

Abstract
The common language that unites all life is carried out by small-molecule chemical signals. These specialized metabolites have evolved to impart diverse cellular and ecological functions, and are broadly applied in medicine, agriculture and nutrition. The rapid accumulation of genomic information has revealed that the metabolic capacity of virtually all organisms is vastly underappreciated. Initially mainly in bacteria and fungi, genome mining technologies were pioneered to accelerate metabolite discovery. Recent efforts are now being expanded to all life forms, including plants, animals and protists, and new integrative omics technologies allow increasingly effective mining of this diversity.
Introduction
Genomically encoded small-molecule chemicals are the common language that unites all life, from single cells to communities of organisms. Whereas many biochemicals are shared among large swaths of the tree of life, some molecules are biosynthesized only by a select subset of organisms and/or are specific to certain ecological niches. These specialized metabolites, also called natural products or secondary metabolites (see glossary, as well as refs.1–3 for in-depth discussions of the definitions of these terms), range in size, shape and complexity, from small terpenes and phosphonates to large and heavily posttranslationally modified gene-encoded peptides. As such, they have often evolved to impart diverse cellular, intraspecies and interspecies functions that perform key roles in physiology and in simple to complex ecosystems. Specialized metabolites provide organisms, from single-cell microorganisms to multicellular plants and animals, with some of their most distinguishing chemical features of color, smell, taste or toxicity. In other words, the blend of specialized metabolites endowed to an organism makes it unique.
Most specialized metabolites have been identified through experimental discovery approaches that take advantage of a chemical or biological feature of the expressed molecule to guide its isolation. Molecules such as penicillin, estradiol and caffeine are just a small selection of nature’s chemical bounty that has had profound societal impact (Figure 1a). Strikingly, the rapid accumulation of genomic and transcriptomic information in recent years has revealed that the metabolic capacity of virtually all organisms is vastly underappreciated, with millions of additional molecules awaiting discovery4–6.
Figure 1. Life’s chemical diversity.
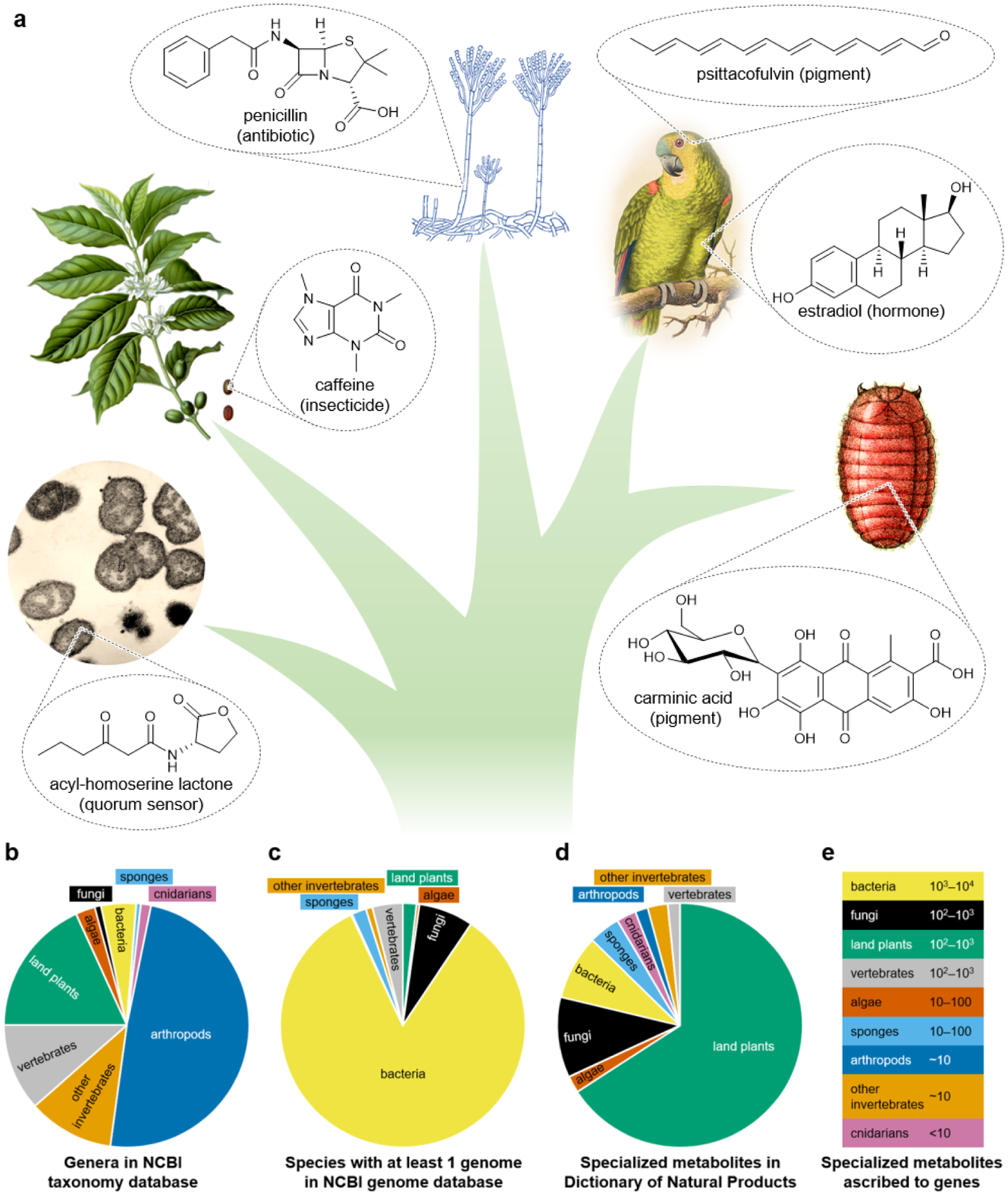
a) Bacteria, fungi, plants and animals produce a wide range of specialized metabolites that help them thrive in their respective environments. There is a large disconnect between (b) the numbers of taxonomic genera in the biosphere (as based on the NCBI taxonomy database), (c) the numbers of genomes available for these species (based on the number of species represented in the NCBI genome database), (d) the numbers of specialized metabolites isolated (based on the number of molecules ascribed to these classes of organisms in the Dictionary of Natural Products) and, (e) the estimated numbers of specialized metabolites that have been linked to genes responsible for their biosynthesis (estimates by the authors). There is likely great potential for discovering new metabolites from animals and protists, and identifying new biosynthetic pathways from plants, animals and protists. Algae includes green, red, and brown algae, diatoms and dinoflagellates. Heterotrophic protists and archaea were not included due to the low number of specialized metabolites isolated from these organisms.
Genome mining seeks to harness gene-based big data methods to expedite the concomitant discovery of specialized metabolites and their biosynthetic genes. With increasing technological improvements in genome sequencing, early mining experiments of relatively simple microbial genomes have been followed in recent years by much more complex genomes and metagenomes of plants, animals and other eukaryotic organisms that organize their biosynthesis genes differently (Figure 1). Additionally, to truly arrive at a deeper understanding of life’s chemistry, genome mining approaches are being developed that provide insight into the functions that these molecules perform in physiology and ecology. Here, we address the why, what, where and how of genome mining and discuss key challenges in figuring out what nature is verbalizing.
Why we mine and what to mine
Historically, specialized metabolites have been isolated and characterized from biological samples collected from the environment or from laboratory-grown organisms whereupon organic extracts of tissues or cells are chemically and biologically analysed. In this way, natural chemicals have been identified, dating back to the year 1803 with the isolation of morphine from opium poppy7. While analytical chemistry tools continue to improve in sensitivity and speed to aid the discovery process8, trends over the past decades have shown a clear deceleration of the discovery of novel structure chemotypes versus the rediscovery of well-known molecular families with subtle chemical modifications9. Genome mining has the potential to change the discovery rate and makes it possible to identify molecules that would otherwise remain under the radar. This is exemplified by the fact that after the publication of the genome sequence of the model organism Streptomyces coelicolor A3(2)10, which had been heavily studied for about half a century already and for which around a dozen (types of) specialized metabolites had been known, genome mining has since led to the discovery of seven additional ones from diverse classes: the nonribosomal peptides coelibactin10 and coelichelin11, the sesquiterpene (+)-epi-isozizaene12, 2-alkyl-4-hydroxymethylfuran-3-carboxylic acids13, the SCO-2138 RiPP14, the polyketide coelimycin P115 and a new set of partially characterised arsenopolyketides16. As such, genome mining has key differences and advantages as compared to the use of analytical techniques alone. First, it can access specialized metabolites that may not be produced under the growth conditions studied. Second, the approach inherently connects any discovered molecules to their biosynthetic genes, allowing for heterologous expression and bulk production. This is particularly significant because many medicinally valuable molecules are isolated from dwindling natural resources or organisms that are difficult to cultivate, and genome sequencing typically requires much less biomass than the quantities that are required for structural elucidation.
The motivations for genome mining have largely tracked those of the natural products community at large: historically, this has primarily been the exploration of life’s biochemical prowess, the understanding of physiology, and the pursuit of therapeutics. In the last century, the first specialized metabolites were linked to their biosynthetic genes, usually from cloned DNA fragments that could be used to complement mutations in these genes17–19. In the 2000s, genome sequencing started to mature, and the biosynthetic logic of some major classes of medicinal natural products, including polyketides, nonribosomal peptides and terpenoids, had been deciphered to some extent. The newly sequenced genomes often harbored homologs of genes encoding the biosynthesis of these classes of compounds, but which had not been associated with a metabolic product. Heterologous expression of these ‘orphan’ biosynthetic genes resulted in the discovery of several novel natural products, including triterpenes from the Arabidopsis genome20 and the hybrid peptide-polyketide aspyridones from the genome of the model filamentous fungus Aspergillus flavus21. Since these proofs of concept, countless new members of established major compound classes have been discovered through genome mining.
Genome mining is also contributing to the ongoing fundamental search for chemical and biosynthetic novelty in nature. Several specialized metabolites harboring chemical moieties unprecedented for their class, such as furanone22,23 and benzo[a]tetraphene24 polyketides, and aminovinylcysteine based ribosomally processed peptides25, were discovered through genome mining. Even among known specialized metabolites, there are numerous structures for which the biosynthetic machinery was only recently elucidated — often through genome mining — such as for the piperazate26, thiotetronate27, oxazolone28,29, isoxazole30, indolyloxazole31, alkyne32,33, N-nitroso34, and diazo35 moieties, polybrominated phenolics from marine bacteria36, plant-like isoquinoline alkaloids in diverse fungi37 and vinca alkaloids from medicinal plants38. As new biosynthetic reactions and structural classes are discovered, our ability to reliably predict orphan genes for novel molecular scaffolds will continue to improve. Still, there are many biochemical scaffolds for which the genetic basis is still completely or mostly a mystery, such as the polycyclic ethers found in dinoflagellates39, or the ladderanes produced by anammox bacteria40,41. There are doubtlessly numerous novel scaffolds not represented among known specialized metabolites that will one day be discovered through genome mining.
Our understanding of ribosomally-synthesized peptides has particularly benefited from the rise of genome mining, thanks to the fact that their structures can often be relatively easily predicted from genomic data. One class of these peptides, the RiPPs (for ribosomally-synthesized and post-translationally modified peptides)42, is particularly noteworthy for its broad distribution across all three domains of life and our growing knowledge of its diversity of peptidic modifications43. Not to be confused with non-ribosomal peptide synthetase products, new structural families of RiPPs continue to be discovered such as the spliceotides44 and epipeptides45 from bacteria, dikaritins46,47 from fungi, and the lyciumins48 from plants. Ribosomally-derived specialized metabolites are not always RiPPs and can range remarkably in size, from small molecules like the pyrroloquinoline alkaloid ammosamide49,50 to small proteins like three-finger toxins from spitting cobras51 and venom proteins from spiders52,53. Similar discovery trends can also be seen in the other major biosynthetic lineages, where the mining of genomes has resulted in a growth of chemical and biochemical knowledge.
What else is there to mine and what happens to genome mining after we have exhaustively identified all specialized metabolite scaffolds? Based on the inventory of known specialized metabolites and those that are already connected to biosynthetic genes, the future remains bright considering the efficiency and breadth of new strategies for genome mining and given the increased extent of resources available for mining. Even in cases where the mining of orphan genes leads to re-discovery of previously reported specialized metabolites, solace comes in the discovery of new enzymes and biosynthetic knowledge that may have biotechnological utility.
In recent years, new motivations for genome mining have emerged from two new areas of research: microbiomes and synthetic biology. In microbiome research, the mining of specialized metabolites and the genes encoding their biosynthesis provides a window into the mechanisms responsible for key phenotypes mediated by the microbiome, such as pathogen suppression54,55 or host immunomodulation56. Moreover, it potentially enables the design of synthetic microbial consortia that can be used as live therapies or biologicals57–59, based on genome-based prediction of the chemical capabilities of individual strains. In synthetic biology, pathways are being mined from genomes mainly as a source of enzymological diversity, which are started to be used as ‘parts’ for metabolic engineering of novel molecules with desirable properties60. This may enable combinatorialization of enzymes61 or even computer-aided design62 to create ‘new-to-nature’ molecules.
Where to mine
Since genome mining is predicated on the availability of omics data, growth in the field has relied on improvements in sequencing technologies. To this day, the majority of genome mining has been conducted on bacterial genomes, which, given their comparatively small size and low repeat content, dominate publicly available genomic databases (Figure 1c). Further simplifying the mining process within bacteria is their propensity to physically cluster genes in operons and biosynthetic gene clusters (BGCs, see Box 1) for cooperative biosynthesis of specialized metabolites. This has allowed researchers to readily formulate hypotheses regarding the biosynthesis of molecules of interest, even in cases where substrates and enzymes have no precedent. For instance, genes clustered with a gene known to be involved in the biosynthesis of a specialized metabolite are often promising candidates to focus experiments that aim to identify other genes involved in its biosynthetic pathway.
Box 1: gene clustering in specialized metabolism.
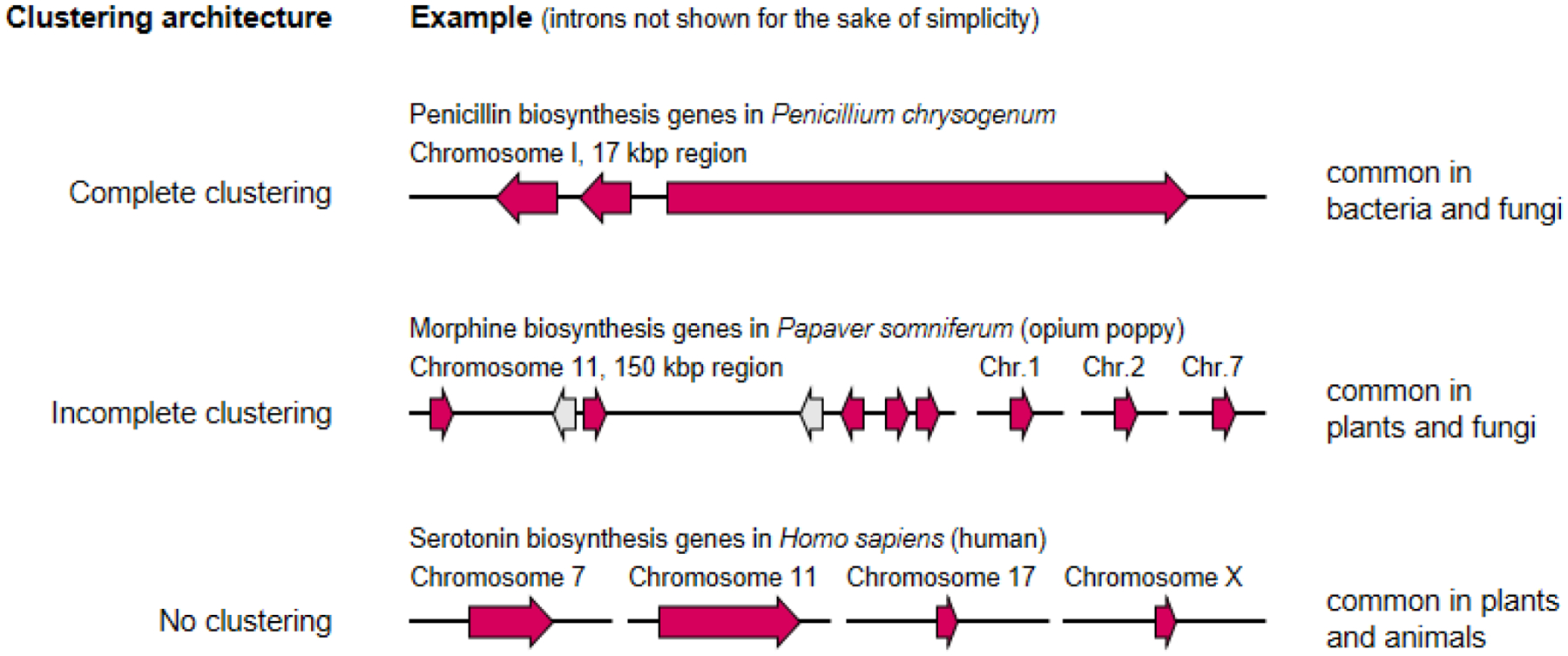
In most organisms, genes involved in specialized metabolic pathways are encoded contiguously on the chromosome in so-called biosynthetic gene clusters (BGCs). The extent to which biosynthetic genes are clustered differs between different taxonomic groups, and specifically between the plant, fungal and bacterial kingdoms, which show increasing degrees of gene clustering. As an illustration, in the model actinomycete bacterium Streptomyces coelicolor, 22 BGCs have been experimentally characterized and linked to products (including two single enzyme-coding genes), and for none of the corresponding pathways is there evidence of being encoded in multiple genomic loci. On the other hand, out of the 23 BGCs experimentally characterized in the model fungus Aspergillus nidulans, at least three pathways have been shown to be split over multiple loci: those for the biosynthesis of austinol / dehydroaustinol194, emericellin195 and nidulanin A196. In the model plant Arabidopsis thaliana, only four pathways have been experimentally shown to be encoded by BGCs: those for the biosynthesis of thalianol, marneral, arabidiol, and tirucalladienol. While several other pathways seem to show partial clustering157,197, the pathways for the biosynthesis of glucosinolates, flavonoids, strigolactones, arabidopyrones, camalexin and 4-hydroxyindole-3-carbonyl nitrile seem to be (almost) devoid of clustering. Still, even in plants, BGCs are an attractive target for pathway discovery, as they provide ‘low-hanging fruits’ that can be straightforwardly identified in genome sequences5. In protists, several examples of BGCs have been reported86,198, while in animals, not much is known about gene clustering. Yet, a recent global synteny network analysis makes clear that gene order in mammals is clearly nonrandom and may have large functional repercussions199.
There are several hypotheses for why the genes for specialized metabolic pathways are clustered on the genome. The four main ones are the following:
Coordinated gene expression. In bacteria, given that transcription and translation occur in the same cellular location, the biophysics of transcriptional regulation favors co-regulation of operons located near the gene encoding a pathway-specific regulator102. In fungi and plants, there is evidence that clustered genes are co-regulated through epigenetic modification of chromosomal regions200,201.
The selfish operon hypothesis. Given that horizontal gene transfer of BGCs, but also their deletion, occurs frequently in bacteria and fungi, the ‘survival’ of BGCs in the biosphere may depend on their ability to spread to other strains and species; clustering may increase chances of genes to be jointly transferred202. This can be supplemented by a ‘persistence hypothesis’, stating that clustered genes are less likely to be interrupted by a segmental duplication and therefore more likely to survive as a unit203.
Avoiding toxic intermediates. According to this hypothesis, clustering of genes is an adaptation against the accumulation of toxic pathway intermediates. Clustering promotes co-inheritance of the entire pathway, so that (sub)lethal genotypes carrying only part of the pathway are avoided204.
Co-adaptation through co-inheritance. Many clusters in plants and fungi have formed in dynamic chromosomal regions as part of evolutionary arms races with competing species205. Especially in sexual organisms, rapid adaptation of pathways may only be possible when co-adapted alleles of the underlying genes are not constantly separated by recombination events. This has recently been proposed to drive repeated and independent evolution of gene clusters encoding phenylpropanoid degradation pathways in fungi206.
Soil microorganisms, and in particular the actinomycetes, were already a popular source of specialized metabolites in the pre-genomic era and were thus obvious targets for early sequencing and mining efforts. The first genomes of Streptomyces, Salinispora and Saccharopolyspora species pre-2008 revealed that the actinomycetes were more metabolically rich than originally thought, with many species dedicating over 10% of their genomic space to the production of dozens of specialized metabolites10,63–65. This trend has now been observed in many other environmental bacteria, especially those with large genomes in excess of 10 Mb. The filamentous marine cyanobacterium Moorea producens, for instance, devotes roughly one-fifth of its genome in this manner66. Due to decreasing costs of bacterial genome sequencing, recent efforts have ballooned in scale to mining 10,000–100,000+ genomes at a time for novel molecules67,68.
The specialized chemistry of uncultivated bacteria that dominate the microbiota of animals, plants and other host organisms has also been examined through genome mining, highlighting the importance of microbial metabolites in mitigating health and disease within their hosts. Whether it be human gut bacteria69, plant rhizosphere microbial communities70, or marine sponge microbiota71, the metagenomic mining of the microbial dark matter of life is quickly revealing that microorganisms are indispensable for the host’s chemical fitness. In cases where there is no living host, such as in soils, seawater and even the air, environmental DNA (eDNA) further reveals the exquisite metabolic capacity of the earth’s microbiota72,73. While attempts to exploit eDNA as a genetic resource for natural product discovery were initiated already two decades ago74, better computational infrastructure such as reference databases75 and profiling software76, as well as massively increased sequencing volumes, have now turned this into a promising technology. Indeed, innovative efforts have now led to the engineered production of drug leads directly from the mining of soil eDNA samples77,78.
Filamentous fungi, such as Aspergillus nidulans and Penicillium chrysogenum, have also long been known to cluster their genes for the biosynthesis of, for example, the antibiotic penicillin or the carcinogenic toxin aflatoxin18,79. While fungi and bacteria share many of the same hallmark secondary metabolic pathways, they also feature distinctive enzymatic reactions such as the reducing iterative polyketide synthases (PKSs) that produce the cholesterol-reducing agent lovastatin80. With their larger genomes, fungi also encode many more BGCs than the most prolific bacteria. The fungus Aspergillus tanneri NIH1004 has 95 BGCs81, setting it up as the most fruitful amongst the fungi.
Long thought to be a uniquely microbial phenomenon, it is now becoming increasingly clear that BGCs are found throughout the tree of life (Box 1). Land plants dwarf all other organisms for known specialized metabolites (Figure 1d). Plant molecules, like the anticancer drug taxol, the plant hormone gibberellin or caffeine (which functions as an insecticide yet is best known as a constituent of coffee and other caffeinated drinks), dominate the literature on specialized metabolism with over 145,000 described molecules. Early experiments connecting plant chemistry and genes relied upon sequencing expressed sequence tag libraries and transcriptomes. In recent years, plant genomics has gained traction, revealing the genomic context of specialized metabolism. The triterpene thalianol in Arabidopsis was one of the first plant compounds for which it was shown that its encoding genes are chromosomally clustered82, yet in a manner very much unlike the bacterial BGCs. Genes within plant BGCs are typically not organized in tight operons but rather with large intergenic regions that can span up to a few hundred kb in stretches, and as such, genes are typically transcribed separately83. Recent plant omic studies have connected genes to the production of iconic opioid, cannabinoid, and vinca alkaloid plant molecules, leading to renewable fermentation opportunities for their robust production38,84,85.
The success of the plant community in connecting genes to specialized chemistry has opened the floodgates to other eukaryotic systems that each harbor distinctive chemistry. For instance, some of the most notorious environmental toxins are produced by diverse marine microalgae. Recently, a BGC was established in the diatom Pseudo-nitschia multiseries for the global production of the amnesic shellfish toxin domoic acid86. By contrast, dinoflagellates produce arguably the largest and most complex chemicals known from nature, polyether toxins such as brevetoxin and maitotoxin87. While biosynthesis genes have yet to be identified for these dinoflagellate compounds — perhaps due to their massive genomes that regularly exceed humans and assemble into liquid crystalline chromosomes88 — the recent assembly of the toxic ~6.4-Gb Amphidinium gibbosum draft genome revealed an abundance of suspected PKS and nonribosomal peptide synthetase (NRPS) genes89. On top of this, the recent reconstruction of hundreds of genomes of plankton species from metagenomic data provides a rich set of unexplored genomic data to mine for specialized metabolic diversity90.
Historically, the anthropocentric bias of biomedical research has led scientists to qualify metabolites isolated from many animals as distinct from bacterial, fungal and plant specialized metabolites. However, a more impartial perspective should recognize that many animal specialized molecules are chemically related to and perform functions similar to their non-animal counterparts. While in some cases, animal-derived specialized metabolites are biosynthesized by specialized microbiome members91,92, the biosynthetic capacities of the animal itself should not be underestimated. Humans, for instance, produce numerous steroid hormones such as estradiol, cortisol and aldosterone, the thyroid hormone triiodothyronine, and even the antiviral ribonucleotide 3ʹ-deoxy-3′,4ʹ-didehydro-CTP93. The recently discovered routes from bird94,95 and mollusc96,97 genomes to produce complex polyketides as well as a novel sesquiterpene biosynthetic pathway from flea beetles98 exemplify the chemical ingenuity of animals themselves in making important molecules key to their fitness and survival. In some cases, such pathways have been horizontally acquired from bacteria, as is evident for the β-lactam antibiotic biosynthetic genes found in the genome of the springtail Folsomia candida99,100, but in most documented cases mentioned above, their biosynthesis seems to have evolved independently, indicating that considerable quantities of distinct chemistry may be discovered though mining animal genomes.
Now that eukaryotic genome sequencing is becoming more routine, we anticipate that genome mining projects will soon extend to all organisms (Box 2). While there have been sporadic reports of specialized biosynthetic genes and gene clusters being functionally elucidated from, for example, the nematode Caenorhabditis elegans101, the fruit fly Drosophila melanogaster102 and the seaweed Digenea simplex103, large swaths of organisms such as arthropods, cnidarians and other invertebrates are understudied for their biosynthetic capacities yet well-known for their specialized chemistry.
Box 2: How much is there to mine?
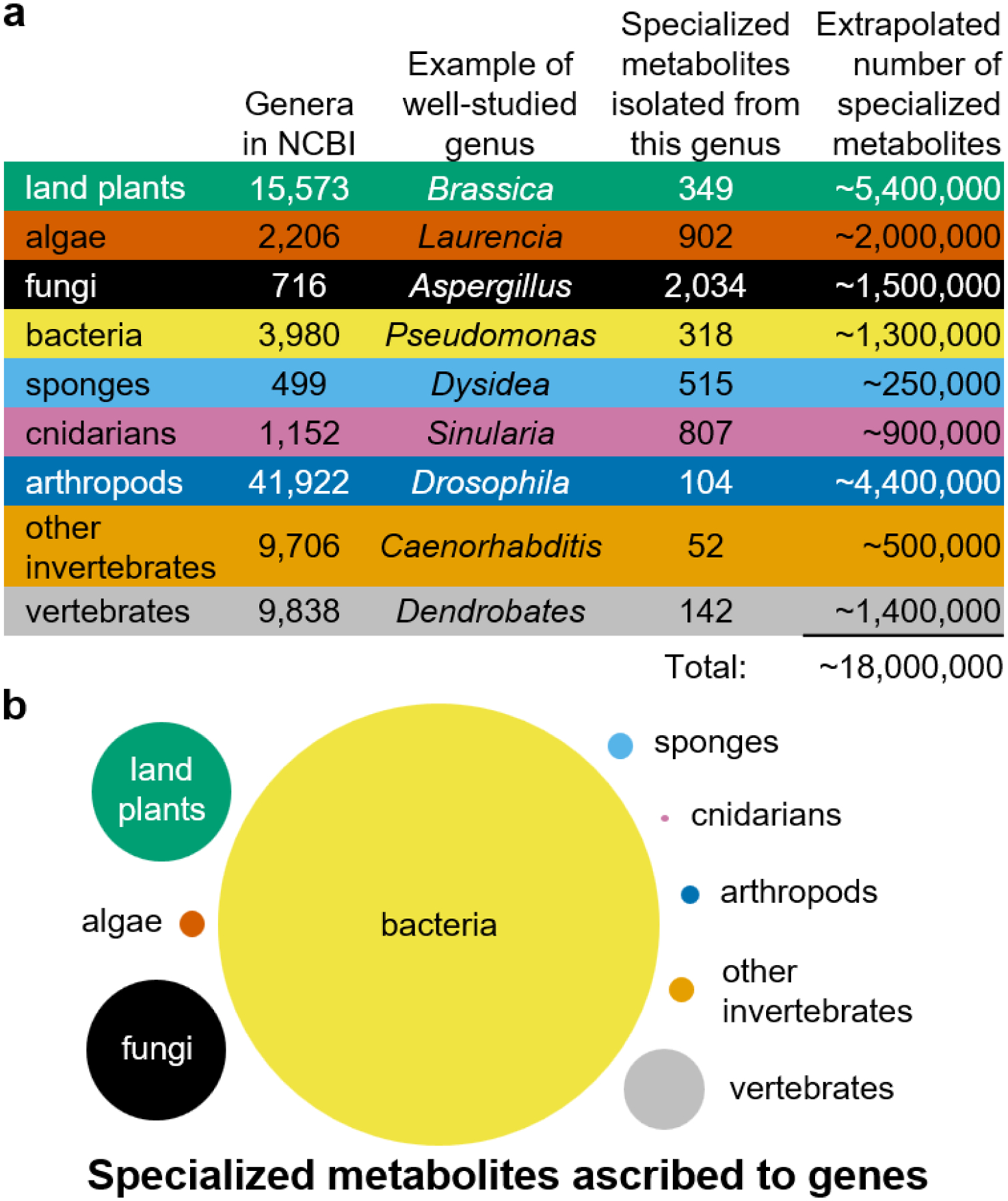
a: Estimating the total number of specialized metabolites by multiplying the number of specialized metabolites reported for a relatively well-studied genus — assumed to be representative — by the number of genera for the type of organism. These could be overestimates because genera may share specialized metabolites, or underestimates because more specialized metabolites may be discovered for the chosen genus or more genera may still be discovered. Number of specialized metabolites were sourced from Natural Product Atlas207 for Pseudomonas and Aspergillus, and from Dictionary of Natural Products for all other genera. This data considers only the isolation source, not whether the specialized metabolite was produced by the host or a microbial symbiont. b: Areas indicate relative numbers of specialized metabolites whose biosynthetic genes have been identified, based on estimates made by the authors.
Both the large diversity of molecules found in nature and the even larger diversity of biosynthetic genes found in genome sequences make it clear that the chemical and enzymological space available to genome mining is vast. Yet, it is difficult to gauge just how vast it is.
Focusing on possibly the most chemically diverse clade of microorganisms, the actinomycetes, Doroghazi et al. have claimed that sequencing a well-chosen set of only ~15,000 actinomycete genomes would reveal virtually all naturally occurring GCFs in this class of bacteria124. They based this statement on extrapolating a rarefaction curve of GCFs, in which sampling had been corrected for phylogeny within the limits of the dataset used. A subsequent study on the diversity of NRPS gene clusters, which included a larger number of genomes and used chemical structure predictions to support family assignments, indicated no signs of saturation around 15,000 genomes, however, suggesting that genome-encoded biosynthetic diversity may be larger than previously estimated, at least for this class of pathways158. Similarly, Schorn et al.208 revisited estimates of biosynthetic diversity based on a study of rare marine actinomycete genomes, which suggested that rarefaction analyses may be too conservative to estimate diversity across the biosphere, as they inherently do not take into account genomes from unsampled ecological niches and taxonomic subgroups.
A rough estimate of the total number of specialized metabolites employed by life can be made based on known biodiversity (Fig. 1b) and metabolic diversity (panel a and Fig. 1d): on the order of tens of millions. Contrasting this to the number of elucidated specialized metabolites (on the order of half a million) suggests we have merely scratched the surface of the biochemical diversity present in the biosphere. Studies on bacteria and fungi support this notion, showing that regardless of the rapid accumulation of known specialized metabolites and associated risks of rediscovery, the absolute numbers of structurally novel specialized metabolites discovered over the past 20 years has remained remarkably steady, at around 150–250 per year9,209.
While the estimates in panel a suggest there is great potential for the discovery of specialized metabolites throughout the whole tree of life, our understanding of their biosynthesis is heavily skewed towards bacteria (panel b), likely due to the greater availability of genomic data for bacteria (Fig. 1c). Even for the relatively well-studied specialized metabolism of bacteria, our understanding of culturable species dwarfs uncultured bacteria. This could be remedied by bringing more bacterial species into culture through new sampling or cultivation strategies210,211, or by expanding metagenomic studies of diverse environments globally, and in turn mining the resulting genomics data. Nevertheless, to spur our understanding of specialized metabolism throughout the whole tree of life, it will similarly be imperative to collect thorough genomic data for a wide variety of eukaryotic organisms.
How to mine — identifying and prioritizing candidates
A range of computational approaches has been developed to automatically identify the sets of genes that encode specialized metabolic pathways across genome sequences (Figure 2). Many of these approaches have originally been developed for bacteria (and sometimes for fungi and plants), but the principles employed have the potential to be extended to other life forms. Below, we review these methodologies and the taxa they support, and what would be required to extend them into new taxonomic spaces.
Figure 2.
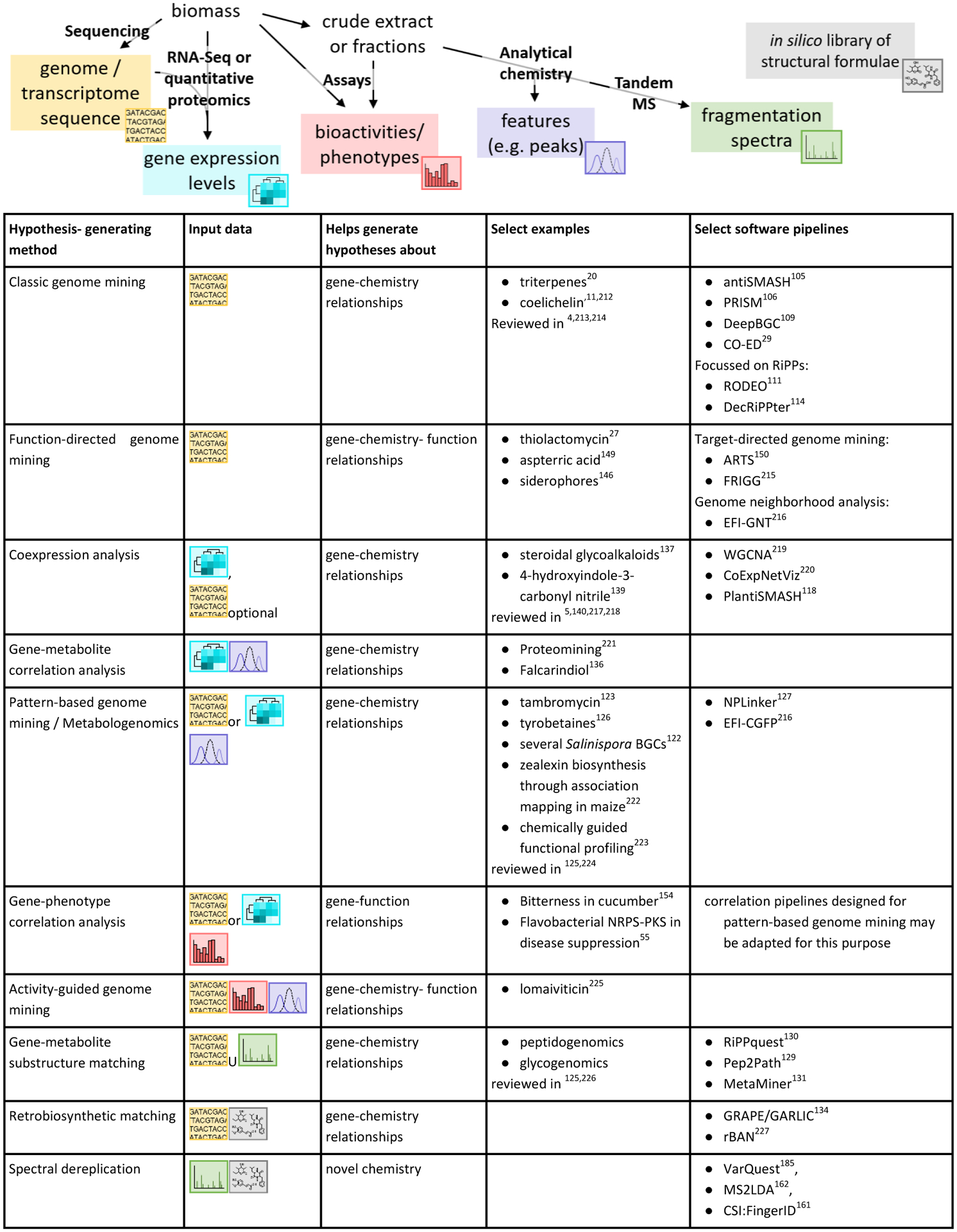
Overview of genome mining technologies that combine genome sequence data with gene expression levels, metabolomic data, biological activity or phenotypic data, and chemical structure data. Each combination has its own strengths and may allow generating hypotheses focused on finding an unknown biosynthetic pathway for an important known molecule, discovering new metabolites with desired biological activities, or identifying potential links between metabolites and the genes and gene clusters that likely encode their biosynthesis.
The physical clustering of enzyme-coding genes in BGCs greatly facilitates the identification of biosynthetic pathways. While BGCs are highly variable in terms of gene content and often strain-specific due to their rapid evolution and frequent horizontal gene transfer104, they often do possess common properties in the form of enzyme families that are responsible for the catalysis of biochemical reactions central to the biosynthesis of entire specialized metabolite compound classes. This feature has made it possible to largely automate the identification of BGCs in genomes. Widely used software tools such as antiSMASH105 and PRISM106 employ profile Hidden Markov Models (pHMMs107) of protein domains to identify gene combinations encoding enzyme families that are signatures for specific pathway types. While both these tools generally provide very similar results, development of antiSMASH has focused more on functional and comparative analyses, while PRISM has specialized in combinatorial predictions of chemical structures that can be used for automated matching with mass-spectral data. The use of pHMMs is very reliable for identifying BGCs encoding many well-established types of biosynthetic machinery such as PKSs, NRPSs and known classes of RiPPs, but risks overlooking less studied and wholly novel classes of BGCs. Probabilistic BGC prediction methods such as ClusterFinder108 (which is also integrated into antiSMASH) and DeepBGC109, or comparative genomics approaches that identify metabolism-associated nonsyntenic blocks of genes between genomes are more likely to detect non-standard BGCs, but have higher false-positive rates. In addition, for RiPPs, specialized tools have emerged for the identification of BGCs encoding the production of distant members of known classes or members of altogether novel classes. Some of these, like BAGEL110, use pHMM-based detection techniques similar to those seen in antiSMASH and PRISM. Others either make use of bait-based approaches (using specific query enzymes to identify loci that contain homologues of it)111,112 or use machine-learning approaches to identify potential precursor-peptide-encoding genes, the hits of which can be prioritized using either metabolomics-based matching113 or comparative genomics to identify operons that are taxon-specific and are therefore deemed to encode a specialized metabolic function114. For publicly available genomes, BGCs identified using antiSMASH can be interactively browsed in online databases such as IMG-ABC115 and antiSMASH-DB116. Recently, it has become clear that in plants, specialized metabolic pathways are sometimes encoded by BGCs83 (Box 1), and specific algorithms have been devised for their detection117,118. However, there are also many examples of pathways in plants that are encoded by sets of genes distributed across multiple chromosomes instead of being located in a single gene cluster. When extending genome mining approaches to unexplored parts of the tree of life, it remains to be seen to what extent genes in these taxa will be clustered. Some recent evidence suggests that the phenomenon of gene clustering also occurs in protists; for example, the domoic acid biosynthetic pathway in the diatom Pseudo-nitzschia multiseries was shown to be encoded by a four-gene gene cluster86. However, gene cluster detection algorithms originally devised for bacteria may require considerable optimization to make them effective for studying protist or animal genomes. Efforts to adapt antiSMASH for detecting BGCs in plants in a new tool called ‘plantiSMASH’118 showed that, for this to be effective, new libraries of pHMMs focused on plant enzymology needed to be constructed, and the algorithm had to be adjusted to account for the considerably larger (and more variable) intergenic regions found in plant genomes106.
Computational predictions often lead to an overabundance of candidate specialized metabolic pathways that could be investigated, necessitating prioritization in some way. Given that the chemical structures of hundreds of thousands of specialized metabolites have been elucidated, a considerable number of these will be responsible for the biosynthesis of known molecules or their closely related variants. Hence, dereplication is required to assess whether molecules and biosynthetic genes are novel compared to those ones that have been discovered and characterized earlier. The simplest way of doing this is based on sequence information: if a BGC of interest is highly similar in sequence to a gene cluster that has been experimentally linked to a known specialized metabolite, it likely codes for the production of the same molecule. In 2015, a community effort established the Minimum Information about a Biosynthetic Gene cluster (MIBiG)75, a data standard and online repository for depositing annotations and metadata on BGCs for which a product has been identified. The antiSMASH pipeline for BGC identification automatically compares each identified BGC against this repository of ~2,000 BGC of known function. When studying large numbers of genomes at once, BGC sequence similarity networks108 can be utilized to identify ‘gene cluster families’ (GCFs) that cluster together with MIBiG reference clusters. The BiG-SCAPE software framework automates the process of generating these networks and facilitates their interactive exploration, which makes it possible to quickly explore the biosynthetic diversity within hundreds or even thousands of prokaryotic genomes at once119. It remains to be seen to which extent this technology is universally applicable across the tree of life. For example, it was recently shown that plant triterpene biosynthetic loci may be highly similar in terms of domain composition, while having evolved independently and leading to divergent chemical outcomes120. These analyses suggest that at least certain categories of biosynthetic pathways in plants through combinatorilization of a limited set of enzyme families, of which the members can have different catalytic activities or regioselectivities. Hence, for pathway types and organisms in which gene evolution is largely decoupled from gene cluster evolution, more automated phylogenetic methods need to be developed to perform comparative analysis at the gene level as well as the gene cluster level. Beyond plants, it should not be excluded that this is the case for other eukaryotic branches of the tree of life as well.
Identification, dereplication and prioritization workflows can be further improved by combining the information from the genome sequence with data obtained from analytical techniques121. For instance, if the same or similar molecules are produced by different organisms, they can be expected to harbor the same or similar biosynthetic genes. Pattern-based genome mining122 (also known as metabologenomic correlation analysis123,124, Figure 3a) correlates patterns of spectral data (most commonly liquid chromatography (LC) mass spectrometry (MS) features) to the presence of homologous biosynthetic genes across strains. This approach (reviewed in detail here125) has mostly been pioneered in bacteria, for which sufficiently large numbers of genomes and metabolomes can be obtained. In a recent metabologenomic correlation study, gene cluster families (GCFs) were linked to a MS network, leading to the discovery of the tyrobetaine metabolites126. Recently, the mathematics behind the association scoring was improved and formalized in a software tool called NPLinker127. The advantage of this technology is that no prior knowledge on biosynthetic mechanisms is required to link molecules to gene clusters, as it is purely based on correlations. A strategy that establishes genomic–metabolomic co-occurrence patterns has great potential to mine the genomes of poorly studied organisms, even when virtually nothing is known about a taxon’s enzymology.
Figure 3. Linking genes to molecules using metabolomics and transcriptomics.
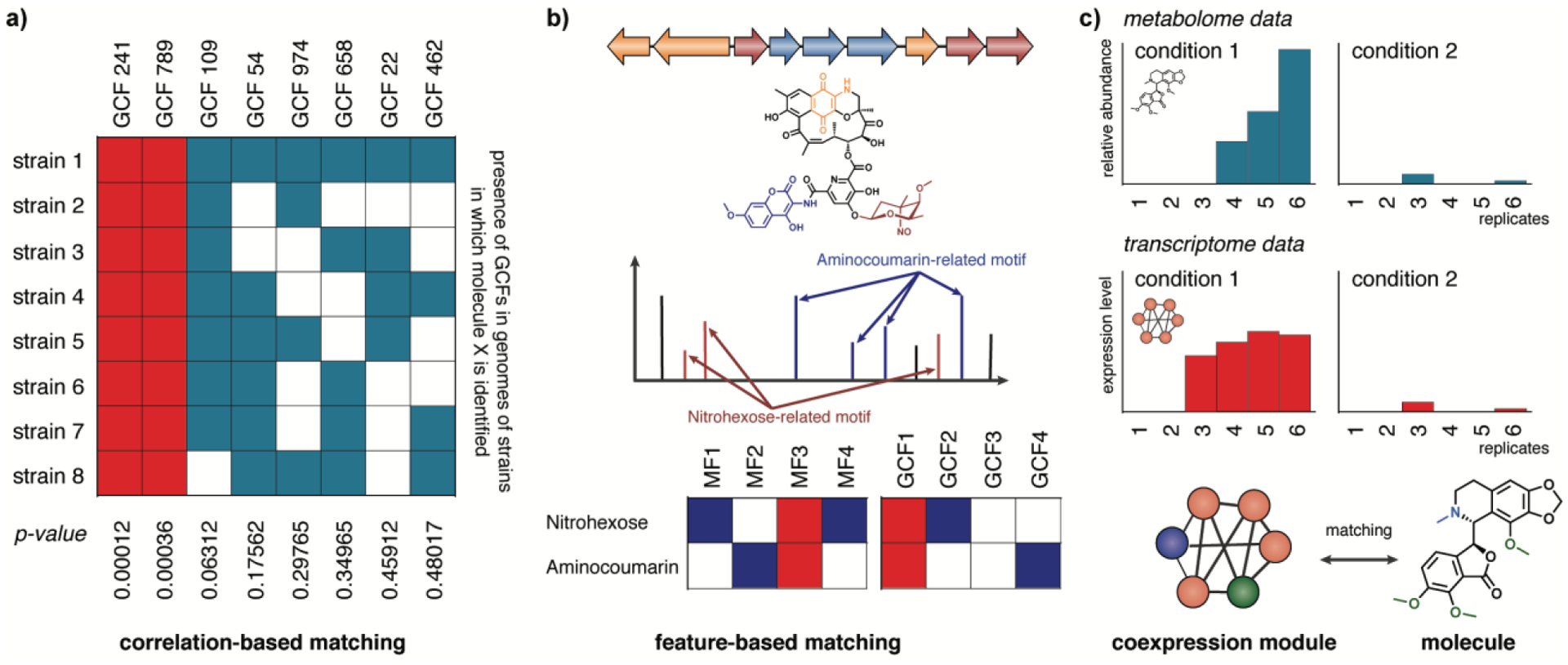
Several approaches have been developed to link metabolites to genes and gene clusters encoding their biosynthesis. a) In bacteria, pattern-based genome mining approaches have been developed that match families of molecules (related by spectral similarity) to gene clusters families (GCFs, related by sequence similarity) through metabologenomic correlation123, which identifies which GCFs co-occur strongly in the same strains where a given metabolite is observed. b) Molecules can also be connected to genes and gene clusters through feature-based matching, in which chemical features (substructures and modifications that are either manually annotated or identified using algorithms that identify motifs in MS/MS data) are linked to genes and gene modules that are known to be responsible for the biosynthesis of such features. c) Transcriptomic data can also be used to identify potential biosynthetic pathways for a molecule of interest by, for example, identifying modules of coexpressed genes whose expression correlates with the presence of a given metabolite across a range of divergent conditions (for example, different biological stresses136).
Another approach that also harnesses analytical chemistry to improve genome mining predictions is the correlation of mass shifts in tandem MS fragmentation patterns to a BGC’s bioinformatically predicted building blocks (Figure 3b). At first, semi-manual approaches were developed that allowed matching of peptides (peptidogenomics14) and glycosylated specialized metabolites (glycogenomics128) to BGCs. More recently, this matching has been automated for peptides in algorithms like Pep2Path129, RiPPquest130 and MetaMiner131. The latter algorithms, which focus on RiPPs, could also be very relevant for finding novel peptidic metabolites in uncharted taxa, as recent evidence is emerging that RiPPs are produced not only by bacteria, but also by fungi132, plants48 and animals133. Going forward, the bigger challenge will be to extend these approaches beyond peptides to specialized metabolites in general125.
Instead of partial structural information from mass spectra, fully elucidated chemical structures can also be used to identify new biosynthetic pathways and aid in dereplication. There are many specialized metabolites for which the chemical structure is known but the biosynthetic genes are not. For drug discovery purposes, this may pose a major problem, given the considerable effort wasted elucidating the chemical structure of a known molecule. Recently, an innovative method called GRAPE/GARLIC was established134 to tackle the puzzle that is ‘connecting genes to molecules’ for polyketides and nonribosomal peptides in an automated fashion: by breaking down known specialized metabolite structures into their biochemical building blocks and retro-biosynthetically matching these with building blocks predicted to be incorporated into molecules based on BGC sequence information, the authors were able to identify thousands of putative matches between gene clusters and molecules. Of around 16,831 BGCs, around 2,500 had best matching scores to reference molecules that were so low that they are very likely to encode the biosynthesis of novel products. While this may seem relatively little, one should consider that the remaining set of ~14,000 BGCs is enriched with many near-copies of BGCs from highly studied taxa for which large numbers of genomes have been sequenced. The retro-biosynthetic principle, while useful, seems largely limited to bacterial polyketides and nonribosomal peptides, and expanding retro-biosynthetic algorithms to other life forms will require considerable expansions of our knowledge of their biosynthetic routes. Training more generic models for enzymatic mechanisms based on large-scale experimental data are needed here, as well as high-throughput assays on ‘enzymatic dark matter’ from unexplored taxa to provide the required training data for such models.
The presence of specialized metabolites can also be correlated to biosynthetic genes’ transcriptional levels in different conditions or across different tissues (Figure 3c). For example, the biosynthetic pathway for ingenol mebutate from Euphorbia plants was unraveled by identifying members of relevant enzyme families that were highly expressed in seeds135. Similarly, another recent study analysed the production of the defense metabolite falcarindiol by tomato across seven different biotic stress treatments, to identify relevant enzyme-coding genes that were upregulated in conditions when increased amounts of the molecule were observed136. This principle seems universally applicable and is widely useful for accelerating genome mining efforts.
Indeed, in plants, coexpression analysis has already been frequently used with success to identify genes that show similar expression patterns across a large number of samples, within the same species or even cross-species137. Often, this is done by using one or more ‘bait’ genes, which are predicted or even known to belong to a pathway of interest, to recruit additional members of that pathway138,139. However, unsupervised approaches are also being developed, which can be used to predict candidate pathways without prior knowledge. These methods rely on detecting coexpressed modules of genes given a set of transcriptomic samples, a procedure for which a range of algorithms is available140. Recently, the identification of coexpression modules was shown to effectively and comprehensively retrieve genes implicated in methionine-derived aliphatic glucosinolate biosynthesis in Arabidopsis thaliana and Brassica rapa141. A key factor in the success of this study was the use of a graph clustering method that allows modules to overlap in their gene content, which makes sense given the fact that plant specialized metabolic enzymes are often promiscuous and may have dual functions in multiple pathways. In general, the advantage of coexpression approaches seems to be that they are generally applicable, also when the genes encoding a pathway of interest are only partially clustered or not clustered at all. Moreover, for eukaryotes with complex genomes that are hard to assemble contiguously, coexpression-based approaches could also be performed on the basis of fragmented genome assemblies or transcriptome assemblies. A challenge for these approaches is how to find the right combination of conditions that distinguishes expression patterns of a pathway of interest most effectively from those of other pathways, without requiring massive amounts of expensive transcriptome sequencing. One possible strategy to do this would be to first generate (targeted or untargeted) metabolome data for a variety of samples, before choosing which samples are prioritized for RNA sequencing. Alternatively, integrative approaches could be developed that leverage structural information from metabolome data (for example, mass shifts and predicted substructures) to help prioritize which sets of coexpressed enzyme-coding genes are most likely responsible for the production of a given metabolite.
How to mine — function-first approaches
No matter how powerful modern genome mining approaches are to identify the genomic basis for chemical diversity, these methods are relatively blind and untargeted — usually, a molecule’s physiological and ecological importance is only considered at the very end, after structural characterization and elucidation of its biosynthetic pathway. Function has traditionally been investigated only in a very narrow sense, that is, by considering hits in activity assays relevant to human health and prosperity, to the neglect of physiological and more subtle ecological functions. Functions such as the arthropod-attracting capabilities of geosmin and 2-methylisoborneol terpenoids from streptomycete bacteria142 or the conferring of heat stress resilience by flavonols by regulating levels of reactive oxygen species143 were only identified decades after these metabolites were structurally characterized. To truly deepen our understanding of the fundamental roles of these molecules in biology and to allow for more targeted approaches to leverage them in, for example, drug discovery, it will be crucial to devise methods to help prioritize biosynthetic pathway candidates based on the specialized metabolite’s predicted function.
A good example of such a ‘function-first’ method, which has already gained traction, is based on the co-localization of genes within the same BGC that are indicative of function. For example, the colocalization of iron transport genes with biosynthetic genes led to the discovery of siderophore molecules, such as coelichelin and salinichelins in bacteria144, and sideretin from plants145 (and this principle has recently been generalized146). The colocalization of resistance genes or duplicated genes resembling antimicrobial targets within BGCs offers a more generalizable approach to the discovery of bioactive molecules with specific cellular targets (Figure 4a). This approach, called target-directed genome mining, was first validated with the rediscovery of the thiolactomycin antibiotics as fatty acid synthase inhibitors from orphan bacterial BGCs that contain an open reading frame predicted to be a resistance gene27, associated with target modification of the FabF fatty acid ketosynthase. Newer studies colocalizing putative target-modifying resistance genes with BGCs to identify compounds with activities against the resistance gene target include the proteasome inhibitor fellutamide B from the fungus Aspergillus nidulans147 and topoisomerase inhibitors pyxidicylines from the myxobacterium Pyxidicoccus fallax An d48148. A clever twist on this resistance gene-guided approach led to the discovery of the fungal sesquiterpenoid aspterric acid as a potent herbicide, by deploying the fungal dihydroxy-acid dehydratase self-resistance gene as a transgene to render plants resistant to aspterric acid149. In order to automate the resistance-based genome mining procedure, a web service called the “Antibiotic Resistant Target Seeker” (ARTS) was developed to identify BGCs containing likely self-resistance genes, suggesting they code for the production of specialized metabolites with specific biological targets150. Intuitively, the approach is probably applicable to any organisms in which biosynthetic pathways are genomically clustered, as long as there is sufficient selective pressure for the resistance genes to co-cluster (through facilitating co-expression and co-inheritance with the pathway). While resistance-based genome mining is a breakthrough as a key function-first strategy, the vast majority of BGCs do not contain self-resistance genes or other genes that unambiguously indicate a specific function. Hence, there is a great need for development of additional strategies to generate hypotheses about the function of the molecules produced by the remaining majority of pathways. We believe that, again, the essence of these approaches will be in combining genomics with other types of data. Below, we outline three possible ways in which this could be achieved.
Figure 4. Function-first genome mining approaches.
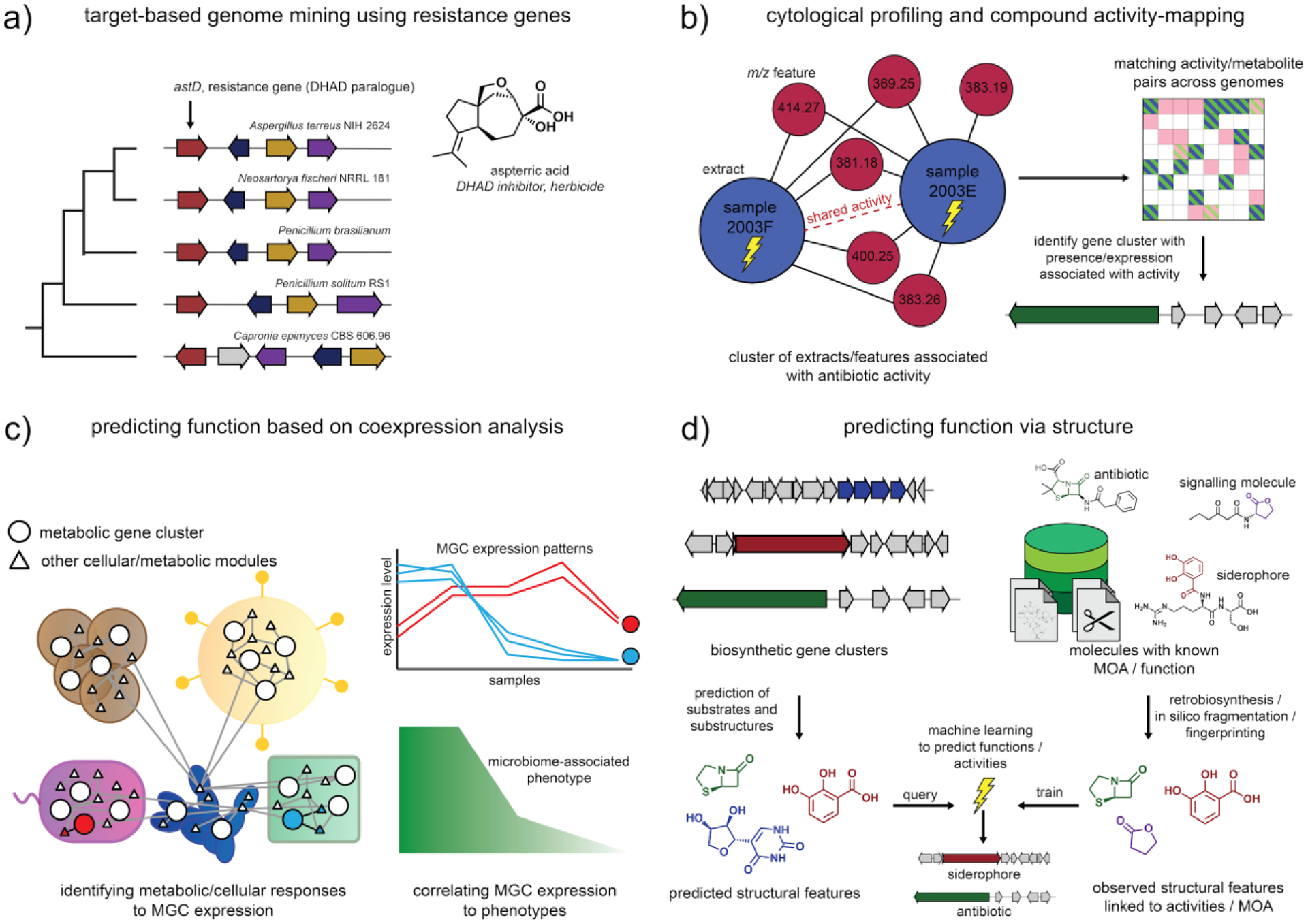
In order to more effectively identify molecules with desired activities, function-first genome mining approaches have been and are being developed. a) In target-based genome mining approaches, self-resistance genes are identified that genomically cluster with the biosynthetic genes. Such self-resistance genes are often resistant copies of a housekeeping gene whose protein product is targeted by the metabolite biosynthesized from the pathway. This provides a way to directly predict the mechanism of action for metabolic products of a subset of gene clusters. b) Cytological profiling can be used to identify the effects that metabolic extracts have on certain cell lines, and compound activity-mapping can identify which underlying mass-spectral features are likely responsible for activities that are shared between extracts. The activities and/or metabolites can then be matched to the presence or expression of genes and gene cluster to identify a candidate biosynthetic route towards the underlying molecule. c) Functions of products of biosynthetic genes and gene clusters can be predicted by looking for coexpression with other genes in the same organism (predicting function based on the guilt-by-association principle) or across organisms (identifying the potential effect that a pathway has on other organisms or on a microbiome-associated phenotype). d) Structural features and substructures that are likely part of the metabolic product of a gene cluster can be predicted in silico; sometimes, these substructures are diagnostic for a certain mechanism of action or biological activity, and machine learning algorithms can be trained to predict these activities based on sets of structural features.
A first possibility would entail correlating genomic information to bioactivities displayed by extracts (Figure 4b). There has already been some success in correlating bioactivities of extracts as determined by cytological profiling151 to untargeted metabolomics of the same extracts using a technique called Compound Activity Mapping152, facilitating the discovery of the quinocinnolinomycins, a new family of specialized metabolites that cause endoplasmic reticulum stress. The obvious next step will be to combine this with genomic and/or transcriptomic data to immediately identify the genes responsible for an activity of interest. Also, when cytological profiling does not give immediate insights into the mode of action of a molecule, it could be complemented with transcriptome analysis of the target cells during exposure. Indeed, machine learning methods have recently been devised that predict pharmacological properties of drug molecules, directly related to the mechanism of action, based on large-scale transcriptional response data153. In principle, this approach would be applicable to any life forms, for which extracts can be made, including plants, many protists, and invertebrates. This could also be done through genome-wide association studies that map phenotypes to genetic variation within a species, as has been successfully practiced to discover the cucurbitacin gene cluster responsible for the bitter taste in cucumber154.
A second way to perform function-first genome mining would be to study the effects of the expression of BGCs on other community members within their native ecosystem, and, optionally, how they relate to emergent properties of such an ecosystem (Figure 4c). This applies primarily to microbial ecosystems and microbiota associated with plant or animal hosts. For example, metatranscriptome data from soil microbial communities were recently used to investigate the ecological roles of BGCs from novel bacterial clades identified through metagenomic binning; coexpression of BGCs with iron starvation response genes or antimicrobial resistance genes thus indicated roles for their products as siderophores or antimicrobials155. This concept could be extended by also looking at coexpression across species, that is, correlating the expression of putative antibiotic biosynthesis BGCs with stress responses in other organisms in the community to identify the likely target organisms. The expression of specific BGCs could also be correlated to microbiome-associated phenotypes156 that a community confers to its host, such as disease suppression or stress resilience, to identify which molecules are likely to be responsible for mediating these phenotypes. In host organisms, such as plants and animals, expression of particular biosynthetic pathways can also be linked to functions by studying the effects on the microbiome composition; for example, a recent study linked specific triterpene pathways to either the promotion or inhibition of specific rhizosphere microbiome community members, which highlighted their specific roles in microbiome modulation157.
A third strategy for function-first genome mining would be combining (sub-)structure prediction from sequence with structure-based prediction of biological activities and macromolecular targets (Figure 4d). Both of these prediction tasks are currently highly prone to error, but significant progress is being made on both fronts, so that a robust platform may become a reality in the not-too-distant future. Several tools are currently emerging that can predict the core scaffolds of key classes of specialized metabolites from sequence information with increasing accuracy and detail105,106,158,159, and several efforts are underway to complement these with additional predictions of tailoring and cyclization reactions106,160. Also, genome-based structure predictions could be integrated with metabolomics-based (sub)structure predictions161,162, which could confirm or guide routes through biochemical reaction space. Based on all these developments, considerable improvements in specialized metabolite structure prediction from genome and metabolome data can be expected in the near future. At the same time, within the field of computational drug discovery, methods are emerging that allow predicting macromolecular targets of drug molecules based on their chemical structures163. For example, the algorithm SPIDER dissects specialized metabolites into pharmacophore-sized fragments and predicts which proteins a compound targets by comparison to a library of 13,695 chemical structures of molecules of known function from the Collection of Bioactive Reference Analogs (COBRA)164. This method successfully predicted polypharmacological features of the macrolide archazolid A. Similarly, in another recent study, a deep learning model was trained that could successfully predict antibiotic activities of molecules with only limited chemical similarity to those used for training165. When, in the future, both sequence-based metabolite structure prediction and structure-based macromolecular target prediction become increasingly accurate, they could be coupled together to predict biological targets directly from gene cluster sequences. The recently published PRISM4 pipeline provides a first step in this direction, using support-vector machines to predict the activities of the molecular products of gene clusters based on their predicted structures166. For the moment, this strategy is likely to be relevant mostly for bacteria and fungi, and to some extent for plants; however, when synthetic biology approaches60 and in vitro expression systems167 increasingly allow experimental characterization of large sets of enzymes from animals and protists, opportunities will likely emerge to apply this strategy in these taxa as well.
Altogether, the biosynthetic gene identification and prioritization is moving towards the incorporation of an increasingly large number of different data types. Moving forward, the pioneering approaches will likely harness an even larger number of data types simultaneously.
How to mine — testing candidates
Fundamentally, there are three types of approaches to identify the metabolic product(s) of a BGC: 1, heterologous expression in a model organism; 2, genetic manipulation of the native host; and 3, in vitro reconstitution.
Heterologous expression involves the cloning (also known as ‘capture’) of a BGC into a plasmid, cosmid or artificial chromosome, possible manipulation of the BGC, transfer into a genetically-tractable heterologous host, and testing for the presence of metabolic products compared to the unmodified heterologous host168–170. When possible, heterologous expression is a highly desired approach, because it enables both facile scale-up of metabolite production for structural elucidation and biological testing, and manipulation of the captured BGC for biosynthetic investigations and analog production. The large size of many BGCs has spurred the development of cloning methods that can capture BGCs directly from genomic DNA, such as transformation-associated recombination (TAR) in yeast171,172, Linear-Linear Homologous Recombination (LLHR) in E. coli173, or PfAgo-based artificial restriction enzymes in vitro174. One benefit of these PCR-free techniques is that it avoids mutation of the BGC, making sequence verification unnecessary. BGCs can also be cloned and assembled using PCR-based techniques, but since sequence verification of large BCGs by Sanger sequencing can be a bottleneck, doing so using next-generation sequencing technologies175 will likely gain popularity.
Heterologous expression has some notable potential challenges: promoters and ribosome-binding sites (RBSs) may not be recognized, genes may require RNA splicing, proteins may require chaperones, post-translational modification or transport to organelles, required metabolic precursors or cofactors may not be present, or the heterologous pathway could encounter metabolic bottlenecks due to non-optimal enzyme stoichiometry. If the pathway’s reactions are impeded to different extents, heterologous production could result in the production of metabolic intermediates or shunt products instead of the “true” specialized metabolite. Conventional wisdom states that employing heterologous hosts that are phylogenetically close relatives to the organism from which the BGC originates improves the chances of success, but exceptions to this dogma are known176. Techniques such as CRAGE177 aim to streamline testing a BGC in a multitude of heterologous hosts, increasing the chances of at least one succeeding. Research dedicated to developing genetic toolkits for a variety of organisms will be crucial to streamline the heterologous expression of BGCs from organisms not closely related to classic model organisms.
Synthetic biology approaches aim to circumvent the aforementioned challenges associated with heterologous expression by ‘refactoring’ the candidate biosynthetic genes and/or engineering heterologous hosts (‘chassis’) optimized for heterologous expression of biosynthetic pathways. Chassis have been developed that provide metabolic precursors and post-translational modifications required for specific classes of specialized metabolism or to inactivate competing metabolic pathways. Refactoring usually entails bringing candidate biosynthetic genes under the control of well-characterized promoters and RBSs, elimination of introns and organellar targeting signals, and codon optimization60. However, gaps in our understanding of these cellular processes—for instance, how codon optimization affects gene expression and protein folding—still limit the rationality of our refactoring efforts. Several streamlined workflows for refactoring candidate biosynthetic genes have been described178,179. The use of combinatorial libraries180 and independently tunable promoters181 can help optimize the stoichiometry of biosynthetic genes in vivo. While fully synthesizing refactored BGCs de novo instead of refactoring captured BGCs is currently still prohibitively expensive for all but the best-funded projects, we expect this practice to become widespread as gene synthesis costs continue to decline.
Alternatively, the candidate gene(s) can be inactivated or repressed in their native host, followed by testing for the loss of, or decrease in the quantity of, a metabolite compared to the wild-type host182. To more thoroughly establish the gene–metabolite link, ideally a genetic complementation experiment should also be carried out182. The biggest drawback to this approach is that it can be difficult or impossible to manipulate genes in non-model organisms, but thankfully this situation is improving thanks to the broad host range of CRISPR–Cas9 technologies. The emergence of CRISPR–Cas9-based ‘microbiome editing’ technologies183,184 has even made it possible to knock out genes in specific members of a complex microbiome.
Reconstitution of the pathway in vitro provides some advantages orthogonal to the in vivo approaches above, such as allowing for easier identification of pathway intermediates, determination of enzyme kinetics and substrate specificities, and quick optimization of the pathway enzyme stoichiometry167. However, in vitro reconstitution can be challenging if the metabolic precursor(s) or order of the enzymes in the metabolic pathway is unknown, or if any of the enzymes are insoluble, unstable or cannot be purified.
Once a metabolite has been identified as being the product of the candidate genes, its identity will need to be established. Depending on the method that was used to select the candidate genes, one may already have a hypothetical structure or chemical class. The act of ‘dereplication’ seeks to quickly identify whether the metabolite is, or is closely related to, any known molecules. Some currently popular approaches to dereplication are based on MS-MS spectral networking (such as GNPS8), MS-MS spectral-substructure matching (such as VarQuest185, MS2LDA162 and CSI:FingerID161) and NMR spectral clustering (such as SMART186), but it is worth remembering that dereplication tools are only as effective as the databases/training data that underlie them. If the molecule is likely novel, structural elucidation will be necessary. Nowadays, this is most commonly achieved through 2D-NMR techniques, with a slow uptick in the application of computer-assisted structure elucidation187 (CASE) technologies. X-ray crystallography (occasionally aided by the crystalline sponge method188), and more recently, microcrystal electron diffraction189, can also provide important insights into challenging structural elucidation problems.
Finally, some recent studies circumvent biological experimentation altogether by chemically synthesizing the predicted products of a BGC190–193. BGCs for RiPPs and non-ribosomally synthesized peptides are particularly amenable to this approach, as the structures of their products are highly predictable and their production can be streamlined through solid-phase peptide synthesis. Although doubt about the true identity of the BGC’s product remains, this approach has yielded molecules with promising biological activities190–192.
Conclusions and future perspectives
The study of the chemistry of life has been brought to a next level by genome mining technologies initially developed in microorganisms. Now that large-scale genome sequencing is expanding to all branches of the tree of life, there is a great opportunity to port and extend genome mining technologies to other life forms and engage in truly global studies of life’s chemistry. At the same time, the microbial field has much to learn from scientists studying humans and mammals, who have been very effective at identifying physiological roles of mammalian specialized metabolites, whereas microbiologists have perhaps focused too much on metabolite functions restricted to inhibiting or killing other organisms. Additionally, plant biologists’ extensive experience using gene expression analysis to link genes to molecules and identify their functions may become incredibly useful to the microbial field to acquire deeper perspectives into the physiological roles of many metabolites that have appeared ‘inert’ for so long. Finally, protists and invertebrates provide an immense uncharted biological diversity that is mostly untapped and likely to yield numerous new and surprising findings. All in all, great potential presents itself in unifying these diverse scientific communities to find common ground between molecules and genes that may have seemed unrelated for so long. This will facilitate arriving at a deeper fundamental biological understanding of the ecological and physiological roles of life’s chemistry, and more effectively leveraging it for the common good in medicine, agriculture and nutrition.
Acknowledgements
This work was supported by the US National Institutes of Health (F32-GM129960 to T.dR. and R01-GM085770 to B.S.M.) and European Research Council Starting Grant 948770-DECIPHER (to M.H.M.). We thank members of the Moore and Medema labs for helpful discussions.
Glossary
The terms ‘secondary metabolites’, ‘natural products’ and ‘specialized metabolites’ are often used interchangeably. Below we attempt to delineate the differences in how we use these terms:
- Secondary metabolite
A metabolite that is not strictly required for growth and development, as opposed to a primary metabolite; often important for the survival of an organism in its environment
- Natural product
A small molecule originating from a living organism or natural source that is often prized for its medicinal properties or other biological activities of utility to humanity
- Specialized metabolite
A natural compound of limited clade- or niche-specific distribution with a specialized role in ecology or physiology
Other terms:
- Biosynthetic gene cluster
(BGC) A set of genes that is physically collocated on the chromosome and together encodes the production, regulation and transport of one or more specific metabolites
- Gene cluster family
(GCF) A set of similar biosynthetic gene clusters across strains or species, the members of which are responsible for the production of the same or very similar metabolites
- RiPP
Ribosomally synthesized and post-translationally modified peptide, biosynthesized through the action of tailoring enzymes on a ribosomally-translated precursor peptide
- Polyketide synthase
(PKS) Enzyme involved in the biosynthesis of polyketide metabolite; some form modular assembly lines of multidomain proteins, while others act as stand-alone enzymes
- Nonribosomal peptide synthetase
(NRPS) Enzyme involved in the polymerization of amino acids or other organic acids into peptide metabolites without involvement of the ribosome
- Profile Hidden Markov model
A computational model, trained on a multiple-sequence alignment of a protein family, used to assess whether other proteins are also part of (or related to) this family
References
- 1.Davies J Specialized microbial metabolites: functions and origins. J. Antibiot 66, 361–364 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Chevrette MG et al. Evolutionary dynamics of natural product biosynthesis in bacteria. Nat. Prod. Rep 37, 566–599 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Erb M & Kliebenstein DJ Plant Secondary Metabolites as Defenses, Regulators, and Primary Metabolites: The Blurred Functional Trichotomy. Plant Physiol 184, 39–52 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziemert N, Alanjary M & Weber T The evolution of genome mining in microbes - a review. Nat. Prod. Rep 33, 988–1005 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Medema MH & Osbourn A Computational genomic identification and functional reconstitution of plant natural product biosynthetic pathways. Nat. Prod. Rep 33, 951–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keller NP Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol 17, 167–180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lockermann G Friedrich Wilhelm Serturner, the discoverer of morphine. Journal of Chemical Education 28, 277–279 (1951). [Google Scholar]
- 8.Wang M et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol 34, 828–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pye CR, Bertin MJ, Lokey RS, Gerwick WH & Linington RG Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. U. S. A 114, 5601–5606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentley SD et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Lautru S, Deeth RJ, Bailey LM & Challis GL Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat. Chem. Biol 1, 265–269 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Lin X, Hopson R & Cane DE Genome mining in Streptomyces coelicolor: molecular cloning and characterization of a new sesquiterpene synthase. J. Am. Chem. Soc 128, 6022–6023 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corre C, Song L, O’Rourke S, Chater KF & Challis GL 2-Alkyl-4-hydroxymethylfuran-3-carboxylic acids, antibiotic production inducers discovered by Streptomyces coelicolor genome mining. Proc. Natl. Acad. Sci. U. S. A 105, 17510–17515 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kersten RD et al. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat. Chem. Biol 7, 794–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Escribano JP et al. Structure and biosynthesis of the unusual polyketide alkaloid coelimycin P1, a metabolic product of the cpk gene cluster of Streptomyces coelicolor M145. Chem. Sci 3, 2716 (2012). [Google Scholar]
- 16.Cruz-Morales P et al. Phylogenomic analysis of natural products biosynthetic gene clusters allows discovery of arseno-organic metabolites in model streptomycetes. Genome Biol. Evol 8, 1906–1916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malpartida F & Hopwood DA Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature 309, 462–464 (1984). [DOI] [PubMed] [Google Scholar]
- 18.Smith DJ, Burnham MKR, Edwards J, Earl AJ & Turner G Cloning and heterologous expression of the penicillin biosynthetic gene cluster from Penicillium chrysogenum. Nature Biotechnology 8, 39–41 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Feitelson JS, Malpartida F & Hopwood DA Genetic and biochemical characterization of the red gene cluster of Streptomyces coelicolor A3(2). J. Gen. Microbiol 131, 2431–2441 (1985). [DOI] [PubMed] [Google Scholar]
- 20.Fazio GC, Xu R & Matsuda SPT Genome mining to identify new plant triterpenoids. J. Am. Chem. Soc 126, 5678–5679 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Bergmann S et al. Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat. Chem. Biol 3, 213–217 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Franke J, Ishida K & Hertweck C Genomics-driven discovery of burkholderic acid, a noncanonical, cryptic polyketide from human pathogenic Burkholderia species. Angew. Chem. Int. Ed Engl 51, 11611–11615 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Biggins JB, Ternei MA & Brady SF Malleilactone, a polyketide synthase-derived virulence factor encoded by the cryptic secondary metabolome of Burkholderia pseudomallei group pathogens. J. Am. Chem. Soc 134, 13192–13195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pidot S, Ishida K, Cyrulies M & Hertweck C Discovery of clostrubin, an exceptional polyphenolic polyketide antibiotic from a strictly anaerobic bacterium. Angew. Chem. Int. Ed Engl 53, 7856–7859 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Claesen J & Bibb M Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides. Proc. Natl. Acad. Sci. U. S. A 107, 16297–16302 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Y-L, He H-Y, Higgins MA & Ryan KS A heme-dependent enzyme forms the nitrogen–nitrogen bond in piperazate. Nature Chemical Biology 13, 836–838 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Tang X et al. Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem. Biol 10, 2841–2849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dassama LMK, Kenney GE & Rosenzweig AC Methanobactins: from genome to function. Metallomics 9, 7–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rond T. de, de Rond T, Asay JE & Moore BS Co-Occurrence of enzyme domains guides the discovery of an oxazolone synthetase. BioRxiv, doi: 10.1101/2020.06.11.147165 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obermaier S & Müller M Ibotenic acid biosynthesis in the fly agaric is initiated by glutamate hydroxylation. Angew. Chem. Int. Ed Engl 59, 12432–12435 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brachmann AO et al. A desaturase-like enzyme catalyzes oxazole formation in Pseudomonas indolyloxazole alkaloids. Angew. Chem. Int. Ed Engl (2021) doi: 10.1002/anie.202014491. [DOI] [PubMed] [Google Scholar]
- 32.Marchand JA et al. Discovery of a pathway for terminal-alkyne amino acid biosynthesis. Nature 567, 420–424 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Zhu X, Liu J & Zhang W De novo biosynthesis of terminal alkyne-labeled natural products. Nat. Chem. Biol 11, 115–120 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Ng TL, Rohac R, Mitchell AJ, Boal AK & Balskus EP An N-nitrosating metalloenzyme constructs the pharmacophore of streptozotocin. Nature 566, 94–99 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waldman AJ & Balskus EP Discovery of a diazo-forming enzyme in cremeomycin biosynthesis. J. Org. Chem 83, 7539–7546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwal V et al. Metagenomic discovery of polybrominated diphenyl ether biosynthesis by marine sponges. Nat. Chem. Biol 13, 537–543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baccile JA et al. Plant-like biosynthesis of isoquinoline alkaloids in Aspergillus fumigatus. Nat. Chem. Biol 12, 419–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caputi L et al. Missing enzymes in the biosynthesis of the anticancer drug vinblastine in Madagascar periwinkle. Science 360, 1235–1239 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Satake M et al. Brevisin: an aberrant polycyclic ether structure from the dinoflagellate Karenia brevis and its implications for polyether assembly. J. Org. Chem 74, 989–994 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Sinninghe Damsté JS et al. Linearly concatenated cyclobutane lipids form a dense bacterial membrane. Nature 419, 708–712 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Rattray JE et al. A comparative genomics study of genetic products potentially encoding ladderane lipid biosynthesis. Biol. Direct 4, 8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arnison PG et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep 30, 108–160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montalbán-López M et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep 38, 130–239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morinaka BI et al. Natural noncanonical protein splicing yields products with diverse β-amino acid residues. Science 359, 779–782 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Freeman MF, Helf MJ, Bhushan A, Morinaka BI & Piel J Seven enzymes create extraordinary molecular complexity in an uncultivated bacterium. Nat. Chem 9, 387–395 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Umemura M et al. Characterization of the biosynthetic gene cluster for the ribosomally synthesized cyclic peptide ustiloxin B in Aspergillus flavus. Fungal Genet. Biol 68, 23–30 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Nagano N et al. Class of cyclic ribosomal peptide synthetic genes in filamentous fungi. Fungal Genet. Biol 86, 58–70 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Kersten RD & Weng J-K Gene-guided discovery and engineering of branched cyclic peptides in plants. Proc. Natl. Acad. Sci. U. S. A 115, E10961–E10969 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jordan PA & Moore BS Biosynthetic pathway connects cryptic ribosomally synthesized posttranslationally modified peptide genes with pyrroloquinoline alkaloids. Cell Chem Biol 23, 1504–1514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ting CP et al. Use of a scaffold peptide in the biosynthesis of amino acid-derived natural products. Science 365, 280–284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kazandjian TD et al. Convergent evolution of pain-inducing defensive venom components in spitting cobras. Science 371, 386–390 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pineda SS et al. Structural venomics reveals evolution of a complex venom by duplication and diversification of an ancient peptide-encoding gene. Proc. Natl. Acad. Sci. U. S. A 117, 11399–11408 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanggaard KW et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun 5, 3765 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu S et al. Competition for iron drives phytopathogen control by natural rhizosphere microbiomes. Nat Microbiol 5, 1002–1010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carrión VJ et al. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 366, 606–612 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Guo C-J et al. Discovery of Reactive Microbiota-Derived Metabolites that Inhibit Host Proteases. Cell 168, 517–526.e18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Santhanam R et al. Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping. Proc. Natl. Acad. Sci. U. S. A 112, E5013–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durán P et al. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175, 973–983.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D’hoe K et al. Integrated culturing, modeling and transcriptomics uncovers complex interactions and emergent behavior in a three-species synthetic gut community. eLife 7, e37090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smanski MJ et al. Synthetic biology to access and expand nature’s chemical diversity. Nature Reviews Microbiology 14, 135–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reed J et al. A translational synthetic biology platform for rapid access to gram-scale quantities of novel drug-like molecules. Metab. Eng 42, 185–193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eng CH et al. ClusterCAD: a computational platform for type I modular polyketide synthase design. Nucleic Acids Res 46, D509–D515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Udwary DW et al. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. U. S. A 104, 10376–10381 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Omura S et al. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. U. S. A 98, 12215–12220 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oliynyk M et al. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat. Biotechnol 25, 447–453 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Leao T et al. Comparative genomics uncovers the prolific and distinctive metabolic potential of the cyanobacterial genus. Proc. Natl. Acad. Sci. U. S. A 114, 3198–3203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ju K-S et al. Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc. Natl. Acad. Sci. U. S. A 112, 12175–12180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shigdel UK et al. Genomic discovery of an evolutionarily programmed modality for small-molecule targeting of an intractable protein surface. Proc. Natl. Acad. Sci. U. S. A 117, 17195–17203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Donia MS et al. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158, 1402–1414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mendes R et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332, 1097–1100 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Wilson MC et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506, 58–62 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Owen JG et al. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc. Natl. Acad. Sci. U. S. A 110, 11797–11802 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Charlop-Powers Z et al. Global biogeographic sampling of bacterial secondary metabolism. Elife 4, e05048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brady SF, Chao CJ, Handelsman J & Clardy J Cloning and heterologous expression of a natural product biosynthetic gene cluster from eDNA. Org. Lett 3, 1981–1984 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Medema MH et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol 11, 625–631 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reddy BVB, Milshteyn A, Charlop-Powers Z & Brady SF eSNaPD: a versatile, web-based bioinformatics platform for surveying and mining natural product biosynthetic diversity from metagenomes. Chem. Biol 21, 1023–1033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hover BM et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat Microbiol 3, 415–422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peek J et al. Rifamycin congeners kanglemycins are active against rifampicin-resistant bacteria via a distinct mechanism. Nat. Commun 9, 4147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Trail F et al. Physical and transcriptional map of an aflatoxin gene cluster in Aspergillus parasiticus and functional disruption of a gene involved early in the aflatoxin pathway. Appl. Environ. Microbiol 61, 2665–2673 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kennedy J et al. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 284, 1368–1372 (1999). [DOI] [PubMed] [Google Scholar]
- 81.Mounaud S et al. Annotated genome sequence of Aspergillus tanneri NIH1004. Microbiology Resource Announcements vol. 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Field B & Osbourn AE Metabolic diversification--independent assembly of operon-like gene clusters in different plants. Science 320, 543–547 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Nützmann H, Huang A & Osbourn A Plant metabolic clusters – from genetics to genomics. New Phytologist vol. 211 771–789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luo X et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 567, 123–126 (2019). [DOI] [PubMed] [Google Scholar]
- 85.Galanie S, Thodey K, Trenchard IJ, Filsinger Interrante M & Smolke CD Complete biosynthesis of opioids in yeast. Science 349, 1095–1100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brunson JK et al. Biosynthesis of the neurotoxin domoic acid in a bloom-forming diatom. Science vol. 361 1356–1358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kita M & Uemura D Marine huge molecules: the longest carbon chains in natural products. Chem. Rec 10, 48–52 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Chow MH, Yan KTH, Bennett MJ & Wong JTY Birefringence and DNA condensation of liquid crystalline chromosomes. Eukaryot. Cell 9, 1577–1587 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beedessee G et al. Integrated omics unveil the secondary metabolic landscape of a basal dinoflagellate. BMC Biol 18, 139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Delmont TO et al. Functional repertoire convergence of distantly related eukaryotic plankton lineages revealed by genome-resolved metagenomics. BioRxiv, doi: 10.1101/2020.10.15.341214 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zan J et al. A microbial factory for defensive kahalalides in a tripartite marine symbiosis. Science 364, eaaw6732 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vaelli PM et al. The skin microbiome facilitates adaptive tetrodotoxin production in poisonous newts. Elife 9, e53898 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gizzi AS et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 558, 610–614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cooke TF et al. Genetic Mapping and Biochemical Basis of Yellow Feather Pigmentation in Budgerigars. Cell 171, 427–439.e21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sabatini M et al. Biochemical characterization of the minimal domains of an iterative eukaryotic polyketide synthase. FEBS J 285, 4494–4511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cutignano A et al. Biosynthesis and cellular localization of functional polyketides in the gastropod mollusc Scaphander lignarius. Chembiochem 13, 1759–66, 1701 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Torres JP, Lin Z, Winter JM, Krug PJ & Schmidt EW Animal biosynthesis of complex polyketides in a photosynthetic partnership. Nat. Commun 11, 2882 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beran F et al. Novel family of terpene synthases evolved from trans-isoprenyl diphosphate synthases in a flea beetle. Proc. Nat. Acad. Sci. U.S.A 113, 2922–2927 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roelofs D et al. A functional isopenicillin N synthase in an animal genome. Molecular Biology and Evolution 30, 541–548 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Suring W, Mariën J, Broekman R, van Straalen NM & Roelofs D Biochemical pathways supporting beta-lactam biosynthesis in the springtail Folsomia candida. Biol. Open 5, 1784–1789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shou Q et al. A hybrid polyketide–nonribosomal peptide in nematodes that promotes larval survival. Nature Chemical Biology 12, 770–772 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Izoré T et al. Drosophila melanogaster nonribosomal peptide synthetase Ebony encodes an atypical condensation domain. Proc. Nat. Acad. Sci. U.S.A 116, 2913–2918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chekan JR et al. Scalable biosynthesis of the seaweed neurochemical, kainic acid. Angew. Chem. Int. Ed Engl 58, 8454–8457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Medema MH, Cimermancic P, Sali A, Takano E & Fischbach MA A systematic computational analysis of biosynthetic gene cluster evolution: lessons for engineering biosynthesis. PLoS Comput. Biol 10, e1004016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blin K et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47, W81–W87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Skinnider MA, Merwin NJ, Johnston CW & Magarvey NA PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res 45, W49–W54 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eddy SR Profile hidden Markov models. Bioinformatics 14, 755–763 (1998). [DOI] [PubMed] [Google Scholar]
- 108.Cimermancic P et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158, 412–421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hannigan GD et al. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res 47, e110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Heel AJ et al. BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res 46, W278–W281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tietz JI et al. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol 13, 470–478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Santos-Aberturas J et al. Uncovering the unexplored diversity of thioamidated ribosomal peptides in Actinobacteria using the RiPPER genome mining tool. Nucleic Acids Res 47, 4624–4637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Merwin NJ et al. DeepRiPP integrates multiomics data to automate discovery of novel ribosomally synthesized natural products. Proc. Natl. Acad. Sci. U. S. A 117, 371–380 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kloosterman AM et al. Expansion of RiPP biosynthetic space through integration of pan-genomics and machine learning uncovers a novel class of lanthipeptides. PLoS Biol 18, e3001026 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Palaniappan K et al. IMG-ABC v.5.0: an update to the IMG/Atlas of Biosynthetic Gene Clusters Knowledgebase. Nucleic Acids Res 48, D422–D430 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Blin K et al. The antiSMASH database version 2: a comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Res 47, D625–D630 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schläpfer P et al. Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol 173, 2041–2059 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kautsar SA, Suarez Duran HG, Blin K, Osbourn A & Medema MH plantiSMASH: automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res 45, W55–W63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Navarro-Muñoz JC et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol 16, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu Z et al. Drivers of metabolic diversification: how dynamic genomic neighbourhoods generate new biosynthetic pathways in the Brassicaceae. New Phytol 227, 1109–1123 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schorn MA et al. A community resource for paired genomic and metabolomic data mining. Nat. Chem. Biol, doi: 10.1038/s41589-020-00724-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Duncan KR et al. Molecular networking and pattern-based genome mining improves discovery of biosynthetic gene clusters and their products from Salinispora species. Chem. Biol 22, 460–471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Goering AW et al. Metabologenomics: correlation of microbial gene clusters with metabolites drives discovery of a nonribosomal peptide with an unusual amino acid monomer. ACS Cent Sci 2, 99–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Doroghazi JR et al. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol 10, 963–968 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.van der Hooft JJJ et al. Linking genomics and metabolomics to chart specialized metabolic diversity. Chem. Soc. Rev 49, 3297–3314 (2020). [DOI] [PubMed] [Google Scholar]
- 126.Parkinson EI et al. Discovery of the tyrobetaine natural products and their biosynthetic gene cluster via metabologenomics. ACS Chem. Biol 13, 1029–1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eldjárn GH et al. Ranking microbial metabolomic and genomic links in the NPLinker framework using complementary scoring functions. BioRxiv, doi: 10.1101/2020.06.12.148205 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kersten RD et al. Glycogenomics as a mass spectrometry-guided genome-mining method for microbial glycosylated molecules. Proc. Natl. Acad. Sci. U. S. A 110, E4407–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Medema MH et al. Pep2Path: automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput. Biol 10, e1003822 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mohimani H et al. Automated genome mining of ribosomal peptide natural products. ACS Chem. Biol 9, 1545–1551 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cao L et al. MetaMiner: a scalable peptidogenomics approach for discovery of ribosomal peptide natural products with blind modifications from microbial communities. Cell Syst 9, 600–608.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vogt E & Künzler M Discovery of novel fungal RiPP biosynthetic pathways and their application for the development of peptide therapeutics. Appl. Microbiol. Biotechnol 103, 5567–5581 (2019). [DOI] [PubMed] [Google Scholar]
- 133.Safavi-Hemami H et al. Modulation of conotoxin structure and function is achieved through a multienzyme complex in the venom glands of cone snails. J. Biol. Chem 287, 34288–34303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dejong CA et al. Polyketide and nonribosomal peptide retro-biosynthesis and global gene cluster matching. Nat. Chem. Biol 12, 1007–1014 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Luo D et al. Oxidation and cyclization of casbene in the biosynthesis of Euphorbia factors from mature seeds of Euphorbia lathyris L. Proc. Natl. Acad. Sci. U. S. A 113, E5082–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jeon JE et al. A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell 180, 176–187.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Itkin M et al. Biosynthesis of antinutritional alkaloids in solanaceous crops is mediated by clustered genes. Science 341, 175–179 (2013). [DOI] [PubMed] [Google Scholar]
- 138.Lau W & Sattely ES Six enzymes from mayapple that complete the biosynthetic pathway to the etoposide aglycone. Science 349, 1224–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rajniak J, Barco B, Clay NK & Sattely ES A new cyanogenic metabolite in Arabidopsis required for inducible pathogen defence. Nature 525, 376–379 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saelens W, Cannoodt R & Saeys Y A comprehensive evaluation of module detection methods for gene expression data. Nat. Commun 9, 1090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wisecaver JH et al. A global coexpression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell 29, 944–959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Becher PG et al. Developmentally regulated volatiles geosmin and 2-methylisoborneol attract a soil arthropod to Streptomyces bacteria promoting spore dispersal. Nat Microbiol 5, 821–829 (2020). [DOI] [PubMed] [Google Scholar]
- 143.Muhlemann JK, Younts TLB & Muday GK Flavonols control pollen tube growth and integrity by regulating ROS homeostasis during high-temperature stress. Proc. Natl. Acad. Sci. U. S. A 115, E11188–E11197 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bruns H et al. Function-related replacement of bacterial siderophore pathways. ISME J 12, 320–329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rajniak J et al. Biosynthesis of redox-active metabolites in response to iron deficiency in plants. Nat. Chem. Biol 14, 442–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Crits-Christoph A, Bhattacharya N, Olm MR, Song YS & Banfield JF Transporter genes in biosynthetic gene clusters predict metabolite characteristics and siderophore activity. Genome Res 31, 239–250 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yeh H-H et al. Resistance gene-guided genome mining: serial promoter exchanges in Aspergillus nidulans reveal the biosynthetic pathway for fellutamide B, a proteasome inhibitor. ACS Chem. Biol 11, 2275–2284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Panter F, Krug D, Baumann S & Müller R Self-resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria. Chem. Sci 9, 4898–4908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yan Y et al. Resistance-gene-directed discovery of a natural-product herbicide with a new mode of action. Nature 559, 415–418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mungan MD et al. ARTS 2.0: feature updates and expansion of the Antibiotic Resistant Target Seeker for comparative genome mining. Nucleic Acids Res 48, W546–W552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nonejuie P et al. Application of bacterial cytological profiling to crude natural product extracts reveals the antibacterial arsenal of Bacillus subtilis. J. Antibiot 69, 353–361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kurita KL, Glassey E & Linington RG Integration of high-content screening and untargeted metabolomics for comprehensive functional annotation of natural product libraries. Proc. Natl. Acad. Sci. U. S. A 112, 11999–12004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Aliper A et al. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm 13, 2524–2530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shang Y et al. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346, 1084–1088 (2014). [DOI] [PubMed] [Google Scholar]
- 155.Crits-Christoph A, Diamond S, Butterfield CN, Thomas BC & Banfield JF Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558, 440–444 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Oyserman BO, Medema MH & Raaijmakers JM Road MAPs to engineer host microbiomes. Curr. Opin. Microbiol 43, 46–54 (2018). [DOI] [PubMed] [Google Scholar]
- 157.Huang AC et al. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364, eaau6389 (2019). [DOI] [PubMed] [Google Scholar]
- 158.Chevrette MG, Aicheler F, Kohlbacher O, Currie CR & Medema MH SANDPUMA: ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 33, 3202–3210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Helfrich EJN et al. Automated structure prediction of trans-acyltransferase polyketide synthase products. Nat. Chem. Biol 15, 813–821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Agrawal P & Mohanty D A machine-learning-based method for prediction of macrocyclization patterns of polyketides and nonribosomal peptides. Bioinformatics (2020) doi: 10.1093/bioinformatics/btaa851. [DOI] [PubMed] [Google Scholar]
- 161.Dührkop K, Shen H, Meusel M, Rousu J & Böcker S Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. U. S. A 112, 12580–12585 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.van der Hooft JJJ, Wandy J, Barrett MP, Burgess KEV & Rogers S Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. U. S. A 113, 13738–13743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rodrigues T, Reker D, Schneider P & Schneider G Counting on natural products for drug design. Nat. Chem 8, 531–541 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Reker D et al. Revealing the macromolecular targets of complex natural products. Nat. Chem 6, 1072–1078 (2014). [DOI] [PubMed] [Google Scholar]
- 165.Stokes JM et al. A deep learning approach to antibiotic discovery. Cell 181, 475–483 (2020). [DOI] [PubMed] [Google Scholar]
- 166.Skinnider MA et al. Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nat. Commun 11, 6058 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Karim AS et al. In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol 16, 912–919 (2020). [DOI] [PubMed] [Google Scholar]
- 168.Zhang JJ, Tang X & Moore BS Genetic platforms for heterologous expression of microbial natural products. Nat. Prod. Rep 36, 1313–1332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Huo L et al. Heterologous expression of bacterial natural product biosynthetic pathways. Natural Product Reports 36, 1412–1436 (2019). [DOI] [PubMed] [Google Scholar]
- 170.Lin Z, Nielsen J & Liu Z Bioprospecting through cloning of whole natural product biosynthetic gene clusters. Front Bioeng Biotechnol 8, 526 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee NCO, Larionov V & Kouprina N Highly efficient CRISPR/Cas9-mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Res 43, e55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yamanaka K et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. U. S. A 111, 1957–1962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fu J et al. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol 30, 440–446 (2012). [DOI] [PubMed] [Google Scholar]
- 174.Enghiad B & Zhao H Programmable DNA-guided artificial restriction enzymes. ACS Synth. Biol 6, 752–757 (2017). [DOI] [PubMed] [Google Scholar]
- 175.Shapland EB et al. Low-Cost, High-Throughput Sequencing of DNA assemblies using a highly multiplexed Nextera process. ACS Synth. Biol 4, 860–866 (2015). [DOI] [PubMed] [Google Scholar]
- 176.Zhang JJ, Tang X, Zhang M, Nguyen D & Moore BS Broad-host-range expression reveals native and host regulatory elements that influence heterologous antibiotic production in Gram-negative bacteria. MBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang G et al. CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria. Nat Microbiol 4, 2498–2510 (2019). [DOI] [PubMed] [Google Scholar]
- 178.Harvey CJB et al. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci Adv 4, eaar5459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Casini A et al. A pressure test to make 10 molecules in 90 Days: external evaluation of methods to engineer biology. J. Am. Chem. Soc 140, 4302–4316 (2018). [DOI] [PubMed] [Google Scholar]
- 180.Smanski MJ et al. Functional optimization of gene clusters by combinatorial design and assembly. Nat. Biotechnol 32, 1241–1249 (2014). [DOI] [PubMed] [Google Scholar]
- 181.Meyer AJ, Segall-Shapiro TH, Glassey E, Zhang J & Voigt CA Escherichia coli ‘Marionette’ strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol 15, 196–204 (2019). [DOI] [PubMed] [Google Scholar]
- 182.Proctor RH, Hohn TM & McCormick SP Restoration of wild-type virulence to Tri5 disruption mutants of Gibberella zeae via gene reversion and mutant complementation. Microbiology 143, 2583–2591 (1997). [DOI] [PubMed] [Google Scholar]
- 183.Rubin BE et al. Targeted genome editing of bacteria within microbial communities. BioRxiv, doi: 10.1101/2020.07.17.209189 (2020). [DOI] [Google Scholar]
- 184.Lam KN et al. Phage-delivered CRISPR-Cas9 for strain-specific depletion and genomic deletions in the gut microbiota. BioRxiv, doi: 10.1101/2020.07.09.193847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gurevich A et al. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat Microbiol 3, 319–327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Reher R et al. A convolutional neural network-based approach for the rapid annotation of molecularly diverse natural products. J. Am. Chem. Soc 142, 4114–4120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Burns DC, Mazzola EP & Reynolds WF The role of computer-assisted structure elucidation (CASE) programs in the structure elucidation of complex natural products. Nat. Prod. Rep 36, 919–933 (2019). [DOI] [PubMed] [Google Scholar]
- 188.Inokuma Y et al. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 495, 461–466 (2013). [DOI] [PubMed] [Google Scholar]
- 189.Danelius E, Halaby S, van der Donk WA & Gonen T MicroED in natural product and small molecule research. Nat. Prod. Rep (2020) doi: 10.1039/d0np00035c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chu J et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol 12, 1004–1006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chu J, Vila-Farres X & Brady SF Bioactive synthetic-bioinformatic natural product cyclic peptides inspired by nonribosomal peptide synthetase gene clusters from the human microbiome. J. Am. Chem. Soc 141, 15737–15741 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chu J et al. Synthetic-Bioinformatic Natural Product Antibiotics with Diverse Modes of Action. J. Am. Chem. Soc 142, 14158–14168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hudson GA, Hooper AR, DiCaprio AJ, Sarlah D & Mitchell DA Structure prediction and synthesis of pyridine-based macrocyclic peptide natural products. Org. Lett 23, 253–256 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lo H-C et al. Two separate gene clusters encode the biosynthetic pathway for the meroterpenoids austinol and dehydroaustinol in Aspergillus nidulans. J. Am. Chem. Soc 134, 4709–4720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sanchez JF et al. Genome-based deletion analysis reveals the prenyl xanthone biosynthesis pathway in Aspergillus nidulans. J. Am. Chem. Soc 133, 4010–4017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Andersen MR et al. Accurate prediction of secondary metabolite gene clusters in filamentous fungi. Proc. Natl. Acad. Sci. U. S. A 110, E99–107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Huang AC et al. Unearthing a sesterterpene biosynthetic repertoire in the Brassicaceae through genome mining reveals convergent evolution. Proc. Natl. Acad. Sci. U. S. A 114, E6005–E6014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shoguchi E et al. A new dinoflagellate genome illuminates a conserved gene cluster involved in sunscreen biosynthesis. Genome Biol. Evol 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhao T & Schranz ME Network-based microsynteny analysis identifies major differences and genomic outliers in mammalian and angiosperm genomes. Proc. Natl. Acad. Sci. U. S. A 116, 2165–2174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bok JW et al. Chromatin-level regulation of biosynthetic gene clusters. Nat. Chem. Biol 5, 462–464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yu N et al. Delineation of metabolic gene clusters in plant genomes by chromatin signatures. Nucleic Acids Res 44, 2255–2265 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lawrence JG & Roth JR Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics 143, 1843–1860 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ballouz S, Francis AR, Lan R & Tanaka MM Conditions for the evolution of gene clusters in bacterial genomes. PLoS Computational Biology vol. 6 e1000672 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.McGary KL, Slot JC & Rokas A Physical linkage of metabolic genes in fungi is an adaptation against the accumulation of toxic intermediate compounds. Proc. Natl. Acad. Sci. U. S. A 110, 11481–11486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Field B et al. Formation of plant metabolic gene clusters within dynamic chromosomal regions. Proc. Natl. Acad. Sci. U. S. A 108, 16116–16121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gluck-Thaler E & Slot JC Specialized plant biochemistry drives gene clustering in fungi. ISME J 12, 1694–1705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.van Santen JA et al. The Natural Products Atlas: an open access knowledge base for microbial natural products discovery. ACS Cent Sci 5, 1824–1833 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Schorn MA et al. Sequencing rare marine actinomycete genomes reveals high density of unique natural product biosynthetic gene clusters. Microbiology 162, 2075–2086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Skinnider MA & Magarvey NA Statistical reanalysis of natural products reveals increasing chemical diversity. Proc. Natl. Acad. Sci. U. S. A 114, E6271–E6272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Thrash JC Culturing the uncultured: risk versus reward. mSystems 4, e00130–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Atanasov AG, Zotchev SB, Dirsch VM, International Natural Product Sciences Taskforce & Supuran, C. T. Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov (2021) doi: 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Challis GL & Ravel J Coelichelin, a new peptide siderophore encoded by the Streptomyces coelicolor genome: structure prediction from the sequence of its non-ribosomal peptide synthetase. FEMS Microbiol. Lett 187, 111–114 (2000). [DOI] [PubMed] [Google Scholar]
- 213.Blin K, Kim HU, Medema MH & Weber T Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform 20, 1103–1113 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Medema MH & Fischbach MA Computational approaches to natural product discovery. Nat. Chem. Biol 11, 639–648 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kjærbølling I, Vesth T & Andersen MR Resistance gene-directed genome mining of 50 species. mSystems 4, e00085–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zallot R, Oberg N & Gerlt JA The EFI web resource for genomic enzymology tools: leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry 58, 4169–4182 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Usadel B et al. Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ 32, 1633–1651 (2009). [DOI] [PubMed] [Google Scholar]
- 218.Serin EAR, Nijveen H, Hilhorst HWM & Ligterink W Learning from co-expression networks: possibilities and challenges. Frontiers in Plant Science 7, 444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tzfadia O et al. CoExpNetViz: comparative co-expression networks construction and visualization tool. Front. Plant Sci 6, 1194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gubbens J et al. Natural product proteomining, a quantitative proteomics platform, allows rapid discovery of biosynthetic gene clusters for different classes of natural products. Chem. Biol 21, 707–718 (2014). [DOI] [PubMed] [Google Scholar]
- 222.Ding Y et al. Genetic elucidation of interconnected antibiotic pathways mediating maize innate immunity. Nat Plants 6, 1375–1388 (2020). [DOI] [PubMed] [Google Scholar]
- 223.Levin BJ et al. A prominent glycyl radical enzyme in human gut microbiomes metabolizes trans-4-hydroxy-l-proline. Science 355, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Soldatou S, Eldjarn GH, Huerta-Uribe A, Rogers S & Duncan KR Linking biosynthetic and chemical space to accelerate microbial secondary metabolite discovery. FEMS Microbiol. Lett 366, fnz142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kersten RD et al. Bioactivity-guided genome mining reveals the lomaiviticin biosynthetic gene cluster in Salinispora tropica. Chembiochem 14, 955–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Mohimani H & Pevzner PA Dereplication, sequencing and identification of peptidic natural products: from genome mining to peptidogenomics to spectral networks. Nat. Prod. Rep 33, 73–86 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ricart E et al. rBAN: retro-biosynthetic analysis of nonribosomal peptides. J. Cheminform 11, 13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]