

Abstract
cGAS, an innate immune sensor of cellular stress, recognizes double‐stranded DNA mislocalized in the cytosol upon infection, mitochondrial stress, DNA damage, or malignancy. Early models suggested that cytosolic localization of cGAS prevents autoreactivity to nuclear and mitochondrial self‐DNA, but this paradigm has shifted in light of recent findings of cGAS as a predominantly nuclear protein tightly bound to chromatin. This has raised the question how nuclear cGAS is kept inactive while being surrounded by chromatin, and what function nuclear localization of cGAS may serve in the first place? Cryo‐EM structures have revealed that cGAS interacts with nucleosomes, the minimal units of chromatin, mainly via histones H2A/H2B, and that these protein–protein interactions block cGAS from DNA binding and thus prevent autoreactivity. Here, we discuss the biological implications of nuclear cGAS and its interaction with chromatin, including various mechanisms for nuclear cGAS inhibition, release of chromatin‐bound cGAS, regulation of different cGAS pools in the cell, and chromatin structure/chromatin protein effects on cGAS activation leading to cGAS‐induced autoimmunity.
Keywords: chromatin, cyclic GMP–AMP synthase, DNA sensing, innate immunity, nucleosome
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Immunology; Structural Biology
This review summarizes mounting evidence for cGAS presence in the cell nucleus and on chromatin, and discusses its possible regulatory significance and how it can be reconciled with its cytoplasmic DNA sensor roles.
A cytosolic DNA sensor in the nucleus
Imagine you are a principal of a medieval village and need to place a guard to listen for approaching villains at night. Would you place the guard at a very quiet spot near the outer wall, or rather in the vibrant and noisy inn at the center of the village? We all know the answer, but if it comes to the vertebrate's cell innate immune system, cGAS, the guard sensing foreign DNA, is strangely situated where all the noise is, i.e., bound to chromatin in the nucleus. Upon sensing DNA, cGAS (for “cyclic GMP–AMP synthase”) catalyzes the production of 2′3′‐cGAMP from the common nucleotide–triphosphate precursors ATP and GTP (Ablasser et al, 2013; Gao et al, 2013; Sun et al, 2013). 2′3′‐cGAMP, a highly stable small molecule with a unique mixed‐phosphodiester linkage, can be transferred between cells and is recognized by the transmembrane receptor protein Stimulator of Interferon Genes (STING STING) (Ablasser et al, 2013; Zhang et al, 2013) (Fig 1). Oligomers formed by activated STING trigger pleiotropic downstream events, including activation of IRF3‐ and NF‐κB‐dependent signaling cascades for production of type I interferons and pro‐inflammatory cytokines, autophagy, and lysosomal cell death (Ishikawa & Barber, 2008; Zhong et al, 2008; Sun et al, 2009; Wu et al, 2013). For more details on cGAS–STING downstream signaling, please refer to (Hopfner & Hornung, 2020).
Figure 1. Simplified scheme of the cGAS–STING pathway and sources of nuclear and external DNA.
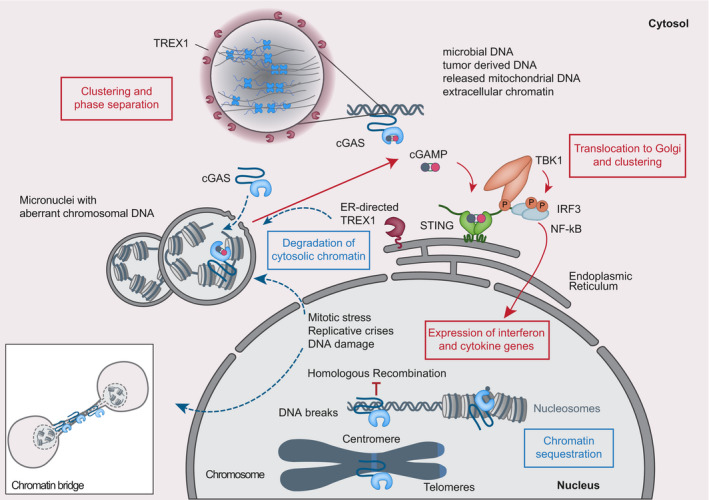
Cytosolic DNA recognition leads to formation of active cGAS via clustering and formation of large liquid–liquid phase‐separated cGAS‐DNA condensates excluding the ER‐directed exonuclease TREX1. Nuclear cGAS is sequestered at chromatin in an inactive state. Active cGAS produces cyclic GMP‐AMP (cGAMP), which binds to STING. STING relocalizes to the perinuclear Golgi and forms a clustered platform on which the TBK1 kinase phosphorylates the transcription factor IRF3. Phosphorylated IRF3 enters the nucleus and along with NF‐κB triggers expression of type I interferon and pro‐inflammatory cytokine genes.
cGAS is a nucleotidyltransferase that consists of an unstructured flexible N‐terminal domain of about 160 amino acids length, followed by a bilobal C‐terminal catalytic domain of the Mab21 fold containing a zinc‐binding dimerization motif (zinc‐thumb, Fig 2A). cGAS is activated by DNA‐dependent dimerization and multimerization (Fig 2B). Here, two DNA binding sites at its catalytic domain, denoted A and B, cooperate to assemble cGAS dimers on two sandwiched DNA molecules. Formation of higher‐order oligomers concentrates cGAS and DNA and stabilizes the active dimer states. Human cGAS additionally possesses a third DNA binding site C, which is involved in providing additional DNA contacts to stabilize higher‐order cGAS–DNA structures, including liquid‐phase condensates (Civril et al, 2013; Li et al, 2013; Zhang et al, 2014; Du & Chen, 2018; Xie et al, 2019) (Fig 1).
Figure 2. Active and chromatin‐sequestered, inhibited states of cGAS.
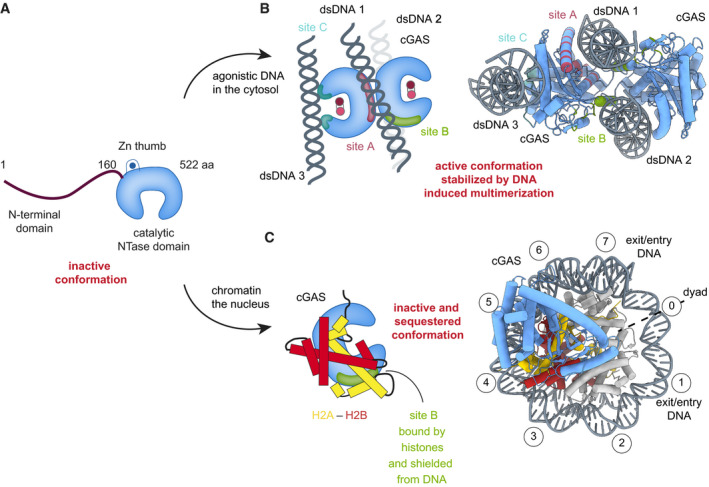
(A) Schematics of cGAS domain architecture. (B) cGAS is activated by agonistic DNA through a process that involves dimer formation, clustering, and induction of an active conformation with restructured active site. DNA interacts with sites A and B, and in human cGAS also with site C (PDB codes 4LEZ, 6EDB). (C) Structure of the mouse cGAS catalytic domain (blue) bound to an NCP (gray with yellow/red H2A/H2B) in disc view (PDB code 7A08). Nucleosomal superhelix locations (SHL) of DNA wrapped around the histone core are shown as encircled numbers. Nucleosomal histones binding to DNA binding site B shields it from DNA interaction and sterically prevents formation of active cGAS dimers.
cGAS efficiently recognizes dsDNA of a minimal length of > 40 bp, making pathogenic foreign DNA as well as endogenous nuclear or mitochondrial DNA potent cGAS agonists (Hopfner & Hornung, 2020). When self‐DNA leaks into the cytosol during cellular stress (such as mitochondrial stress, DNA damage, mitotic arrest, or senescence), or is present in form of cytosolic micronuclei, cGAS is activated leading to a state of sterile inflammation (Härtlova et al, 2015; West et al, 2015; de Oliveira Mann & Kranzusch, 2017; Glück et al, 2017; Harding et al, 2017; Mackenzie et al, 2017; Yang et al, 2017; Ablasser & Chen, 2019; Zierhut et al, 2019). In a healthy cell, however, cGAS is kept inactive even in cellular events that directly expose it to self‐DNA, such as mitosis, when cGAS associates with chromatin directly after nuclear envelope breakdown or remains in the form of post‐mitotic persistent nuclear cGAS pools bound to chromatin (Yang et al, 2017; Zierhut et al, 2019).
The question how cGAS autoreactivity to, e.g., genomic DNA is prevented arose as soon as cGAS was discovered as the cytosolic DNA sensor upstream of STING. First described as a cytosolic protein (Sun et al, 2013), cGAS was soon found to actually be present in both cytosol and nucleus (Orzalli et al, 2015). However, the lack of tools to study endogenous cGAS, combined with the lack of understanding of physiological roles of cGAS in the nucleus, kept the field in a state of controversy. In the meantime, nuclear cGAS has been implicated in a plethora of related functions, from canonical roles such as recognition of herpesviral DNA or HIV infection within the nucleus to non‐canonical roles in genome surveillance and inhibition of DNA repair (Orzalli et al, 2015; Lahaye et al, 2018; Liu et al, 2018; Jiang et al, 2019). Recent findings indicate that cGAS is even sequestered in the nucleus through highly salt‐resistant interactions with chromatin (Volkman et al, 2019). These tight interactions involving DNA binding site B in cGAS are important for maintaining nuclear cGAS inactive and avoiding autoreactivity in the cell. In cells, cGAS can bind to nucleosomes with high affinity and without being activated (Lahaye et al, 2018; Zierhut et al, 2019). However, cGAS can be activated when incubated in vitro with synthetic or extracted cellular chromatin, indicating that nucleosomal linker DNA may act as activating ligand in vitro and suggesting that additional cellular mechanisms are in place to keep cGAS suppressed in the nucleus (Mackenzie et al, 2017).
Nuclear cGAS is inhibited by the acidic patch of the nucleosome
A recent collection of six publications, reporting a total of eleven cryo‐electron microscopy structures of cGAS bound to the nucleosome, revealed how nuclear cGAS is tethered to and kept inactive by chromatin (Boyer et al, 2020; Cao et al, 2020; Kujirai et al, 2020; Michalski et al, 2020; Pathare et al, 2020; Zhao et al, 2020). These studies, using human nucleosome core particles (NCPs) reconstituted on the synthetic “Widom 601” DNA‐positioning sequence, were facilitated by the earlier demonstration that the structurally more accessible cGAS catalytic domain is sufficient for nuclear localization and tethering to chromatin (Volkman et al, 2019), allowing removal of the ~150 aa unstructured N‐terminal stretch known to induce aggregation and liquid–liquid phase condensation.
cGAS turned out to be tightly anchored to a nucleosome surface known as the “acidic patch”, a conserved region at the interface between histones H2A‐H2B serving as interaction site for many chromatin‐bound proteins (Fig 2C) (McGinty & Tan, 2016). A highly conserved arginine residue (R241mcGAS/R255hcGAS) in vertebrate cGAS—the so‐called arginine anchor—tightly binds to the canonical H2A acidic‐patch residues (E61, D90, E92; Fig 3). Further, cGAS–NCP interactions include equally well‐conserved residues located in two described tethering loops, containing the arginine anchor R241mcGAS/R255hcGAS as well as R222mcGAS/R236hcGAS, and a second interface located at cGAS DNA binding site B, where some DNA‐contacting residues have been repurposed for protein–protein interactions with histones (Figs 2B and C, and 4B). The extent of the interaction surface between cGAS and the nucleosome's histone core, approximately 880 Å2, explains how nucleosomes can bind cGAS with low nanomolar affinity, surpassing cGAS' affinity for agonistic dsDNA by 10‐ to 100‐fold. In fact, NCPs are able to inhibit cGAS in vitro even in the presence of free agonistic DNA. The mechanism of cGAS inhibition by chromatin becomes clear when comparing the available structures of active DNA‐bound cGAS dimer with NCP‐bound cGAS (Fig 2B), revealing at least two elements simultaneously contributing to inhibition: DNA binding site B is blocked by its interactions with histones H2A‐H2B, and binding of free DNA to site A is sterically hindered by nearby nucleosomal DNA at nucleosomal superhelix location (SHL) 6/7, preventing formation of the active cGAS dimer. As DNA binding to both site A and B is required for cGAS dimerization and subsequent conformational changes into its catalytic active state, cGAS is hereby kept in an inactive conformation bound to chromatin.
Figure 3. Recognition of the nucleosome acidic patch by cGAS and other binders via an arginine anchor.
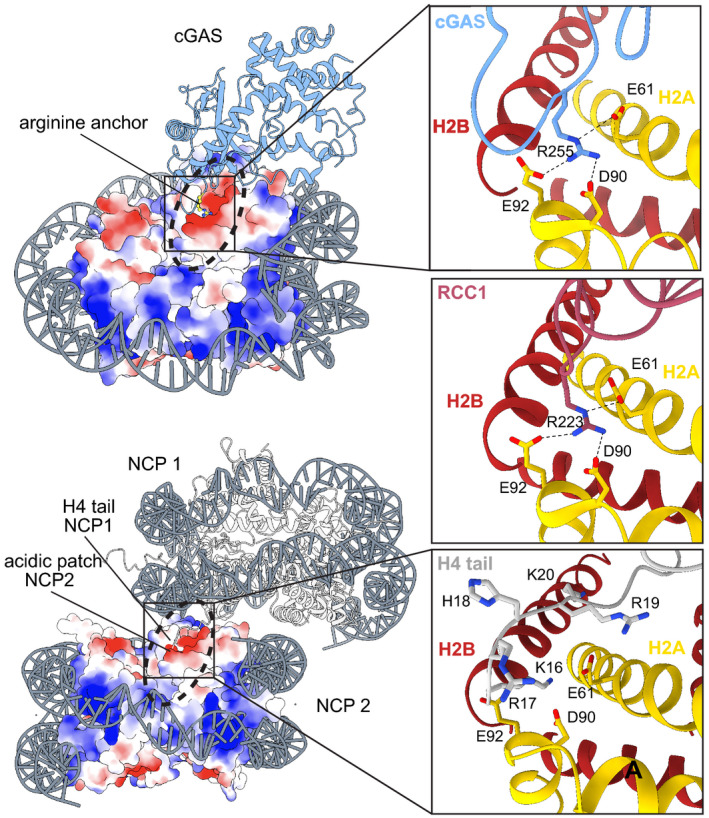
Top: Surface electrostatics representation of the cGAS–nucleosome complex (PDB code 7A08) and close‐ups of different arginine anchor interactors with the acidic patch at the interface between histones H2A‐H2B (yellow, red). cGAS (blue, PDB code 7A08), RCC1 (pink, PDB code 3MVD). Bottom: Surface electrostatics representation of two stacked NCPs and close‐up of H4 tail interactions from NCP1 with NCP2 (gray, PDB code 1AOI).
Figure 4. Stoichiometries of mouse and human cGAS–nucleosome complexes and their interaction sites.
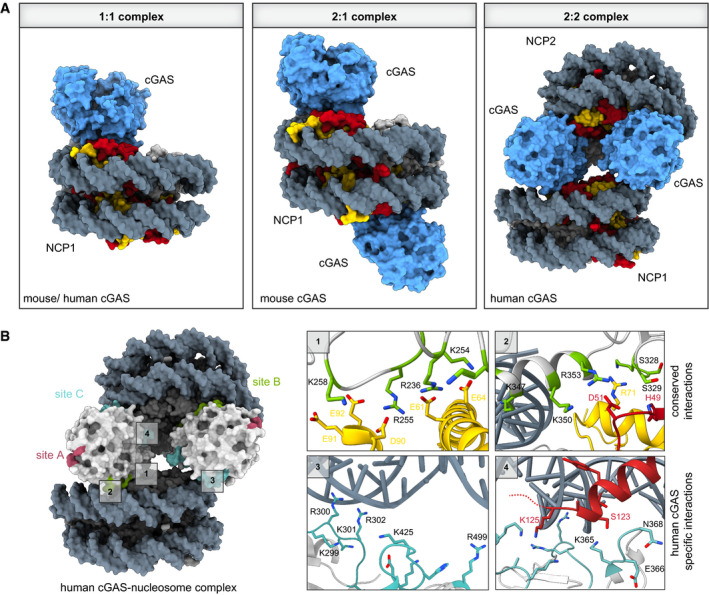
(A) Comparison of cGAS–nucleosome complex stoichiometries from different cryo‐electron microscopy structures. While mouse cGAS is found in 1:1 (PDB code 7A08) or 2:1 complexes with NCPs (PDB code 6XJD), human cGAS exhibits a preferred 2:2 stoichiometry (PDB code 7COM). (B) Close‐ups of four distinct cGAS–nucleosome interaction interfaces involving DNA binding sites B and C (PDB code 7COM and 6Y5D). Human cGAS DNA binding site A (pink), site B (green), and site C (cyan) are depicted. Close‐ups 1 and 2 show the conserved residues in site B involved in anchoring cGAS to the acidic patch, and close‐ups 3 and 4 show the human‐specific cGAS site C residues involved in interactions with nucleosomal DNA of NCP2.
While the new reports all agree on cGAS sequestration being mainly mediated by the acidic patch, they also reveal significant species‐specific differences involving additional interaction surfaces between cGAS and the nucleosomal DNA. Six mouse cGAS–NCP and five human cGAS–NCP structural analyses yielded complexes with stoichiometries of either 1:1, 2:1, 1:2, or 2:2 (Fig 4A). Each NCP has two acidic patches related by a twofold symmetry, and since cGAS binding to the acidic patch does not induce apparent conformational changes to the histone core, each NCP can bind two cGAS monomers to form a 2:1 complex. Notably, structures of human cGAS‐nucleosome complexes showed preferential 2:2 stoichiometry, with two cGAS protomers sandwiched between two nucleosomes (Fig 4B). Here, the two cGAS protomers connect two NCPs, resulting in multimers of stacked NCPs. On the other hand, 1:2 stoichiometry in human cGAS‐NCP structures can be attributed to one of two cGAS monomers missing in some of the particles of such cGAS–NCP stacks, due to its flexibility. Stacking of NCPs by the human cGAS protein is achieved by the previously described interactions of cGAS DNA binding site B to the acidic patch and SHL5.5 of the proximal NCP, with additional interactions of cGAS DNA binding site C to nucleosomal DNA at position SHL3 and SHL4 of the distal NCP. The interaction with the distal NCP is mainly mediated by residues in the less conserved KRKR‐loop (K285hcGAS, K299hcGAS, R300hcGAS, K301hcGAS, and R302hcGAS) of human cGAS, with potential additional contributions of residues on the KKH‐loop (K427hcGAS, K248hcGAS, and H429hcGAS). These positively charged residues are only partially conserved in mouse cGAS, explaining the species‐specific structural differences. Interestingly, residues on a nearby conserved β‐hairpin loop in human cGAS (K365hcGAS, E366hcGAS, G367hcGAS, N368hcGAS, and G369hcGAS) are in close proximity to the C‐terminal tail of histone H2B of the distal NCP (Fig 4B). It may thus be worth testing if histone tails, and their post‐translational modifications, also play a role in cGAS–NCP interactions, and whether this might be species‐specific. In addition, the question arises whether cGAS‐induced NCP stacking, along with its ability to compact DNA into a higher‐order state (Jiang et al, 2019), affects chromatin structure in vivo, and whether tethered cGAS can also regulate or impact on functional properties of chromatin.
Implications of chromatin structure for cGAS and autoimmunity
Hierarchical packaging of eukaryotic DNA into chromatin plays a central role in the regulation of DNA‐associated cellular processes, such as transcriptional regulation or the DNA damage response. The ‘primary structure’ of chromatin results from the organization of the genome into nucleosomes, where 147–165 bp of nucleosomal DNA are wrapped around the histone octamer consisting of two copies each of the four core histones (H2A, H2B, H3, and H4). This so‐called beads‐on‐a‐string first basic layer of organization does, however, not reflect the actual state of chromatin under physiological conditions. While the exact local three‐dimensional organization of chromatin in the cell remains an area of intense research and might be subject to regulation and modulation, a paradigm in the field for chromatin folding and condensation suggests that with increasing salt concentrations, an array of nucleosomes folds into a 10 nm‐diameter filament, followed by formation of a 30 nm‐diameter fiber‐like structure (Hansen, 2002; Krietenstein & Rando, 2020). These two states of intra‐nucleosomal interactions can be regulated in vitro by ionic strength and are mainly mediated by interactions of the H4 N‐terminal tail from one nucleosome with the acidic patch of the adjacent nucleosome (Fig 3) (Arya & Schlick, 2009). This implies that inhibition of cGAS by the nucleosome acidic patch should be sensitive to the chromatin organization state, i.e., that the level of chromatin condensation should dictate the availability and accessibility of cGAS‐binding acidic patch surfaces on chromatin.
Chromatin can be further divided into at least two very distinct states of organization. Euchromatin represents the “expressed” state, being less condensed and present at transcriptionally active genes that are marked by histone H3 and H4 acetylation and H3K4 methylation (H3K4me). In contrast, heterochromatin, which represents the “repressed” state and is required for proper nuclear architecture, is highly condensed and concentrated in pericentromeric and telomeric regions, which in turn are enriched in repetitive DNA sequences such as satellite DNA and transposons (Allshire & Madhani, 2018). Hallmarks of heterochromatin structure are histone H3 trimethylation modifications (H3K27me3 and H3K9me3) and the associated heterochromatin protein 1 (HP1), which forms a dimer bridging two H3K9‐methylated nucleosomes and thus further increases compaction. Condensation of heterochromatin is fundamental for maintaining genome stability as the compact structure of heterochromatin restricts or controls activities such as transcription, replication, recombination, and DNA damage response signaling, especially in regions with highly repetitive sequences (Janssen et al, 2018).
Mapping the chromatin binding sites of overexpressed GFP‐NLS‐cGAS and transgenically expressed GFP‐cGAS in human and murine dendritic cells, respectively, revealed that nuclear cGAS is enriched on centromeric satellite and LINE DNA repeat elements (Gentili et al, 2019). Interestingly, cGAS localization on chromatin does not correlate with open chromatin marks such as H3K27Ac but instead with repressive H3K9me enriched on pericentromeric heterochromatin, and with the centromeric histone H3 variant CENP‐A, suggesting that cGAS preferably localizes to densely packed regions in heterochromatin. In addition, while overexpressed cGAS is distributed throughout the nucleus, endogenous cytosolic cGAS accessing the nuclear compartment in the course of mitotic nuclear envelope rupture forms peripheral cGAS foci. Which mechanisms control cGAS distribution in the nucleus remains to be determined, as well as how this distribution may change depending on the cell state, e.g., infection or cell cycle progression. Another interesting question is how nuclear cGAS is kept away from active sites of transcription, including nucleosome‐free/nucleosome‐depleted regions (NFRs). NFRs are found in transcriptional regulatory regions such as promoters, enhancers, or terminators, while gene‐coding regions generally have a higher and more regular nucleosome occupancy. Given the heterogeneous structure and organization of chromatin in the nucleus, and there is the counterintuitive notion that at least in vitro, compact but not open chromatin is a substrate stimulating cGAS activity (Mackenzie et al, 2017; Michalski et al, 2020), details of cGAS activation by chromatin states need clarification (Fig 5A–C). For instance, levels of nuclear cGAS might require tight regulation as to not exceed the number of freely available nucleosomal binding sites (considering that there are also numerous other acidic‐patch binders present; Fig 3). Consistent with this, induced chromatin decondensation/decompaction through histone deacetylase inhibition or cellular HMGN5 overexpression was found to suppress activation of experimentally elevated nuclear cGAS levels (Guey et al, 2020).
Figure 5. Effects of chromatin structure and chromatin bound proteins on cGAS activation.
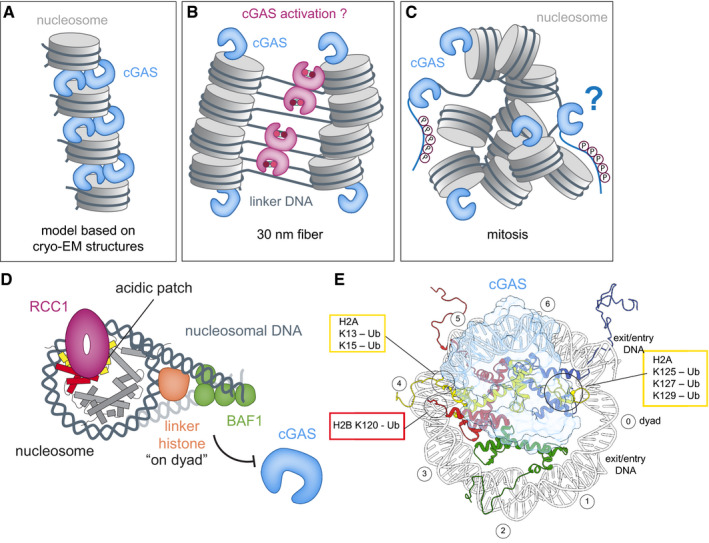
(A–C) Schematics of cGAS (blue) bound to nucleosomes (gray) based on cryo‐EM structure models (A), 30 nm‐fiber condensed‐chromatin model (B), and oligomers during mitosis (C). Accessibility of acidic patches determines whether cGAS is sequestered away from free linker DNA. During mitosis, cGAS is additionally inhibited through hyperphosphorylation of its N‐terminal region. (D) Chromatin‐binding factors, such as RCC1, BAF1, and linker histones, alter chromatin structure and compete with cGAS for binding sites. (E) Relation of important histone ubiquitylation sites on the nucleosome (disc view) to the cGAS‐binding site B.
The stoichiometry of available chromatin sites for cGAS sequestration is particularly imbalanced during cell division. At the onset of vertebrate mitosis, breakdown of the nuclear envelope allows the recruitment of both the nuclear as well as cytosolic pool of cGAS onto chromosomes, disabling any potential regulatory mechanisms based on cellular cGAS compartmentalization (Zierhut et al, 2019). In addition, chromosomes undergo global compaction during mitotic entry resembling the described in vitro state of liquid‐like phase condensates (Batty & Gerlich, 2019) (Fig 5C). This is partly due to free Mg2+ ion levels rising during mitosis to concentrations that are sufficient to promote chromatin condensation (and thus subsequent cGAS activation) in vitro (Maeshima et al, 2018). Other mechanisms contributing to mitotic chromatin compaction involve chromatin binding factors, interactions of flexible histone tails with DNA, and mitosis‐associated changes in histone tail post‐translational modifications such as deacetylation. Addressing this conundrum, a mechanism for cGAS inhibition during mitosis involves cell cycle‐dependent hyperphosphorylation of its N‐terminal region (Zhong et al, 2020; Li et al, 2021). The evenly distributed negative charges of phosphorylated serine and threonine residues along the N‐terminal regions of cGAS specifically interfere with its recognition of nuclear DNA and selectively suppress its activity during mitosis. Moreover, there are additional controls of STING downstream signaling during cell division, such as mitotic Golgi vesiculation, which may further inhibit STING activation by limiting its trafficking after cGAMP binding (Uhlorn et al, 2020).
The opposite effect is observed when cGAS is activated by chromatin for instance in micronuclei. Micronuclei are aberrant nuclear structures containing chromatin fragments surrounded by a nuclear envelope. They form after mitosis as result of a range of cell division defects, such as mitotic errors, impaired chromosome segregation, or DNA damage. These events lead to the appearance of so‐called chromatin bridges and lagging chromosomes, which then recruit their own nuclear envelope to generate micronuclei. cGAS is recruited to micronuclei upon rupture of their nuclear envelope (Bartsch et al, 2017; Dou et al, 2017; Glück et al, 2017; Harding et al, 2017; Mackenzie et al, 2017; Yang et al, 2017; Bakhoum et al, 2018), resulting in the exposure of micronuclear chromatin to the cytosolic compartment and in recruitment and activation of the cytosolic cGAS pool. One reason for the activation of cGAS by micronuclear chromatin might be that the limited amount of accessible nucleosome acidic patches they contain are insufficient to sequester the whole cytosolic pool of cGAS, allowing free cGAS activation by nearby free chromosomal DNA. In this context, the DNA 3′‐5′ exonuclease TREX1 was found as a critical regulator of cytosolic DNA sensing in chromosomally unstable cells, degrading micronuclear DNA when bound to the endoplasmic reticulum (ER) (Mohr et al, 2021). In contrast to the degradation of micronuclear DNA by ER‐associated TREX1, cytosolic immunostimulatory DNA bound to cGAS forms phase‐separated liquid–liquid droplets that limit its degradation by TREX1 (Zhou et al, 2021). Other possible mechanisms differentiating cGAS‐activating potential of micronuclear chromatin vs intact‐nuclei chromatin would be potential aberrant chromatin structures in micronuclei recognized by cGAS, or yet to be identified mechanisms or factors for cGAS inhibition present in nuclei but absent from micronuclei. Nevertheless, there is so far no direct biochemical evidence that aberrant chromatin in micronuclei cannot just recruit, but also activate cGAS—recent work in fact implicates co‐occurring chromatin bridges, rather than micronuclei, as the actual source of cGAS activation after drug‐induced mitotic errors (preprint: Flynn et al, 2021). Another example of cytosolic chromatin activating cGAS is neutrophil extracellular traps (NETs), which are released by neutrophils during a specific form of regulated cell death called NETosis. These structures, composed of chromatin and antimicrobial molecules, are phagocytosed by macrophages and translocate into the cytoplasm, where they activate cGAS (Apel et al, 2021). As in the case of micronuclei, NETs expose aberrant chromatin structures to an excess of cytosolic cGAS, again possibly exhibiting too few acidic patches to inhibit cGAS. Indeed, fragmentation of NETs is required for cGAS activation in vitro, whereas digestion of their extranucleosomal DNA results in cGAS inactivation, suggesting that decompaction of chromatin exposing extranucleosomal DNA is required for cGAS activation in the cytosol.
What other mechanisms for cGAS inhibition exist in the nucleus?
Chromatin organization and structure are affected by several chromatin‐bound proteins. Well‐known influencers of chromatin structure dynamics are linker histones such as H1 and H5, which stabilize interactions of additional 20‐bp linker DNA with the nucleosome periphery to form the chromatosome (Fig 5D). Linker histones are known to drive further compaction of chromatin by stabilizing folded and oligomeric states in vitro and by promoting heterochromatin formation in vivo (Dorigo et al, 2004; Fan et al, 2005; Lu et al, 2009; Song et al, 2014). Linker histones may affect cGAS inhibition by chromatin on the one hand via further compaction of chromatin, reducing the number of accessible acidic patches for cGAS sequestration, and on the other hand through hindrance of cGAS activation by restricting access to linker DNA. In any case, binding of linker histones to nucleosomal DNA inhibits cGAS activity in vitro more strongly than chromatin lacking linker histones (Uggenti et al, 2020). Roles of histones in limiting cGAS activation in the nucleus are further observed in patients with the type I interferonopathy Aicardi–Goutières syndrome (AGS). Here, biallelic mutations in LSM11 and RNU7‐1, encoding components of the replication‐dependent histone pre‐mRNA‐processing complex, result in misprocessing of canonical histone transcripts in this subset of AGS patients and thus specifically disturb linker histone stoichiometries, while maintaining core histone levels (Uggenti et al, 2020). The altered levels of linker histones lead to cGAS redistribution within the nucleus, including more frequent nuclear membrane herniations containing increased cGAS concentrations, followed by an enhanced cGAS‐dependent type I interferon signature. Taken together, histones play a key role in suppressing autoreactivity to nuclear self‐DNA by cGAS sequestration via the histone H2A–H2B interface and the additional requirement for linker histones bound to chromatin to avoid autoimmunity. Further investigation is required to fully understand how changes in nuclear linker histone stoichiometries and distinct linker histone variants affect nuclear cGAS distribution and how cGAS localization in turn relates to its activation in the nucleus.
Linker histones are not the only proteins restricting accessibility of cGAS to nuclear DNA, as the barrier‐to‐autointegration factor 1 (BAF1) can also dynamically outcompete cGAS for DNA binding upon acute loss of nuclear envelope integrity (Guey et al, 2020; Ma et al, 2020) (Fig 5D). BAF1 is a small (10 kDa) chromatin‐bound protein essential for mitosis, nuclear assembly, and the DNA damage response (Haraguchi et al, 2001; Margalit et al, 2005; Haraguchi et al, 2008; Jamin & Wiebe, 2015). BAF1 depletion in cells results in increased nuclear envelope ruptures, leading to the recruitment of cytosolic cGAS, formation of cGAS foci at the nuclear entry site, and subsequently a robust interferon response. Thus, BAF1 binding to DNA at sites of nuclear envelope rupture provides an additional mechanism to avoid cGAS autoreactivity toward nuclear self‐DNA, especially when the cytosolic cGAS pool gains access to nuclear DNA.
In addition to protein–protein interactions, RNA can also outcompete dsDNA for binding to cGAS, as protection of quiescent hematopoietic stem cells from exhaustion due to cGAS activity depends on the presence of a circular RNA (termed cia‐cGAS) in the cell nucleus (Xia et al, 2018). In this case, cGAS inhibition relies on the high nuclear abundance and stronger cGAS‐binding affinity of cia‐cGAS RNA compared to self‐DNA, preventing cGAS activation by nuclear DNA and subsequent immune response induction. Finally, possible post‐translational modifications of cGAS or histones are another aspect to consider regarding regulation of cGAS activity in the nucleus. Notably, a so‐far unidentified post‐translational modification specific for a portion of chromatin‐bound cGAS and disappearing upon DNA stimulation could represent one such mechanism (Michalski et al, 2020), but further investigation is required to identify this modification and understand its role in cGAS regulation.
How is cGAS released from chromatin?
The tight interaction of cGAS with chromatin raises the question to what extent regulated dissociation is necessary in the context of, e.g., cellular infections. While the cGAS–nucleosome complex is, with a dissociation half‐life of about 70 s, quite dynamic for allowing rapid redistribution onto free DNA, the latter cannot efficiently compete and release cGAS from the acidic patch of the nucleosome (Michalski et al, 2020). Nucleo‐cytosolic distribution of cGAS might be sufficient to allow sensing of cytosolic DNA, but recognition of agonistic nuclear DNA in the context of homologous recombination or infections presumably needs protein cofactors or post‐translational modifications on either histones or cGAS to prevent nucleosomal competition. For instance, some of the described sumoylation and ubiquitination sites in cGAS (e.g., K335mcGAS) (Cui et al, 2017) have the potential to prevent nucleosome tethering by steric hindrance and may play a role in regulating chromatin binding of cGAS.
Histone tails are well known for their functions in nucleosome dynamics and recruitment of co‐factors to chromatin. Deletion of N‐terminal tails from all four core histones did not affect cGAS binding to the nucleosome or its inhibition, suggesting that sequestration by the acidic patch is the dominant mechanism of inhibition (Boyer et al, 2020). However, histone tails and possible modifications thereof can play a major role in the regulation of chromatin‐bound cGAS, such as the facilitation of cGAS release. Modifications in close proximity of the cGAS‐nucleosome binding site, or modifications on spatially close histone tails, could potentially displace cGAS or prevent its sequestration by the acidic patch. Comparing the available structures of ubiquitinated nucleosomes, possible cGAS‐regulating histone modifications include ubiquitination of histone H2A at sites K13, K15 or K125, K127, K129, and H2B ubiquitination on K120 (Fig 5E) (Wilson et al, 2016; Anderson et al, 2019; Worden et al, 2019). These histone modifications are known to be important in the DNA damage response, where each of them has different physiological consequences, such as promoting non‐homologous end joining (NHEJ) or homologous recombination (HR) (Uckelmann & Sixma, 2017). Ubiquitination of H2BK120, for instance, stimulates chromatin relaxation, which in turn facilitates recruitment of factors required for DNA damage checkpoint activation and DNA repair initiation (Schwertman et al, 2016). Another modification is S139 phosphorylation of variant histone H2A.X (creating the γH2A.X mark), which was found to interact with cGAS and to facilitate its recruitment to the site of DNA damage (Liu et al, 2018). However, further experimental evidence is required to understand how cGAS activity and its localization on chromatin is regulated by these modifications upon DNA damage.
The release of cGAS from chromatin could also be directed by competition with other chromatin co‐factors interacting with the acidic patch. The acidic patch is a highly negatively charged narrow groove in the nucleosome core, known as its principal protein‐docking region utilized by numerous nucleosome interactors such as regulator of chromosome condensation 1 (RCC1, Fig 3) (Makde et al, 2010), latency‐associated nuclear antigen (LANA) (Barbera et al, 2006), interleukin‐33 (IL‐33) (Roussel et al, 2008), silent information regulator 3 (SIR3) (Armache et al, 2011) and high‐mobility group nucleosomal binding domain 2 (HMGN2) (Kato et al, 2011). cGAS has to compete with the H4 tail and other proteins for the acidic patch, in particular since, e.g., RCC1 has a much higher abundance in the cell than cGAS (Hein et al, 2015).
How is cGAS localization regulated in the cell?
cGAS localization to both the cytosol and the nucleus has now been established in different cell types including fibroblasts and keratinocytes (Orzalli et al, 2015), primary immune cells (Gentili et al, 2019; Jiang et al, 2019; Volkman et al, 2019), and cancer cells (Liu et al, 2018). However, distribution of these two cGAS protein pools in the cell differs depending on determinants such as cell‐type, cell differentiation or cell density (Yang et al, 2017; Jiang et al, 2019; Volkman et al, 2019). In differentiated primary mouse macrophages for instance, almost 95% of cGAS protein in the cell is localized to the nucleus (Volkman et al, 2019). While the cytosolic cGAS protein pool is important for immune signaling against aberrant dsDNA in the cytosol, the nuclear cGAS pool is kept inactive, suggesting that cGAS enzymatic activity is an exclusive function of cytosolic cGAS. It remains unclear what the contributions are of nuclear vs the cytosolic pools of cGAS protein in activation of the cGAS–STING pathway during, e.g., infection. Is the cytosolic cGAS fraction at steady‐state enough to elicit an immune response against the evading pathogen or must predominantly nuclear cGAS relocate to the cytosol in order to fight infection? This also raises the question of what potential upstream cellular stress signals influence cGAS localization.
Use of the cGAS arginine anchor mutant R255EhcGAS, in order to release cGAS from its tight sequestration by chromatin in cells, has revealed that cGAS remains nuclear even when not bound to the nucleosomal acidic patch, indicating the requirement for an active mechanism of cGAS translocation to the cytosol (Michalski et al, 2020). Part of this localization could be the propensity of cGAS to bind genomic DNA, as a cGAS DNA‐binding site mutant (C396A/ C397A) has a reduced nuclear presence (Jiang et al, 2019). In addition, both the N‐terminal domain of cGAS, containing amino acids 1–160, and the C‐terminal catalytic domain 161–522 can spontaneously accumulate in the nucleus, suggesting that at least two independent nuclear localization signals are present in cGAS (Gentili et al, 2019). These nuclear localization signals are opposed by a recently discovered functional NES on the C‐terminal catalytic domain (169–174) of cGAS, which is required for translocation of cGAS into the cytosol (Sun et al, 2021). Here, the cGAS NES is required for cGAS export to the cytosol upon DNA stimulation and for the subsequent interferon response, suggesting that the available cytosolic cGAS fraction prior to stimulation is insufficient to elicit a robust immune response to dsDNA (Sun et al, 2021).
Why is cGAS in the nucleus in the first place?
The biologically most interesting still unanswered question surrounding nuclear cGAS is why cGAS is kept there in the first place. One possible explanation is that the nucleus is merely a reservoir to keep cytosolic cGAS levels low, yet enabling the cell to replenish or increase cytosolic levels in a regulated fashion without the need for slow transcriptional programs. Alternatively, since most DNA viruses shown to be antagonized by the cGAS–STING pathway are known to replicate exclusively in the nucleus while shielding their genome from detection when in the cytosol, cGAS might have nuclear DNA‐sensing functions as well. In this context, several studies have focused on how cGAS exerts its canonical functions as an antiviral pathogen recognition receptor in the nucleus, facilitated by other nuclear immune factors or auxiliary proteins (Orzalli et al, 2015; Lahaye et al, 2018). Interestingly, nuclear DNA viruses and retroviruses, such as HSV‐1, organize their genomes into nucleosomes by appropriating eukaryotic histones and hijacking the host nucleosome assembly machinery (Lieberman, 2008; Oh et al, 2015). Some large DNA viruses even encode histone homologs themselves, and two recent cryo‐EM structures of these viral nucleosome‐like particles revealed their structural similarity to eukaryotic counterparts, including partial conservation of the acidic patch (preprint: Liu et al, 2021; Valencia‐Sánchez et al, 2021). The HSV‐1 viral genome is packaged into nucleosomes already 1 h after infection and subsequent nuclear entry (Kent et al, 2004), indicating that wrapping of the viral genome into nucleosomes could represent a novel viral immune evasion mechanism. In this case, the virus uses inhibition of cGAS by the acidic patch as a strategy to overcome immune recognition in the nucleus when its genome first gets exposed for replication in the host.
On the other hand, evidence is now accumulating for additional, STING‐independent non‐canonical functions of cGAS in the nucleus, including roles in tumor progression by inhibiting DNA repair, or as decelerator of DNA replication forks suppressing replication‐associated DNA‐damage, and even in regulation of histone modifications in response to an RNA virus infection (Liu et al, 2018; Jiang et al, 2019; Chen et al, 2020; Cui et al, 2020). DNA damage‐induced lesions such as DNA double‐strand breaks (DSB) can be repaired via two major pathways: NHEJ, which ligates broken DNA ends regardless of homology, and HR, which uses an undamaged DNA template to repair the break while retaining the original sequence. Two independent studies recently found that nuclear cGAS selectively inhibits DSB repair by HR, thus exacerbating genome instability leading to tumorigenesis and cell death (Liu et al, 2018; Jiang et al, 2019). Both studies agree that the catalytic domain of cGAS is sufficient for inhibiting HR and that cGAS DNA‐binding properties and nuclear localization are required, but not its catalytic activity. However, two alternative mechanisms for how cGAS inhibits HR have been proposed: (i) poly‐ADP‐ribose (PAR)‐mediated interaction between cGAS and PARP1, which in turn impairs the interaction of PARP1 with the DSB repair factor Timeless (Liu et al, 2018), and (ii) cGAS‐dependent compaction of template DNA into higher‐order, ladder‐like, phase‐separated structures that prevent RAD51‐ssDNA filaments from invading the dsDNA template, therefore inhibiting D‐loop formation underlying HR (Jiang et al, 2019). Since the in vitro studies were done in absence of nucleosomes, it is yet unclear whether DNA ends per se could overcome nucleosomal inhibition of DNA binding by cGAS, requiring additional recruitment mechanisms to sites of DNA damage. Furthermore, since pathway choice between NHEJ and HR is substantially regulated at the level of chromatin, selective inhibition of HR by cGAS may also proceed via cGAS‐nucleosome interactions. Further clarification is needed to understand the role of nuclear cGAS beyond its actions as an innate immune sensor.
Conclusion
Research on the biology of nuclear cGAS is just in the beginning. Surveillance of genomic instability or cellular stress by detecting changes in chromatin structure or inhibition of HR leading to the elimination of cells under acute genomic stress represent possible primordial functions of cGAS and could provide an explanation for its evolutionary conservation prior to the emergence of interferon‐based immunity. Thus, cGAS might be strategically positioned as a guard in the village's noisy inn, and not only as sequestered “prisoner”. There it could sense upcoming plots or detect intruders that mingle among the local villagers, i.e., cell stress, before they can cause major damage. Predominant localization of cGAS to the nucleus of the cell could indicate that this guard's function is to first keep the village safe from within and as a second task to protect it against threats from the outside.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
The authors thank S. Michalski for discussions. We acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG) (Project‐ID 369799452—TRR237 and Gottfried Wilhelm Leibniz‐Prize to K.‐P.H.) and the Cancer Research Institute (Eugene V. Weissman Fellowship to C.C.O.M.). Figures were prepared using UCSF ChimeraX (Goddard et al, 2018). Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2021) 40: e108293.
Contributor Information
Carina C de Oliveira Mann, Email: mann@genzentrum.lmu.de.
Karl‐Peter Hopfner, Email: hopfner@genzentrum.lmu.de.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K‐P, Ludwig J, Hornung V (2013) cGAS produces a 2′‐5′‐linked cyclic dinucleotide second messenger that activates STING. Nature 498: 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Schmid‐Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V (2013) Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503: 530–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Chen ZJ (2019) cGAS in action: expanding roles in immunity and inflammation. Science 363: eaat8657 [DOI] [PubMed] [Google Scholar]
- Allshire RC, Madhani HD (2018) Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19: 229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CJ, Baird MR, Hsu A, Barbour EH, Koyama Y, Borgnia MJ, McGinty RK (2019) Structural basis for recognition of ubiquitylated nucleosome by Dot1L methyltransferase. Cell Rep 26: 1681–1690.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel F, Andreeva L, Knackstedt LS, Streeck R, Frese CK, Goosmann C, Hopfner K‐P, Zychlinsky A (2021) The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci Signal 14: eaax7942 [DOI] [PubMed] [Google Scholar]
- Armache K‐J, Garlick JD, Canzio D, Narlikar GJ, Kingston RE (2011) Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 Å resolution. Science 334: 977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya G, Schlick T (2009) A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments. J Phys Chem A 113: 4045–4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J‐A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NKet al (2018) Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553: 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera AJ, Chodaparambil JV, Kelley‐Clarke B, Joukov V, Walter JC, Luger K, Kaye KM (2006) The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311: 856–861 [DOI] [PubMed] [Google Scholar]
- Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, Spehlmann ME, Frey N, Saftig P, Chalaris Aet al (2017) Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet 26: 3960–3972 [DOI] [PubMed] [Google Scholar]
- Batty P, Gerlich DW (2019) Mitotic chromosome mechanics: how cells segregate their genome. Trends Cell Biol 29: 717–726 [DOI] [PubMed] [Google Scholar]
- Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, Zhang Q (2020) Structural basis of nucleosome‐dependent cGAS inhibition. Science 370: 450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D, Han X, Fan X, Xu R‐M, Zhang X (2020) Structural basis for nucleosome‐mediated inhibition of cGAS activity. Cell Res 30: 1088–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, Levine AS, Zou L, Chen Z, Lan L (2020) cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv 6: eabb8941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner K‐P (2013) Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498: 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Yu H, Zheng X, Peng R, Wang Q, Zhou Y, Wang R, Wang J, Qu B, Shen Net al (2017) SENP7 potentiates cGAS activation by relieving SUMO‐mediated inhibition of cytosolic DNA sensing. PLoS Pathog 13: e1006156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Yu Q, Chu L, Cui Y, Ding M, Wang Q, Wang H, Chen Y, Liu X, Wang C (2020) Nuclear cGAS functions non‐canonically to enhance antiviral immunity via recruiting methyltransferase Prmt5. Cell Rep 33: 108490 [DOI] [PubMed] [Google Scholar]
- Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ (2004) Nucleosome arrays reveal the two‐start organization of the chromatin fiber. Science 306: 1571–1573 [DOI] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Zet al (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550: 402–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Chen ZJ (2018) DNA‐induced liquid phase condensation of cGAS activates innate immune signaling. Science 361: 704–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, Stein A, Woodcock CL, Skoultchi AI (2005) Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123: 1199–1212 [DOI] [PubMed] [Google Scholar]
- Flynn PJ, Koch PD, Mitchison TJ (2021) Chromatin bridges, not micronuclei. Activate cGAS after drug‐induced mitotic errors in human cells. bioRxiv 10.1101/2021.02.02.429360 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney B, Zillinger T, Serganov A, Liu Y, Jones R, Hartmann Get al (2013) Cyclic [G (2′, 5′) pA (3′, 5′) p] is the metazoan second messenger produced by DNA‐activated cyclic GMP‐AMP synthase. Cell 153: 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentili M, Lahaye X, Nadalin F, Nader GP, Lombardi EP, Herve S, De Silva NS, Rookhuizen DC, Zueva E, Goudot C (2019) The N‐terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep 26: 2377–2393.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang T‐W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19: 1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018) UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guey B, Wischnewski M, Decout A, Makasheva K, Kaynak M, Sakar MS, Fierz B, Ablasser A (2020) BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science 369: 823–828 [DOI] [PubMed] [Google Scholar]
- Hansen JC (2002) Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct 31: 361–392 [DOI] [PubMed] [Google Scholar]
- Haraguchi T, Koujin T, Segura‐Totten M, Lee KK, Matsuoka Y, Yoneda Y, Wilson KL, Hiraoka Y (2001) BAF is required for emerin assembly into the reforming nuclear envelope. J Cell Sci 114: 4575–4585 [DOI] [PubMed] [Google Scholar]
- Haraguchi T, Kojidani T, Koujin T, Shimi T, Osakada H, Mori C, Yamamoto A, Hiraoka Y (2008) Live cell imaging and electron microscopy reveal dynamic processes of BAF‐directed nuclear envelope assembly. J Cell Sci 121: 2540–2554 [DOI] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548: 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtlova A, Erttmann SF, Raffi FAM, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JAet al (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti‐microbial innate immunity. Immunity 42: 332–343 [DOI] [PubMed] [Google Scholar]
- Hein M, Hubner N, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak I, Weisswange I, Mansfeld J, Buchholz Fet al (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163: 712–723 [DOI] [PubMed] [Google Scholar]
- Hopfner K‐P, Hornung V (2020) Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol 21: 501–521 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455: 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamin A, Wiebe MS (2015) Barrier to Autointegration Factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr Opin Cell Biol 34: 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Colmenares SU, Karpen GH (2018) Heterochromatin: guardian of the genome. Annu Rev Cell Dev Biol 34: 265–288 [DOI] [PubMed] [Google Scholar]
- Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, Sohn J, Sung P, Gekara NO (2019) Chromatin‐bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J 38: e102718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, van Ingen H , Zhou B‐R, Feng H, Bustin M, Kay LE, Bai Y (2011) Architecture of the high mobility group nucleosomal protein 2‐nucleosome complex as revealed by methyl‐based NMR. Proc Natl Acad Sci USA 108: 12283–12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent J, Zeng P‐Y, Atanasiu D, Gardner J, Fraser N, Berger S (2004) During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol 78: 10178–10186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietenstein N, Rando OJ (2020) Mesoscale organization of the chromatin fiber. Curr Opin Genet Dev 61: 32–36 [DOI] [PubMed] [Google Scholar]
- Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, Hirai S, Funabiki H, Kurumizaka H (2020) Structural basis for the inhibition of cGAS by nucleosomes. Science 370: 455–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, Zueva E, Maurin M, Nadalin F, Knott GJet al (2018) NONO detects the nuclear HIV capsid to promote cGAS‐mediated innate immune activation. Cell 175: 488–501.e22 [DOI] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P (2013) Cyclic GMP‐AMP synthase is activated by double‐stranded DNA‐induced oligomerization. Immunity 39: 1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Huang T, Du M, Chen X, Du F, Ren J, Chen ZJ (2021) Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 371: eabc5386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman PM (2008) Chromatin organization and virus gene expression. J Cell Physiol 216: 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan Ret al (2018) Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563: 131–136 [DOI] [PubMed] [Google Scholar]
- Liu Y, Toner CM, Philippe N, Jeudy S, Zhou K, Bowerman S, White A, Edwards G, Abergel C, Luger K (2021) Melbournevirus‐encoded histone doublets are recruited to virus particles and form destabilized nucleosome‐like structures. bioRxiv 10.1101/2021.04.29.441998 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Wontakal SN, Emelyanov AV, Morcillo P, Konev AY, Fyodorov DV, Skoultchi AI (2009) Linker histone H1 is essential for Drosophila development, the establishment of pericentric heterochromatin, and a normal polytene chromosome structure. Genes Dev 23: 452–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Qian W, Bambouskova M, Collins PL, Porter SI, Byrum AK, Zhang R, Artyomov M, Oltz EM, Mosammaparast Net al (2020) Barrier‐to‐autointegration factor 1 protects against a basal cGAS‐STING response. MBio 11: e00136‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C‐A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch Aet al (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K, Matsuda T, Shindo Y, Imamura H, Tamura S, Imai R, Kawakami S, Nagashima R, Soga T, Noji Het al (2018) A transient rise in free Mg2+ ions released from ATP‐Mg hydrolysis contributes to mitotic chromosome condensation. Curr Biol 28: 444–451.e6 [DOI] [PubMed] [Google Scholar]
- Makde RD, England JR, Yennawar HP, Tan S (2010) Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467: 562–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalit A, Segura‐Totten M, Gruenbaum Y, Wilson KL (2005) Barrier‐to‐autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc Natl Acad Sci USA 102: 3290–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, Tan S (2016) Recognition of the nucleosome by chromatin factors and enzymes. Curr Opin Struct Biol 37: 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski S, de Oliveira Mann CC , Stafford CA, Witte G, Bartho J, Lammens K, Hornung V, Hopfner K‐P (2020) Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587: 678–682 [DOI] [PubMed] [Google Scholar]
- Mohr L, Toufektchan E, von Morgen P , Chu K, Kapoor A, Maciejowski J (2021) ER‐directed TREX1 limits cGAS activation at micronuclei. Mol Cell 81: 724–738.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Sanders IF, Chen EZ, Li H, Tobias JW, Isett RB, Penubarthi S, Sun H, Baldwin DA, Fraser NW (2015) Genome wide nucleosome mapping for HSV‐1 shows nucleosomes are deposited at preferred positions during lytic infection. PLoS One 10: e0117471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Mann CC , Kranzusch PJ (2017) cGAS conducts micronuclei DNA surveillance. Trends Cell Biol 27: 697–698 [DOI] [PubMed] [Google Scholar]
- Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM (2015) cGAS‐mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci USA 112: E1773–E1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, Kempf G, Weiss J, Kozicka Z, Guey B (2020) Structural mechanism of cGAS inhibition by the nucleosome. Nature 587: 668–672 [DOI] [PubMed] [Google Scholar]
- Roussel L, Erard M, Cayrol C, Girard JP (2008) Molecular mimicry between IL‐33 and KSHV for attachment to chromatin through the H2A–H2B acidic pocket. EMBO Rep 9: 1006–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertman P, Bekker‐Jensen S, Mailand N (2016) Regulation of DNA double‐strand break repair by ubiquitin and ubiquitin‐like modifiers. Nat Rev Mol Cell Biol 17: 379 [DOI] [PubMed] [Google Scholar]
- Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu R‐M, Zhu P, Li G (2014) Cryo‐EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344: 376–380 [DOI] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z (2009) ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 106: 8653–8658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Huang Yu, Mei S, Xu F, Liu X, Zhao F, Yin L, Zhang Di, Wei L, Wu Cet al (2021) A nuclear export signal is required for cGAS to sense cytosolic DNA. Cell Rep 34: 108586 [DOI] [PubMed] [Google Scholar]
- Uckelmann M, Sixma TK (2017) Histone ubiquitination in the DNA damage response. DNA Repair 56: 92–101 [DOI] [PubMed] [Google Scholar]
- Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El‐Daher M‐T, Rice GI, Dhir S, Wheeler AP, Dhir A (2020) cGAS‐mediated induction of type I interferon due to inborn errors of histone pre‐mRNA processing. Nat Genet 52: 1–9 [DOI] [PubMed] [Google Scholar]
- Uhlorn BL, Gamez ER, Li S, Campos SK (2020) Attenuation of cGAS/STING activity during mitosis. Life Sci Alliance 3: e201900636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia‐Sánchez MI, Abini‐Agbomson S, Wang M, Lee R, Vasilyev N, Zhang J, De Ioannes P, La Scola B, Talbert P, Henikoff S (2021) The structure of a virus‐encoded nucleosome. Nat Struct Mol Biol 28: 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Cambier S, Gray EE, Stetson DB (2019) Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 8: e47491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DAet al (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Benlekbir S, Fradet‐Turcotte A, Sherker A, Julien J‐P, McEwan A, Noordermeer SM, Sicheri F, Rubinstein JL, Durocher D (2016) The structural basis of modified nucleosome recognition by 53BP1. Nature 536: 100–103 [DOI] [PubMed] [Google Scholar]
- Worden EJ, Hoffmann NA, Hicks CW, Wolberger C (2019) Mechanism of cross‐talk between H2B ubiquitination and H3 methylation by Dot1L. Cell 176: 1490–1501.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Wang S, Ye B, Du Y, Li C, Xiong Z, Qu Y, Fan Z (2018) A circular RNA protects dormant hematopoietic stem cells from DNA sensor cGAS‐mediated exhaustion. Immunity 48: 688–701.e7 [DOI] [PubMed] [Google Scholar]
- Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, Patel DJ (2019) Human cGAS catalytic domain has an additional DNA‐binding interface that enhances enzymatic activity and liquid‐phase condensation. Proc Natl Acad Sci USA 116: 11946–11955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ (2017) cGAS is essential for cellular senescence. Proc Natl Acad Sci USA 114: E4612–E4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP containing mixed phosphodiester linkages is an endogenous high‐affinity ligand for STING. Mol Cell 51: 226–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, Chen ZJ (2014) The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch‐like conformational changes in the activation loop. Cell Rep 6: 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, West AP, Liu WR, Li P (2020) The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587: 673–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang Y‐Y, Li Y, Diao F, Lei C, He X, Zhang Lu, Tien Pet al (2008) The adaptor protein MITA links virus‐sensing receptors to IRF3 transcription factor activation. Immunity 29: 538–550 [DOI] [PubMed] [Google Scholar]
- Zhong L, Hu M‐M, Bian L‐J, Liu Y, Chen Q, Shu H‐B (2020) Phosphorylation of cGAS by CDK1 impairs self‐DNA sensing in mitosis. Cell Discovery 6: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Mohr L, Maciejowski J, Kranzusch PJ (2021) cGAS phase separation inhibits TREX1‐mediated DNA degradation and enhances cytosolic DNA sensing. Mol Cell 81: 739–755.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo J‐D, Carroll T, Funabiki H (2019) The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178: 302–315.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]