

Abstract
Over half of the world’s population resides in areas at risk for dengue virus infection. A vaccine will be pivotal to controlling spread. However, the only licensed vaccine, Dengvaxia, has been shown to increase the risk of severe disease among a subset of individuals. Vaccine efforts are hampered by a poor understanding of antibody responses, including those generated by vaccines, and whether antibody titers can be used as a marker of protection from infection or disease. Here we present the results of an ancillary study to a phase III vaccine study (N=611). All participants received three doses of either Dengvaxia or placebo and followed for six years. We performed neutralization tests on annual samples and during confirmed dengue episodes (N=16,508 total measurements). We use mathematical models to reconstruct long-term antibody responses to vaccination and natural infection, and identify subclinical infections. There were 87 symptomatic infections reported. We estimated a further 351 subclinical infections. Cumulative vaccine efficacy was positive for both subclinical and symptomatic infection although the protective effect of the vaccine was concentrated to the first three years following vaccination. After accounting for post-vaccination antibody titers, we found no difference between the risk of infection or disease between placebo and vaccine recipients, suggesting that antibody titers are a good predictor of both protection and disease risk.
Introduction
The four dengue virus serotypes (DENV1-4) cause 50 million symptomatic infections each year1. The development of a vaccine is considered essential to the global strategy to combat dengue. However, the global rollout of the only licensed vaccine, Dengvaxia, has stopped due to increased risk of hospitalisation in dengue-inexperienced individuals who were vaccinated compared to unvaccinated controls, which only became apparent post licensure2,3. In the Philippines, the Dengvaxia license has been permanently revoked. The failure to recognise the weakness in the vaccine at an earlier time point has highlighted our poor understanding of dengue immunopathogenesis and potentially problems in the design of phase III dengue trials4.
A key uncertainty that remains is the role of antibodies in determining dengue disease risk5,6. Dengue studies have previously highlighted that immune responses generated from an infection provide temporary cross-protection (lasting from 6 months-2 years) to heterologous serotypes but appear to subsequently increase the risk of severe disease7,8. Previous analyses in cohort participants have demonstrated that the antibody titer elicited by natural infection at a moment in time is associated with both the underlying risk of infection and whether individuals who do become infected develop severe disease or not5,6. However, the relevance of these findings to vaccine-induced immunity remains unclear. It is unknown whether the antibody response to Dengvaxia is comparable to natural infection, and provides comparable levels of protection from infection and disease9. A major complication in answering these questions is that most infections are subclinical and not detected in phase III trials that only measure symptomatic disease10. These subclinical infections nevertheless change the underlying antibody titers within an individual, changing their risk of future infection and disease. In order to help obtain a more mechanistic understanding of dengue vaccines we need to characterise the long-term dynamics of vaccine-induced antibodies and compare them to those generated from natural infection. We also need to measure the effect of vaccines on the risk of infection rather than just symptomatic disease. These analyses require the long-term follow-up of vaccinated individuals and placebo controls that exceed the normal durations of Phase III trials.
Here we present the results of a cohort made up of participants from a Phase III Dengvaxia trial in Cebu, Philippines (N=611, 417 vaccine recipients, 194 placebo recipients, mean age of 8 years at baseline, age range: 2-14y) that were followed for over six years following their third dose (Table S1). Plaque reduction neutralization tests (PRNTs) were conducted on blood samples collected annually and during symptomatic infections throughout this period11,12. Individuals were either recruited prior to their first dose (N=112) or after the third dose of the vaccine or placebo (N=499). We identify baseline serostatus in placebo recipients using neutralization titers in pre-vaccination sera where this was available (N=32) or post-dose three sera where it was not (N=192). To identify baseline serostatus among vaccine recipients we use the results of an NS1 serology assay that can discriminate between vaccine and natural infection-derived immunity13. We find 98% agreement between the NS1 method and direct measurement in pre-vaccination titer among the 80 individuals where both were measured (this assumes indeterminate results from the NS1 assay were all seropositive, Table S2). We use a mathematical modelling framework that uses parametric approaches to estimate how antibody titers respond to both vaccination and infection to reconstruct individual antibody responses over the course of the follow-up and in parallel identify subclinical infections (Figure 1A–C, Figure S1). We specifically capture the dynamics of post-vaccine and post-infection antibody responses, and allow for differences by prior immune status. This data and analytical approach allow us to answer key unanswered questions about the comparability of antibody dynamics from vaccines and natural infection and the long-term vaccine efficacy for both subclinical and symptomatic infections.
Figure 1. Antibody responses and detected infections during follow-up.
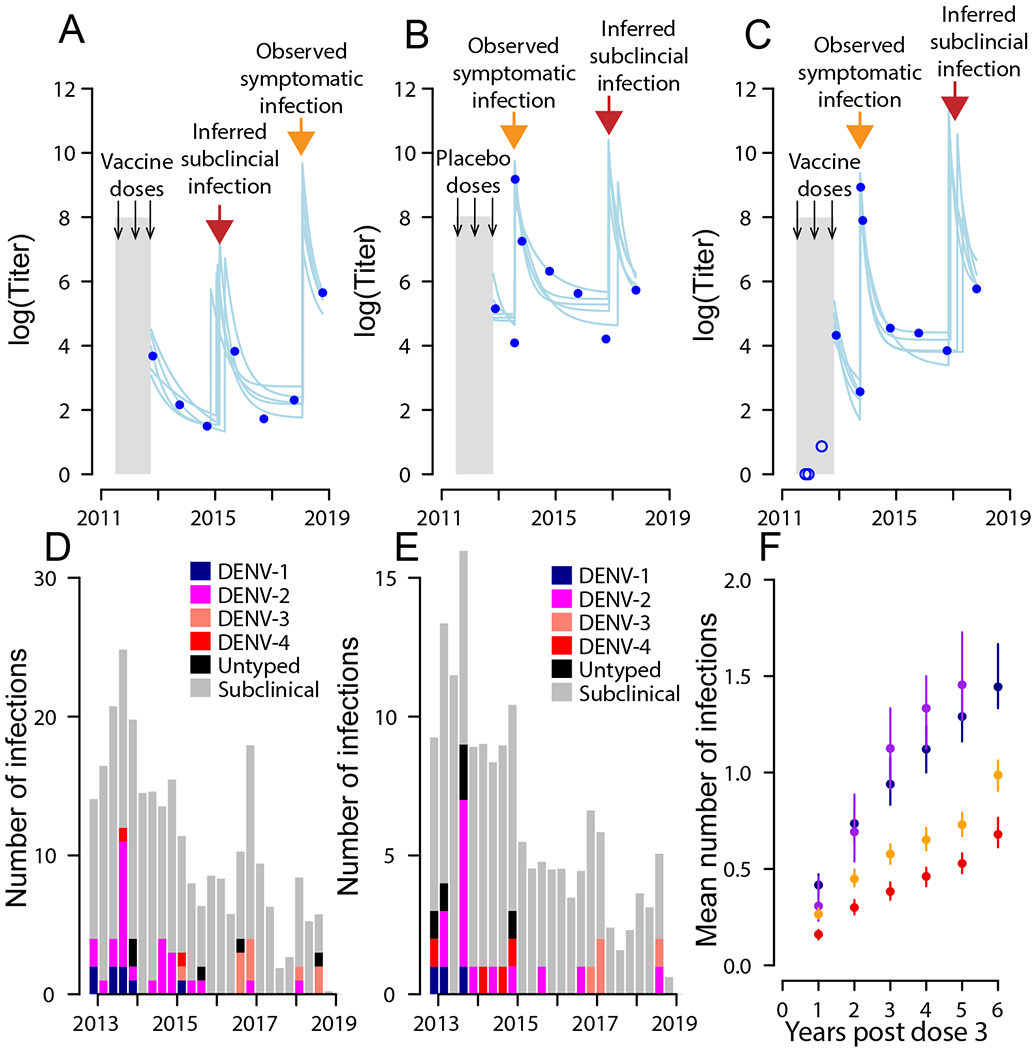
a–c, Measured (dots) and modeled (lines) antibody titers for three individuals. Two individuals were vaccine recipients (a,c) and one received a placebo (b). The orange arrows represent symptomatic infection events, and the red arrows represent inferred subclinical infections. Pre-dose three titers are available for c and are shown as blue open circles. d,e Distribution of symptomatic and subclinical infections over time in vaccine recipients (d) and placebo recipients (e). f, The mean cumulative number of infections (symptomatic plus subclinical) for recipients by vaccination status (vacc., vaccinated; plac., placebo) and baseline serostatus (−ve, negative; +ve, positive). The uncertainty bars represent 95% bootstrap confidence intervals from repeated reconstructions of individuals’ infection histories (n = 100).
Results
Infection and disease burden in the six years following vaccination
Overall, 90% of individuals with pre-vaccination samples were seropositive at vaccination, highlighting the substantial burden of infection in this community. In the six years post the third dose of the vaccine, there were 87 symptomatic infections (12 DENV1, 43 DENV2, 16 DENV3, 6 DENV4, 10 where the serotype was unknown), of which 9 led to hospitalisation (Table S3). There were an additional 53 symptomatic infections in between the first and the third dose of the vaccine. The attack rate of symptomatic infections post dose 3 was 0.025 per year in the vaccine arm and 0.031 per year in the placebo arm (p-value for no difference of 0.018). In addition, we estimate there were 351 subclinical infections (95%CI: 331-373), representing 19.9% of all infections (95%CI: 18.9%-20.8%) with a subclinical attack rate of 0.097/yr in the vaccine arm and 0.126/yr in the placebo arm (Figure 1D–E) (p-value for no difference of <0.001). We find that the total number of infections over the study differed by both vaccine status and baseline serostatus, with seronegative placebo recipients experiencing an average of 0.31 (95%CI: 0.21-0.43) infections per year, compared to 0.25 (95%CI: 0.19-0.30) for seronegative vaccine recipients, 0.15 (95%CI: 0.12-0.17) for seropositive placebo recipients and 0.11 for seropositive vaccine recipients (95%CI: 0.09-0.12) (Figure 1F).
Vaccine efficacy to clinical and subclinical infection
We find that for baseline seropositive individuals, the vaccine efficacy for symptomatic infections was 61.0% (95%CI: 29.6-86.5) in the year following the third dose. This fell to a cumulative vaccine efficacy for 39.4% (95%CI: 0.2-63.0) after six years, where we consider all symptomatic infections over the six year period (Figure 2A). For subclinical infections, the cumulative vaccine efficacy falls from 50.3% (95%CI: 9.2-70.0) to 36.2% (95%CI: 12.2-49.6) over the same time range (Figure 2B). Our results are similar to those estimated across the Phase III trial sites in Latin America and Asia in the year following the third dose (vaccine efficacy of 41.7% to subclinical infection)14. When considering vaccine efficacy over discrete windows of two years rather than cumulatively over the study period, we find that there was no significant protection from symptomatic infection after 3 years and no protection from subclinical infection after 5 years (Figure 2C–D). This highlights that the protective efficacy of the vaccine is concentrated in the early years post vaccination. Some individuals were infected in between their first and third doses. We find that cumulative vaccine efficacy to symptomatic infection from the first dose was only slightly higher than when calculated post dose three (63.6%, 95%CI: 45.7-79.0 up to one year post dose three and 53.5%, 95%CI: 37.2-70.1 after six years) (Figure S2). Our findings are robust to uncertainties in the determination of baseline serostatus (Figure S3). As there were very few individuals that were seronegative at baseline (49 individuals), we could not estimate the vaccine efficacy in this population. Our findings of reduced vaccine efficacy after three years suggest the introduction of a booster dose after the completion of the primary vaccination series may prolong the duration of protection. This strategy has been used elsewhere. In particular, a booster dose was introduced for the Japanese encephalitis vaccine (Imojev) that is based on the same chimerivax technology as Dengvaxia following similar observations of waning immunity15.
Figure 2. Vaccine efficacy for individuals seropositive at baseline.
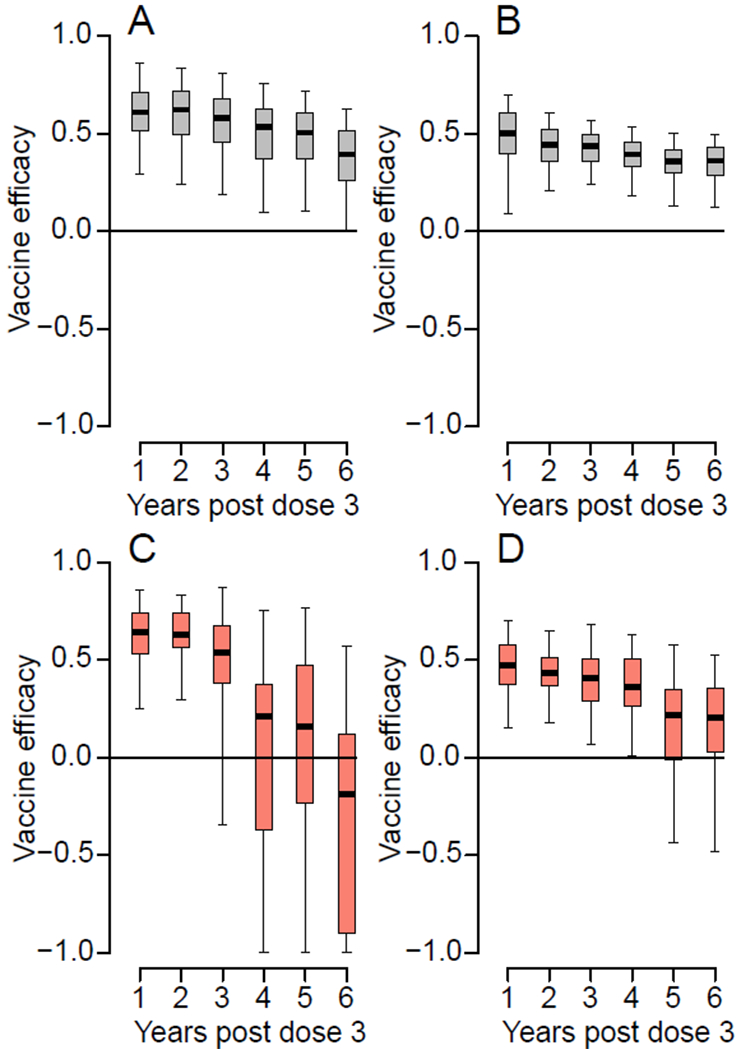
(A) Cumulative vaccine efficacy for symptomatic infection by year. (B) Cumulative vaccine efficacy for subclinical infection by year. (C) Vaccine efficacy for symptomatic infection by year over two-year rolling windows. The estimate is plotted at the maximum of the window (e.g., the vaccine efficacy over years 3-4 post dose three is plotted at year 4). (D) Cumulative vaccine efficacy for subclinical infection by year over two-year rolling windows. In each panel, the boxplot represents the mean, the interquartile range and 2.5 and 97.5 percentiles of the estimated vaccine efficacy from repeated infection history reconstructions (N=100).
Comparing post infection and post vaccine antibody dynamics
Post vaccination, the antibody titers of previously seronegative individuals rapidly declined (Figure 3A). However, this was only apparent once we removed titer measurements following post-vaccine symptomatic and subclinical infections. Ignoring these post-vaccine infections would give a false appearance of stable titers in these individuals (dotted line in Figure 3A). In seropositive individuals, the rise and decline in titers from vaccination was less drastic. Natural infection resulted in higher initial titers compared to vaccination (comparison of peak titers in Figure 3B versus those in Figure 3A), irrespective of vaccination or serostatus (Table S4). Antibody titers decayed following infection with a stable setpoint antibody load reached after one year, consistent with what has previously been observed with dengue antibody titers measured through hemagglutination inhibition assays5. In particular, among baseline seropositive individuals, we observe negligible difference in the mean antibody responses following natural infection in vaccinated as compared to placebo recipients. Serotype-specific antibody responses have been proposed to play an important role in individual-level risk of infection and disease, and could potentially help explain the differential efficacy by serotype for Dengvaxia2,16. We find that measured antibody responses for DENV1-3 are very similar to each other, and DENV-4 are consistently lower than the other serotypes (Figure 3C). The difference between serotypes is of the same magnitude in the year following vaccination events as in the year following infection events, suggesting that any observed differences in serotype-specific titers are principally driven by the virus used in the PRNT assay (Table S5) rather than from biologically relevant effects.
Figure 3. Antibody titer response following vaccination or infection.
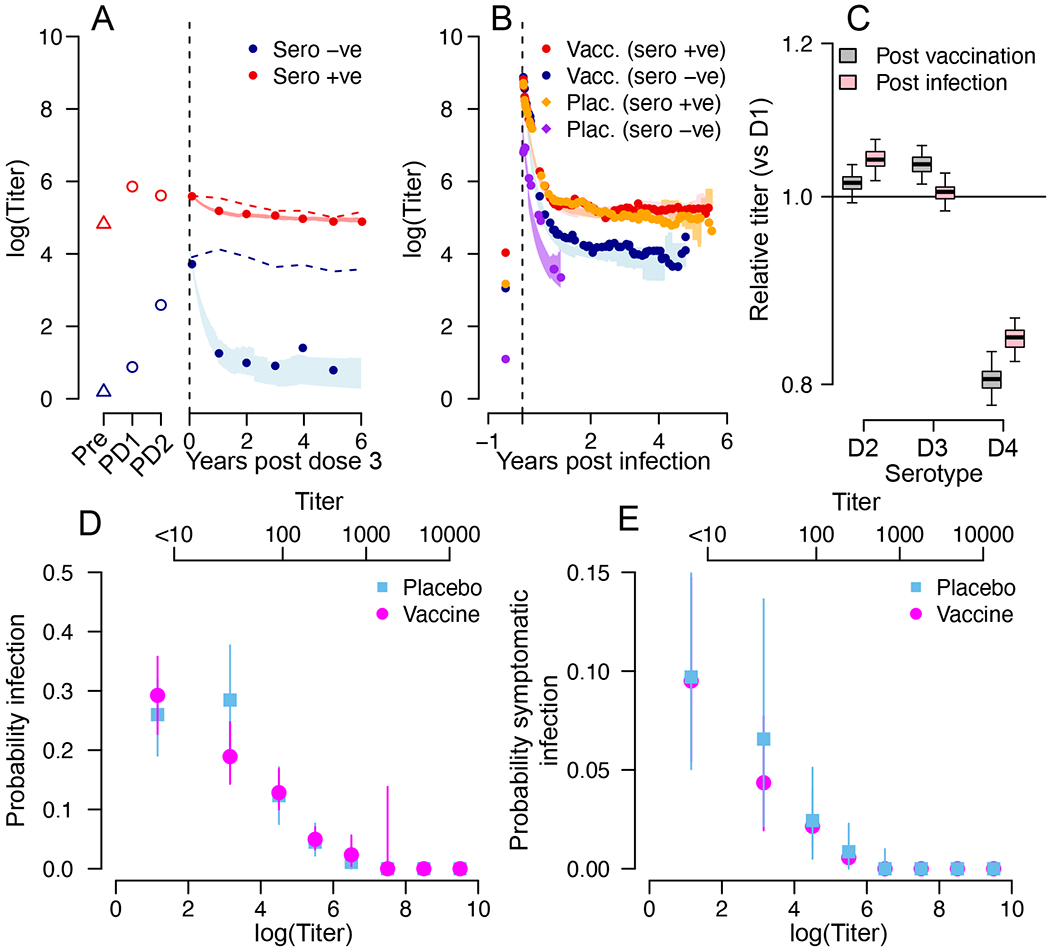
(A) Mean titers following vaccination and prior to an infection for those seronegative at baseline (blue) and seropositive at baseline (red). The lines represent the mean estimate and the shaded blue and red areas represent 95% credible intervals from the model. The dashed lines are the mean measured titers when subclinical infections are not removed. ‘Pre’ is pre-dose 1, ‘PD1’ is post dose 1, ‘PD2’ is post dose 2. (B) Mean titers following symptomatic infection for placebo recipients seronegative at baseline (purple), placebo recipients seropositive at baseline (orange), vaccine recipients seronegative at baseline (blue) and vaccine recipients seropositive at baseline (red). The lines represent the mean estimate and the shaded areas represent 95% credible intervals from the model. (C) Mean titer in the year following vaccination comparing each serotype to DENV-1 (black) and mean titer in the year following symptomatic infection comparing each serotype to DENV-1 (red). Each boxplot represents the mean, the interquartile range and 2.5 and 97.5 percentiles of the estimated difference in mean titer from repeated infection history reconstructions (N=100). (D) Mean and 95% confidence intervals for the probability of infection (subclinical or symptomatic) as a function of antibody titer for vaccine recipients (green) and placebo recipients (brown) from repeated infection history reconstructions (N=100). (E) Mean and 95% confidence intervals for the probability of symptomatic infection as a function of antibody titer for vaccine recipients (green) and placebo recipients (blue) from repeated infection history reconstructions (N=100). For (D) and (E) antibody titers on both a linear scale (top axis) and on a natural logarithmic scale (bottom axis) are provided.
Relationship between antibody titer and infection risk
Using our reconstructed antibody titers, we explore the relationship between an individual’s titer on a particular day and their risk of infection (Figure 3D) and symptomatic infection (Figure 3E). We find that there is a strong decline in the probability of both infection and disease as titers increase, with very low risk of infection or disease for individuals with titers >1:400, reducing to essentially no risk for individuals with titers >1:1000. Across the study subjects, 19% had mean titers of >1:400 and 4% had mean titers of >1:1000. Once we account for the same antibody titer, we show that there is no difference in the probability of infection or disease between vaccine and placebo recipients. Our findings suggest that once an individual reaches a setpoint antibody load (typically reached around a year following infection) of greater than 1:400, they should be at a limited risk of future infection. These findings are consistent with antibodies directly providing protection or enhancing disease risk through the presence of sub-neutralizing antibodies. Antibodies may also be correlated with other immune functions, which are driving individual profiles. Our results are specific to the reference viruses used in the neutralization assay in this study (Table S5). Using different reference viruses could shift the titers associated with protection either up or down17.
We assess the longer-term survival from infection and disease as a function of both baseline serostatus and vaccination status. Among individuals that were seropositive at baseline, we found that vaccine recipients had a lower risk of infection (Figure 4A) and disease (Figure 4B) than placebo recipients. We find that after their third dose, it took 3.3 years (95%CI: 2.2, 5.4) for 50% of placebo recipients to have experienced their first post-vaccination infection, whereas this took 5.3 years (95%CI: 4.2, >6) for vaccine recipients. We find that post dose three antibody titer appears key to determining infection and disease survival risk, irrespective of vaccination status (Figure 4C–D). The time for half of the individuals to experience an infection for individuals who had a low antibody titer (<1:50) following their third dose was 2.0 years (95%CI: 0.8,4.5), compared to 4.0 years (95%CI: 3.1-5.1) for those with medium titers (1:50 - 1:400) and >6 years for those with high titers (>1:400). In each case, there was no difference in risk between vaccine and placebo recipients once adjusting for post dose three titer. We also observe a strong effect of post dose three titer on the risk of symptomatic disease, with again limited difference between vaccine and placebo recipients (Figure 4D). We find no significant difference between vaccine and placebo recipients who were seronegative at baseline in their survival from infection or disease with a median time to the first infection being 1.4 years for the vaccine recipients and 1.2 years for the placebo recipients (Figure S4). This lack of a signal is likely driven by the small number of seronegative individuals in this cohort.
Figure 4. Time-to-event analysis.
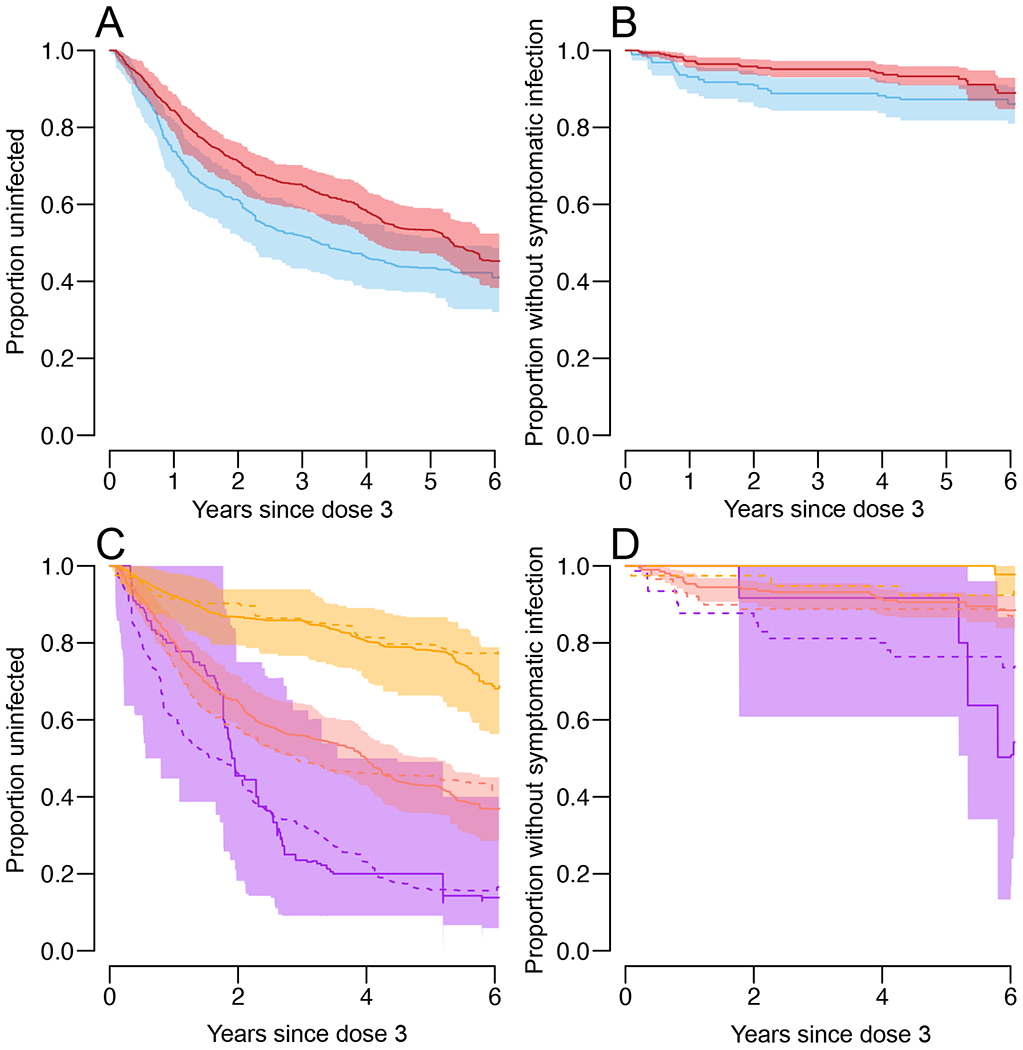
a,b, Proportion of baseline seropositive individuals who have not had an infection (subclinical or symptomatic) (a) or a symptomatic infection (b) after dose 3, by vaccination status (vaccination, red; placebo, blue). c,d, Proportion of baseline seropositive individuals who have not had an infection (subclinical or symptomatic) (c) or a symptomatic infection (d) after dose 3, by vaccination and titer status. Placebo, dotted lines; vaccination, solid lines; low titer (<1:50), purple; medium titer (1:50–1:400), salmon; and high titer (>1:400), orange. All panels represent mean and 95% confidence intervals from repeated infection history reconstructions (n = 100).
Discussion
By combining detailed long-term measurements of antibodies from a vaccine cohort with analytical tools that can reconstruct full antibody trajectories for each individual, we have been able to compare the antibody responses between vaccinated and unvaccinated individuals, and the association with subsequent infection and disease risk among individuals that were seropositive at baseline. As there were only a small number of seronegative individuals recruited into the study, we did not have the statistical power to explore the relationship between titer and infection risk for these individuals. It remains unclear where the antibodies generated in seronegative individuals by the vaccine are qualitatively different from those generated in seropositive individuals. Serological titers linked to protection and risk may also differ across settings, depending on the strain-specific infection history of the population as well as the specific assay protocol used. Despite these limitations, this work has demonstrated that antibody titers are a good marker of future infection and disease risk. Further, while the Dengvaxia vaccine does not generate the same magnitude of neutralizing titers as natural infections, the post-vaccination titer can be used as a predictor of future risk. These approaches, including the tracking of quantitative antibody titers, and analyses at baseline, will be applicable to other vaccine studies and should be applied in future trials.
Methods
Ethical review
This study was approved by the ethical review boards of Walter Reed Army Institute of Research, Chong Hua Hospital, and the University of Florida. Informed consent was obtained from all participants as well as their parents/guardians.
Study setting
The study was conducted in Cebu city, an urban setting in the Philippines with a population size of 920,000. Cebu, much like the rest of the country has experienced a high force of infection from dengue virus for decades. Cebu was chosen by Sanofi Pasteur as a site for their Phase III trial of Dengvaxia.
Cohort design
All individuals who participated in the Phase III trial (ClinicalTrials.gov Identifier: NCT01373281) in the Cebu city site were invited to participate in a separate ancillary study. This ancillary study was not considered a clinical trial and therefore not registered in a clinical trials database. The only inclusion criteria was having participated in the Phase III trial and the only exclusion criteria was unwillingness to participate. We attempted to recruit all Phase III trial participants to provide a comprehensive assessment as possible of the immune dynamics following vaccination in the underlying cohort. The primary objective of the ancillary study was to establish a repository of blood samples from vaccine trial participants, therefore, the target for enrollment was determined principally by logistical considerations regarding collection and processing of specimens as well as the overall limit to enrollment in the parent trial. Recruitment occurred at two time points - prior to the first dose and 1 month after the third dose of the vaccine or placebo. Subjects enrolled prior to vaccination had blood samples collected at enrollment and one month after each vaccine dose. Subjects enrolled after vaccination had a blood sample collected at ancillary study enrollment. Following recruitment individuals were followed up for events of symptomatic disease. This active disease surveillance consisted of weekly telephone contacts and home visits for subjects who could not be contacted by telephone. Individuals reporting an acute febrile illness had acute and convalescent blood draws taken, which were then tested using PCR and IgG/IgM ELISAs. Symptomatic individuals with a positive PCR result or with an IgM rise were considered confirmed infections. In addition to blood draws during illness events, there were annual blood draws collected from all participants.
As some individuals were only recruited into the ancillary study after they had received their third dose of the vaccine, the ancillary study was unable to identify disease events that occurred in the year long period between the first and third dose of the vaccine. However, all individuals, irrespective of whether they were recruited into the ancillary study prior to their first vaccination dose or after their third dose, were actively followed by Sanofi Pasteur for disease in the underlying Phase III trial for the year-long period between their first and third doses. This separate dataset therefore provides disease events in the subset of individuals that were only recruited in the ancillary study after they had received their third dose.
Antibody testing
Plaque reduction neutralisation tests (PRNTs) were conducted on all sera from annual blood draws and on the acute and convalescent blood draws in confirmed infections11. The analysis is conducted using PRNT50 titers. The lowest serum dilution was 1:10.
Baseline serostatus
Most individuals were recruited after their third dose, which, for vaccine recipients, will have changed their antibody titers. We therefore used different strategies to determine baseline serostatus for placebo and vaccine recipients. For placebo recipients, we considered individuals to be baseline seronegative if they had no neutralization titers (<1:10) to any serotype in either the pre-dose one sera where this was available (N=32) and in the post-dose three sera where it was not (N=162). This assumes no change in serostatus had occurred in the time between the first and the third dose of the placebo. For vaccine recipients, we used the results of an NS1 serology assay on the first post dose 3 samples. This assay can discriminate between vaccine and natural infection-derived immunity13. Note that this approach will not work for all dengue vaccines. It has been useful for Dengvaxia because the YFV backbone continued in the vaccine strain results in no DENV NS1 response. A subset of vaccinated individuals had indeterminate results from this assay (52 individuals). In a subset of vaccinated individuals (N=80) we also have pre-vaccination sera available. We note 98% agreement in the assigning of serostatus using the NS1 assay versus using the neutralization titers if indeterminate results are all considered as seropositive. We therefore consider all individuals with indeterminate results to be seropositive at baseline. In calculating the vaccine efficacy, we conducted sensitivity results where we varied how indeterminate results were considered.
Data available for each individual
We used data from individuals that had at least one annual blood draw (N=611). For each individual, we had their date of birth, the dates of any confirmed symptomatic dengue events, the dates of recruitment, when they left the cohort and annual blood draws, their vaccination status and the dates of the three doses, baseline serostatus, and PRNT50 titer to each of the four dengue serotypes for each confirmed illness event and the annual blood draws.
Characterizing titer dynamics following vaccination and symptomatic infection
We characterize the mean PRNT50 antibody titer across all four serotypes for different time windows following vaccination. We separately consider those seropositive and those seronegative at baseline. We include all antibody measurements from all individuals that had PRNTs conducted on sera in the time window of interest (<60days, 60days-6m, 6m-1.5y, 1.5y-2.5y, 2.5y-3.5y, 3.5y-4.5y, 4.5y-5.5y, >5.5y). In the main analysis we exclude titers from individuals that had an infection event (either subclinical or symptomatic) post vaccination and prior to the blood draw. We also repeat the analysis where we include all titer measurements, irrespective of the presence of intermediary infections.
We also characterize the mean PRNT50 antibody titer across all four serotypes for the same time windows (<60days, 60days-6m, 6m-1.5y, 1.5y-2.5y, 2.5y-3.5y, 3.5y-4.5y, 4.5y-5.5y, >5.5y) following symptomatic infections. We again separately consider those seropositive and those seronegative at baseline and exclude titers from individuals that had an infection event (either subclinical or symptomatic) post the infection event and prior to the blood draw.
Antibody responses post vaccination and symptomatic infection by serotype
In a separate analysis, we assess whether there were substantial differences in titers by serotype when comparing vaccinated with naturally infected individuals. To do this, we calculate the mean PRNT for each serotype separately across all individuals that had blood draws in the year following vaccination and the mean PRNT for each serotype separately across all individuals that had blood draws in the year following symptomatic infections (again excluding readings where an infection event occurred prior to the blood draw). We then calculate the relative titer for each serotype, relative to that for DENV1.
Mathematical model to reconstruct antibody titers
We developed a mathematical model that reconstructs the daily mean antibody titer (i.e., the average titer across all four serotypes when the titers are placed on a natural logarithmic scale) for each individual during their time in the cohort. This approach is based on efforts that reconstructed titers in a dengue cohort in Thailand5.
Notation
We follow and extend the notation from the original paper that presented this approach5. For each individual i and the total historic infection events prior to time t as ni(t). We use an indicator, ψ = 1…ni(t), to index the infection number. The time point of each infection is captured by τIi,ψ. The times of all previous infections prior to time t is Hi(t). For individuals who received a vaccine, the time point of each infection is captured by τVi,ψ. To capture the antibody measurements, we use NAi to mark the total number of blood draws an individual had, with π = 1…NAi used to index each blood draw and τAi,π used to capture the time of each blood draw. Ai,π is the true mean antibody titer across the four serotypes at blood draw πand A*i,π represents what is actually measured. Λi (t) is the cumulative force of infection on individual i up to time t.
Overall approach
We consider that prior to any dengue vaccination or infection, individuals have no antibody titers to dengue. This assumes negligible impact of infection by other flaviviruses and vaccination by other flavivirus vaccines. Following an infection or vaccination event, antibody titers will immediately rise and subsequently decay to a new level. This reflects the assumption that each antibody rise is made up of both a temporary rise of short-lived antibodies (that subsequently disappear) and a permanent rise of long-lived antibodies. Future infections will result in further rises and decays. Our ability to detect subclinical infections is based on changes in neutralization titers. Infections that do not result in a change in neutralization titers or only a very transient change would be missed. It remains unclear how common such infections are.
Modelling permanent titer rises
We assume that the magnitude of the permanent rise in titers depends on whether it was triggered by vaccination or natural infection and by the baseline serostatus of the individual. We model the permanent rise in titers from an infection event, QIi(ψ)as:
Where a gamma distributed random effect with mean when individual i was a vaccine recipient and was seronegative at baseline, when they were a vaccine recipient that was seropositive at baseline and when they were placebo recipient. There were insufficient seronegative placebo recipients to break this last group by serostatus. The variance of the gamma distribution had a parameter ωv, irrespective of the serostatus of the individual.
For individuals that were vaccinated and were seronegative at baseline, we model the overall permanent rise in titers over the three doses of the vaccine as:
Where is a gamma distributed random effect with mean parameter The variance of the gamma distribution had the same parameter ωv.
For the majority of individuals, we do not have the antibody titer for individuals prior to vaccination. Therefore, for seropositive individuals, we cannot discriminate between permanent antibody titers generated by the vaccine from permanent titers that were present prior to vaccination. We therefore cannot estimate both baseline titers and permanent rise in titers from vaccination in the model. Therefore, for individuals that were seropositive at baseline, the permanent rise in titers from vaccination was forced to be zero. This means that any permanent rise in titers from the vaccine will be captured in the baseline titers.
This value was also forced to be 0 for unvaccinated individuals.
Modelling temporary titer rises
We assume that the temporary antibodies will decay over time. The initial magnitude of the short-lived titers and the rate of decay will depend on whether it was triggered by vaccination or natural infection and by the baseline serostatus of the individual.
We model the temporary rise in short-lived titers from a natural infection event as:
Where is a gamma distributed random effect that captures the instantaneous rise in temporary titers from the most recent infection prior to time t with mean parameter when individual i was a vaccine recipient and was seronegative at baseline, with mean parameter when individual i was a vaccine recipient and was seropositive at baseline and when individual i was a placebo recipient. The variance parameter was γVin all instances. is a gamma distributed random effect that captures the decay in temporary titers from the most recent infection with mean parameter δImand variance parameter δv.
For individuals that were vaccinated, as we do not have titers post the first and second doses for most individuals, we model the overall impact of the three doses using a single rise and decay. For time periods where there have been no subsequent natural infections following the third dose, we model the temporary rise in short-lived titers generated from the vaccine as:
Where is a gamma distributed random effect that captures the instantaneous rise in temporary titers from the vaccination with mean parameter when individual i was seronegative at baseline and with mean parameter when individual i was seropositive at baseline. The variance parameter was γV (the same parameter as for natural infections) in both instances. δVi is a gamma distributed random effect that captures the decay in temporary titers from the vaccination with mean parameter when the individual was seronegative at baseline and when they were seropositive. The variance parameter δv (the same parameter as for natural infections).
For individuals that were not vaccinated or for individuals that were vaccinated in time periods where there has been a subsequent natural infection following the third dose, we assume there were no temporary titers:
Overall trajectory
Assuming that the permanent rises from vaccination and natural infection are additive and that the titers from natural infection or vaccination are lost following subsequent infections, the mean antibody titer at blood draw k for an individual i is:
Modelling infection histories
We assume that each individual can get infected up to four times (reflecting the four serotypes), irrespective of their vaccination status. We assume a different force of infection by year so that for a time t, the force of infection is:
Where the parameter represents the mean daily force of infection per serotype in 2012 (the year after which individuals had received their third dose) and β|t|is the relative force of infection in year /t/ as compared to 2012.
The contribution to the likelihood for an individual i can be broken down into periods prior to an infection and periods with an infection. We use a daily time step. Each day during which no infection occurs, the contribution to the likelihood for serotype s is where there have been no more than 365 days have passed since an infection by any serotype (this time window prevents more than one infection between two sequential blood draws) and the individual has not been previously infected with that serotype and the contribution to the likelihood is zero otherwise. For each infection that occurs at day t, the contribution to the likelihood is . While we cannot be certain that there was not more than one infection within any 365 day period, this appears unlikely as the mean duration of temporary cross-protection between serotypes has been estimated as two years7.
Use of data augmentation in the context of imperfect observation
Under conditions of full observation, the probability of an individual’s life-course of infection for an individual up to the time point of a blood draw is:
Where τIi,0is individual’s i date of birth and Ti is the time at which they had their final blood draw. This assumes the same force of infection across the four serotypes.
Full infection histories are not observed. There will have been infections prior to entry and subclinical infections would not have been identified through the surveillance system.
For titers generated from infections prior to recruitment, we estimate a baseline titer Ai(t0)that represents the mean titer at the point of recruitment. Individuals that are seronegative at baseline are given a baseline titer of 0 (i.e., this is not estimated). To incorporate subclinical infections during the study we use a Bayesian data augmentation approach, as previously described, where the timing and serotypes of subclinical infections are considered as nuisance parameters. Through this approach, we ensure that no individual is infected during the study period more than once by the same serotype and that there are no successive infections within a 365 day window.
Finally, underlying assay variability may mean that the measured antibody titer is different to the underlying true antibody titer. We assume that the measured titer (A*i,π) is normally distributed with mean equal to the true antibody titer (Ai,π) and standard deviation, σ, where σ is a parameter that is estimated.
MCMC
As previously described, we use a Markov chain Monte Carlo approach to fit the model parameters and the augmented data5.
Step 1: The model parameters are updated using Metropolis-Hastings. For the parameters that capture the dynamics of antibodies following natural infection the likelihood is calculated using titers that were measured between 30 days before and 365 days after the detected infections. Similarly, when updating the parameters that determine the dynamics of titers post vaccination we only include titers measured in the year following vaccination.
Step 2: Among those with symptomatic infections, the rise in titers may not be the same day as the day of symptom onset. We therefore use an independence sampler to update the day of titer rise. At each iteration, the day of the titer rise was updated for 100 randomly chosen symptomatic infections using a uniform distribution ranging from 10 days before to 10 days after the day of symptom onset.
Step 3: The infection day is updated for randomly selected subclinical infections (N=300).
Step 4: The baseline titer is updated for randomly selected seropositive individuals (N=300) using a uniform distribution (range: 0-10).
Step 5: We use reversible jump–MCMC to add and remove unobserved infections. To add undetected infections we follow the following algorithm:
Randomly select individual.
Draw a candidate date for the infection event using a uniform distribution (range: day of dose three to day of final blood draw).
Draw titer dynamic responses for the new infection.
Update the date of infections to include this additional infection
For the removal of undetected infections, we:
Randomly select individual.
If that individual has undetected infections, randomly choose one of their infections as a candidate for removal.
Update the date of infections to include the removal of this infection
Estimation of impact of titer on infection and disease
To estimate the probability of infection given an individual’s titer we use 100 augmented datasets that have the full infection histories of each individual and their daily titers. For each augmented dataset, we combine all person-time of individuals that have titers within different bins (<2.3, 2.3-3,3-4,4-5,5-6,6-7,7-8,8-9,>9 on a log scale where 2.3 represent the lower limit of detection, equivalent to <10 on a linear scale). We exclude all person-time from individuals that had an infection within the prior year and therefore were not able to have a subsequent infection in the model, due to temporary cross-protective antibodies. Considering placebo and vaccine recipients separately, we calculate the proportion of all person-time that had an infection event in each of 100 infection history reconstructions. We annualize all the probabilities using the expression 1-exp(−365x) where x is the daily probability of infection. To explore the probability of symptomatic disease, we repeat the analysis, however we only consider detected symptomatic infections in the numerator. To incorporate uncertainty, we use a bootstrap approach where for each infection history reconstruction, we resample all the person-time with replacement and recalculate the probabilities of infection and disease. We use the 2.5 and 97.5 percentiles from the resultant distribution.
Vaccine efficacy
We calculate cumulative vaccine efficacy from both post dose 3 for up to six years and post dose 1 for up to seven years where we consider symptomatic disease as the outcome. We also calculate cumulative vaccine efficacy from post dose 3 when subclinical infection is the outcome. As we do not have blood draws for most individuals during the first year of the study we could not assess vaccine efficacy from post dose 1 where subclinical infection is the outcome. Finally, we calculate vaccine efficacy using 2-year rolling windows for both symptomatic disease and subclinical infection of the vaccine. Given the importance of baseline serostatus, we separately consider those seronegative at baseline with those seropositive at baseline. The vaccine efficacy equation we use is:
Where Incvacc(t)is the incidence prior to time t among vaccine recipients and is calculated as:
Where NInf(t) is the number of infections prior to time t and PT(t) is the person time prior to time t.
Adjustment for baseline serostatus misclassification
Where there are no pre-dose 1 samples available, serostatus in placebo recipients was assigned using post 3 samples. Vaccine recipients were assigned a serostatus using the results of the NS1 assay run on post dose 3 samples. As some vaccine and placebo recipients may have been seronegative prior to dose 1 but became subsequently infected prior to the third dose, they would be incorrectly classified as baseline seropositive. To adjust for this uncertainty, we use data from the 112 individuals who also provided sera prior to dose 1. Of individuals where baseline samples were available, 68 were vaccinated and considered baseline seropositive as per the NS1 assay, however, one (1.4%) of these were in fact seronegative according to the PRNT assay from the baseline examples. Five additional individuals had an indeterminate reading, all of which were seropositive according to the baseline samples. In addition, 27 were placebo recipients and considered baseline seropositive from the PRNT data, however, 3 of these (11.1%) were in fact seronegative according to the baseline samples. This would suggest that 1.4% of vaccinated individuals that are assigned a seropositive label were in fact seronegative and 11.1% of placebo individuals that are assigned a seropositive label were in fact seronegative. To incorporate this uncertainty, for each reconstructed dataset we use a Bernoulli random draw to decide whether each vaccinated and placebo individual with a seropositive label as defined at post dose 3 was in fact seronegative at baseline, where the mean of the Bernoulli distribution is taken from that calculated from resampling the individuals where the true baseline status is known. This approach incorporates sampling uncertainty. We also conduct a sensitivity analysis where no adjustment is made and obtain consistent results (Figure S3B). 57 vaccinated individuals had an indeterminate baseline serostatus. Of these 5 were in the subset of individuals where baseline samples were also available. All of these individuals were seropositive at baseline. We therefore considered all individuals with an indeterminate serostatus to be seropositive at baseline. However, we also considered a sensitivity analysis where 20% of these individuals were in fact seronegative and obtained consistent results (Figure S3C).
Time to event analysis
We use Kaplan-Meier curves to separately estimate the time to first infection, and first symptomatic infection following the final dose of the vaccine/placebo as a function of baseline serostatus and vaccination status. In addition, to explore the importance of post dose 3 titer on the survival function, for baseline seropositive individuals, we recalculate the Kaplan-Meier curves separately for individuals that had a low titer (defined as <1:50) following vaccine/placebo in the first blood draw post the final dose of the vaccine/placebo, a medium titer (defined as 1:50-1:400) and a high titer (defined as >1:400). In each analysis, to incorporate uncertainty we estimate the survival functions from each of 100 augmented datasets and present the 2.5%, median and 97.5% quantiles of the resulting distribution.
Note that in the survival analysis we consider the first infection following the third dose. In the vaccine efficacy we consider all infections (irrespective if they are the first or not), which means the two analyses are not strictly comparable.
Supplementary Material
Acknowledgements.
Funding. This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (grant numbers P01AI034533, 5R01AI114703-05), the U.S. Military Infectious Diseases Research Program and the European Research Council (grant number. 804744). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Code availability. C++ code used in this study is available at https://github.com/pdgcam/DengueTiters.git.
Publisher's Disclaimer: Disclaimer. Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the authors, and are not to be construed as official, or as reflecting true views of the National Institutes of Health, Department of the Army or the Department of Defense. The investigators have adhered to the policies for protection of human subjects as prescribed in AR 70–25.
Competing statements: The authors declare no competing interests.
Data availability.
Data used for this project is available at https://github.com/pdgcam/DengueTiters.git. To preserve anonymity date information has been removed. Instead all time periods, including all dates of illness and dates of blood draws, have been replaced with days since enrollment.
References
- 1.Stanaway JD et al. The global burden of dengue: an analysis from the Global Burden of Disease Study 2013. Lancet Infect. Dis. 16, 712–723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hadinegoro SR et al. Efficacy and Long-Term Safety of a Dengue Vaccine in Regions of Endemic Disease. N. Engl. J. Med 373, 1195–1206 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Wilder-Smith A et al. Deliberations of the Strategic Advisory Group of Experts on Immunization on the use of CYD-TDV dengue vaccine. Lancet Infect. Dis 19, e31–e38 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Moi ML, Takasaki T & Kurane I Human antibody response to dengue virus: implications for dengue vaccine design. Trop. Med. Health 44, 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salje H et al. Reconstruction of antibody dynamics and infection histories to evaluate dengue risk. Nature 557, 719–723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katzelnick LC et al. Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reich NG et al. Interactions between serotypes of dengue highlight epidemiological impact of cross-immunity. J. R. Soc. Interface 10, 20130414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabin AB Research on dengue during World War II. Am. J. Trop. Med. Hyg 1, 30–50 (1952). [DOI] [PubMed] [Google Scholar]
- 9.Ferguson NM et al. Benefits and risks of the Sanofi-Pasteur dengue vaccine: Modeling optimal deployment. Science 353, 1033–1036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halstead SB Dengue. Lancet 370, 1644–1652. [DOI] [PubMed] [Google Scholar]
- 11.Russell PK, Nisalak A, Sukhavachana P & Vivona S A plaque reduction test for dengue virus neutralizing antibodies. J. Immunol 99, 285–290 (1967). [PubMed] [Google Scholar]
- 12.Thomas SJ et al. Dengue plaque reduction neutralization test (PRNT) in primary and secondary dengue virus infections: How alterations in assay conditions impact performance. Am. J. Trop. Med. Hyg 81, 825–833 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sridhar S et al. Effect of Dengue Serostatus on Dengue Vaccine Safety and Efficacy. N. Engl. J. Med 379, 327–340 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Olivera-Botello G et al. Tetravalent Dengue Vaccine Reduces Symptomatic and Asymptomatic Dengue Virus Infections in Healthy Children and Adolescents Aged 2–16 Years in Asia and Latin America. Journal of Infectious Diseases vol. 214 994–1000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chotpitayasunondh T et al. Post-licensure, phase IV, safety study of a live attenuated Japanese encephalitis recombinant vaccine in children in Thailand. Vaccine 35, 299–304 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Fried JR et al. Serotype-specific differences in the risk of dengue hemorrhagic fever: an analysis of data collected in Bangkok, Thailand from 1994 to 2006. PLoS Negl. Trop. Dis 4, e617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katzelnick LC et al. Dengue viruses cluster antigenically but not as discrete serotypes. Science 349, 1338–1343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used for this project is available at https://github.com/pdgcam/DengueTiters.git. To preserve anonymity date information has been removed. Instead all time periods, including all dates of illness and dates of blood draws, have been replaced with days since enrollment.