

Abstract
Background:
Prostate cancer (PCa) phenotypes vary from indolent to aggressive. Molecular subtyping may be useful in predicting aggressive cancers and directing therapy. One such subtype involving deletions of chromodomain helicase DNA binding protein 1 (CHD1), a tumor suppressor gene, are found in 10–26% of PCa tumors. In this study, we evaluate the functional cellular effects that follow CHD1 deletion.
Methods:
CHD1 was knocked out (KO) in the non-tumorigenic, human papillomavirus 16 (HPV16)-immortalized prostate epithelial cell line, RWPE-1, using CRISPR/Cas9. In vitro assays such as T7 endonuclease assay, western blot, and sequencing were undertaken to characterize the CHD1 KO clones. Morphologic and functional assays for cell adhesion and viability were performed. To study expression of extracellular matrix (ECM) and adhesion molecules, a real-time (RT) profiler assay was performed using RWPE-1 parental, non-target cells (NT2) and CHD1 KO cells.
Result:
Compared to parental RWPE-1 and non-target cells (NT2), the CHD1 KO cells had a smaller, rounder morphology and were less adherent under routine culture conditions. Compared to parental cells, CHD1 KO cells showed a reduction in ECM and adhesion molecules as well as a greater proportion of viable suspension cells when cultured on standard tissue culture plates and on plates coated with laminin, fibronectin or collagen I. CHD1 KO cells showed a decrease in the expression of secreted protein acidic and rich in cysteine (SPARC), matrix metalloproteinase 2 (MMP2), integrin subunit alpha 2 (ITGA2), integrin subunit alpha 5 (ITGA5), integrin subunit alpha 6 (ITGA6), fibronectin (FN1), laminin subunit beta-3 precursor (LAMB3), collagen, tenascin and vitronectin as compared to parental and NT2 cells.
Conclusion:
These data suggest that in erythroblast transformation specific (ETS) fusion-negative, phosphatase and tensin homolog (PTEN) wildtype PCa, deletion of CHD1 alters cell-cell and cell-matrix adhesion dynamics, suggesting an important role for CHD1 in the development and progression of PCa.
Keywords: cell adhesion, CHD1, integrins, laminin, prostate cancer
Introduction
Prostate cancer (PCa) is the most common malignancy diagnosed in men, with approximately 191,930 new cases and 33,330 deaths predicted in 2020.1 PCa is biologically complex and heterogeneous; while a large proportion of PCas are slow growing and indolent, some patients diagnosed with low risk, localized PCa experience disease recurrence and progression even with aggressive treatment.2,3 Efforts to understand the mechanisms driving this heterogeneity have been undertaken to optimize patient risk stratification. By doing so, clinicians can reduce overtreatment of low-risk patients and identify those at higher risk for relapse.4
Classifying the genomic spectrum of prostate cancer has been one approach to defining PCa subsets. Common genomic alterations observed in PCa include the deletion of tumor suppressors and the prostate specific homeobox transcription factor NK3 homeobox 1 (NKX3-1), copy number alterations of MYC, androgen receptor variants, and oncogenic fusions involving the E26 transformation-specific (ETS) family of transcription factors.
The deletion of the tumor suppressor phosphatase and tensin homolog (PTEN) is frequently detected in aggressive or castrate resistant PCas.4 The loss of PTEN leads to the activation of phosphoinositide 3-kinase (PI3K)/ protein kinase B (AKT)/ mechanistic target of rapamycin (mTOR) signaling, promoting cell growth and inhibition of autophagy.5 When the PI3K/AKT/mTOR pathway is inhibited, the Ras/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway may be activated, further stimulating tumor propagation and metastasis.6
ETS fusions, seen in 50–70% of PCa tumors, are characterized by the rearrangement of an ETS gene family member such that it is used at its 5ⶀ end to the untranslated region of other genes that differ in their prostate specificity and androgen responsiveness. The most common of these fusions is transmembrane protease, serine 2-ETS-related gene (TMPRSS2-ERG). In this fusion, overexpression of ERG is driven by the prostate specific, androgen-dependent promoter element of TMPRSS2, which encodes a transmembrane protease.7 ETS rearrangements are known to co-occur with loss of the tumor suppressor PTEN, which can lead to increased expression of ETS transcription factors that contribute to oncogenesis due to upregulated transcription of genes needed for cellular migration.8 Although other novel and potentially actionable fusions have been reported, such as those involving friend leukemia integration 1 (FLI1), BRAF, RAF1, PIK3 catalytic subunit alpha/beta (PIK3CA/B) and R-spondin-2-precursor (RSPO2, they occur far less frequently than those involving ETS gene family members.9
In the absence of PTEN loss or ETS gene fusions, the molecular processes that initiate and support progression of PCa are less clear.10–12 At the epigenetic level, distinct methylation profiles have been identified in ETS fusion positive and fusion negative tumors, leading to up- or down-regulation of distinct genes potentially responsible for the tumor phenotype, including enhancement of cell proliferation and migration.13 At the genomic level, next-generation sequencing has identified recurrent molecular changes in ETS fusion-negative PCa that include speckle-type POZ protein (SPOP) mutations in exon 6 or 7 causing strong downregulation of SPOP (seen in 6–15% of PCa), serine protease inhibitor Kazal-type 1 (SPINK1) overexpression (10%), mitogen-activated protein kinase kinase kinase 7 (MAP3K7) deletions (18–38%), and chromo-domain helicase DNA binding protein-1 (CHD1) deletions/non-synonymous mutations (15–27%). SPOP mutations often co-occur with CHD1 deletion.14 However, the exact contribution of these molecular changes in the initiation and progression of PCa is not yet well understood. Elucidating these molecular mechanisms could potentially enhance current patient risk stratification schema and guide treatment options.
Genome-wide, allele-specific analysis of homozygous gene deletions (HODs) in primary PCa revealed that CHD1 is the second most frequent HOD and was accompanied with hemi- or homozygous deletions primarily affecting 2q, 5q and 6q.15 CHD1 is a chromatin remodeling factor that impacts many cellular processes in the embryo and adult. CHD1 facilitates various aspects of DNA transcription through its ability to keep DNA in an open and transcriptionally active state via ATP-dependent assembly, shifting, and removal of nucleosomes from DNA.16,17 CHD1 is also a substrate recognition component of the histone acetylation18 complex, known as Spt-Ada-Gcn5-acetyltransferase (SAGA), which regulates transcription through Pol-II and the Pol-I transcription termination step. CHD1 is also associated with histone deacetylase (HDAC) activity and the modulation of pre-mRNA splicing.19 Finally, CHD1 is required for the maintenance of pluripotency in embryonic stem cells20 and the endothelial to hematopoietic transition during embryonic development.21 These roles of CHD1 were supported by a recent report describing how CHD1 specifically aids in homologous recombination-mediated DNA repair of double strand breaks.22 The findings suggest the importance of CHD1 in maintaining genomic stability and that CHD1 deletion leads to specific genomic alterations that may initiate PCa.
Materials and methods
This study was granted approval by the Rutgers Electronic Institutional Review Board (eIRB) with approval number # CR00011175. No human subjects were used in this research.
Cell culture
Normal prostate epithelial cell line (RWPE-1), and prostate cancer cell lines PC3, DU145, VCaP, LNCaP, C4-2 and NCI H660 were obtained from the American Type Culture Collection and grown according to recommended culture conditions. RWPE-1 cells were grown in keratinocyte serum-free medium (K-SFM) (Life Technologies, 10724-011).
Generation of CHD1 knock out
To knock out the CHD1 in RWPE-1, the Sigma all-in-one lentiviral-CRISPR format containing gRNA, Cas9, puromycin, and GFP elements in the vector pLV-U6g-EPCG (The Genome Editing Core Facility, Rutgers Child Health Institute of New Jersey, New Brunswick, NJ, USA) was used. A CHD1 SgRNA sequence with one off-target site was chosen from exon 3 to generate CHD1-pLV-U6g-EPCG (202-ATCAAGCCTCATCTAATAGCGG). pLV-U6g-EPCG was used as the non-target control plasmid (LVP-NT). In a 10 cm dish, 293T cells were transfected with helper plasmids and co-transfected with either CHD1-pLV-U6g-EPCG or pLV-U6g-EPCG. At 24 h, the medium containing lentiviral particles was collected. 106 RWPE-1 cells in a 6 cm dish were infected with either LVP-202 or LVP-NT particles in a total volume of 3 ml at two different ratios (2:1 and 2.25:0.75, K-SFM: viral suspension). K-SFM medium was replaced at 24 h. At 48 h post-infection, cells were single cell sorted for green fluorescent protein (GFP) in 96 well plates.
T7 endonuclease assay
After cell number expansion, genomic DNA was isolated, and genomic polymerase chain reaction (PCR) of the CRISPR-CHD1 target region was set up with two sets of primers (CHD1 Ex3a For: tctaccctcactggagaccatt/CHD1, Ex3a Rev: TCTGTGGAACTCAACACACCA, and CHD1 Ex3b For: AAAACAAGAAATCCTAGGTGCCAA/CHD1, Ex3b Rev: GCCTCTTCACCTCACACTGAT). The cycling conditions used were 94°C/2 min, 31 cycles of 94°C/20 s, 60°C/20 s, and 72°C/30 s, 72°C/3 min, and cooling to 4°C. To generate heteroduplexes, 5 μl of the genomic PCR product was transferred to a fresh tube and subjected to touch down PCR conditions of 95°C/5 min, 95°C to 85°C at 2°C/s, 85°C to 25°C at 0.1°C/s, and cooling to 4°C. 1 μl of T7 endonuclease (NEB) was then added to the tube along with 1 μl NEB2 buffer and 2 μl water. The mixture was incubated at 37°C for 15 min, and 2 μl of 0.5M of ethylenediamine tetraacetic acid (EDTA) were added. 10 μl of the heteroduplexes were visualized after separation on a 2% agarose gel containing ethidium bromide.
Western blot analysis
Cells were grown to 80% confluency and lysed with cell lysis buffer (Cell Signaling #9803) on ice for 30 min. Lysate was cleared by centrifuging at 10,000 rpm for 10 min and collecting the supernatant. 30 μg of protein lysate was separated on 4–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel, transferred to nitrocellulose membranes using the iBlot transfer apparatus (Invitrogen). Primary antibodies used included glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology Inc., 25778), CHD1 (Novus Biologicals, NB100-60411), focal adhesion kinase (FAK) (Cell Signaling Technologies or CST, 3285P), phosphorylated extracellular signal regulated kinase (pErk) (CST, 4370S), total Erk (CST, 9102S), phosphorylated protein kinase B (pAKT) (CST, 4060S), total AKT (CST, 72), SPARC (CST, 8725), pMEK 1/2 (mitogen-extracellular signal-regulated kinase 1/2) (CST, 9154), total MEK 1/2 (CST, 9126), tenascin C (CST, 12221), and vitronectin (CST, 60896). Secondary antibodies included horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (Calbiochem, 401315). Clarity™ enhanced chemiluminescent (ECL) reagent (Bio-Rad, 102030712) was used as a substrate for western blots. Images were obtained with a Bio-Rad imager, and signals were quantitated using Bio-Rad Image Lab software.
RT-profiler assay
Complimentary DNA (cDNA) was made from RNA isolated from RWPE-1, nontarget control (NT2), Cr2, Cr16, and Cr21 cells and ECM and adhesion molecules. RT2 profiler PCR array (Qiagen, PAHS-013Z) was used to analyze gene expression. The PCR array consisted of 84 related genes including cell adhesion molecules such as transmembrane receptors, cell-cell adhesion proteins, extracellular matrix molecules such as basement membrane elements, collagens, and ECM structural components, ECM proteases, ECM protease inhibitors, and other ECM molecules. All samples were tested in duplicate. Data analysis determined fold changes in gene expression (ΔΔ Ct).
SPARC and MMP2 validation
RWPE-1, NT2, Cr2, Cr16, and Cr21 cells were grown to 80% confluency. The culture supernatant medium was used to detect secreted human SPARC and total MMP2 proteins with enzyme-linked immunoassay (ELISA) (Quantikine ELISA for Human SPARC, R&D Systems DSP00; Quantikine ELISA for Total MMP-2, R & D systems MMP200) according to the manufacturer’s instructions. Proteins levels in the conditioned medium was calculated in ng/ml, based on a standard curve generated with pure human SPARC and MMP2.
Growth of cells in plates coated with various matrices
To study cell adhesion properties, 3 × 104 cells each of RWPE-1, NT2, Cr2, Cr16, and Cr21 were seeded onto culture dishes in 2 ml of K-SFM medium including regular (Costar, 3516), collagen I coated (Corning Biocoat Cellware, 354400), fibronectin coated (Corning Biocoat Cellware, 354457), and laminin coated 35 mm dishes (Corning Biocoat Cellware, 354458). On day 3, fresh K-SFM was added to the cultures. On day 12, suspended cells and adherent cells were quantified for each type of plate. To do so, the medium was collected from each dish, centrifuged at 1200 rpm for 5 min, and then aspirated. Pelleted cell clusters and cells adhered to each plate type were trypsinized with 500 μl of 0.0025% Trypsin-EDTA4 for the same length of time. Trypsin-EDTA was neutralized with 500 μl of 0.1% Soybean Trypsin Inhibitor in Dulbecco’s phosphate-buffered saline (DPBS) (ATCC, 30-2104). The number of viable cells and percent viability in the 1 ml suspension was determined using the Vi-Cell counter (Beckman Coulter Life Sciences).
In vivo experiments
Animal housing and experiments were conducted under IACUC Protocol Number 113-016-2 in accordance with institutional guidelines for humane animal treatment and complied with relevant legislation. 5 × 105 cells each of RWPE-1, NT2, Cr2, Cr16, and Cr21 were resuspended in 100 ul of complete K-SFM, mixed with matrigel (Corning, 344248), and injected subcutaneously into the right and left flanks of four nude mice. Tumor growth was evaluated at 20 weeks. Tissue from the site of injection was dissected and stained with haemotoxylin and eosin (H&E).
Immunohistochemistry (IHC) was performed using VENTANA anti-p63 antibody (Roche Diagnostics) as a basal cell marker and anti-CK8/18 antibody (Leica Biosystems) as a luminal cell marker. Images of sections stained with H&E, p63, and CK8/18 antibodies were visualized using the Axioskop microscope (Zeiss) and NIS-Elements (Nikon) imaging software.
Immunofluorescence (IF) was performed using anti-laminin-332 (Abcam, ab14509) as well as a secondary antibody conjugated to Alexa Fluor (Invitrogen), which fluoresces red at 594 nm. Staining of tissue and spheroids was performed by Tissue Analytical Services (Rutgers Cancer Institute of New Jersey). IF slides were digitized at 20× magnification using a Olympus versus 120 Whole Slide Scanner (Biomedical Informatics Shared Resource of the Rutgers Cancer Institute of New JerseP30CA072720) and visualized using OlyVIA software (Olympus Life Science).
Spheroid culture
For spheroid number
Solid spheroid cultures were generated by gently mixing 800 cells with 40 μl of growth factor-reduced, phenol red-free matrigel (Corning, 356231) in a well. Incubation at 37°C for 30 min allowed the matrigel to solidify. Then, 1 ml of K-SFM was added to the well, which was then reheated to 37°C. The medium was changed every third day. Spheroids were counted with the aid of a microscope on day 10.
For spheroid limiting dilution assay
Determination of spheroid (formation) frequencies from each cell line was determined by adding 5, 10, 25, 50, 100 and 250 cells/well of a round bottom, low adhesion 96-well plate. 18 wells per dilution were plated for each group of cells and maintained at 37°C and 5% CO2. Once a week, half of the medium (100 µl) was carefully removed from each well, and an equal volume of fresh complete K-SFM medium was added. Wells were evaluated for spheroid formation daily for 12 days with an inverted microscope, where a minimum of at least 10 cells defined a spheroid. Spheroid frequency data analysis was performed using the extreme limiting dilution analysis (ELDA) website (https://bioinf.wehi.edu.au/software/elsd/index.html).23
For spheroid staining
Solid spheroid cultures were set up by gently mixing 5000 cells with 40 μl of matrigel (Corning, 356231) in a well. After the matrigel solidified, it was coated with complete K-SFM and incubated at 37°C for 12 day. The medium was changed every third day. Paraffin-embedding was performed using a modification of the method described by Pinto et al.24 Briefly, the medium was removed from the well, and 4% paraformaldehyde was added and incubated at room temperature for 30 min. Subsequently, the paraformaldehyde was removed, and 70% alcohol was added to the well. Alcohol was then removed, after which the matrigel containing the spheroids was removed and placed onto a thin layer of histogel. Another layer of molten histogel was added on top of the spheroids. This mixture was allowed to dry for a couple of minutes before being embedded into paraffin blocks. Sections of these paraffin blocks were stained with H&E as well as anti-p63, anti-CK8/18, and anti-laminin-332 antibodies.
Results
CHD1 protein expression is varied in prostate cell lines
Western blot detecting CHD1 showed variable protein expression among prostate cell lines [Figure 1(a)]. The prostate cancer cell line, DU145, and normal prostate epithelial cell line, RWPE-1, showed high expression of CHD1 protein. In contrast, the prostate cancer lines, VCaP, LNCaP, and PC3 expressed lower levels of CHD1. NCI-H660, a neuroendocrine cell line, expressed the lowest levels of CHD1. We chose the normal prostate epithelial cell line, RWPE-1, to knockout CHD1.
Figure 1.

Generation of RWPE-1 CHD1 KO line. (a) Western blot showing CHD1 expression across a panel of prostate cancer cell lines (NCI H660, C4-2, LNCaP, VCap, DU145 and PC3) and the normal prostate epithelial cell line, RWPE-1. CHD1 expression is highest in RWPE-1 as compared to the cancer cell lines which show variable levels of CHD1. (b) Top panel: Gel run image representative of genomic PCR products from clones targeted with sgRNA designed for the CHD1 gene (Cr2, Cr13, Cr16, Cr21) and with the non-target vector control (NT1, NT2, NT6, NT15). The Exon 3b forward and reverse primers used, amplified a 1 kb region in exon 3 of the CHD1 gene that includes the sgRNA sequence upstream of the PAM sequence and the Cas 9 target sequence. Once Cas9 enzyme makes sgRNA/PAM specified cuts in the genomic DNA, the DNA repair process of nonhomologous end joining can cause insertions and or deletions in the DNA leading to presence of heteroduplexes along the DNA double strand. T7 endonuclease is known to nick dsDNA in heteroduplex regions. Bottom panel: Gel run of products resulting from digestion of the 1 kb genomic PCR product with T7 endonuclease, following denaturation and reannealing. Cr2, Cr13, Cr16 and Cr21 show the additional low molecular weight fragments representative of heteroduplex formation resulting from insertions/deletions. NT1, NT2, NT6 and NT15 show only a 1 kb product indicating that SgRNA/PAM based specific Cas9 digestion has not taken place in the non-target controls. (c) Western blot confirming CHD1 expression in NT2, NT6, and NT15 and loss of expression in Cr2, Cr16, and Cr21. Cr13 showed expression of CHD1. (d) Clones were subsequently confirmed as wildtype or mutant through Sanger sequencing. RWPE-1 and NT2 showed the wildtype CHD1 sequence. Cr2 and Cr21 showed an insertion of T and A, respectively, shown in boxes. Cr21 showed a 3-base deletion, demonstrated as dashes. Cr16 demonstrated a 10-base deletion, shown as dashes.
CHD1, chromodomain helicase DNA binding protein 1; dsDNA, double-stranded DNA; NT, non-target cells; PCR, polymerase chain reaction; PAM, protospacer adjacent motif; sgRNA, small guide RNA.
Abolition of CHD1 expression in RWPE-1 cells by CRISPR/Cas 9 gene knock-out strategy
As described, RWPE-1 cells were transduced with either LVP-202, containing a small guide RNA (sgRNA) against the target region in exon 3 of CHD1, or LVP-NT. Positive clones were selected by GFP-based single cell sorting. The T7 endonuclease assay confirmed that Cas9 had targeted the exon 3 of CHD1. Genomic PCR with exon 3b forward/reverse primers amplified the 1 kb target region [Figure 1(b)]. The T7 endonuclease assay showed that CHD1-targeted clones Cr2, Cr13, Cr16, and Cr21 had the higher molecular weight genomic PCR product along with two other smaller fragments [Figure 1(b)]. The results were reconfirmed with an additional set of genomic PCR primers.
CHD1 expression analysis by western blot showed that while NT2, NT6, and NT15 showed CHD1 protein expression, the CHD1-targeted clones, Cr2, Cr16, and Cr21 showed loss of CHD1 expression [Figure 1(c)]. Genomic PCR products from Cr2, Cr16, and Cr21 were cloned into a TOPO TA vector, and individual colonies were sequenced. A representation of CHD1 sequences in the Cas9 target region from RWPE-1 including NT2, Cr2, Cr16, and Cr21 shows insertions and deletions in CHD1-targeted clones [Figure 1(d)], which include a single base insertion (Cr2), a 15-base deletion (Cr16), and a single base insertion and three base deletion (Cr21).
CHD1 KO cells show change in morphology
Under 2D culture conditions in standard tissue culture plates, parental RWPE-1 cells and nontarget control NT2 cells have a spindle, epithelioid morphology, while the CHD1 KO cells Cr2, Cr16, and Cr21 are smaller and rounder (Figure 2). In early, less dense cultures, CHD1 KO cells Cr2, Cr16, and Cr21 were observed to grow in a widespread manner, while RWPE-1 and NT2 cells grew more compactly after plating (image not shown). At time of passage, CHD1 KO clones were observed to adhere to regular tissue culture cell ware in 24–48 h, whereas the parental RWPE-1 and NT2 cells adhered in 4–6 h.
Figure 2.

Morphological changes in CHD1 KO cells 20× magnification phase contrast images of the parent, RWPE-1, non-target control, NT-2, and the RWPE-1 CHD1 KO lines, Cr2, Cr16, and Cr21 cultured on standard adherent tissue culture plates. RWPE-1 and NT2 cells have a spindly epithelioid morphology, while the CHD1 KO cells are rounder and smaller.
CHD1, chromodomain helicase DNA binding protein 1; KO, knockout.
CHD1 KO cells show lowered expression of adhesion molecules and ECM proteins
ECM and adhesion molecules expressed in RWPE-1, NT2, and CHD1 KO clones (Cr2 and Cr21) were analyzed using an RT2 profiler PCR array. Figure 3(a) shows genes that changed more than two-fold in CHD1 KO cells compared to parental RWPE-1 cells. ITGA2 was down-regulated, while ITGA4 is up-regulated in CHD1 KO cells. Integrin ligands such as collagen (COL16A1, COL4A2, COL5A1, and COL6A2),4 and the basement membrane component, LAMB3, were downregulated in CHD1 KO cells. The most dramatically down-regulated ECM components in CHD1 KO cells were MMP2, SPARC, and vitronectin (VTN). Other ECM genes that were down-regulated included contactin 1 (CNTN1), ECM1, MMP3, TIMP3, and tenascin C (TNC), whereas MMP12 and selectin L (SELL) were up-regulated.
Figure 3.

Expression of extracellular matrix proteins and adhesion molecules in CHD1 KO cells. (a) Graph demonstrating the expression of genes that were altered at least two-fold in the NT2 and CHD1 KO lines, Cr16 and Cr21, compared to RWPE-1. ITGA2 was down-regulated, while ITGA4 is up-regulated in CHD1 KO cells. The integrin ligands, collagen (COL16A1, COL4A2, COL5A1 and COL6A2), FN1, and the laminin component, LAMB3, are downregulated in CHD1 KO cells. The most down-regulated ECM components in the CHD1 KO cells were MMP2, SPARC and VTN. The ECM proteins ITGA4, MMP12, and SELL were up regulated. (b) Plot depicting levels of secreted SPARC protein as detected by ELISA. High levels of SPARC are secreted by RWPE-1 compared to the CHD1 KO lines (**p < 0.01). NT2 cell lines secreted higher levels of SPARC than RWPE-1 cell lines (*p < 0.05). Error bars represent standard deviation. (c) Top panel: Western blot showing levels of SPARC in 30 μg of lysate from RWPE-1, NT2, Cr2, Cr16, and Cr21. SPARC is detectable in RWPE-1 and NT2 but not in the CHD1 KO clones. Bottom panel: loading control. (d) Plot depicting levels of secreted MMP2 protein as detected by ELISA. High levels of MMP2 are secreted by RWPE- 1 compared to the CHD1 KO lines (**p < 0.01). NT2 cell lines secreted higher levels of MMP2 than RWPE-1 cell lines (*p < 0.05). Error bars represent standard deviation. (e) Western blot showing levels of TNC (top panel), VTN (middle panel) and GAPDH (lower panel) in the five cell lines. Cr2 and Cr21 express low levels of TNC compared to RWPE-1 and NT2, while Cr16 expresses TNC levels similar to the parental lines. Cr2 and Cr21 express lower levels of VTN than RWPE-1, while Cr16 expresses similar levels. Bar graphs below show levels of TNC (left) and VTN (right) relative to GAPDH in the five lines.
CHD1, chromodomain helicase DNA binding protein 1; ECM, extracellular matrix; ELISA, enzyme-linked immunosorbent assay; FN1, fibronectin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ITGA2/4, integrin subunit alpha 2/4; KO, knockout; LAMB3, laminin subunit beta-3 precursor; MMP2/12, matrix metalloproteinase 2/12; NT2, non-target cells; SPARC, secreted protein acidic and rich in cysteine; TNC, tenascin; VTN, vitronectin.
RT2 profiler PCR array results for SPARC and MMP2 proteins were further validated by ELISA. In the three CHD1 KO clones, Cr2, Cr16, and Cr21, SPARC and MMP2 were secreted to significantly lesser levels when compared to RWPE-1 and NT2 cells [Figures 3(b) and (d)].
On western blot, SPARC was detected in RWPE-1 and NT2 whole cell lysates but was undetectable in CHD1 KO clones [Figure 3(c)]. TNC and VTN levels were also lower in the protein lysates from KO lines compared to lysates from parental lines [Figure 3(e)].
CHD1 KO cells show altered cell adhesion properties
Since CHD1 KO cells expressed lower levels of integrins and their ligands collagen type I (COLL I), FN1, and laminin,25 we examined how that affected cell adhesion to coated plates. RWPE-1, NT2, Cr2, Cr16, and Cr21 cells were plated onto standard and COLL I, FN1, and LN-coated plates. On day 3, RWPE-1 and NT2 were observed to have fewer cells in suspension than CHD1 KO clones (image not shown). The percent viability of cells on day 13 was similar between adherent and non-adherent cells from all plates [Figure 4(a), left and right]. All five cell lines showed similar numbers of viable adherent cells on standard, FN1, and LN plates. However, Cr16 and Cr21 showed significantly more viable adherent cells than RWPE-1, NT2, and Cr2 on COLL I-coated plates [Figure 4(b), left]. In addition, Cr16 and Cr21 showed significantly more viable suspended cells than all other lines [**p < 0.01, Figure 4(b), right]. An analysis of viable suspended cells relative to total viable cells showed that CHD1 KO cell lines have more viable cells in suspension than RWPE-1 and NT2 on FN1 and laminin coated plates [Figure 4(c)].
Figure 4.

CHD1 KO cells show more viable suspended cells than either RWPE-1 or NT2. (a) Plots depicting percent viability of cells growing in either an adherent (left) or suspended (right) fashion. In all groups, all cell lines showed similar viability. Percent viability of adherent cells was higher than that of suspended cells. Plots represent data from biological triplicates. Error bars represent standard deviation. (b) Plots demonstrating the number of viable cells from each of the five cell lines growing in either an adherent (left) or suspended (right) fashion on coated plates. Viable adherent cell numbers were not significantly different among the five lines on standard, laminin or fibronectin coated places. More Cr16 and Cr21 cells adhered on collagen I coated plates than RWPE-1, NT2, or Cr2 (**p < 0.01). In addition, more Cr16 and Cr21 cells grew in suspension compared to RWPE-1, NT2, and Cr2 on all coated plates tested (**p < 0.01). Error bars represent SD. (c) Plot depicting percent of viable suspended cells in total viable cells on different plates. Cr2, Cr16, and Cr21 had the most viable suspended cells on fibronectin and laminin coated plates. Error bars represent SD
CHDI, chromodomain helicase DNA binding protein 1; KO, knockout; NT2, non-target cells; SD, standard deviation.
Reduced pAKT signaling in CHD1 KO cells
Levels of pAKT, pErk and pMEK in lysates from parental and CHD1 KO cells grown on regular plates were evaluated with western blots (Figure 5). While RWPE-1, NT2, Cr2, Cr16, and Cr21 expressed similar amounts of total Erk, Cr2 expressed slightly less pErk compared to the others [Figure 5(a), left]. However, there was no significant difference in the ratio of pErk to Erk levels between cell lines [Figure 5(a), right]. Compared with the CHD1 KO clones, RWPE-1 and NT2 express high levels of pAKT [Figure 5(b), left]. While pAKT/AKT levels were not significantly different between RWPE-1 and NT2, there was a significant difference between RWPE-1 and the CHD1 KO lines [Figure 5(b), right]. Finally, there was no difference in expression of pMEK or pMEK/MEK levels between RWPE-1 and the CHD1 KO lines (Figure 5c).
Figure 5.
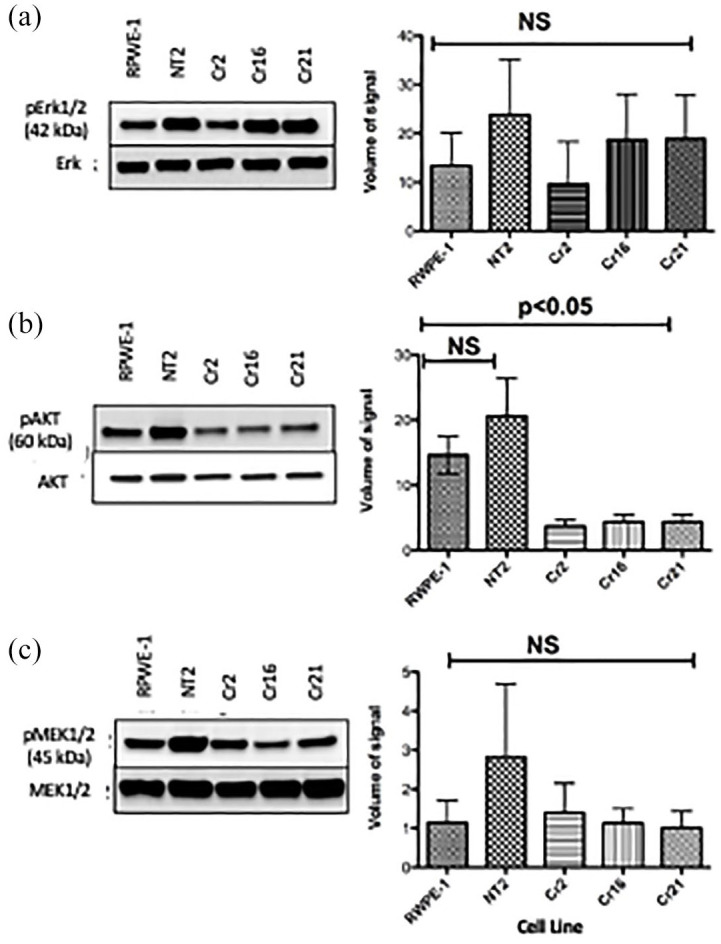
CHD1 KO cells Cr2, Cr16, and Cr21 show decreased pAKT levels. (a) Left: Western blot showing similar pErk expression in all cell lines. Right: Plot demonstrating pErk/Erk signal volume. There was no significant difference in pErk/Erk between cell lines. Error bars represent standard deviation. (b) Left: Western blot demonstrating that CHD1 KO lines express significantly lower pAKT levels than RWPE-1 and NT2 cell lines. Right: Plot depicting greater levels of pAKT/AKT in RWPE-1 and NT2 cell lines than CHD1 KO cell lines. Error bars represent SD. (c) Left: Western blot showing similar levels of pMEK in the five cell lines. Right: Plot depicting pMEK/MEK signal volume. There was no significant difference in pMEK/MEK between cell lines. Error bars represent standard deviation.
CHD1, chromodomain helicase DNA binding protein 1; KO, knockout; pAKT, protein kinase B; pERK, protein extracellular signal-regulated kinase; pMEK, protein mitogen-activated protein kinase; NT2, non-target cells; SD, standard deviation.
Spheroids/acini derived from CHD1 KO cells secrete a compromised basal lamina in both matrigel cultures and in vivo
RWPE-1, NT2, Cr2, Cr16, and Cr21 cells grown in matrigel formed both regular (compact) spheroids as well as irregular spheroids [Figure 6(a)]. Despite plating an equal number of viable cells at the start of the spheroid growth experiment, CHD1 KO lines produced more irregular spheroids than wildtype lines [Figure 6(b)]. The number of compact spheroids was not affected. Further analysis, based on limiting dilutions, showed that the KO cells required fewer cells than either RWPE-1 or NT2 to form a spheroid [Figure 6(c)]. Under microscopic analysis, cells in the spheroids from RWPE-1, NT2, Cr2, Cr16, and Cr21 lines expressed the basal cell marker p63 (Figure 7, row 1), whereas spheroids from all five lines are negative for the luminal cell marker CK8/18 (images not shown). Figure 7, row 2, shows the fluorescent red laminin-332 stain of spheroids from all five cell lines. RWPE-1 and NT2 spheroids demonstrate bright red fluorescence. However, very low levels of laminin-332 were expressed in the CHD1 KO spheroids, as evidenced by the minimal expression of red fluorescence in the Cr2, Cr16, and Cr21 spheroids. In addition, compact spheroids from all lines displayed laminin-332 stain on the outer edge of the spheroid whereas irregular spheroids demonstrated laminin-332 stain within the cluster.
Figure 6.

Generation of spheroids/acini in matrigel cultures. (a) Compact and irregular spheroids generated from RWPE-1, NT2, Cr2, Cr16, and Cr21 cell lines after being mixed with matrigel and grown for 10 days. (b) Plot depicting the number of compact and irregular spheroids from the five cell lines. All lines produced more irregular than compact spheroids. CHD1 KO lines generated more irregular spheroids than parental cells (*p < 0.05, **p < 0.01). Data is from biological triplicates, and error bars represent SD. (c) Plot showing the number of cells that need to be plated to form an acinus from each cell line. RWPE-1 requires the largest number of cells, while Cr21 requires the fewest. Error bars represent SD.
CHD1, chromodomain helicase DNA binding protein 1; KO, knockout; NT2, non-target cells; SD, standard deviation.
Figure 7.

Evaluation of p63 and laminin-332 expression in spheroids/acini derived from CHD1 KO cells in both matrigel cultures and in vivo. (Row a) p63 (basal cell component) staining of RWPE-1, NT2, Cr2, Cr16, and Cr21 spheroids grown in Matrigel. All spheroid cells expressed p63. (Row b) Laminin-332 (basal lamina component) staining of the spheroids in row A under IF. All spheroids displayed the laminin-332 stain in the outer periphery when compact and irregular staining when irregularly shaped. Spheroids from RWPE-1 and NT2 lines are brightly stained with laminin-332, while CHD1 KO lines show significantly dimmer staining. (Row c) H&E staining of tissue from nude mice subcutaneously injected with RWPE-1, NT2, Cr2, Cr16, and Cr21 cells. All cell lines formed acini of regular and irregular shapes. Acini from CHD1 KO cells are larger, more irregular, and more dispersed than RWPE-1 and NT2. (Row d) Laminin-332 staining of the acini in row C under IF. RWPE-1 and NT2 acini demonstrated bright staining, while Cr2, Cr16, and Cr21 cell lines display significantly less staining. White bars represent 100 μm.
CHD1, chromodomain helicase DNA binding protein 1; H&E, hematoxylin and eosin; KO, knockout.
When grown in nude mice, RWPE-1, NT2, Cr2, Cr16 and Cr21 cells did not form overt tumors but showed different growth characteristics. Using H&E staining, we observed that RWPE-1 cells formed fewer and more regular acini; in contrast, NT2 cell lines grew acini with extensively keratinized centers. The KO cells formed larger, irregular shaped acini that were more dispersed throughout the tissue (Figure 7, row 3). Staining of acinar basal laminae with anti-laminin-332 showed prominent red fluorescence on the outer edges of acinar structures in RWPE-1 and NT2 lines. Similar to the in vitro CHD1 KO spheroids, in vivo acini of CHD1 KO cells demonstrated low levels of red fluorescent laminin-332 expression (Figure 7, row 4). In addition, in vivo, KO cells formed larger acini with thick basal cell layers, as indicated by DAPI and p63 stains (image not shown). More specifically, Cr21 cell lines grew large acini with very irregular edges and keratinized centers. Laminin-332 stain was weakest at the irregular edges. Acini from all cell lines stained positive for basal cell marker p63 and negative for luminal cell marker CK8/18 (images not shown).
Discussion
Prostate cancer tumors have been classified into different molecular subtypes based on exome and whole genome sequencing of tumors. Two of the most common subtypes include ETS fusion-positive, CHD1 positive (ETS+CHD1+) and ETS fusion-negative, CHD1 negative (ETS−CHD1−) PCa.26,27 CHD1 is thought to be a major contributor to genomic stability and gene transcription, as it plays a role in chromothripsis, DNA repair, and DNA methylation.19,22,28 Epigenetic profiling has shown that of 27,000 significant CpG sites analyzed, 3103 sites in 1962 genes showed at least a 10% difference between ETS fusion-positive and negative PCa tumors.13 In recent years, epigenetic and sequencing studies of ETS fusion-positive and negative PCa genomes have revealed key differences between the two that could be attributed to their CHD1 status. Genomic rearrangements in Ets−CHD1− PCa genomes were found to be predominantly intrachromosal, compared to Ets+CHD1+ genomes which were found to be mostly interchromosomal.29
In our study, we knocked out CHD1 in the nontumorigenic prostate epithelial cell line, RWPE-1, which is ETS fusion negative, PTEN positive, and highly expresses CHD1.30,31 Analysis of the CHD1 KO clones showed altered cell adhesion properties and morphology, a reduction in basal pAKT levels, and a reduction in laminin, as compared with the nontarget control clone, NT2, and parental RWPE-1 cells.
Our analysis of ECM and adhesion molecule gene expression showed significant downregulation of ITGA2. Integrins are heterodimeric proteins, consisting of α and β subunits, expressed on the cell surface which act as receptors for ECM proteins. Alterations of integrin expression levels in cancer cells correlate with changes in invasiveness and tumor progression.32 In mouse models, integrin α2β1 is critical for the suppression breast cancer metastasis in vivo; in contrast, its re-expression in breast cancer cells have been demonstrated to abrogate the malignant phenotype.33,34 In immunohistochemical analysis of prostatic adenocarcinomas, integrin α2 was found to be downregulated in 70% of primary tumors but paradoxically frequently upregulated in corresponding lymph node metastases.35 In our study, CHD1 KO cells demonstrated decreased expression of integrin α2 and two of its ligands, as well as downregulation of other ECM proteins. This finding may contribute to the increased number of non-adherent cells observed in the culture. Growth of CHD1 KO cells on plates coated with different matrices showed the same anchorage-independence property. The ability of cells to be anchorage-independent and survive the anoikis process are two important hallmarks of tumor cells.36 Our results indicate that CHD1 may contribute to both events. More CHD1 KO cells survive than parental or nontarget cells in suspension. The increased survival of non-adherent cells and the spreading of sparsely-plated 2D cultures of CHD1 KO cells indicate a role for CHD1 in cell-ECM and cell-cell interactions.
It has been shown previously that protein levels of integrin β1C, an inhibitor of cell proliferation, are reduced in prostate carcinoma compared to normal prostate cells.37 For normal cells, adhesion to the ECM is an important cue for cell survival processes mediated via focal adhesion kinase activation and phosphorylation of AKT.36 In the absence of cell-ECM interactions, anchorage-dependent cells undergo anoikis by caspase 8-integrin-mediated cell death. Cells can escape apoptosis by suppressing integrin expression. If CHD1 loss downregulates integrins, integrin-mediated cell-ECM adhesion may be abolished, allowing cells to spread to distant niches and permitting tumor progression.38 Once the cells arrive, they can adapt to the new niche by changing the repertoire of integrins secreted, allowing for cell adhesion.
In the current study, there was no change in the levels of phosphorylated ERK or MEK when comparing CHD1 KO cell lines with RWPE-1 cells. In contrast, levels of phosphorylated AKT were significantly reduced in the 2D culture model. Harma et al.39 observed increased phosphorylation of AKT for multiple prostate cell lines, including RWPE-1 cells at 2 weeks in the 3D culture models as compared to 2D cultures. Our finding of reduced AKT phosphorylation requires further evaluation to determine the interplay between CHD1 and AKT signaling, culture model type, and the phenotypes presented herein.
A meta-analysis of 18 gene array datasets looking at transition from normal to localized PCa and from localized to metastatic PCa showed that genes for cell adhesion, tight junction signaling, and integrin signaling were downregulated in both transitions. Along with the integrins, their ligands also tended to be downregulated. Levels of integrin ligands including COL4A6, COL13A1, FGB, COL19A1, COL18A1, COL14A1, and COL1A2, negatively correlated with Gleason score.40 In the current study, CHD1 KO cells demonstrated a downregulation of integrin ligands, suggesting that CHD1 loss could trigger tumorigenesis or contribute to a more aggressive phenotype.
We also observed a downregulation of SPARC and MMP2 in CHD1 KO cells. Previous studies in animal models suggest that SPARC and MMP2 play a role in cell adhesion. Knockdown of SPARC decreased cell-cell adhesion in post gastrula development in Xenopus laevis.41 Furthermore, SPARC deletion in transgenic adenocarcinoma of mouse prostate (TRAMP) mice led to enhanced development and progression of cancer.42 In addition, our data demonstrate upregulation of ITGA4, MMP12, and SELL, three proteins involved in cell invasion and tumorigenesis,43–45 all of which could lead to increased motility of CHD1 KO cells.
An earlier study of a lentiviral mediated short hairpin RNA (shRNA) knock down of CHD1 in RWPE-1 and another nontumorigenic prostate epithelial cell line, OPNC2, reported an increase in cellular invasion. In addition, in OPCN2, increased clonogenicity was observed.30 A similar CHD1 knock down approach in mouse prostate epithelial cells (MPECs) also showed an increase in clonogenicity, invasiveness, and survivability. However, renal grafting of these cells into mice resulted in no tumors.15 In these studies, CHD1 was only partially knocked out; in contrast, in our study, CHD1 was completely knocked out, allowing the KO cells to form more acini that were larger and more spread out in the subcutaneous layer, compared to RWPE-1 and NT2 lines. This observation suggests that the deletion of CHD1 and the ensuing downregulation of cell adhesion proteins might cause these cells to spread.
Laminin is a major ECM protein component of the basement membrane and consists of various combinations of different α, β, and γ chains. Thus laminin-332 consists of α3, β3 and γ2 chains. In our study, CHD1 KO cells showed a downregulation of laminin β3 (LAMB3), and subsequently, very weak staining with laminin-332, which potentially could compromise the basement membrane structure. Moreover, in some acini, particularly those from Cr21, the laminin-332 stain was discontinuous. In vivo data from this study must be interpreted with caution due to limited number of observations. In a recent study of spheroids from an integrin β4 knock down in RWPE-1 cells, the laminin-332 layer was found to be discontinuous, thus facilitating an invasive budding phenotype. This phenotype is comparable to the abnormal luminal invasion seen in high grade prostate intraepithelial neoplasia.46 CHD1 deletion leads to downregulation of ECM and adhesion molecules, which in turn, allows cells to detach from the basement membrane and potentially metastasize. Loss of CHD1 in fusion negative tumors could mediate escape from anoikis, and we speculate that this may be an initial step of tumorigenesis.
Conclusion
Deletion of CHD1 in ETS fusion negative and PTEN wild type normal prostate epithelial cells downregulates the levels of adhesion molecules and ECM proteins, causing alterations in cell-cell and cell-matrix interactions. This dysregulation leads to compromise of the basal lamina, potentially allowing cells to move out of acini into the surrounding tissue; as a result, they are able to contribute to the initiation and progression of prostate cancer.
Acknowledgments
The authors would like to thank the Genome Editing Core Facility of the Rutgers Child Health Institute of New Jersey for the lentiviral CRISPR constructs, the Tissue Analytical Services of the Rutgers Cancer Institute of New Jersey for H&E and IHC staining of slides, and the Biomedical Informatics Shared Resource of the Rutgers Cancer Institute of New Jersey (P30CA072720) for the digital scanning of IF slides.
Footnotes
Conflict of interest statement: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Eric A. Singer receives support from Astellas/Medivation. Kim M. Hirshfield currently works at MRL, Merck & Co., Inc. The other authors declare no conflicts of interest.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH P30CA072720, UM1, C.R. Bard Foundation, and the Walter and Louise Sutcliffe Foundation.
ORCID iD: Alexandra L. Tabakin  https://orcid.org/0000-0003-4046-2966
https://orcid.org/0000-0003-4046-2966
Contributor Information
Aparna Kareddula, Department of Medicine, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Daniel J. Medina, Department of Medicine, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Whitney Petrosky, Department of Medicine, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Sonia Dolfi, Department of Medicine, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Irina Tereshchenko, Department of Medicine, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Kelly Walton, Department of Medicine/Division of Medical Oncology, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Hana Aviv, Department of Pathology and Laboratory Medicine, Rutgers -Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Evita Sadimin, Section of Urologic Pathology, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA.
Alexandra L. Tabakin, Section of Urologic Oncology, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA
Eric A. Singer, Section of Urologic Oncology, Rutgers Cancer Institute of New Jersey/Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA
Kim M. Hirshfield, MRL, Merck & Co., Inc., 2000 Galloping Hill Rd, Kenilworth, NJ, USA.
References
- 1.National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: prostate cancer, https://seer.cancer.gov/statfacts/html/prost.html (2020, accessed 1 November 2020).
- 2.Freedland SJ, Humphreys EB, Mangold LA, et al. Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. JAMA 2005; 294: 433–439. [DOI] [PubMed] [Google Scholar]
- 3.Kupelian PA, Mahadevan A, Reddy CA, et al. Use of different definitions of biochemical failure after external beam radiotherapy changes conclusions about relative treatment efficacy for localized prostate cancer. Urology 2006; 68: 593–598. [DOI] [PubMed] [Google Scholar]
- 4.Tabakin AL, Sadimin ET, Tereshchenko I, et al. Correlation of prostate cancer CHD1 status with response to androgen deprivation therapy: a pilot study. J Genitourin Disord 2018; 2: 1006. [PMC free article] [PubMed] [Google Scholar]
- 5.Shorning BY, Dass MS, Smalley MJ, et al. The PI3K-AKT-mtor pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci 2020; 21: 4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler DE, Marlein C, Walker HF, et al. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 2017; 8: 56698–56713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM.Recurrent gene fusions in prostate cancer. Nat Rev Cancer 2008; 8: 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollenhorst PC, Ferris MW, Hull MA, et al. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev 2011; 25: 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015; 161: 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai C, Wang H, He HH, et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest 2013; 123: 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunderfranco P, Mello-Grand M, Cangemi R, et al. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS One 2010; 5: e10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun C, Dobi A, Mohamed A, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 2008; 27: 5348–5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geybels MS, Alumkal JJ, Luedeke M, et al. Epigenomic profiling of prostate cancer identifies differentially methylated genes in TMPRSS2:ERG fusion-positive versus fusion-negative tumors. Clin Epigenetics 2015; 7: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012; 44: 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu W, Lindberg J, Sui G, et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene 2012; 31: 3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skene PJ, Hernandez AE, Groudine M, et al. The nucleosomal barrier to promoter escape by RNA polymerase II is overcome by the chromatin remodeler Chd1. Elife 2014; 3: e02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ocampo J, Chereji RV, Eriksson PR, et al. The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res 2016; 44: 4625–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura K, Tsuzuki T, Kato M, et al. Prognostic value of intraductal carcinoma of the prostate in radical prostatectomy specimens. Prostate 2014; 74: 680–687. [DOI] [PubMed] [Google Scholar]
- 19.Siggens L, Cordeddu L, Ronnerblad M, et al. Transcription-coupled recruitment of human CHD1 and CHD2 influences chromatin accessibility and histone H3 and H3.3 occupancy at active chromatin regions. Epigenetics Chromatin 2015; 8: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaspar-Maia A, Alajem A, Polesso F, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 2009; 460: 863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh FM, Lizama CO, Wong P, et al. Emergence of hematopoietic stem and progenitor cells involves a Chd1-dependent increase in total nascent transcription. Proc Natl Acad Sci U S A 2015; 112: E1734–E1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kari V, Mansour WY, Raul SK, et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep 2016; 17: 1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Y, Smyth GK.ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009; 347: 70–78. [DOI] [PubMed] [Google Scholar]
- 24.Pinto MP, Jacobsen BM, Horwitz KB.An immunohistochemical method to study breast cancer cell subpopulations and their growth regulation by hormones in three-dimensional cultures. Front Endocrinol (Lausanne) 2011; 2: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindskog M, Laurell A, Kjellman A, et al. A randomized phase II study with ilixadencel, a cell-based immune primer, plus sunitinib versus sunitinib alone in synchronous metastatic renal cell carcinoma. J Clin Oncol 2020; 38(Suppl. 5): 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012; 487: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shtivelman E, Beer TM, Evans CP.Molecular pathways and targets in prostate cancer. Oncotarget 2014; 5: 7217–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belden WJ, Lewis ZA, Selker EU, et al. CHD1 remodels chromatin and influences transient DNA methylation at the clock gene frequency. PLoS Genet 2011; 7: e1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baca SC, Prandi D, Lawrence MS, et al. Punctuated evolution of prostate cancer genomes. Cell 2013; 153: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang S, Gulzar ZG, Salari K, et al. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene 2012; 31: 4164–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bello D, Webber MM, Kleinman HK, et al. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis 1997; 18: 1215–1223. [DOI] [PubMed] [Google Scholar]
- 32.Desgrosellier JS, Cheresh DA.Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010; 10: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramirez NE, Zhang Z, Madamanchi A, et al. The α2β1 integrin is a metastasis suppressor in mouse models and human cancer. J Clin Invest 2011; 121: 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zutter MM, Santoro SA, Staatz WD, et al. Re-expression of the alpha 2 beta 1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc Natl Acad Sci U S A 1995; 92: 7411–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonkhoff H, Stein U, Remberger K.Differential expression of alpha 6 and alpha 2 very late antigen integrins in the normal, hyperplastic, and neoplastic prostate: simultaneous demonstration of cell surface receptors and their extracellular ligands. Hum Pathol 1993; 24: 243–248. [DOI] [PubMed] [Google Scholar]
- 36.Guadamillas MC, Cerezo A, Del Pozo MA.Overcoming anoikis– pathways to anchorage-independent growth in cancer. J Cell Sci 2011; 124: 3189–3197. [DOI] [PubMed] [Google Scholar]
- 37.Perlino E, Lovecchio M, Vacca RA, et al. Regulation of mRNA and protein levels of beta1 integrin variants in human prostate carcinoma. Am J Pathol 2000; 157: 1727–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gassmann P, Haier J.The tumor cell-host organ interface in the early onset of metastatic organ colonisation. Clin Exp Metastasis 2008; 25: 171–181. [DOI] [PubMed] [Google Scholar]
- 39.Harma V, Virtanen J, Makela R, et al. A Comprehensive panel of three-dimensional models for studies of prostate cancer growth, invasion and drug responses. PLoS One 2010; 5: e10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorlov IP, Byun J, Gorlova OY, et al. Candidate pathways and genes for prostate cancer: a meta-analysis of gene expression data. BMC Med Genomics 2009; 2: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huynh MH, Zhu SJ, Kollara A, et al. Knockdown of SPARC leads to decreased cell-cell adhesion and lens cataracts during post-gastrula development in Xenopus laevis. Dev Genes Evol 2011; 220: 315–327. [DOI] [PubMed] [Google Scholar]
- 42.Said N, Frierson HF, Chernauskas D, et al. The role of SPARC in the TRAMP model of prostate carcinogenesis and progression. Oncogene 2009; 28: 3487–3498. [DOI] [PubMed] [Google Scholar]
- 43.Chung IC, Chen LC, Chung AK, et al. Matrix metalloproteinase 12 is induced by heterogeneous nuclear ribonucleoprotein K and promotes migration and invasion in nasopharyngeal carcinoma. BMC Cancer 2014; 14: 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wen X, Wang H, Chai P, et al. An artificial CTCF peptide triggers efficient therapeutic efficacy in ocular melanoma. Mol Ther Oncolytics 2020; 18: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie J, Yang P, Lin HP, et al. Integrin α4 up-regulation activates the hedgehog pathway to promote arsenic and benzo[α]pyrene co-exposure-induced cancer stem cell-like property and tumorigenesis. Cancer Lett 2020; 493: 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang M, Nagle RB, Knudsen BS, et al. A basal cell defect promotes budding of prostatic intraepithelial neoplasia. J Cell Sci 2016; 130: 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]