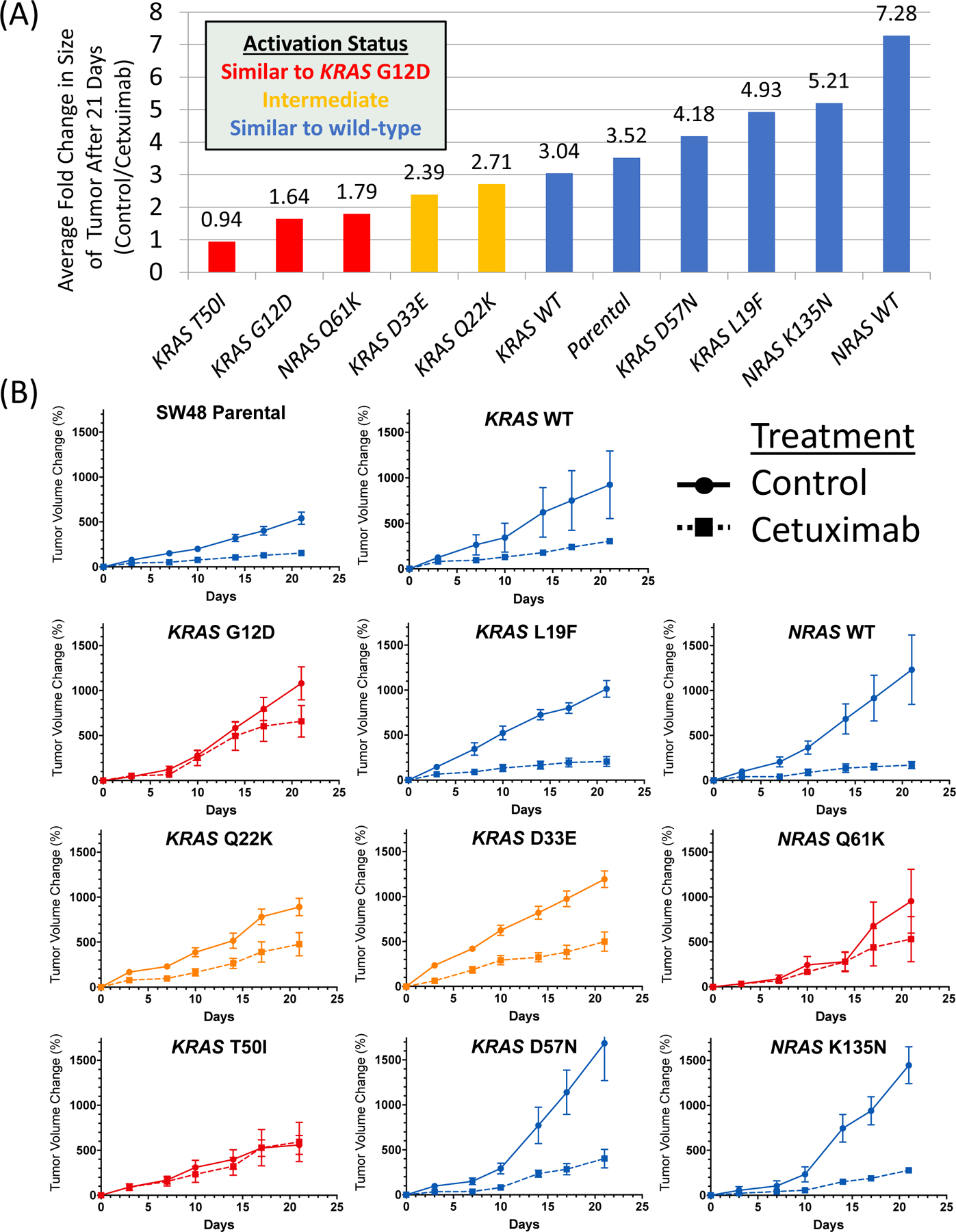
Impact of cetuximab treatment on tumor volume in mouse xenograft models derived from SW48 isogenic clones carrying select RAS mutations. (A) Fold change in size of xenograft at the end of 21 days of cetuximab treatment relative to control and (B) tumor size during 21-day treatment cycle with cetuximab or control (mean +/− standard error) for each tested variant.
When we compared xenografts grown in the absence of cetuximab, we noted no difference in tumor volume change when comparing individual mutations to each other and wild-type transduced controls with a One-Way ANOVA with correction for multiple comparisons. We also performed a Western blot assessing RAS expression and a RAS-GTP pulldown of isogenic lines (Supplemental Figure 3) which demonstrated comparable expression and that all lines were more active than negative control and showed a range of activity that reflected other functional assays except for the NRAS K135N line, which was activating in the pulldown but not in any other assay. Mutations with all 4 assays are summarized in Supplemental Table 3.
Impact of Functional Status on Clinical Outcomes
We next assessed the prevalence and clinical impact of FACT assay characterized variants among patients from our epidemiologic cohorts with clinical outcomes. As seen in Supplemental Figure 1, most patients had a functionally active variant with activity similar to the KRAS G12D activating mutant. Overall survival did not differ in patients with a mutation occurring above vs below 60% relative activity (HR 1.38, 95% CI 0.84–2.27, P=0.13) but showed trends towards being worse in the less active variants. We reviewed the variants making up this group and noted that all but one of the patients included in the group with RAS mutation activity <60% of control were NRAS, which likely drove this difference in outcome. We chose 60% based on the histogram in supplemental Figure 1A, but subsequently recreated histograms split by gene and created new cut points specific to KRAS (80%) and NRAS (60%) and found that neither KRAS (HR 0.97, 95% CI 0.83–1.14, P=0.73) nor NRAS (HR 1.14, 95% CI 0.68–1.92, P=0.60) signaling activity impacted OS. Unfortunately, the population with minimally active RAS variants was small so cut points are quite high and we were unable to determine if atypical variants with functional activity near wild-type had prognosis closer to wild-type patients. When assessing RAS activity as a linear prognostic variable in a Cox-regression model, there was no association with OS in univariate (P=0.94) or multivariate models (P=0.41) that controlled for age, sex, primary location, and MSI status.
Discussion:
With increased use of more comprehensive sequencing beyond hot-spot annotation, we increasingly find variants of unknown significance. These mutations pose a challenge for clinicians to interpret. Here we present a landscape of RAS mutations in CRC across 9485 patients with tissue and blood sequencing demonstrating a 1.2% prevalence of atypical RAS mutations and provide functional characterization across 4 orthogonal platforms. We demonstrate that NRAS mutations are associated with worse prognosis than KRAS mutations, however atypical NRAS variants appear more likely to lack increased functional activity than atypical KRAS alterations. Though some atypical RAS variants do not appear to increase signaling activity (18/57), many resulted in signaling between wild-type and activating control (23/57) and a subset were more activating than known activating mutations, such as KRAS G12D (16/57) (Figure 3). Additionally, the majority of highly recurrent atypical variants do activate RAS dependent signaling. Thus, in addition to having functional annotation, the landscape and prevalence data in Supplemental Table 1 and 2 provide further information about which variants may have clinical significance as recurrent alterations are more likely to have functional and clinical relevance.
While we tried to provide a comprehensive annotation of the prevalence and functional significance of as many mutations as possible, there will always be newly discovered uncommon alterations and it was not pragmatic to perform functional validation on all variants. We focused on recurrent alterations as they affect the most patients and by nature of the fact that they are recurrent, are more likely to have oncogenic significance. The atypical variants KRAS L19F, Q22K, D33E, and T50I warrant special attention as they occur at frequencies greater than KRAS codon 59 or NRAS codon 117/146 variants which are currently in standard of care guidelines for clinical testing(6). While KRAS L19F increased MAPK signaling in the FACT assay, it did not appear to transform cells in the Ba/F3 assay or xenograft models. Although categorizing variants as “active” or “inactive” is convenient, it overlooks the fact that variants, even in the same gene, may have different effects on cellular signaling and survival that manifest in different ways. Although KRAS L19F increased signaling, the presence of this variant alone may not cause resistance to anti-EGFR therapy or cause transformation in the Ba/F3 assay. Our results mirror prior work that showed increased RAS pathway signaling with L19F, however limited oncogenic transforming potential(20). This variant has not been previously assessed for effects on anti-EGFR therapy and the observation that our work shows increased signaling but continued response to cetuximab would suggest a potential intermediate phenotype and highlights the benefit of orthogonal functional annotation. Using a single assay in isolation may not detect nuanced responses or differing signaling profiles. The FACT assay is focused on ERK translocation to the nucleus, however this may not be sufficient to determine that a variant is clinically relevant if taken in isolation. Based on this, KRAS L19F mutant may represent a functional neomorph that retains the aspects of RAS activity assessed in the FACT assay (translocation of ERK to the nucleus) but does not retain aspects of KRAS signaling that may be required to mediate transformation (or cetuximab resistance). This may explain other discordant findings across assays, such as the discordant RAS-GTP pulldown for NRAS K135N suggesting increased signaling, despite the other 3 assays showing the variant does not cause ERK translocation, transformation or cetuximab resistance.
This idea that atypical variants need to be considered within the context of other alterations is also seen in atypical BRAF mutations. The BRAF D594F mutation is a kinase-dead variant that can hyper-activate ERK phosphorylation in the presence of concurrent RAS alterations but lacks this activity in RAS wild-type models(21). A recent review of 163 patients with atypical RAS mutations by Pietrantonio et al. showed high rates of co-occurring RAS/BRAF and NF1 mutations in 30% and 12% (respectively) of cases with atypical RAS variants(22). Although we had a small number of patients with atypical variants in our clinical cohort, we did note a higher rate of atypical RAS variants co-occurring with BRAF alterations (P<0.0001), however this may be spurious due to sample size. We have seen similar findings when reviewing ERBB2 mutations in mCRC, where concurrent PIK3CA mutations were 2–4x as common in tumors with ERBB2 mutations than in tumors with ERBB2 wild-type tumors(23). Even among well conserved and highly prevalent mutations in RAS, variants may have different evolutionary fitness. Winters et al. introduced a library of mutations in KRAS codon 12 and 13 and showed through barcoded sequencing that variants within the same codon had surprising diversity in fitness based on ability to establish tumors(24). These findings all suggest that both individual mutations and the genomic context within which they occur are important to understand when considering targeted therapeutic strategies.
Separately, our finding of worse outcomes in patients with NRAS mutations helps add further support to the prognostic relevance of these mutations in light of mixed prior findings. In the MRC COIN trial, a pooled analysis of five German trials, and an Italian retrospective cohort, there was no difference in outcome between KRAS and NRAS variants(25–27). There were only 38, 39, and 47 NRAS mutant cancers noted in these studies, respectively, limiting statistical power. Contrary to this, Cercek et al showed in a larger study (N=87 NRAS mutant tumors) that NRAS mutations showed a trend towards worse survival than KRAS mutations (P=0.05)(28). Our study was larger than these prior reports and showed a statistically significant better OS in KRAS compared to NRAS alterations (Figure 2, HR 0.75, 95%CI 0.58–0.97, P=0.012). There are different isoform specific signaling pathways of KRAS, NRAS, and HRAS, that drive differential expression through Raf-1 and Pi3K and their differing distribution for different cancer histologies suggests these genes impact biology differently(29,30).
Our study must be interpreted in the context of several limitations. While we aimed to provide orthogonal validation across Ba/F3 and xenograft models for a selection of RAS variants (Supplemental Table 3), it was not practical to functionally validate all 114 variants assessed with 3 assays as this would have required enormous expenditure beyond our resource capacity. Despite this, positive controls, such as the KRAS P34R and T58I which were chosen due to prior characterization in hereditary cancer syndromes showed matched activity in the FACT assay and most atypical variants were highly concordant across platforms. We also acknowledge that while we did see substantial differences in growth following cetuximab noted in the xenograft work performed, the SW48 cell lines using for our isogenic clone generation contains a MEK Q56P mutation which may impact sensitivity to EGFR inhibitors (31). Although the gold-standard would be to treat patients with each atypical variant with anti-EGFR agents and assess response in prospective trials, the rarity of these variants makes that impractical. As well, most of our sequencing results came from large tertiary centers which may not be representative. Future efforts to support multi-institutional data-sharing from community and tertiary centers could address this gap while also providing a large population to better describe prevalence and clinical implications of uncommon variants. This would help confirm the prognostic and clinical implications of individual atypical variants which was challenging in our study due to small sample size for individual variants.
Despite these limitations, this work demonstrated atypical variants are present in 1.2% of patients with mCRC, a rate higher than previously reported, and provides best available evidence to guide patient care when one of these variants is found(22). We demonstrate that NRAS mutations are associated with a worse prognosis than KRAS alterations and identify KRAS L19F, Q22K, D33E, and T50I as more prevalent than many guideline included RAS variants. These variants should be considered for testing in patients with mCRC as part of standard care in future testing and alternate non-anti-EGFR antibody treatments should be prioritized where available if a KRAS Q22K, D33E, or T50I variant is identified.
Supplementary Material
Statement of Translational Relevance:
Mutations in KRAS/NRAS (RAS) predict lack of benefit from anti-EGFR therapy in metastatic colorectal cancer (mCRC). However, it is unclear if all RAS mutations have similar impact and atypical mutations exist beyond those that standard guidelines recommend testing. We reviewed 9485 patients and identified 1.2% of patients with atypical mutations outside standard guidelines. Although most atypical mutations were rare, some occurred more frequently than variants in current guidelines. Atypical variants were associated with survival similar to other RAS mutations (worse than wild-type survival) and NRAS variants were associated with worse survival than KRAS. We functionally characterized 114 variants with an in-vitro cell-based assay and provide orthogonal validation using Ba/F3 transformation and mouse xenograft models. Guideline cited variants all increased kinase activity, however there were additional atypical variants including KRAS L19F, Q22K, D33E, and T50I that appear both prevalent and relevant variants for consideration as additions to standard guidelines in mCRC.
Acknowledgements:
JL was a member of the UBC Clinician Investigator program, the recipient of a CAMO Fellowship, the ASCO - J. Edward Mahoney Foundation Young Investigator Award, and the RCPSC Detweiler Fellowship. SK is the recipient of NIH R01 (CA 1184843 & CA 187238) and CCSG grants (CA016672) that supported this research. We are grateful to the MDA Moon Shots program for research funding that supported this work.
Supporting Grants:
CCSG CA016672, NIH R01 CA187238, NIH R01 CA184843, ASCO Young Investigator Award and MD Anderson Moon Shots
Footnotes
Disclosures: BM, GT, and OZ were employees of Novellus during this project. JX and JLM were employees of Caris Life Sciences during this project.
References:
- 1.Peeters M, Kafatos G, Taylor A, Gastanaga VM, Oliner KS, Hechmati G, et al. Prevalence of RAS mutations and individual variation patterns among patients with metastatic colorectal cancer: A pooled analysis of randomised controlled trials. Eur J Cancer. 2015;51:1704–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26049686 [DOI] [PubMed] [Google Scholar]
- 2.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. [DOI] [PubMed] [Google Scholar]
- 3.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. [DOI] [PubMed] [Google Scholar]
- 4.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25:1346–55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24718886 [DOI] [PubMed] [Google Scholar]
- 5.Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24024839 [DOI] [PubMed] [Google Scholar]
- 6.Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular biomarkers for the evaluation of colorectal cancer: Guideline from The American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017;35:1453–96. [DOI] [PubMed] [Google Scholar]
- 7.Loree JM, Dowers A, Tu D, Jonker DJ, Edelstein DL, Quinn H, et al. Expanded low allele frequency RAS and BRAF V600E testing in metastatic colorectal cancer as predictive biomarkers for cetuximab in the randomized CO.17 trial. Clin Cancer Res. 2021;27:52–9. Available from: https://clincancerres.aacrjournals.org/content/27/1/52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-bianchi A, Tu D, Siena S, et al. Association of KRAS p . G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. J Am Med Assoc. 2010;304:1812–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20978259 [DOI] [PubMed] [Google Scholar]
- 9.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D Tumor Mutations With Outcome in Patients With Metastatic Colorectal Cancer Treated With First-Line Chemotherapy With or Without Cetuximab. J Clin Oncol. 2012;30:3570–7. Available from: 10.1200/JCO.2012.42.2592 [DOI] [PubMed] [Google Scholar]
- 10.Segelov E, Thavaneswaran S, Waring PM, Desai J, Robledo KP, Gebski VJ, et al. Response to cetuximab with or without irinotecan in patients with refractory metastatic colorectal cancer harboring the KRAS G13D mutation: Australasian gastro-intestinal trials group ICECREAM study. J Clin Oncol. 2016;34:2258–64. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27114605 [DOI] [PubMed] [Google Scholar]
- 11.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45:D777–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27899578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schubbert S, Bollag G, Lyubynska N, Nguyen H, Kratz CP, Zenker M, et al. Biochemical and Functional Characterization of Germ Line KRAS Mutations. Mol Cell Biol. 2007;27:7765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, Bollag G, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–6. Available from: 10.1038/ng1748 [DOI] [PubMed] [Google Scholar]
- 14.Cohen-Saidon C, Cohen AA, Sigal A, Liron Y, Alon U. Dynamics and Variability of ERK2 Response to EGF in Individual Living Cells. Mol Cell. Elsevier Ltd; 2009;36:885–93. Available from: 10.1016/j.molcel.2009.11.025 [DOI] [PubMed] [Google Scholar]
- 15.Hong DS, Morris VK, El Osta B, Sorokin AV, Janku F, Fu S, et al. Phase 1B Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAF V600E Mutation. Cancer Discov. 2016; Available from: 10.1158/2159-8290.CD-16-0050 [DOI] [PMC free article] [PubMed]
- 16.Golbstein J, Tocker Y, Sharivkin R, Tarcic G, Vidne M. A novel high-throughput multispectral cell segmentation algorithm. Commun Comput Inf Sci. 2017;723:754–66. [Google Scholar]
- 17.Palacios R, Steinmetz M. IL3-dependent mouse clones that express B-220 surface antigen, contain ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell. 1985;41:727–34. [DOI] [PubMed] [Google Scholar]
- 18.Kong K, Ng PK-S, Scott KL, Kong K, Ng PK-S, Scott KL. Ba/F3 transformation assays. Oncotarget. Impact Journals; 2017;8:35488–9. Available from: http://www.oncotarget.com/fulltext/17828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng PKS, Li J, Jeong KJ, Shao S, Chen H, Tsang YH, et al. Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell. Cell Press; 2018;33:450–462.e10. Available from: https://pubmed.ncbi.nlm.nih.gov/29533785/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith G, Bounds R, Wolf H, Steele RJC, Carey FA, Wolf CR. Activating K-Ras mutations outwith hotspot codons in sporadic colorectal tumours-implications for personalised cancer medicine. Br J Cancer. 2010;102:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. Cell Press; 2010;140:209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pietrantonio F, Yaeger R, Schrock AB, Randon G, Romero-Cordoba S, Rossini D, et al. Atypical RAS Mutations in Metastatic Colorectal Cancer. JCO Precis Oncol. 2019;1–11. [DOI] [PMC free article] [PubMed]
- 23.Loree JM, Bailey AM, Johnson AM, Yu Y, Wu W, Bristow CA, et al. Molecular landscape of ErbB2/ErbB3 mutated colorectal cancer. J Natl Cancer Inst. 2018;110:1409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winters IP, Chiou SH, Paulk NK, McFarland CD, Lalgudi PV, Ma RK, et al. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat Commun. Springer US; 2017;8. Available from: 10.1038/s41467-017-01519-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–14. Available from: 10.1016/S0140-6736(11)60613-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modest DP, Ricard I, Heinemann V, Hegewisch-Becker S, Schmiegel W, Porschen R, et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants - pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol. 2016;32:1–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27358379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schirripa M, Cremolini C, Loupakis F, Morvillo M, Bergamo F, Zoratto F, et al. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. Int J Cancer. 2015;136:83–90. [DOI] [PubMed] [Google Scholar]
- 28.Cercek A, Braghiroli MI, Chou JF, Hechtman JF, Kemeny N, Saltz L, et al. Clinical features and outcomes of patients with colorectal cancers harboring NRAS mutations. Clin Cancer Res. 2017;23:4753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res. 2020; 80 (14): 2969–2974. Available from: 10.1158/0008-5472.CAN-19-3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273:24052–6. [DOI] [PubMed] [Google Scholar]
- 31.Jing C, Li H, Du Y, Cao H, Liu S, Wang Z, et al. MEK inhibitor enhanced the antitumor effect of oxaliplatin and 5-fluorouracil in MEK1 Q56P-mutant colorectal cancer cells. Mol Med Rep. 2019;19:1092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies M a., et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J Clin Oncol. 2015;33:2753–62. Available from: 10.1200/JCO.2014.60.4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen K, Meric-Bernstam F, Zhao H, Zhang Q, Ezzeddine N, Tang LY, et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin Chem. 2015;61:544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818–31. Available from: 10.1158/2159-8290.CD-17-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Network, Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond J a., et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. Nature Publishing Group; 2012;487:330–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22810696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. The Authors; 2016;15:857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24:3539–49. Available from: http://clincancerres.aacrjournals.org/ [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.