

Summary
Coupling assay for transposase-accessible chromatin sequencing (ATAC-seq) with microfluidic separation and cellular barcoding has emerged as a powerful approach to investigate chromatin accessibility of individual cells. Here, we define a protocol for constructing single-cell ATAC-seq libraries from maize seedling nuclei and the preliminary computational steps for assessing data quality. This protocol can be readily adapted to other plant species or tissues with minor changes to reveal chromatin accessibility variation among individual cells.
For complete details on the use and execution of this protocol, please refer to Marand et al. (2021).
Subject areas: developmental biology, genomics, molecular biology, plant sciences, sequence analysis, sequencing, single cell
Graphical abstract
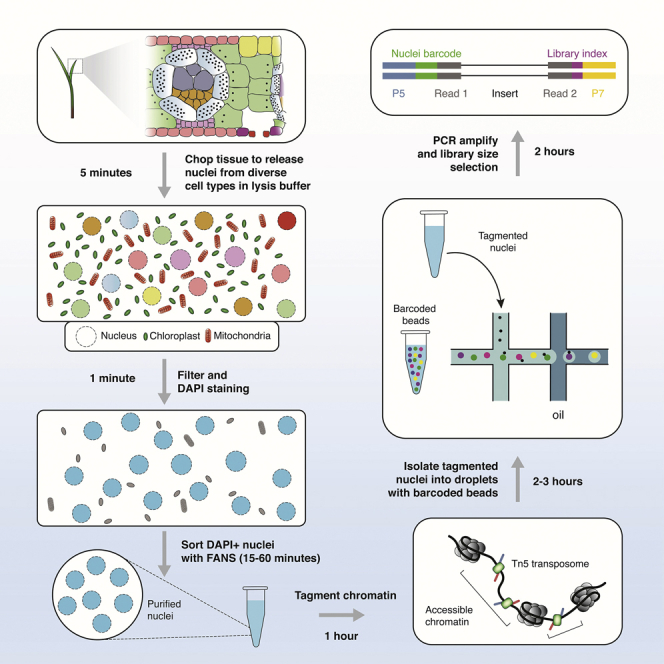
Highlights
-
•
Protocol to construct scATAC-seq libraries from plant samples
-
•
Computational pipeline to analyze scATAC-seq data
-
•
Example code to identify distinct cell states in Zea mays
Coupling assay for transposase-accessible chromatin sequencing (ATAC-seq) with microfluidic separation and cellular barcoding has emerged as a powerful approach to investigate chromatin accessibility of individual cells. Here, we define a protocol for constructing single-cell ATAC-seq libraries from maize seedling nuclei and the preliminary computational steps for assessing data quality. This protocol can be readily adapted to other plant species or tissues with minor changes to reveal chromatin accessibility variation among individual cells.
Before you begin
Experimental design
-
1.
In the following protocol, we describe the development of a single-cell ATAC-seq (scATAC-seq) library using freshly harvested Zea mays (maize) seedlings as an example. The steps described below can be applied to any plant species or tissue that is capable of producing intact nuclei suspensions (Lu et al., 2019; Ricci et al., 2019). In addition to 7-day old seedlings, this protocol has been successfully employed in axillary buds, pistillate inflorescence, staminate inflorescence, crown roots and embryonic roots of Zea mays, as well as the primary root of Arabidopsis thaliana. We detail nuclei isolation using fluorescence-activated nuclei sorting (FANS) but note that traditional wash and centrifugation steps may be taken for laboratories without access to cytometrical instruments with an increased likelihood of organeller contamination (Lu et al., 2017; Zhong et al., 2021).
-
2.
For experiments aiming to compare samples, such as different genotypes, case/control treatments, et cetera, it is imperative to construct all scATAC-seq libraries simultaneously to avoid batch effects (Lafzi et al., 2018). Currently, commercial microfluidic chips (10× Genomics) allow for up to eight samples to be processed in parallel. It is also important to consider that at least one biological replicate per sample is highly recommended for all experimental designs. For experimental designs with more than four samples, biological replicates should be constructed in a separate session to remediate technical effects. We stress that biological replicates are critical for establishing reproducibility of the tested conditions.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Tris base | Sigma-Aldrich | Cat No. 77861 |
| EDTA | Sigma-Aldrich | Cat No. E9884 |
| Spermine | Sigma-Aldrich | Cat No. 85590 |
| KCl | Sigma-Aldrich | Cat No. P9333 |
| NaCl | Sigma-Aldrich | Cat No. S7653 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat No. M6250 |
| Triton X-100 | Sigma-Aldrich | Cat No. X100 |
| 4′,6-Diamidino-2-phenylindole | Sigma-Aldrich | Cat No. D8417 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat No. A9418 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell ATAC Library and Gel Bead Kit v1.1 | 10× Genomics | Cat No. 1000176 |
| Deposited data | ||
| Raw and processed data: scATAC-seq from multiple organs of Zea mays | Marand et al., 2021 | GEO: GSE155178 |
| Experimental models: Organisms/strains | ||
| Maize: B73 | Maize COOP | PI: 550473 |
| Software and algorithms | ||
| Cellranger-atac v1.2 | Cell Ranger ATAC (10× Genomics) | https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/what-is-cell-ranger-atac |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Socrates | Marand et al. 2021 | https://github.com/plantformatics/Socrates |
| Other | ||
| Single-edge razor blade | Fisher Scientific | Cat No. 12-640 |
| Petri dishes | Fisher Scientific | Cat No. FB0875713 |
| Hemacytometer | Sigma-Aldrich | Cat No. Z359629 |
| 40 μm Mini Cell Strainer | pluriSelect | Cat No. 43-10040-40 |
| Beckman Coulter MoFlo Astrios EQ flow cytometer | Beckman Coulter | N/A |
| Sterile preservative free flow cytometry solution | BioSure | Cat No. 1027 |
| Leica TR600 Epifluorescence Microscope | Leica | N/A |
Materials and equipment
For quality analysis of single-cell data, researchers should have R version 4.0 or greater installed on a personal computer, server, or High Performance Computing (HPC) system.
Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 7.5 (1 M) | 15 mM | 750 μL |
| EDTA (0.5 M) | 2 mM | 200 μL |
| Spermine (1 M) | 0.5 mM | 25 μL |
| KCl (3 M) | 80 mM | 1670 μL |
| NaCl (5 M) | 20 mM | 200 μL |
| 2-ME (14.3 M) | 15 mM | 20 μL |
| TritonX-100 (15%) | 0.15% | 500 μL |
| ddH20 | n/a | 46.685 mL |
| Total | n/a | 50 mL |
Collection buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 7.5 (1 M) | 15 mM | 750 μL |
| Spermine (1 M) | 0.5 mM | 25 μL |
| KCl (3 M) | 80 mM | 1670 μL |
| NaCl (5 M) | 20 mM | 200 μL |
| 2-ME (14.3 M) | 15 mM | 20 μL |
| ddH20 | n/a | 47.335 mL |
| Total | n/a | 50 mL |
Lysis and Collection buffers can be stored at 4°C for several months without Spermine and 2-ME, which should be added the day of the experiment. Spermine stock should be stored at −20°C while all the remaining stocks can be stored at 2°C. For optimal results, prepare the Lysis and Collection Buffers fresh the same day as nuclei isolation experiments.
CRITICAL: 2-ME is a potential toxin and Spermine is a skin corrosive/irritant. Both should be handled under a fume hood for safety.
Alternatives: TritonX-100 can be substituted for other non-ionic detergents such as NP-40 (final concentration: 0.5%) or Tween-20 (0.5%).
Step-by-step method details
Sorting intact plant nuclei
Timing: [0.5–1 h]
Profiling chromatin accessibility requires isolating nuclei from heterogenous tissues (Minnoye et al., 2021). Tn5 can tagment organellar genomes with high efficiency due to a lack of nucleosomes on chloroplast and mitochondrial DNA (Lu et al., 2017). The following steps are designed to remove cellular and organellar debris and enrich for single nuclei. The gating strategy illustrated here has been optimized for maize nuclei, users may need to adjust the gating strategy specific to their organism. Growth and sampling conditions for published scATAC-seq data from maize and Arabidopsis thaliana tissues can be found in the STAR Methods section in Marand et. al.
-
1.
Approximately 3–4 B73 maize V1 seedlings (7–10 day old) were placed on a petri dish on ice and saturated with 500 mL of chilled Lysis buffer (Figures 1A and 1B).
-
2.
Using a sterilized single-edge razor blade, seedlings were chopped for 2 min to break the cell wall and release nuclei into solution (Figure 1C).
-
3.
The aqueous nuclei slurry was then filtered through a 40 μm cell strainer (Figure 1D).
Optional: Two layers of sterilized miracloth can be used in place of a 40 μm cell strainer.
-
4.
1 μL of DAPI (1 mg/mL) was added for every 200 μL of lysis buffer recovered (e.g., 2 μL DAPI for 400 μL of lysis buffer post filtering).
-
5.Sorting was performed on a Moflo Astrios EQ running sterile preservative free flow cytometry solution.
-
a.The instrument was set up with a 70 μm tip, sheath pressure at 60 psi and sample pressure at 60.1 psi.
-
b.Nuclei were run between 200 and 1000 events/second depending on their concentration.
-
c.Nuclei were first gated on log scaled forward scatter (FSC) versus log scaled side scatter (SSC) to eliminate debris (Figure 2A).
-
d.All subsequent data were collected in linear mode.
-
e.DAPI width versus DAPI area gating was used to eliminate doublets (Figure 2B).
-
f.The next gate was on a graph depicting green fluorescence vs. DAPI Area to eliminate any green fluorescing material (Figure 2C).
-
g.The final gate was on DAPI area selecting the first strong peak and all subsequent peaks depending on the downstream experiment (Figure 2D).
-
h.Sorting was performed in Purity 1 drop mode and triggered on DAPI fluorescence.
-
a.
-
6.
A total of 120,000 nuclei were collected across four 1.5 mL microcentrifuge catch tubes (30,000 nuclei in 200 μL of collection buffer, per tube) and immediately placed on ice.
Figure 1.

Sample preparation
(A) Seven-day old B73 maize leaf roll on pitri dish.
(B) Samples are saturated with 500 μL nuclear lysis buffer.
(C) Homogenized leaf tissue following two minutes of chopping in nuclear lysis buffer.
(D) Filtered lysate containing DAPI, nuclei, chloroplast, and mitochondrial contamination prior to flow sorting.
Figure 2.

Flow sorting gating strategy
(A) Forward scatter versus side scatter to eliminate debris.
(B) DAPI width versus DAPI area to remove nuclear aggregates.
(C) Green fluorescence versus DAPI area to exclude green fluorescing material.
(D) Distribution of events by DAPI area to collect nuclei at various stages of the cell cycle and ploidy.
Quantification and evaluation of nuclei quality
This step describes the evaluation of nuclei quality and concentration prior to generating scATAC-seq libraries.
-
7.
Four tubes containing nuclear suspensions (∼230 μL in each tube) were centrifuged at 500 RCF for 5 min at 4°C in swinging bucket centrifuge.
-
8.
The supernatant was discarded, leaving approximately 10 μL in the bottom of each tube.
-
9.
Nuclei pellets were resuspended in the remaining 10 μL of collection buffer by pipetting (10-times) and pooled in a single 1.5 mL centrifuge tube (total volume ∼ 40 μL).
-
10.Nuclei concentration and quality were evaluated on a hemacytometer under a fluorescent microscope (Figure 3).
-
a.The total number of nuclei can be determined by multiplying the concentration by the sample volume.
-
a.
-
11.
Input nuclei for the 10× Genomics scATAC-seq solution are required to be suspended in diluted nuclei buffer. To achieve this, nuclei were once again pelleted in a swinging bucket centrifuge at 500 RCF for 5 min at 4°C.
-
12.
The supernatant was discarded with a P200 filter pipette tip, leaving approximately 10 μL of collection buffer.
-
13.
The final 10 μL of collection buffer was discarded using a P10 filter pipette tip.
-
14.
Nuclei were resuspended in 10 μL of diluted nuclei buffer (10× Genomics) by 10-times pipetting with wide-bore P20 filtered tips.
Optional: An additional check of nuclei concentrations and quality can be performed using 1–2 uL of DAPI-stained nuclei with a hemacytometer under a fluorescent microscope. A loss of ∼50% of total nuclei is common during centrifugation.
-
15.
The concentration of nuclei was then adjusted to 3,200 nuclei per μL using diluted nuclei buffer (10× Genomics).
Figure 3.

Evaluating DAPI-stained nuclei quality under a fluorescent microscope
(A) 20× magnification of DAPI-stained nuclei via epifluorescence (top) and bright field (bottom) microscopy. The scale bar indicates 20 μm.
(B) Intact nuclei at 40× magnification via epifluorescence (top) and bright field (bottom) microscopy.
(C) Nuclei with intermediate ruptured nuclear envelopes at 40× magnification via epifluorescence (top) and bright field (bottom) microscopy.
(D) Lysed nuclei at 40× magnification via epifluorescence (top) and bright field (bottom) microscopy.
Single-cell ATAC-seq library preparation
-
16.
scATAC-seq libraries were then prepared following instructions from the manufacturer (10× Genomics) using 5 μL of nuclei in diluted nuclei buffer (∼16,000 nuclei). See the manual at https://assets.ctfassets.net/an68im79xiti/2NEwsG0Yu3RuxvtQiWXZo3/1e939394fa43a4f0bd88d79383833b16/CG000209_Chromium_NextGEM_SingleCell_ATAC_ReagentKits_v1.1_UserGuide_RevE.pdf
Notes for preparing single-cell ATAC-seq libraries
-
17.Maintaining nuclei integrity is one of the main factors for successfully preparing scATAC-seq libraries.
-
a.To avoid any nuclei damage, incubate the samples at 4°C (thermocycler) or on ice after the isothermal incubation period. It is important to have the Master Mix prepared as soon as the tagmentation is performed (usually 2 min before the reaction finishes).
-
b.Keep in mind that if the sample number is less than eight it is necessary to load the unused chip wells with 50% glycerol. An easy solution is to load unused wells while the tagmentation reaction is being performed (5–10 min before the reaction finishes).
-
c.For optimum loading, wait at least 30 s before adding glycerol in each sequential row.
-
a.
-
18.Optimal droplet formation on the Chromium microfluidics chip H requires a predictable flow rate to generate gel beads in emulsion (GEMs).
-
a.Any condition that impairs GEM formation, such as bubbles in the sample/master mix or gel beads well, should be avoided.
-
b.Slowly adding reagents and samples to the bottom of each well helps prevent bubble formation.
-
a.
-
19.
During double-sided size selection, the volumetric ratio of SPRIselect to the sample library specified in the manual (74 μL SPRIselect to 130 μL of library) ensures proper selection of amplified fragments by removing primer dimers and excessively large fragments. Collecting more sample volume than the recommended 130 μL in the manual may negatively affect library quality and recovery.
Expected outcomes
The quality of the scATAC-seq library preparation can be evaluated by assessing the library concentration via Qubit or qPCR and the distribution of fragment lengths using a fragment analyzer (Figure 4A) (Buenrostro et al., 2013, Buenrostro et al., 2015). In general, good quality scATAC-seq libraries will have concentrations greater than 5 ng/μL and exhibit a high proportion of nucleosome-free fragments (∼220bp) and periodic enrichment of fragments corresponding to mono-nucleosomes (∼330bp) and di-nucleosomes (∼500bp). Libraries lacking nucleosome-protected fragments or with low library concentration yields may indicate lysis of the nuclear envelope prior to tagmentation or a low recovery of nuclei (Figure 4B) (Troubleshooting 3).
Figure 4.

Fragment distributions of scATAC-seq libraries
(A) High quality scATAC-seq library. LM; Lower marker (1 bp). UM; Upper marker (6,000 bp).
(B) Poor quality scATAC-seq library. RFU; relative fluorescence units.
Following library sequencing and computational quality control/data processing, a successful scATAC-seq experiment will generally result in 40–60% of input nuclei passing quality control thresholds (example: 15,000 input nuclei yielding 6,000–9,000 high-quality nuclei). Several quality metrics, such as the number of unique Tn5 insertions per nucleus, percentage of reads mapping near transcription start sites (TSSs), and the Fraction of Reads in Peaks (FRiPs) should be assessed – keeping in mind that the distribution of these metrics are highly dependent on the species and organ or tissue-type. As a loose rule, nuclei should have at least 1,000 unique Tn5 insertions, fraction of reads near (1 or 2-kb depending on genome size and or number of genes) TSSs greater than 0.2, and FRiPs greater than 0.1. To establish reproducibility, biological replicates should be constructed and processed independently, and joined following identification of barcodes representing nuclei. Reproducible chromatin accessibility signals can be determined by assessing nuclei similarity according to the biological replicate of origin on the UMAP embedding. Louvain clusters containing approximately equal proportions of two or more biological replicates are generally regarded as reproducible. Additional details and an example of quality control and analytical steps can be found below.
Quantification and statistical analysis
The follow steps outline initial processing and quality control for Zea mays seedling scATAC-seq data generated by Marand et al. (Figure 5). For tutorials and example code for evaluating data quality and clustering with Socrates (Marand et al. 2021), see the following link: https://github.com/plantformatics/Socrates/
-
1.
FASTQ files were generated using cellranger-atac mkfastq (v3.0) with default parameters.
-
2.
FASTQ files were then trimmed and aligned to maize B73 AGP v4 reference genome using cellranger-atac count (v1.2) to obtain an unfiltered BAM file containing all sequenced reads with corrected barcodes appended to the “CB” tag.
-
3.
Uniquely and properly paired mapped reads were extracted using samtools with the “-q 10 -f 3” parameters and by removing reads with more than one alignment in the “XA” tag added by BWA mem (a part of the cellranger-atac count pipeline).
Optional: Researchers may wish to adjust the “-q” parameter depending on the species. We opt to use mapping quality of 10 as a threshold to avoid removing reads derived from regions associated with the whole-genome duplication event that occurred ∼5–12 Mya in maize (Schnable et al., 2011).
-
4.
PCR and optical duplicates were removed using picardtools MarkDuplicates with the following parameters “REMOVE_DUPLICATES=true BARCODE_TAG=CB”.
-
5.
The filtered BAM file was converted to BED (Quinlan and Hall, 2010) format representing base-resolution Tn5 integration sites by adjusting the 5′ coordinates of forward and reverse aligned reads by +4/−5, respectively (Column 1 = chromosome, Column 2 = start position, Column 3 = end position, Column 4 = barcode, Column 5 = strand).
-
6.
Unique Tn5 insertions were retained for each barcode using the UNIX tool uniq.
-
7.Barcodes passing quality thresholds (unique Tn5 insertions per nucleus, percent Tn5 insertions within 2-kb of TSSs, and FRIPs) were identified using the R package, Socrates v0.0.0.9 (example code can be found here: https://github.com/plantformatics/plant_scATAC_STAR_Protocol) (Marand et al., 2021) and are hereafter referred to as nuclei (Troubleshooting 4).
-
a.Socrates uses MACS2 (Zhang et al., 2008) to identify Accessible Chromatin Regions (ACRs) that depends on species-specific information. Researchers should take care to ensure that the parameters selected are appropriate for their species of interest.
-
b.The effective genome size, which represents the proportion of mappable sequence, is generally 60–80% of the total genome size. For maize, the effective genome size was set to 1.6e9.
-
c.Spline-fitting identified nuclei as barcodes with more than 2,348 unique Tn5 insertions (Figure 5A).
-
d.Nuclei with greater than 20% of Tn5 insertions within 2-kb of TSSs and 30% of Tn5 insertions with ACRs were retained as intact nuclei for further analysis (Figure 5A).
-
e.A sparse binary matrix composed of ACRs (rows) by nuclei (columns) was generated by Socrates using the “convertSparseData” function for further processing.
-
a.
-
8.
Following data quality evaluation and processing, nuclei with similar genome-wide chromatin accessibility profiles were clustered into groups by first filtering low-frequency features and nuclei with less than 1,000 accessible features (Figure 5B).
-
9.
Finally, Louvain clusters of nuclei representing putative cell types were identified as using the succession of “regModel”, “reduceDims”, and “callClusters” functions a part of Socrates (Figure 5C) (Troubleshooting 5).
Figure 5.

Quality control and initial scATAC-seq data processing
(A) Identification of high-quality barcodes using spline-fitting and signal enrichment at TSSs and ACRs. FRiPs; Fraction reads in peaks.
(B) Sparse matrix filtering to remove low frequency features (1-kb bins) and nuclei with few accessible features.
(C) Visualization of nuclei similarity colored by graph-based clusters (Louvain) of genome-wide chromatin accessibility profiles.
Limitations
This protocol has not yet been assessed from samples with low nuclei counts (less than 60,000 starting nuclei). Researchers working with small tissues (such as embryos) may need to pool multiple samples or optimize the nuclei extraction procedure to yield sufficient nuclei counts for scATAC-seq library construction. In addition, some samples and species may require adjusting the concentration and/or the type of detergent used, particularly if nuclei appear over-lysed with very few intact nuclei.
Troubleshooting
Problem 1
Few intact nuclei (Step 10).
Potential solution
An abundance of lysed nuclei can occur at several steps of the protocol. Depending on the species (and the composition of the nuclear membrane), increasing or decreasing the detergent concentration can result in fewer lysed nuclei. Adjusting the sorting speed and sheath pressure can also be helpful to reduce the occurrence of unwanted nuclear lysis.
Problem 2
Excessive nuclear aggregation (Step 10).
Potential solution
Nuclear aggregation after sorting can occur when the collection buffer values are low, or nuclei suspensions are highly concentrated. Increasing the collection volume and adding between 1%–5% Bovine Serum Albumin can aid to lower aggregation in nuclei suspensions. Researchers should aim to produce nuclei suspensions at a concentration below 4,000 nuclei per μL for species with nuclei similar in size to maize (<10 μm).
Problem 3
Lack of nucleosome-protected fragments (Expected Outcomes).
Potential solution
The loss of periodic fragment sizes corresponding to nucleosome-free and nucleosome-protected fragments can arise from multiple scenarios. The most common explanation is a lack of intact nuclei prior to tagmentation; lysed nuclei will result in chromatin accessibility profiles that resemble gDNA when sequenced. If nuclei preparations are largely intact, another possibility is excessive PCR cycles (over-amplification of small fragments) or insufficient extension times during library amplification.
Problem 4
Few nuclei passing quality control thresholds (Quantification and Statistical Analysis).
Potential solution
The selection of quality control thresholds is highly dependent on the species, quality of the reference genome and the tissue/organ used in the experiment. As such, the percent TSS and FRiP score thresholds presented above should be taken as guidelines. In the case where few nuclei pass a minimum Tn5 insertion threshold, the knee plot, such as in Figure 5A, should be heavily scrutinized. A reasonable threshold can be set based on the distribution, rather than a predetermined heuristic cut-off. For example, in Figure 5A, a possible cut-off for Tn5 insertions per nucleus can be set to the top of the “knee”, at approximately 630 Tn5 insertions per nucleus (log10[2.8]). It is advisable to ensure a cut-off that results in the same or fewer nuclei than were loaded onto the device. For low percent TSS and FRiP scores, researchers may again set thresholds based on the observed distributions, such as removing nuclei below 1 standard deviation. Ultimately, the quality of nuclei can be established by visualizing accessibility of known marker genes. Nuclei with low read depth, percent TSS and FRiP scores may still exhibit highly discriminative chromatin accessibility profiles representing distinct cell identities. Thus, cell-type-specific marker genes should always be evaluated follow data processing regardless of the observed data quality metrics.
Problem 5
Lack of cluster separation following dimensionality reduction (Quantification and Statistical Analysis).
Potential solution
The absence of underlying structure has several possible explanations. The first is an absence of heterogeneity in the studied cell population. This possibility can be ruled out by visualizing chromatin accessibility profiles of known marker genes of expected cell types to determine if heterogeneity among nuclei is observed on the embedding. Such a possibility can be confirmed by performing clustering to force substructure followed by differential chromatin accessibility analysis. An absence of marked chromatin differences is evidence for a lack of heterogeneity among nuclei within the sample. A second possibility is an overabundance of doublets due to overloading the microfluidics chip. While it may be tempting to load more nuclei than the manufacturer’s recommendation, an abundance of barcodes containing multiple nuclei adds substantial noise to the data set. In particular, barcodes containing multiple nuclei with different chromatin and cell states typically fall within an intervening space between distinct cell-type clusters on the manifold. If researchers have loaded more than the recommended ∼16,000 nuclei, extra care should be taken to remove as many predicted doublets as possible using pre-existing computational solutions, such as DoubletFinder, Scrublet, or ArchR (Granja et al., 2021; McGinnis et al., 2019; Wolock et al., 2019). A third possibility is an unsuccessful isolation of intact nuclei. Heavily degraded and broken nuclei lose chromatin structure demarcating cell identity. Analysis of scATAC-seq as a pseudobulk ATAC-seq data set (by aggregating reads from all nuclei) is highly informative for evaluating the data quality from a traditional viewpoint. An absence of clear regions of enrichment at TSSs and coverage plots resembling whole-genome shotgun sequencing are indications that the majority of nuclei were broken prior to loading the chip or the tagmentation step has failed.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Robert J. Schmitz (schmitz@uga.edu).
Materials availability
This study did not generate new unique materials.
Acknowledgments
We would like to thank the Center for Tropical and Emerging Global Diseases Shared Resource Laboratory for providing access to flow cytometry instruments. This study was funded with support from the NSF (IOS-1856627, IOS-1802848, and IOS-2026561) and the UGA Office of Research to R.J.S. and an NSF postdoctoral fellowship in biology (DBI-1905869) to A.P.M.
Author contributions
A.P.M. and R.J.S. designed the study. A.P.M., X.Z., and P.A.B.d.R. devised the nuclei isolation and scATAC-seq protocols. J.N. designed the FANS gating strategy and operated the flow cytometer. All authors wrote and approved the final text.
Declaration of interests
R.J.S. is a co-founder of Request Genomics, LLC, a company that provides plant epigenomics services. The remaining authors declare no competing interests.
Data and code availability
Data can be found online at https://www.sciencedirect.com/science/article/abs/pii/S0092867421004931
Code example can be found at https://github.com/plantformatics/plant_scATAC_STAR_Protocol
Tutorials and documentation for Socrates can be found at https://github.com/plantformatics/Socrates/
References
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109:21 29 21–21 29 29. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafzi A., Moutinho C., Picelli S., Heyn H. Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies. Nat. Protoc. 2018;13:2742–2757. doi: 10.1038/s41596-018-0073-y. [DOI] [PubMed] [Google Scholar]
- Lu Z., Hofmeister B.T., Vollmers C., DuBois R.M., Schmitz R.J. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017;45:e41. doi: 10.1093/nar/gkw1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Marand A.P., Ricci W.A., Ethridge C.L., Zhang X., Schmitz R.J. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants. 2019;5:1250–1259. doi: 10.1038/s41477-019-0548-z. [DOI] [PubMed] [Google Scholar]
- Marand A.P., Chen Z., Gallavotti A., Schmitz R.J. A cis-regulatory atlas in maize at single-cell resolution. Cell. 2021 doi: 10.1016/j.cell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnoye L., Marinov G.K., Krausgruber T., Pan L., Marand A.P., Secchia S., Greenleaf W.J., Furlong E.E.M., Zhao K., Schmitz R.J. Chromatin accessibility profiling methods. Nat. Rev. Methods Primers. 2021;1:10. doi: 10.1038/s43586-020-00008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci W.A., Lu Z., Ji L., Marand A.P., Ethridge C.L., Murphy N.G., Noshay J.M., Galli M., Mejía-Guerra M.K., Colomé-Tatché M. Widespread long-range cis-regulatory elements in the maize genome. Nat. Plants. 2019;5:1237–1249. doi: 10.1038/s41477-019-0547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable J.C., Springer N.M., Freeling M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U S A. 2011;108:4069–4074. doi: 10.1073/pnas.1101368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock S.L., Lopez R., Klein A.M. Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Feng S., Duttke S.H., Potok M.E., Zhang Y., Gallego-Bartolome J., Liu W., Jacobsen S.E. DNA methylation-linked chromatin accessibility affects genomic architecture in Arabidopsis. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2023347118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data can be found online at https://www.sciencedirect.com/science/article/abs/pii/S0092867421004931
Code example can be found at https://github.com/plantformatics/plant_scATAC_STAR_Protocol
Tutorials and documentation for Socrates can be found at https://github.com/plantformatics/Socrates/