

Abstract
SARS‐CoV‐2 is a newly emerged coronavirus that caused the global COVID‐19 outbreak in early 2020. COVID‐19 is primarily associated with lung injury, but many other clinical symptoms such as loss of smell and taste demonstrated broad tissue tropism of the virus. Early SARS‐CoV‐2–host cell interactions and entry mechanisms remain poorly understood. Investigating SARS‐CoV‐2 infection in tissue culture, we found that the protease TMPRSS2 determines the entry pathway used by the virus. In the presence of TMPRSS2, the proteolytic process of SARS‐CoV‐2 was completed at the plasma membrane, and the virus rapidly entered the cells within 10 min in a pH‐independent manner. When target cells lacked TMPRSS2 expression, the virus was endocytosed and sorted into endolysosomes, from which SARS‐CoV‐2 entered the cytosol via acid‐activated cathepsin L protease 40–60 min post‐infection. Overexpression of TMPRSS2 in non‐TMPRSS2 expressing cells abolished the dependence of infection on the cathepsin L pathway and restored sensitivity to the TMPRSS2 inhibitors. Together, our results indicate that SARS‐CoV‐2 infects cells through distinct, mutually exclusive entry routes and highlight the importance of TMPRSS2 for SARS‐CoV‐2 sorting into either pathway.
Keywords: Coronavirus, COVID‐19, protease, SARS‐CoV‐2, virus entry
Subject Categories: Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
SARS‐CoV‐2 infects cells through mutually‐exclusive, pH‐independent versus pH‐dependent entry routes, whose distinction is governed by processing protease expression in target cells.

Introduction
The Coronaviridae is a large viral family, consisting of several hundred members, that constitutes the order Nidovirales along with Arteriviridae and Roniviridae (Modrow et al, 2013). To date, four coronaviruses (CoVs) have been identified as leading causes of the common cold in humans (Paules et al, 2020). Three other CoVs that cause severe respiratory disease have emerged in the human population as a result of spillover events from wildlife during the last two decades (Hartenian et al, 2020). Severe acute respiratory syndrome (SARS)‐CoV and Middle East respiratory syndrome (MERS)‐CoV were first isolated in China in 2002 and Saudi Arabia in 2011, respectively (Hartenian et al, 2020). The most recent CoV, SARS‐CoV‐2, is responsible for CoV‐induced disease 2019 (COVID‐19), which became a pandemic in early 2020. As of May 24, 2021, more than 167 million human cases of COVID‐19 have been reported with at least 3.4 million deaths.
Similar to other CoVs, SARS‐CoV‐2 has enveloped, roughly spherical particles, with a diameter between 90 and 110 nm (Caly et al, 2020; Ke et al, 2020; Matsuyama et al, 2020). The viral genome consists of one single‐stranded positive‐sense RNA segment that replicates in the cytosol and encodes four structural proteins. Three transmembrane proteins are embedded in the viral envelope and are exposed at the virion surface, namely the large glycoprotein S, the membrane protein M, and the envelope protein E (Hartenian et al, 2020). The nucleoprotein NP binds to the genomic RNA to form nucleocapsid structures inside viral particles. In the viral envelope, glycoprotein S forms spike‐like projections up to 35 nm in length that are responsible for virus attachment to host cells and penetration by membrane fusion (Turonova et al, 2020).
Although SARS‐CoV‐2 has been the subject of intense research since the beginning of 2020, our current understanding of cell entry remains essentially derived from studies on SARS‐CoV‐1 and other CoVs (Hartenian et al, 2020). SARS‐CoV‐2 has been shown to rely on ACE2 (Hoffmann et al, 2020b), heparan sulfates (Clausen et al, 2020), and neuropilin‐1 (Daly et al, 2020) at the cell surface for infection. Inhibitor studies support the possibility that the virus enters endosomal vesicles and relies on vacuolar acidification for the infectious entry process (Hoffmann et al, 2020b; Ou et al, 2020; Wang et al, 2020). As with many other CoVs, there is intense debate as to whether SARS‐CoV‐2 enters host cells from the plasma membrane directly or through intracellular compartments.
To gain access to the cytosol, enveloped viruses must fuse their envelope with the cell membrane. Several classes of viral fusion proteins are known to mediate this process, each with their own molecular specificities [reviewed in (Harrison, 2015)]. Structural studies categorized the SARS‐CoV‐2 S protein as a class‐I viral fusion protein, within the same group as those from other CoVs, human immunodeficiency virus, and influenza A virus (IAV) (Lai et al, 2017; Walls et al, 2020; Wrapp et al, 2020). Cryo‐electron microscopy showed that the S protein forms homotrimers on the surface of the SARS‐CoV‐2 particle, in which the viral fusion subunits are buried (Ke et al, 2020; Walls et al, 2020; Wrapp et al, 2020). The activation of class‐I viral fusion proteins usually involves proteolytic processing, and membrane fusion is triggered by interactions with cell receptors and sometimes endosomal acidification. Activation and priming are irreversible steps, and class‐I viral fusion proteins act only once (Harrison, 2015). In the case of SARS‐CoV‐2, endosomal acidification appears to be nonessential for spike‐mediated fusion of the host membrane with the viral envelope (Buchrieser et al, 2020). However, why SARS‐CoV‐2 infection is sensitive to the perturbation of endosomal acidification remains unclear.
Several proteases have been proposed to prime and activate the S protein (Bestle et al, 2020; Tang et al, 2021), a step prior to virus fusion and infection. Furin is a calcium‐dependent serine endoprotease that is widely expressed in diverse tissues. It has been proposed to cleave the S protein at site S1/S2 (Bestle et al, 2020; Coutard et al, 2020; Hoffmann et al, 2020a), most likely when viral progeny exit infected cells. The cleavage produces two subunits, S1 and S2. S1 contains a receptor‐binding domain, and S2 is the membrane fusion effector. Additional proteolytic cleavage in the S2 subunit at the site S2′ occurs during virus entry to trigger fusion of the viral envelope with the host cell membrane. Transmembrane serine protease 2 (TMPRSS2), a cell surface trypsin‐like protease (Choi et al, 2009), and cathepsin L, an endolysosomal cysteine protease (Mohamed & Sloane, 2006), have both been proposed to be involved in cleavage at the S2′ site (Bestle et al, 2020; Hoffmann et al, 2020b; Liu et al, 2020; Matsuyama et al, 2020; Shang et al, 2020). Nevertheless, the timing and dynamics of these proteolytic cleavages and their potential roles in SARS‐CoV‐2 activation, fusion, and entry remain poorly characterized.
SARS‐CoV‐2 primarily targets cells of the lung epithelium but is also found in many other epithelial tissues as it spreads throughout the host. Epithelia cells express ACE2, TMPRSS2, and cathepsin L, and these different cellular factors are likely to differentially influence the cell entry mechanisms of SARS‐CoV‐2 in a specific manner. To determine whether SARS‐CoV‐2 enters epithelial cells from intracellular compartments and whether endosomal acidification is involved, we analyzed the proteolytic processing, dependence on low pH for infection, intracellular trafficking, and membrane fusion of the virus in various epithelial cell types. The results showed that SARS‐CoV‐2 shares with other CoVs the ability to use different host cell proteases and distinct entry pathways to infect target cells. Unlike SARS‐ and MERS‐CoV, SARS‐CoV‐2 uses mutually exclusive routes to enter cells, and TMPRSS2 is critical for the sorting of the virus into either pathway.
Results
Characterization of the SARS‐CoV‐2 life cycle in Caco‐2 and Vero cells
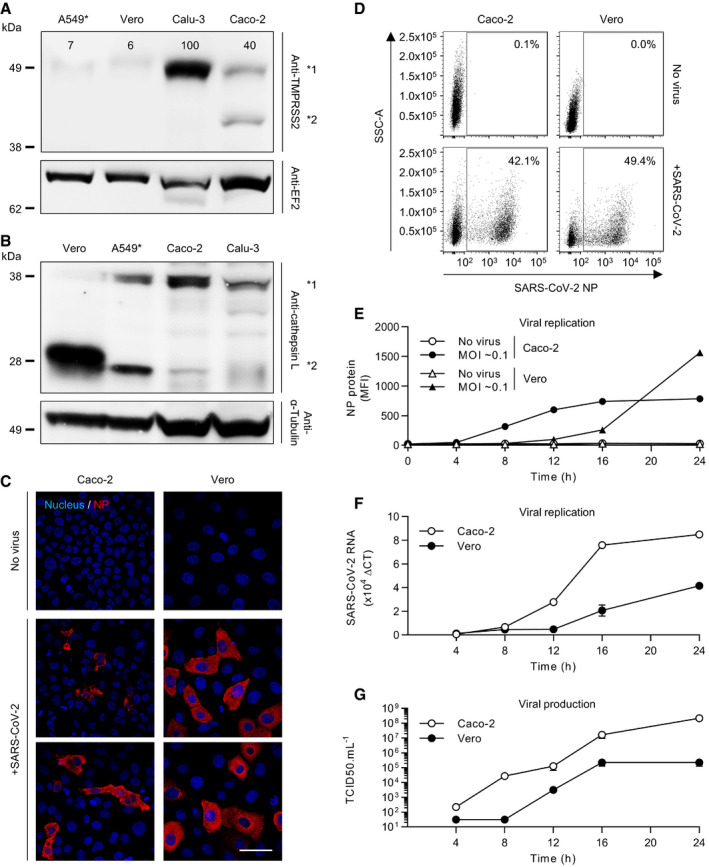
Many epithelial cell types have been reported to support productive SARS‐CoV‐2 infection (Hoffmann et al, 2020b), and both TMPRSS2 and cathepsin L in target cells have been implicated in the proteolytic processing of the viral S protein (Bestle et al, 2020; Hoffmann et al, 2020b; Liu et al, 2020; Matsuyama et al, 2020; Shang et al, 2020). We selected four epithelial cell lines that are known to support SARS‐CoV‐2 infection, i.e., Calu‐3, Caco‐2, A549, and Vero cells (Hoffmann et al, 2020b). A549 cells are intrinsically poorly infectable by SARS‐CoV‐2 due to the absence of the SARS‐CoV‐2 receptor ACE2 (Hoffmann et al, 2020b). As such, we used A549 cells stably overexpressing ACE2 (A549*) (Steuten et al, 2021). When cell lysates were subjected to SDS–PAGE and Western blotting, we found that TMPRSS2 was effectively expressed in Calu‐3 cells and to a lesser extent in Caco‐2 cells (Fig 1A), corroborating results from other groups (Zecha et al, 2020; Steuten et al, 2021). TMPRSS2 was seen as a band of approximately 50 kDa in both Calu‐3 and Caco‐2 cells. A second band of approximately 42 kDa was observed in Caco‐2 cells, which represents a cleaved form of TMPRSS2 (Chen et al, 2010). Regardless of the presence of TMPRSS2, cathepsin L (from 25 to 31 kDa) and its inactive form, i.e., procathepsin L (35 to 41 kDa), were present in all the cell lines (Fig 1B). However, the conversion of procathepsin L to cathepsin L appeared to be significantly higher in Vero cells than in the three other cell lines.
Figure 1. Quantification of SARS‐CoV‐2 infection.
-
A, BCells were lysed and analyzed by SDS–PAGE and Western blotting under nonreducing conditions (A) or reducing conditions (B). TMPRSS2 levels are expressed as percentages of TMPRSS2 levels in Calu‐3 cells normalized to levels of EF2. *1 indicates TMPRSS2 (A) and procathepsin L (B), and *2 shows cleaved TMPRSS2 (A) and cathepsin L (B). A549*, ACE2‐expressing A549 cells; EF2, elongation factor 2.
-
CVero and Caco‐2 cells were infected with SARS‐CoV‐2 at MOIs of 0.2 for 8 h. Infected cells were then permeabilized and immunostained for the intracellular SARS‐CoV‐2 nucleoprotein (NP, red). Nuclei were stained with Hoechst (blue) before imaging by fluorescence confocal microscopy. Scale bars: 100 µm.
-
DVero and Caco‐2 cells were exposed to SARS‐CoV‐2 at MOIs of 0.3 and 0.2, respectively, and harvested 16 h later. After fixation and permeabilization, infected cells were stained with the primary mAb against NP. Infection was analyzed by flow cytometry. SSC‐A = side scatter‐area.
-
EInfection of Vero and Caco‐2 cells was monitored over 24 h using the same flow cytometry‐based assay used for the experiment shown in panel D. Infection is given as the total fluorescence associated with the NP protein‐positive cells. MFI, mean fluorescence intensity. n = 3.
-
FSARS‐CoV‐2 mRNA levels were quantified by qRT–PCR in both Vero and Caco‐2 cells infected at MOIs of 0.3 and 0.4, respectively, for up to 24 h. n = 3.
-
GSupernatants from infected cells were collected during the time course in F and assessed for the production of new infectious viral particles using a TCID50 assay on naïve Vero cells. n = 3–6.
Data information: Images are representative of at least three independent experiments. (E, F) Results are representative of three independent experiments and expressed as mean ± standard error of mean (SEM) of three biological replicates.
Source data are available online for this figure.
To address how the presence or absence of TMPRSS2 influences SARS‐CoV‐2 infectious penetration and how endosomal acidification contributes to the process, we aimed to compare cell lines expressing or not expressing this protease. To this end, we first defined the timing for a single round of infection in the selected cell lines. Calu‐3 and Caco‐2 served as TMPRSS2‐positive (TMPRSS2+) cells, and A549* and Vero cells served as TMPRSS2‐nonexpressing (TMPRSS2−) cells. The susceptibility of Caco‐2 and Vero cells to SARS‐CoV‐2 at a multiplicity of infection (MOI) of 0.2 was assessed 8 h post‐infection (hpi) by fluorescence microscopy after immunostaining with a mouse monoclonal antibody (mAb) against the intracellular viral nucleoprotein NP (Fig 1C). The results showed that 10% of Caco‐2 cells were positive for NP while 35% of Vero cells were infected (Fig 1C).
To quantify infection more accurately, we then performed flow cytometry analysis of Caco‐2 and Vero cells infected with SARS‐CoV‐2 at different MOIs (Fig 1D and E). The fluorescence increased over time and reached a plateau within 16 hpi in Caco‐2 cells (Fig 1E and Appendix Fig S1), showing that the signal detected in the flow cytometry‐based assays corresponded to viral replication and not to the input particles. These kinetics were in agreement with real‐time quantitative reverse transcription PCR (qRT–PCR) monitoring over time of the SARS‐CoV‐2 genome (Fig 1F).
To evaluate the production and release of de novo infectious viral particles, we infected Caco‐2 and Vero cells and quantified virus production up to 24 hpi by a 50% tissue culture infective dose assay (TCID50). Infectious progeny viruses were found to be released from infected cells as early as 8–12 hpi (Fig 1G). Virus replication kinetics and de novo virus release were found to be similar in Calu‐3 and A549* cells. Altogether, our analysis revealed that SARS‐CoV‐2 completes one round of infection, from virus binding and entry to replication and release of de novo infectious particles, within 8 h in Caco‐2 cells and in a somewhat longer window in Vero cells, between 8 and 12 h. In all further experiments, as we aimed to characterize SARS‐CoV‐2 entry mechanisms, we limited our assays to 8 hpi. In addition, as our cell lines differed in their sensitivity to SARS‐CoV‐2 infection, we used different MOIs for each cell line allowing the infection of approximately 20% of the cells. This range of infection generally avoids saturation of cells and thus allows detection of potential inhibitory or enhancing effects of a perturbant.
SARS‐CoV‐2 makes a differential use of host cell proteases for infectious penetration
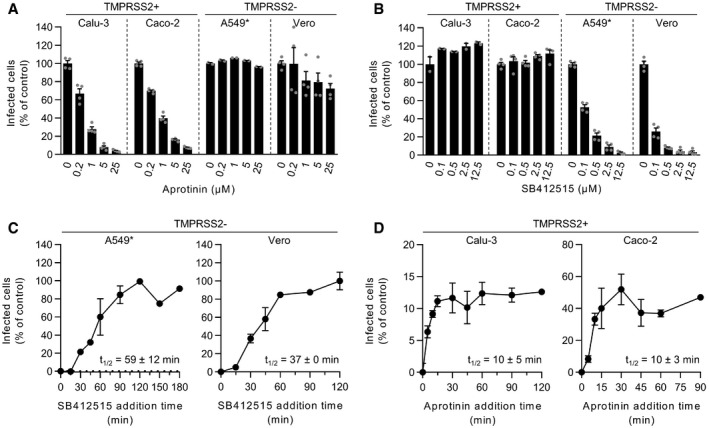
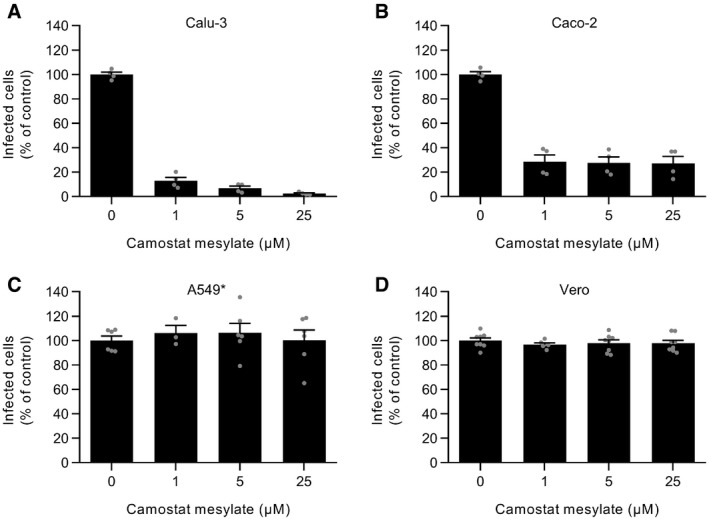
To evaluate the role of the cell surface TMPRSS2 and endolysosomal cathepsin L proteases in the entry mechanisms of SARS‐CoV‐2, we used aprotinin and SB412515, respectively, to selectively inhibit the two proteases. As expected, no noticeable effect was observed when aprotinin was added to TMPRSS2− cells (A549* and Vero cells) prior to infection (Fig 2A and Appendix Fig S2). In agreement with previous work (Bojkova et al, 2020), we observed that aprotinin reduced SARS‐CoV‐2 infection in a dose‐dependent manner in cells that expressed TMPRSS2 (Calu‐3 and Caco‐2 cells) (Fig 2A). Similar results were obtained with camostat mesylate (Fig EV1, EV2, EV3, EV4), a more specific and potent inhibitor of TMPRSS2 than aprotinin, also known to block SARS‐CoV‐2 infection (Hoffmann et al, 2020b). Conversely, SB412515 effectively prevented the infection of cells lacking TMPRSS2 (Vero and A549* cells) in a dose‐dependent manner but had no effect on SARS‐CoV‐2 infection of Calu‐3 and Caco‐2 cells (Fig 2B and Appendix Fig S3). The observation that aprotinin interfered with SARS‐CoV‐2 infection in Calu‐3 and Caco‐2 cells indicated that, in the event that TMPRSS2 was blocked, cathepsin L would not take over and subsequently process SARS‐CoV‐2. Of note, the protease inhibitors, and all other drugs used in this work, were evaluated in a range of concentrations for which no cytotoxicity was detected, as shown by a quantitative assay measuring the release of lactate dehydrogenase into the extracellular medium upon cell death and lysis (Appendix Fig S4A–D).
Figure 2. SARS‐CoV‐2 makes differential use of host cell proteases for infectious penetration.
-
A, BCells were pretreated with the indicated concentrations of aprotinin (A) and SB412515 (B), which are inhibitors of TMPRSS2 and cathepsin L, respectively. Infection of Calu‐3, Caco‐2, A549*, and Vero cells with SARS‐CoV‐2 at MOIs of 0.3, 0.4, 0.2, and 0.3, respectively, was achieved in the continuous presence of the drug. Infected cells were quantified by flow cytometry as described in Fig 1D, and data were normalized to samples where inhibitors had been omitted. n = 2–4 biological replicates.
-
C, DSARS‐CoV‐2 particles were bound to A549* and Vero cells (MOIs 0.2 and 0.3, respectively) (C) or Calu‐3 and Caco‐2 cells (0.6 and 0.5, respectively) (D) on ice for 90 min and subsequently warmed rapidly to 37°C to allow infectious penetration. SB412515 (10 µM, C) or aprotinin (30 µM, D) was added at different times postwarming to block further proteolytic activation. Infection was analyzed by flow cytometry, and data were normalized to samples where protease inhibitors had been omitted. n = 2.
Data information: (A, B) Data are expressed as mean ± SEM from two independent experiments. (C, D) Results are representative of 2–3 independent experiments and expressed as mean ± SEM of two biological replicates.
Source data are available online for this figure.
Figure EV1. Camostat mesylate blocks SARS‐CoV‐2 infection in TMPRSS2+ cells.
-
A–DCalu‐3, Caco‐2, A549*, and Vero cells were pretreated with camostat mesylate, which selectively inhibits TMPRSS2, at the indicated concentrations and subsequently infected with SARS‐CoV‐2 (MOI ˜0.3, 0.4, 0.2, and 0.3, respectively) in the continuous presence of the drug. The proportion of infected cells was quantified by flow cytometry as described in Fig 1D, and data were normalized to that from control samples where camostat mesylate had been omitted. n (A and B) = 4 and n (C and D) = 3–8.
Data information: Data are expressed as mean ± SEM from 2 to 3 independent experiments.
Source data are available online for this figure.
Figure EV2. NH4Cl interferes with SARS‐CoV‐2 replication in Calu‐3 and Caco‐2 cells.
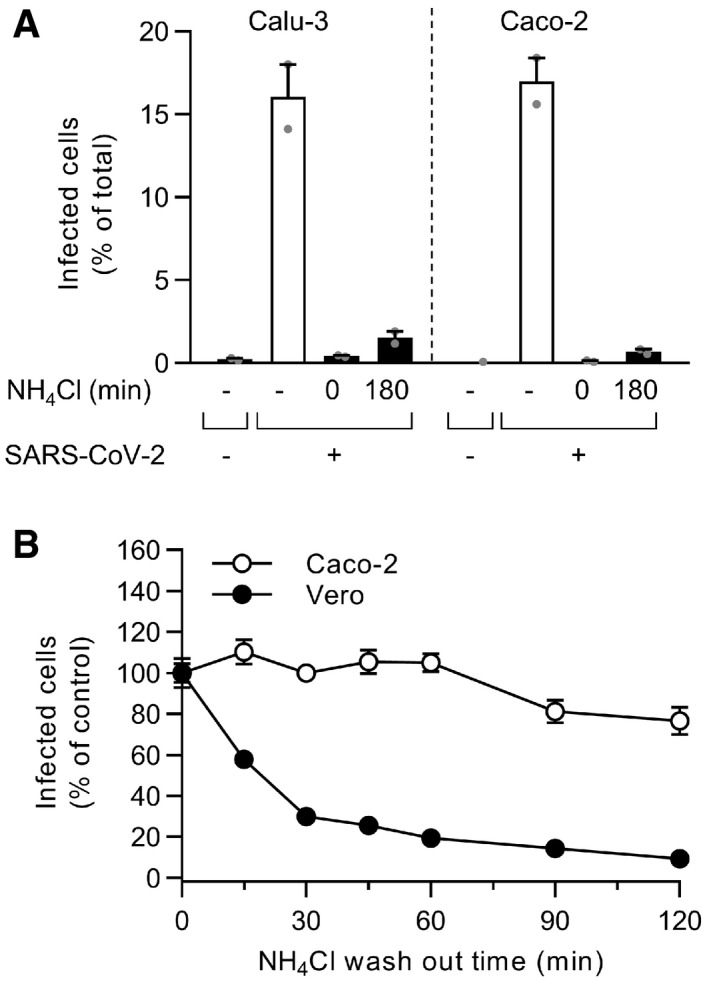
- The controls of Fig 3E are shown here. Briefly, binding of SARS‐CoV‐2 to Calu‐3 or Caco‐2 cells (MOI ˜0.6 and 0.5) was synchronized on ice for 90 min. Subsequently, cells were rapidly shifted to 37°C to allow penetration. NH4Cl (75 mM) was added 3 hpi to neutralize endosomal pH. Infection was analyzed by flow cytometry, and raw data are shown. n = 1–2.
- After the synchronization of SARS‐CoV‐2 binding to Caco‐2 and Vero cells (MOI ˜0.5 and 0.3, respectively) on ice for 90 min, NH4Cl (50 mM) was added and then washed out at the indicated time points to allow endosomal acidification and the acid‐dependent step of SARS‐CoV‐2 infectious penetration. Infection was analyzed by flow cytometry, and values were normalized to those from samples for which NH4Cl was removed at t0. n = 2.
Data information: Results are representative of two independent experiments and expressed as mean ± SEM from two biological replicates.
Source data are available online for this figure.
Figure EV3. The presence of serum during biosynthesis does not influence SARS‐CoV‐2 infectious entry pathways.
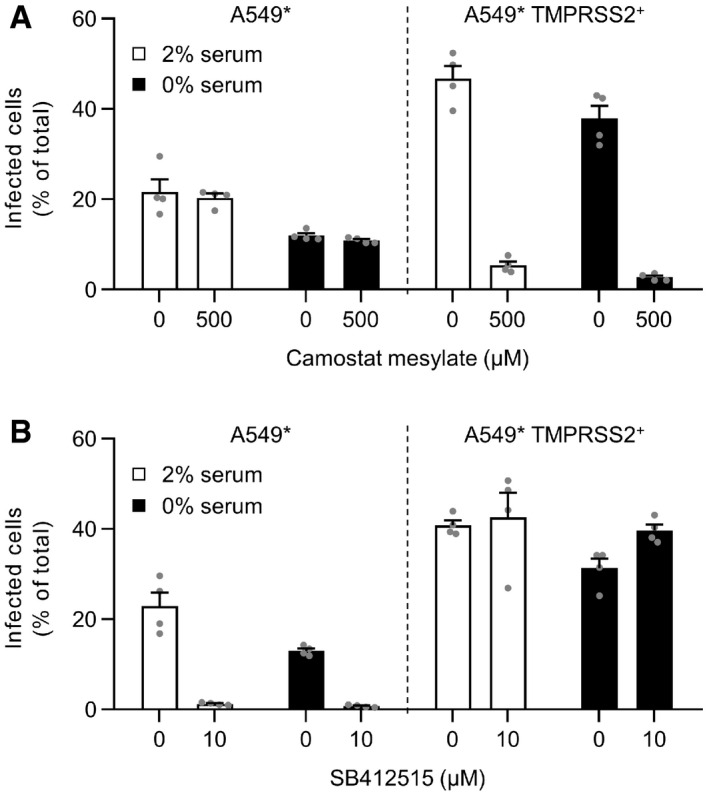
-
A, BA549* cells expressing or lacking TMPRSS2 were pretreated with camostat mesylate at 500 µM (A) or SB412515 at 10 µM (B) and subsequently infected with SARS‐CoV‐2 (MOI of 0.2) produced in the presence or absence of serum. Infection was performed in the continuous presence of the drugs. Infected cells were quantified by flow cytometry as described in Fig 1D, and data were normalized to samples where inhibitors had been omitted. n = 4.
Data information: Data are expressed as mean ± SEM from two independent experiments.
Source data are available online for this figure.
Figure EV4. SARS‐CoV‐2‐mediated cell–cell fusion in TMPRSS2+ A549* cells.
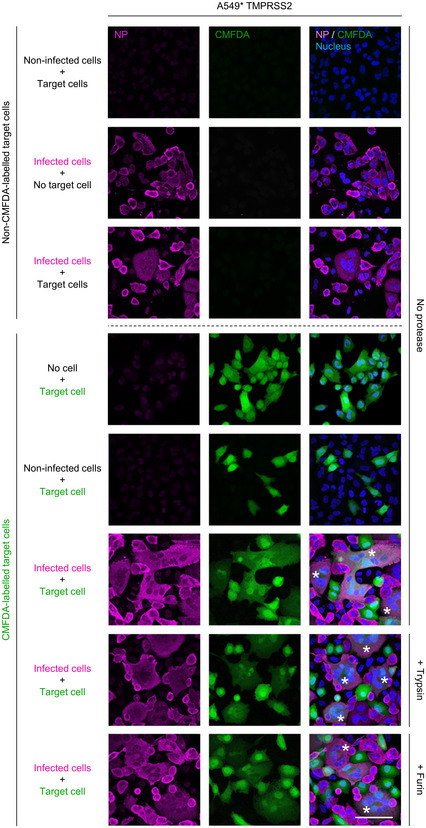
TMPRSS2+ A549* cells were first infected with SARS‐CoV‐2 at an MOI of ˜0.1 for 24 h and cocultured for 5 h along with target cells, which were not infected with SARS‐CoV‐2 but had been prestained with CMFDA, a cytosolic green dye. Cells were subsequently treated with trypsin or furin for 5 min at 37°C and left to coculture for an additional hour at 37°C. After fixation, nuclei were stained with Hoechst (blue), and infected cells were subjected to immunofluorescence staining against SARS‐CoV‐2 nucleoprotein (magenta). Samples were imaged by confocal fluorescence microscopy. White stars indicate syncytia with at least four nuclei. Scale bar: 100 µm.
Data information: Images are representative of three independent experiments.
We next determined the kinetics of the cathepsin L‐ and TMPRSS2‐dependent SARS‐CoV‐2 entry process. Cells were incubated with viruses at a low MOI (0.6<) on ice and rapidly shifted to 37°C to allow virus entry and protease activity. Cathepsin L and TMPRSS2 inhibitors were added at different times after warming to prevent further activation and penetration of the virus. In other words, we determined the time at which inhibition of SARS‐CoV‐2 activation was no longer possible, which resulted in an increase in infection. In both TMPRSS2− cell lines (A549* and Vero cells), the SB412515 add‐in time course revealed that activation by cathepsin L and the subsequent infectious penetration of SARS‐CoV‐2 started after a 15‐min lag and reached a half‐maximal level (t1/2) within 40–60 min (Fig 2C). Evidently, exposure of individual viruses to cathepsin L occurred nonsynchronously during a time span of 15–90 min after warming. The aprotinin add‐in time course showed that productive penetration was much faster in TMPRSS2+ cells (Calu‐3 and Caco‐2 cells) (Fig 2D). The t 1/2 of activation by TMPRSS2 was reached within 5–10 min in both cell lines. Taken together, our observations demonstrated more rapid activation and penetration of SARS‐CoV‐2 in cells expressing TMPRSS2 compared with those in which infection depends on cathepsin L.
TMPRSS2 governs SARS‐CoV‐2 dependence on low pH for infectious entry
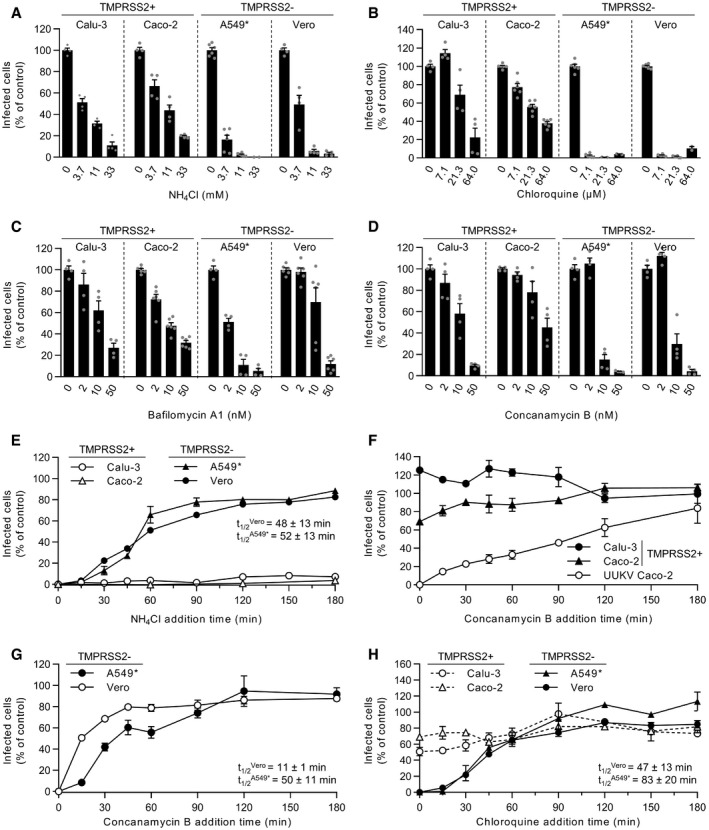
Recent reports have indicated that SARS‐CoV‐2 infection is sensitive to weak lysosomotropic bases that neutralize vacuolar pH such as ammonium chloride (NH4Cl) and chloroquine (Hoffmann et al, 2020b; Ou et al, 2020; Wang et al, 2020). However, TMPRSS2 is active at the cell surface under neutral pH conditions (Choi et al, 2009), unlike cathepsin L, which requires the low‐pH environment typical of endolysosomes (Mohamed & Sloane, 2006). To assess the importance of endosomal acidification for infectious entry in cells expressing (Caco‐2 and Calu‐3) and lacking (A549* and Vero) TMPRSS2, cells were exposed to SARS‐CoV‐2 in the presence of increasing amounts of NH4Cl or chloroquine. Our results showed that both weak bases induced a dose‐dependent inhibition of infection regardless of cell type and of TMPRSS2 expression (Fig 3A and B). However, the dose required to inhibit 50% of SARS‐CoV‐2 infection (IC50) was found to be significantly lower in cells lacking TMPRSS2 than in cells that expressed the protease; this difference reached 200‐fold for chloroquine (Table 1).
Figure 3. SARS‐CoV‐2 infection depends on endosomal acidification.
-
A–DCells were pretreated with endosomal pH‐interfering drugs at the indicated concentrations and subsequently infected with SARS‐CoV‐2 as described in Fig 2A and B in the continuous presence of NH4Cl (A), chloroquine (B), bafilomycin A1 (C), or concanamycin B (D). The proportion of infected cells was quantified by flow cytometry as described in Fig 1D, and data were normalized to control samples that were not treated with inhibitors. n = 2–6 biological replicates.
-
EBinding of SARS‐CoV‐2 to Calu‐3, Caco‐2, A549*, and Vero cells (MOIs of 0.5, 0.6, 0.2, and 0.3, respectively) was synchronized on ice for 90 min. Subsequently, cells were rapidly shifted to 37°C to allow penetration. NH4Cl (50 mM for A549* and Vero cells, and 75 mM for Calu‐3 and Caco‐2 cells) was added at the indicated times to neutralize endosomal pH and block the acid‐dependent step of SARS‐CoV‐2 infectious penetration. The proportion of infected cells was analyzed by flow cytometry, and data were normalized to that from control samples that had not been treated with NH4Cl. n = 2.
-
F, GSame as in (E) but using concanamycin B (50 nM) instead of NH4Cl. Uukuniemi virus (UUKV) was used at a MOI of 150 to control the efficiency of concanamycin B to neutralize endosomal pH in Caco‐2 cells. n = 2.
-
HSame as in (E) but using chloroquine instead of NH4Cl. n = 2.
Data information: (A‐D) Data are expressed as mean ± SEM from at least two independent experiments. (E‐H) Results are representative of three independent experiments and expressed as mean ± SEM of two biological replicates.
Source data are available online for this figure.
Table 1.
Half‐maximal inhibitory concentration (IC50) of inhibitors against SARS‐CoV‐2.
| IC50 ± SEM | TMPRSS2+ | TMPRSS2− | ||
|---|---|---|---|---|
| Calu‐3 | Caco‐2 | A549* | Vero | |
| Aprotinin (µM) | 0.4 ± 0.1 | 0.6 ± 0.0 | x | x |
| Bafilomycin A1 (nM) | 16.3 ± 6.6 | 10.4 ± 2.3 | 2.0 ± 0.6 | 18.6 ± 5.5 |
| Camostat mesylate (nM) | 72.9 ± 25.8 | 806 ± 481 | x | x |
| Chloroquine (µM) | 50.1 ± 24.4 | 27.4 ± 2.8 | 0.3 ± 0.0 | 0.2 ± 0.1 |
| Concanamycin B (nM) | 12.2 ± 5.8 | 50.3 ± 30.4 | 6.0 ± 1.2 | 8.6 ± 2.2 |
| MG‐132 (nM) | 670 ± 205 | 5,249 ± 2,129 | 4.4 ± 1.4 | 16.4 ± 5.6 |
| NH4Cl (mM) | 4.4 ± 0.9 | 7.9 ± 2.4 | 2.2 ± 0.1 | 2.5 ± 0.6 |
| SB412515 (nM) | x | x | 125.7 ± 29.9 | 36.9 ± 10.9 |
To validate the observation that TMPRSS2+ cells were less dependent on endosomal acidification for SARS‐CoV‐2 infection, we used bafilomycin A1 and concanamycin B, which are inhibitors of vacuolar‐type proton‐ATPases (vATPases). Incubation of cells with increasing amounts of the two drugs resulted in a dose‐dependent inhibition of SARS‐CoV‐2 infection (Fig 3C and D). Importantly, the inhibition produced by 10 nM bafilomycin A1 or concanamycin B in TMPRSS2+ cells (Caco‐2 and Calu‐3) was marginal, and the decrease in infection did not surpass 50–80% at 50 nM concanamycin B. For comparison, infection with Uukuniemi virus (UUKV), a late‐penetrating virus that relies on low pH in late endosomes (LEs) for penetration (Lozach et al, 2010), is strongly inhibited in the presence of 2–10 nM concanamycin B or bafilomycin A1 (Lozach et al, 2010). From these results, it was evident that, similar to the case for weak lysosomotropic bases, SARS‐CoV‐2 infection was significantly less sensitive to vATPase inhibitors in TMPRSS2+ cells (Caco‐2 and Calu‐3) than in cells lacking the protease (Vero and A549* cells) (Table 1).
SARS‐CoV‐2 can use two distinct routes to enter and infect target cells
Our results suggested that SARS‐CoV‐2 infection relied more on endosomal acidification in cells devoid of TMPRSS2 than cells expressing the protease. To further investigate this phenomenon, we first determined the kinetics of the acidification step required for the infectious penetration of SARS‐CoV‐2 into TMPRSS2− cells. We took advantage of the fact that the neutralization of endosomal pH is nearly instantaneous upon the addition of NH4Cl to the extracellular medium (Ohkuma & Poole, 1978). For these assays, virus particles were first allowed to attach to A549* and Vero cells on ice. Virus entry was then synchronized by switching the cells rapidly to 37°C, and NH4Cl was added at different times after the temperature switch. To ensure that viral penetration is completely abolished after adding NH4Cl, a concentration of up to 75 mM was used (Fig EV2A). In A549* and Vero cells, viruses had completed the NH4Cl‐sensitive step 15 min after cell warming, and t 1/2 was reached within 50 min (Fig 3E). Overall, the kinetics of SARS‐CoV‐2 acid‐activated penetration closely resembled the time course of cathepsin L‐dependent activation in the absence of TMPRSS2 (Fig 2C).
To examine how long SARS‐CoV‐2 remains acid‐activable in TMPRSS2− cells, we followed the reverse approach of adding NH4Cl. This assay relies on the fact that the neutralization of endosomal pH by NH4Cl is reversible after washing. Virus binding to Vero cells was synchronized at low MOI (˜0.3) on ice, and cells were rapidly shifted to 37°C in the presence of NH4Cl before the weak base was washed out at varying times. In other words, we determined the time at which SARS‐CoV‐2 acid activation was no longer possible. In Vero cells, infection decreased by 70% during the first 30 min and then more slowly until it reached a 90% decrease after 2 h (Fig EV2B), which was the exact opposite of the NH4Cl addition approach (Fig 3E). Together, the NH4Cl add‐in and wash‐out kinetics indicated that SARS‐CoV‐2 infectivity decreases sharply in TMPRSS2− cells if the virus is not allowed to enter the cytosol rapidly by acid‐activated penetration, most likely, from endosomal vesicles.
In Calu‐3 and Caco‐2 cells, both of which express TMPRSS2, it was not possible to determine the timing of the acid‐requiring step. We failed to detect SARS‐CoV‐2‐infected cells when NH4Cl was added, even if the addition occurred several hours after transferring the cells from 4 to 37°C (Fig 3E). In samples where NH4Cl was omitted, however, infection was readily detectable with 17% of Calu‐3 and Caco‐2 cells infected (Fig EV2A), suggesting that the weak base interferes with SARS‐CoV‐2 replication in these two cell lines. To clarify whether NH4Cl blocks virus entry or replication in these cells, we next assessed SARS‐CoV‐2 infection in our wash‐out assay. Neutralization of endosomal pH with NH4Cl in Caco‐2 cells had no noticeable effect on SARS‐CoV‐2 infection for the first hour after warming (Fig EV2B), when the virus had normally already penetrated TMPRSS2+ cells (Fig 2D). In this assay, viral replication was not affected, most likely because the weak base was washed out early enough to avoid side effects. Altogether, these results suggest that SARS‐CoV‐2 does not depend on endosomal acidification for infectious entry into TMPRSS2+ cells, but rather that NH4Cl disrupts Calu‐3 and Caco‐2 cell‐specific functions that are important for SARS‐CoV‐2 replication. NH4Cl not only neutralizes the intracellular pH but also alters all endosomal, lysosomal, and trans‐Golgi network functions that are acid dependent (Helenius, 2013).
As an alternative method to alter endosomal pH, we added the vATPase inhibitor concanamycin B to cell‐bound virus, instead of NH4Cl, at different times after warming. The time course showed that infectious entry of UUKV began later than 15 min and had not reached a maximum 2 h after cell warming (Fig 3F). In marked contrast, SARS‐CoV‐2 infection was virtually insensitive to the concanamycin B addition as early as a few seconds after shifting TMPRSS2+ cells to 37°C (Fig 3F). As expected, SARS‐CoV‐2 passed the concanamycin B‐sensitive step in TMPRSS2− cells within less than 15 min, and infectious entry reached a plateau value after 45 min, somewhat faster than in cells treated with NH4Cl (Fig 3G). This difference in sensitivity to endosomal pH may be because concanamycin B interferes not only with endosomal functions that are acid dependent but also, indirectly, with the maturation of endosomes. However, unlike NH4Cl, it was apparent that concanamycin B had no adverse effect on SARS‐CoV‐2 replication in any of these experiments. Neutralization of endosomal pH by chloroquine leads to similar kinetics of SARS‐CoV‐2 infection as concanamycin B (Fig 3H).
Taken together, these results strongly suggest that SARS‐CoV‐2 can use two different routes to enter and infect target cells, i.e., a fast pH‐independent route in TMPRSS2+ cells (Figs 2D and 3F and H, and EV2B) and a slow acid‐activated route in cells lacking TMPRSS2 (Figs 2C and 3E, G and H).
TMPRSS2 drives the pH‐independent entry of SARS‐CoV‐2
To correlate the presence of TMPRSS2 with the viral entry pathway, we examined SARS‐CoV‐2 infection in the same cell line expressing or lacking the protease. To this end, we stably expressed TMPRSS2 in the TMPRSS2− A549* cells and confirmed the overexpression by SDS–PAGE and Western blotting (Fig 4A) (Steuten et al, 2021). As expected, the TMPRSS2 inhibitor, camostat mesylate, did not inhibit SARS‐CoV‐2 infection when it was added to the parental A549* cells prior to infection (Fig 4B). However, we found that camostat mesylate reduced SARS‐CoV‐2 infection in a dose‐dependent manner in TMPRSS2‐overexpressing A549* cells (Fig 4B). Conversely, the cathepsin L inhibitor (SB412515) efficiently prevented infection of regular A549* cells but had no effect on infection of A549* cells that expressed TMPRSS2 (Fig 4C).
Figure 4. TMPRSS2 drives pH‐ and cathepsin L‐independent SARS‐CoV‐2 entry.
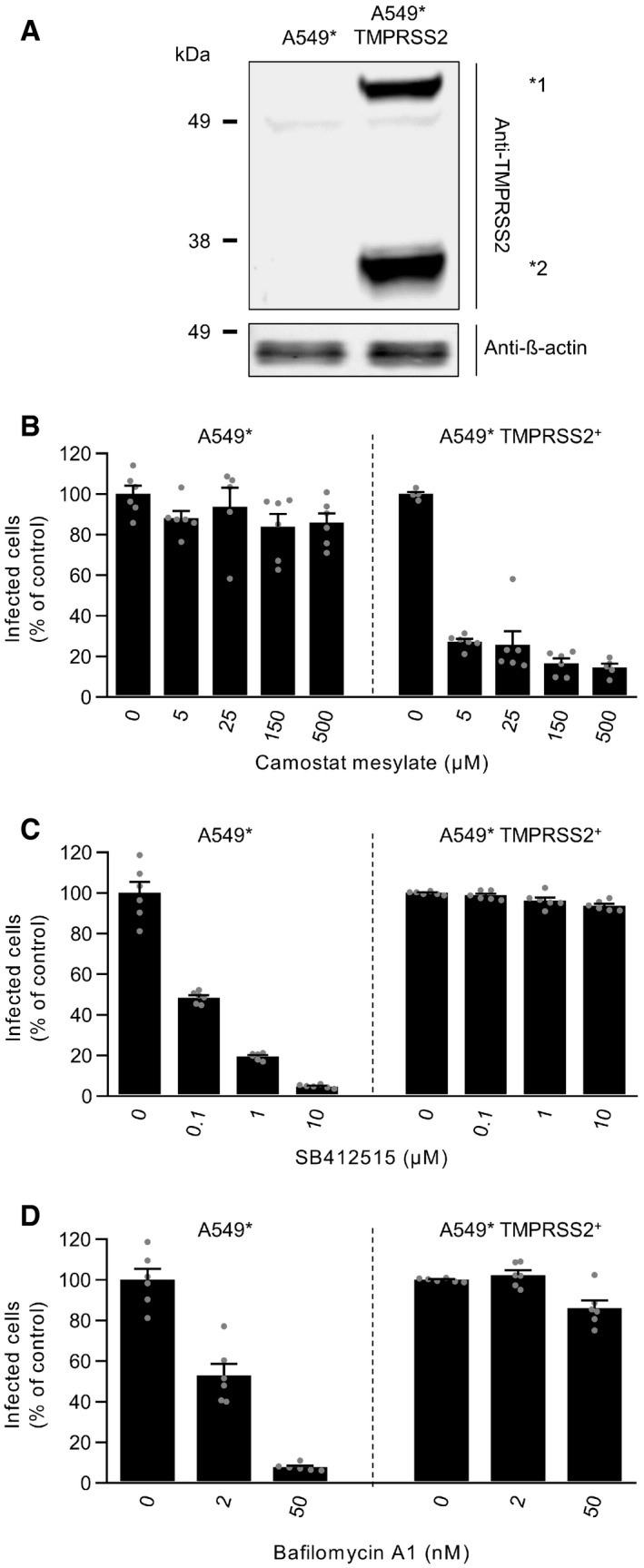
-
ATMPRSS2‐overexpressing and parental A549* cells were lysed and subjected to SDS–PAGE and Western blot analysis under reducing conditions. *1 and *2 indicate the full‐length and cleaved forms of TMPRSS2, respectively.
-
B–DA549* cells expressing or lacking TMPRSS2 were pretreated with the indicated concentrations of camostat mesylate (B), SB412515 (C), and bafilomycin A1 (D). Infection with SARS‐CoV‐2 (MOI ˜0.2) was achieved in the continuous presence of the drug. Infected cells were quantified by flow cytometry as described in Fig 1D, and data were normalized to samples where inhibitors had been omitted. n = 5–6 biological replicates.
Data information: Images are representative of at least three independent experiments. Data are all expressed as mean ± SEM from 3 independent experiments.
Source data are available online for this figure.
Our results indicated that endosomal acidification is required for SARS‐CoV‐2 infection of TMPRSS2− cells but not for infection of cells expressing TMPRSS2 (Fig 3E–H). To evaluate the role of TMPRSS2 in the dependence of SARS‐CoV‐2 on low pH for entry, A549* cells expressing or lacking TMPRSS2 were infected in the presence of bafilomycin A1 to neutralize endosomal pH. As anticipated, infection of TMPRSS2‐ A549* cells with SARS‐CoV‐2 decreased dramatically with increasing concentrations of bafilomycin A1 (Fig 4D). In contrast, there was no noticeable effect of bafilomycin A1 on infection of TMPRSS2+ A549* cells. This confirmed that SARS‐CoV‐2 infection does not rely on endosomal acidification when TMPRSS2 is expressed. Overall, TMPRSS2 appears as the major determinant of the fast pH‐independent route taken by SARS‐CoV‐2 to enter and infect TMPRSS2+ cells.
SARS‐CoV‐2 relies on endolysosomal maturation for infection of TMPRSS2− cells
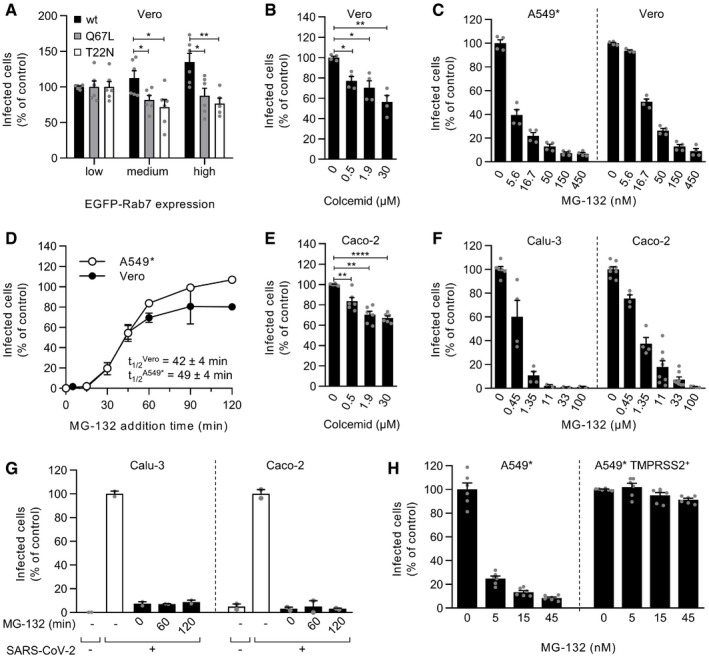
The timing of acid‐dependent and protease‐activated steps suggested that SARS‐CoV‐2 penetration might occur through endolysosomes in cells devoid of TMPRSS2 and from the plasma membrane or early endosomes (EEs) in TMPRSS2+ cells. To determine whether SARS‐CoV‐2 requires the endolysosomal compartments for the productive infection of TMPRSS2− cells, we exploited the small GTPase Rab7a, which is a key player in LE maturation and function. TMPRSS2− Vero cells were transfected with DNA plasmids encoding the wild‐type (wt), dominant‐negative (Rab7a T22N), and constitutively active (Rab7a Q67L) forms of Rab7a tagged with enhanced green fluorescent protein (EGFP) prior to infection with SARS‐CoV‐2. Transfected cells exhibiting different levels of EGFP expression (low, medium, and high) were selected and then analyzed for infection. Increasing expression of wt Rab7a facilitated SARS‐CoV‐2 infection. In contrast, increasing the expression of either Rab7a mutant, which perturbates the maturation of newly formed LEs (Lozach et al, 2010; Lozach et al, 2011a), resulted in an about 20–30% decrease in infection at the highest expression (Fig 5A).
Figure 5. SARS‐CoV‐2 relies on late endosomal maturation for infection.
-
AEGFP‐Rab7a wild‐type (wt), Q79L (constitutively active mutant), and T22N (dominant‐negative mutant) were transiently expressed in Vero cells. The cells were then infected with SARS‐CoV‐2 at an MOI of ˜0.3. Using flow cytometry, cell populations with levels of EGFP‐Rab7a expression varying by roughly one‐log increments were selected, and the proportion of infected cells within each population was quantified at 8 hpi. Data were normalized to that of the cell population with the lowest EGFP‐Rab7a intensity. Unpaired t‐test with Welch’s correction was applied. *P < 0.05; **P < 0.01. n = 5–6 biological replicates.
-
B, CVero and A549* cells were pretreated with colcemid (B) or MG‐132 (C) at the indicated concentrations and subsequently infected with SARS‐CoV‐2 (MOIs ˜0.3 and 0.2, respectively) in the continuous presence of inhibitors. Infection was analyzed by flow cytometry, and data were normalized to samples where inhibitors had been omitted. n = 3–4 biological replicates. Unpaired t‐test with Welch’s correction was applied. *P < 0.05; **P < 0.01; ****P < 0.0001.
-
DSARS‐CoV‐2 particles were bound to A549* and Vero cells (MOIs of 0.2 and 0.3, respectively) on ice for 90 min and then switched rapidly to 37°C to allow infectious penetration. MG‐132 (3.7 µM) was added to cells at the indicated times to block further late endosomal maturation. Infection was analyzed by flow cytometry, and data were normalized to that of control samples that had not been treated with MG‐132. n = 2.
-
EAs in panel B but using a MOI of 0.4 and Caco‐2 cells instead of Vero cells. n = 6 biological replicates.
-
FAs in panel C but using a MOI of 0.3 and 0.4 and Calu‐3 and Caco‐2 cells. n = 2–8 biological replicates.
-
GThe timing of the MG‐132‐sensitive step during SARS‐CoV‐2 infectious entry into Calu‐3 and Caco‐2 cells was assayed as detailed in D but using 60 µM MG‐132 for Caco‐2 cells and MOIs of 0.5 and 0.6, respectively. n = 2.
-
HA549* cells expressing or lacking TMPRSS2 were pretreated with MG‐132 at the indicated concentrations and subsequently infected with SARS‐CoV‐2 (MOI ˜0.2) in the continuous presence of the drug. The proportion of infected cells was quantified by flow cytometry as described in Fig 1D, and data were normalized to that from control samples for which MG‐132 had been omitted. n = 6 biological replicates.
Data information: (A to C, E, F, and H) data are expressed as mean ± SEM from 2‐4 independent experiments. (D and G) Results are representative of at least two independent experiments and expressed as mean ± SEM of two biological replicates.
Source data are available online for this figure.
The analysis relies on the assumption that the different Rab7a forms exhibit similar levels of expression; however, we found a 1.4‐fold higher and a 0.7‐fold lower infection in cells with a similar and high expression of wt Rab7a and either mutant of the small GTPase, respectively (Fig 5A). This comparison revealed a 50% inhibitory effect of the two Rab7a mutants on SARS‐CoV‐2 infection, indicating that virus fusion is hampered in cells with late endosomal vesicles expressing Rab7a T22N or Q67L. This inhibitory effect was very significant as late endosomal maturation is hard to completely abolish by only targeting Rab7a (Huotari & Helenius, 2011). In general, disruption of LE functions rarely leads to a complete block in infection by viruses relying on LEs for penetration (Khor et al, 2003; Quirin et al, 2008; Lozach et al, 2010). Taken together, the combination of increased and decreased infection with increasing levels of Rab7a wt and mutants, respectively, suggested that proper maturation of LEs is mandatory for cathepsin L‐dependent infectious entry of SARS‐CoV‐2.
LE maturation relies on microtubule‐mediated transport to the nuclear periphery and proteasome activity (Lozach et al, 2010; Lozach et al, 2011a). Treatment of Vero cells with colcemid, a drug that interferes with microtubule polymerization, resulted in a 30–45% decrease in infection (Fig 5B). Additionally, late endosomal penetration of IAV and UUKV has been shown to be sensitive to free ubiquitin depletion produced by the proteasome inhibitor MG‐132 (Khor et al, 2003; Lozach et al, 2010). Therefore, to determine whether free ubiquitin was required for SARS‐CoV‐2 infection, A549* and Vero cells were treated with MG‐132. The results showed that SARS‐CoV‐2 infection was strongly inhibited in the presence of MG‐132 in both cell lines (Fig 5C). The calculated IC50 (4–17 nM) confirmed that MG‐132 interfered with the cathepsin L‐mediated SARS‐CoV‐2 entry route with high efficiency (Table 1).
To determine the kinetics of the MG‐132‐sensitive step in the entry process, we followed the same experimental procedure used to determine the kinetics of endosomal acidification‐dependent and cathepsin L‐mediated activation of SARS‐CoV‐2 (Figs 2C and 3E) but with MG‐132 instead of protease inhibitor and NH4Cl. Briefly, viruses were bound to A549* and Vero cells at a low MOI on ice, rapidly switched to 37°C, and then treated with MG‐132 at different timepoints. After a 15‐min lag, infectious penetration occurred asynchronously between 30 and 60 min, reaching t 1/2 within 40–50 min (Fig 5D). This time course was consistent with endolysosomal maturation, which usually lasts 30–60 min (Huotari & Helenius, 2011). Altogether, these results show that cathepsin L‐dependent SARS‐CoV‐2 infection depends on endolysosome maturation in TMPRSS2− A549* and Vero cells.
Inhibitors of LE maturation, i.e., colcemid and MG‐132, also reduced SARS‐CoV‐2 infection in a dose‐dependent manner in TMPRSS2+ cells (Calu‐3 and Caco‐2 cells) (Fig 5E and F). Although the inhibition was significant, the decrease in infection caused by colcemid was barely 25–30% (Fig 5E). The IC50 values of MG‐132 were one to three logs higher in TMPRSS2+ cells than in TMPRSS2− cells (Fig 5F and Table 1). As shown in Fig 5G, infection was not readily detectable in Calu‐3 and Caco‐2 cells when MG‐132 was added at 2 hpi. Additionally, MG‐132 had no effect on SARS‐CoV‐2 infection of TMPRSS2‐overexpressing A549* cells (Fig 5H). This result contrasted with the infection of parental A549* cells, which decreased with increasing amounts of drug (Fig 5H). Taken together, these data suggested that the TMPRSS2‐dependent SARS‐CoV‐2 entry pathway does not rely on LE maturation.
Activation of SARS‐CoV‐2 is incomplete after its release from producer cells
Furin expression in the producer cells and TMPRSS2 expression at the surface of target cells are believed to mediate SARS‐CoV‐2 activation through proteolytic processing of the S protein. Hence, we first evaluated the cleaved spike content in viral particles after biosynthesis. Authentic infectious viruses were analyzed by SDS–PAGE and Western blot using a primary antibody recognizing the S2 segment (S2) of the SARS‐CoV‐2 spike. A strong band corresponding to the full‐length SARS‐CoV‐2 spike, which includes S1 and S2 segments (S0), was detected at 160 kDa (Fig 6A). A second band, although of lower intensity, was clearly visible at 75 kDa, which corresponds to S2. With a lower extent, additional bands were discernible above 160 kDa, which likely correspond to dimeric and trimeric forms of the SARS‐CoV‐2 spike. Upon semi‐quantifying the intensities of the S0 and S2 bands, the ratio of cleaved spike (S2) to the total of spike, i.e., uncleaved plus cleaved (S0 + S2), was found to be one third [(ρ) = S2/S0 + S2] (Fig 6B). These results were consistent with prediction models based on the SARS‐CoV‐2 spike structure (Wrobel et al, 2020).
Figure 6. Proteolytic processing triggers SARS‐CoV‐2 membrane fusion.
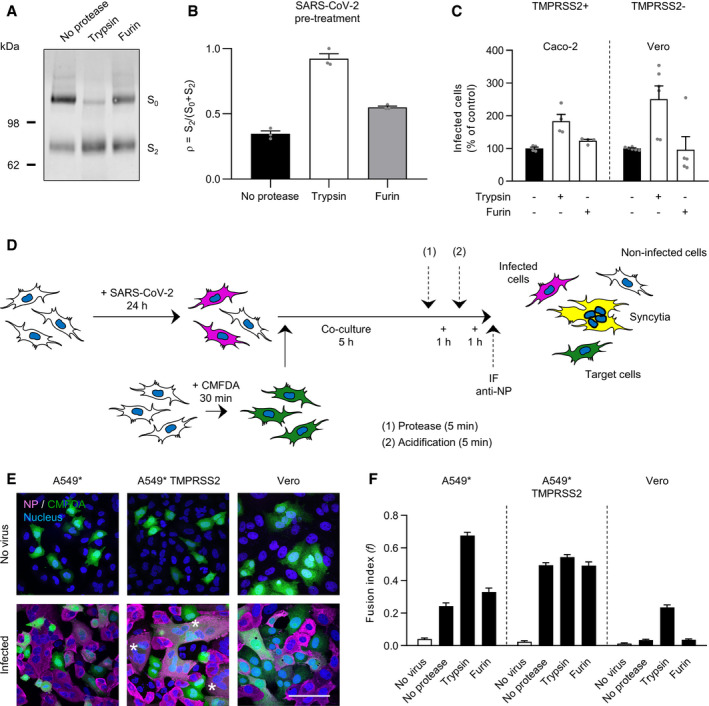
- SDS–PAGE and Western blot analysis of SARS‐CoV‐2 particles treated with trypsin or furin under reducing conditions. S was detected using a rabbit antibody against the SARS‐CoV‐2 S2 region.
- SARS‐CoV‐2 regions S0 (uncleaved) and S2 (cleaved) were semiquantified from (A), and the proportion of S2 on viral particles is shown as (ρ) = S2/S0+S2. n = 3.
- SARS‐CoV‐2 was subjected to pretreatment with trypsin and furin for 15 min at 37°C prior to infection of Caco‐2 and Vero cells (MOIs of 0.4 and 0.3, respectively). Infected cells were quantified by flow cytometry as described in Fig 1D. Data were normalized to samples not pretreated with trypsin. n = 4–6 biological replicates.
- Depicts the cell–cell fusion model employed in this study. (1) or (2) indicates treatment with either exogenous proteases or low‐pH buffers; CMFDA, cytosolic green dye; IF anti‐NP, immunofluorescence staining against the SARS‐CoV‐2 nucleoprotein.
- A549* expressing or lacking TMPRSS2 cells and Vero cells were first infected with SARS‐CoV‐2 at MOIs of 0.1 and 0.2, respectively, for 24 h and then cocultured for 5 h along with target cells, not infected but prestained with CMFDA, a cytosolic green dye. Cells were subsequently treated with trypsin or furin for 5 min at 37°C and left to coculture for an additional hour at 37°C. After fixation, nuclei were stained with Hoechst (blue), and infected cells were subjected to immunofluorescence staining against SARS‐CoV‐2 nucleoprotein (magenta). Samples were ultimately imaged by confocal fluorescence microscopy. White stars indicate syncytia with at least four nuclei. Scale bars: 100 µm.
- Images of microscope fields obtained in (E) were quantified [A549*: n (no virus) = 30, n (no protease) = 80, n (trypsin) = 80, and n (furin) = 60; A549* TMPRSS2: n (no virus) = 30, n (no protease) = 80, n (trypsin) = 79, and n (furin) = 60; Vero: n (no virus) = 40, n (no protease) = 112, n (trypsin) = 114, and n (furin) = 60]. The fusion index is given as f = 1 – [(number of cells in a field after fusion)/(number of nuclei)].
Data information: Images are representative of three independent experiments. (B) Data are expressed as mean ± SEM from three independent experiments. (C) Data are expressed as mean ± SEM from 2 to 3 independent experiments. (F) Results are expressed as mean ± SEM of three independent experiments.
Source data are available online for this figure.
Next, we assessed the efficiency of exogenous furin and trypsin in processing S0 into S2 on viral particles. In this assay, trypsin was used to mimic TMPRSS2 at the cell surface as the two enzymes are closely related and both belong to the group of trypsin‐like proteases. The use of exogenous cathepsin L was excluded because the enzyme is active only at pH ˜5, which would have made it impossible to distinguish the effects of low pH from those of proteolytic cleavage. After treatments with the exogenous proteases, samples were analyzed by Western blot using the primary antibody against S2. Although the presence of serum in our virus preparations, both trypsin and furin proteolytically processed the full‐length form of spike S0 into S2, but with a striking difference in efficiency (Fig 6A). Trypsin treatment greatly reduced the relative intensity of S0, which resulted in an increase in the intensity of S2, with a (ρ) value reaching almost 1.0, while furin treatment had a more modest impact as the (ρ) value increased from 0.3 to 0.5 (Fig 6B). To confirm that the presence of serum had no impact on protease cleavage of spike, A549* cells expressing or lacking TMPRSS2 were infected with SARS‐CoV‐2 produced in the presence or absence of serum. Results showed that the presence of serum did not impact SARS‐CoV‐2 infection and cleavage (Fig EV3A and B).
To determine whether increased proteolytic processing of the S protein results in enhanced SARS‐CoV‐2 activation and infection, viral particles were subjected to exogenous furin and trypsin and then added to Caco‐2 and Vero cells. Using our flow cytometry‐based assay, we found that infection increased as much as 2‐ to 3‐fold following SARS‐CoV‐2 proteolytic preprocessing by trypsin whereas the pre‐exposure of particles to furin had no apparent effect (Fig 6C). Overall, the gain in infection appeared to correlate with greater conversion of S0 into S2 in SARS‐CoV‐2 particles. Taken together, these data indicated that proteolytic processing of spike proteins and activation of SARS‐CoV‐2 were largely incomplete after release of the virus from producer cells.
Proteolytic processing is both sufficient and necessary for SARS‐CoV‐2 fusion
To relate the proteolytic processing of the SARS‐CoV‐2 spike with the viral fusion mechanisms, we then evaluated the capacity of the virus to mediate cell–cell fusion, i.e., the formation of syncytia (‘‘fusion‐from‐within’’) (Fig 6D). Similar systems have recently been described for SARS‐CoV‐2, and previously, for unrelated viruses (Bratt & Gallaher, 1969; Buchrieser et al, 2020; Papa et al, 2021). Briefly, cells were first infected with SARS‐CoV‐2 and then cocultured along with fresh cells, not infected and prestained with the cytosolic green dye CMFDA. CMFDA has the advantage of freely passing through membranes, but once inside the cells, the dye is no longer able to cross the plasma membrane, thus preventing leakage to neighboring cells. Immunofluorescence staining against the viral nucleoprotein allowed for the distinction between infected and target cells. Confocal images clearly showed infected cells in magenta, target cells in green, and the syncytia that result from the fusion of the two in white, i.e., magenta plus green (Figs 6E and EV4, Appendix Figs S5 and S6).
The infection of parental A549* and Vero cells led to the formation of a marginal number of syncytia, and all were small, with only two‐three nuclei (Fig 6E, Appendix Figs S5 and S6). In contrast, large syncytia with six or more nuclei were observed upon infection of TMPRSS2+ A549* cells (Figs 6E and EV4). The extent of cell–cell fusion was confirmed with a fusion (f) index that expresses the average number of fusion events per original mononucleated cell (White et al, 1981). The (f) index reaches 1 when all the nuclei in the microscope field are present in a single cell, and the value is 0 when all cells have one nucleus each. The (f) index ranged from 0.04 to 0.24 in Vero and A549* cells and reached 0.5 in TMPRSS2+ A549* cells, i.e., a 2‐ to 13‐fold higher compared to that obtained with TMPRSS2− target cells (Fig 6F). Combined, our results confirmed that the presence of TMPRSS2 on target cells promotes SARS‐CoV‐2‐mediated syncytia formation (Buchrieser et al, 2020), most likely by achieving the proteolysis of the S protein on the surface of infected cells.
Using our cell–cell fusion model, we then wanted to determine whether furin and trypsin can also complete the proteolytic processing of the SARS‐CoV‐2 spike for fusion. A549* and Vero cells are both negative for TMPRSS2 and thus represent a convenient model for monitoring proteolytic activation of cell surface spike proteins by exogenous proteases. Trypsin treatment of infected A549* and Vero cells cocultured with CMFDA+ target cells resulted in the formation of large syncytia with multiple nuclei (Appendix Figs S5 and S6), similar to those observed after infection of TMPRSS2+ A549* cells (Figs 6E and EV4). In contrast, no difference was observed between furin‐ and mock‐treated‐infected A549* and Vero cells, among which the only cells with more than one nucleus were actively dividing cells (Appendix Figs S5 and S6). Additionally, the fusion index in A549* and Vero cells was significantly increased under trypsin treatment compared with mock‐ and furin‐treated cells (Fig 6F). Of note, neither furin nor trypsin treatment enhanced syncytia formation upon infection of TMPRSS2+ A549* cells (Figs 6F and EV4). Our data collectively indicated that furin is ineffective in completing spike activation and that proteolytic cleavage is both sufficient and necessary for SARS‐CoV‐2 membrane fusion.
Endosomal acidification is required for endolysosomal proteases priming viral fusion
SARS‐CoV‐2 does not rely on endosomal acidification to enter TMPRSS2+ cells, which suggests that the virus does not rely on low pH for membrane fusion but solely for the activation of cathepsin L in cells lacking TMPRSS2. To pursue this possibility, we first assessed whether it was possible to inactivate the virus by applying acidic buffers prior to infection. In such an assay, the virus undergoes a transition toward the postfusion state at the optimal pH. If the transition is irreversible, the spike protein is no longer able to fuse with target‐cell membranes, and thus, the viral particles are rendered noninfectious. With this approach, we found that more than 80% of viruses remained infectious in Caco‐2 and Vero cells after exposure to buffers at pH ˜6 for 10 min (Fig 7A). Semliki forest virus (SFV) is an early‐penetrating virus that has a class II viral fusion glycoprotein with an irreversible priming step that is triggered at a pH‐activation threshold of 6.2 (Lozach et al, 2010). In contrast to SARS‐CoV‐2, the infectiousness of low pH‐pretreated SFV was reduced by 60–70% after exposure to pH ˜6.0 (Fig 7B). We next investigated the influence of low pH on SARS‐CoV‐2 fusion in our cell–cell fusion assay. The formation of syncytia and the (f) index did not differ significantly when cells were treated with low‐pH or neutral buffers regardless of the presence of TMPRSS2 (Fig 7C).
Figure 7. Low pH does not inactivate SARS‐CoV‐2.
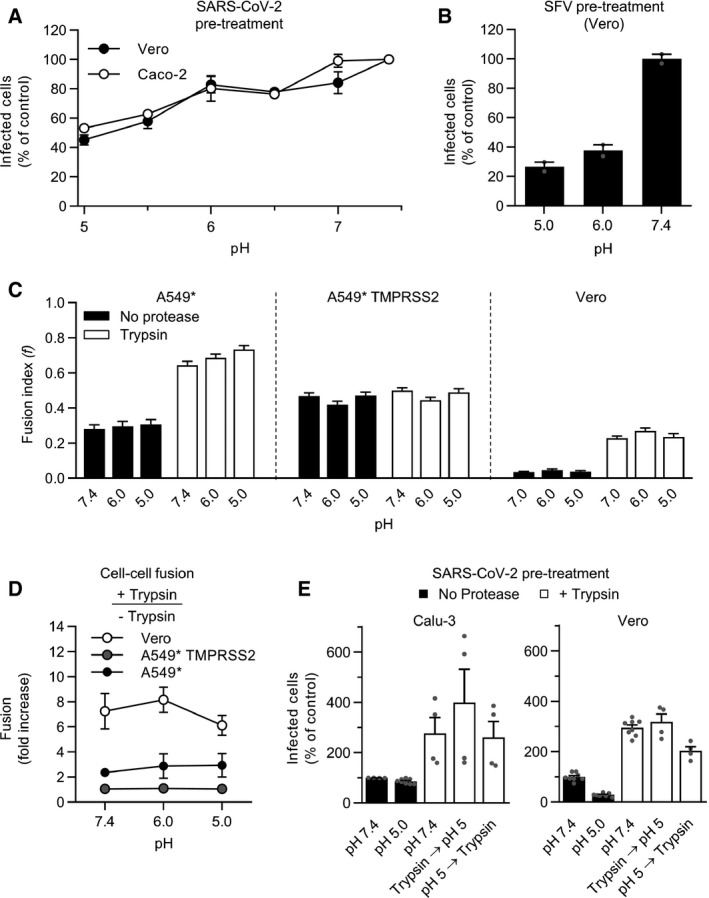
-
A, B(A) SARS‐CoV‐2 and (B) Semliki forest virus (SFV) particles were pretreated at the indicated pH for 10 min at 37°C. Viruses were subsequently neutralized with buffer at pH ˜7.4 and allowed to infect Caco‐2 (MOI ˜0.4) and Vero cells (MOI of 0.3 and 0.8, respectively). Infected cells were then immunostained against the NP protein and SFV E2 protein, respectively, and analyzed by flow cytometry. Data are normalized to that of samples pretreated with buffers at pH ˜7.4. n = 2.
-
CSamples were prepared as described in Fig 6E but, after trypsin treatment, infected cells were subjected to buffers at the indicated pH for 5 min at 37°C and left to recover for an additional hour at 37°C. Images of microscope fields (50 < n < 120) were quantified, and fusion index was calculated as in Fig 6F. n > 50 microscope fields were analyzed.
-
DCell–cell fusion after trypsin treatment according to pH. The fusion is given as the ratio of the values obtained for trypsin‐treated samples to those obtained for untreated samples. n = 3–4.
-
ESARS‐CoV‐2 particles (MOI of 0.3) were first subjected to trypsin treatment for 15 min at 37°C and then exposed to buffers at the indicated pH for 10 min at 37°C, and vice versa. Calu‐3 and Vero cells were then infected at a MOI of 0.3 and analyzed by flow cytometry as described in Fig 1D. n = 4–8 biological replicates.
Data information: (A, B) Results are representative of at least two independent experiments and expressed as mean ± SEM of two biological replicates. (C and D) Results are expressed as mean ± SEM of three independent experiments. (E) Data are expressed as mean ± SEM from 2 independent experiments.
Source data are available online for this figure.
Our results support a model in which endosomal acidification is not essential for SARS‐CoV‐2 membrane fusion, but SARS‐CoV‐2 infection relies on low pH for cathepsin L‐dependent infection in cells lacking TMPRSS2. Therefore, we tested the possibility that acidic pH is required for the activation of the endolysosomal proteases that trigger SARS‐CoV‐2 fusion. In such a scenario, the S proteins that are already primed by proteases should no longer require low pH for fusion. Indeed, we found that the fusion index was not increased when trypsin treatment was followed by exposure to a decrease in pH of 7.4 to 5 (Fig 7C and D), the latter value being typical of the luminal pH of endolysosomes (Lozach et al, 2011a).
To confirm that SARS‐CoV‐2 membrane fusion is independent of low pH, viral particles were then exposed to buffers with pH ˜5 and subsequently subjected to proteolytic cleavage by trypsin. Our results revealed that SARS‐CoV‐2 infectivity was preserved when viral particles were exposed to the low‐pH buffer prior to trypsin treatment, whereas those of virus particles that were solely exposed to acidic pH were not infective (Fig 7E). The infectivity also remained preserved when the virus was first subjected to trypsin and then acidified. Taken together, the results showed that endosomal acidification does not play a role in SARS‐CoV‐2 membrane fusion, whether it occurs before or after the proteolytic processing of viral particles. In addition, our results strongly suggested that the potential pH‐induced conformational changes in the SARS‐CoV‐2 spikes were neither irreversible nor detrimental for viral fusion.
To directly test whether endosomal acidification is needed for the host cell proteases that prime SARS‐CoV‐2 fusion, and not for the fusion mechanisms themselves, we assessed whether preactivated viral particles remain dependent on endosomal acidification for infectious entry. For this purpose, the proteolytic processing of the virus particles was achieved with trypsin and thermolysin prior to the infection of A549* and Vero cells. Thermolysin is known to prime the surface glycoprotein of Ebola virus in the same way as cathepsin L (Schornberg et al, 2006) and therefore was used here to mimic cathepsin L. Thermolysin was chosen as it has the advantage to function at neutral pH. To disrupt the acid‐dependent endolysosomal proteases, the infection was carried out in the continuous presence of 50 nM bafilomycin A1. As A549* and Vero cells do not express TMPRSS2, this assay allowed us to directly test the impact of extracellular protease‐activated viral particles. As reported above (Fig 3C), infection with untreated viral particles was severely impeded when proton pumps were blocked in the absence of TMPRSS2 (Fig 8A). In stark contrast, the protease‐preactivated viral particles remained infectious in the presence of bafilomycin A1 (Fig 8A).
Figure 8. SARS‐CoV‐2 no longer requires endosomal acidification after proteolytic processing.
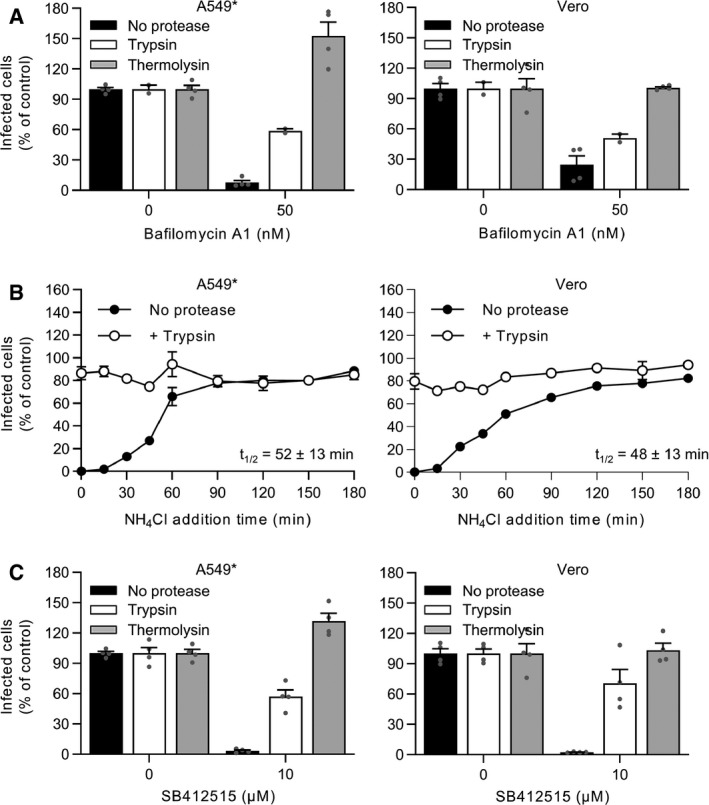
- Trypsin‐ or thermolysin‐activated SARS‐CoV‐2 was allowed to infect A549* and Vero cells at MOIs of 0.2 and 0.3, respectively, in the continuous presence of bafilomycin A1. Infection was quantified by flow cytometry, and data were normalized to that from control samples not exposed to the inhibitor. n = 2–4 biological replicates.
- Binding of trypsin‐activated SARS‐CoV‐2 to A549* and Vero cells (MOI ˜0.2 and 0.3, respectively) was synchronized on ice for 90 min. Subsequently, the cells were rapidly shifted to 37°C to allow penetration. NH4Cl (50 mM) was added at the indicated time to neutralize endosomal pH and block the acid‐dependent step of SARS‐CoV‐2 infectious penetration. Infected cells were analyzed by flow cytometry, and data were normalized to samples where NH4Cl had been omitted. n = 2.
- SARS‐CoV‐2 was first treated with trypsin or thermolysin and then allowed to infect A549* and Vero cells (MOIs ˜0.2 and 0.3) in the continuous presence of SB412515 (cathepsin L inhibitor). Infected cells were quantified by flow cytometry as described in Fig 1D, and data were normalized to samples where SB412515 had been omitted. n = 4 biological replicates.
Data information: (A and C) data are expressed as mean ± SEM from two independent experiments. (B) Results are representative of two independent experiments and expressed as mean ± SEM of two biological replicates.
Source data are available online for this figure.
The capacity of SARS‐CoV‐2 to infect A549* and Vero cells upon proteolytic activation, despite the absence of functional endolysosomal proteases, was confirmed using NH4Cl. As expected, in our synchronized infection assay, untreated particles became NH4Cl insensitive at 50 min postentry (Figs 8B and 3E). However, when the viral particles were pretreated with trypsin, no sensitivity to NH4Cl was observed (Fig 8B). These results strongly supported the model that the virus is no longer dependent on endosomal acidification for infection once activated by proteolytic cleavage.
Finally, we wanted to see whether SARS‐CoV‐2 requires the activity of endolysosomal proteases for infectious entry after its activation. To this end, trypsin and thermolysin were used to preactivate viral particles, and infection of A549* and Vero cells was performed in the presence of the cathepsin L inhibitor SB412515. As expected, the cathepsin L inhibitor completely abolished infection when viruses were not preactivated with proteases (Figs 2B and 8C). Conversely, preactivation of SARS‐CoV‐2 with thermolysin fully restored infection of A549* and Vero cells in the presence of SB412515 (Fig 8C). Trypsin treatment was also found to rescue infection, however with a lower extent (i.e., 50–70%) (Fig 8C). Taken together, these results suggest that TMPRSS2− and cathepsin L‐dependent penetration pathways require differential proteolytic processing of the SARS‐CoV‐2 protein S.
Altogether, our data show that SARS‐CoV‐2 resembles other CoVs in that its entry depends on diverse host cell proteases. It can use two distinct routes; either TMPRSS2 mediates the pH‐independent penetration of the virus from on or near the cell surface or, alternatively, the virus is transported to endolysosomes, where low pH activates cathepsin L, which in turn primes viral fusion and penetration.
Discussion
The infectious entry process of CoVs is complex (Hartenian et al, 2020). Several host cell proteases can prime the CoV spike (S) protein for viral membrane fusion, but it is not yet known whether these mechanisms require specific proteases or a coordinated, spatiotemporal combination of multiple proteases. The importance of endosomal acidification in the productive penetration of all CoVs is also a matter of debate. Furin, TMPRSS2, and cathepsin L have all been implicated in CoV activation for entry (Bestle et al, 2020; Hoffmann et al, 2020b; Liu et al, 2020; Matsuyama et al, 2020; Shang et al, 2020), and agents elevating endosomal pH, such as chloroquine, have been reported to interfere with infection (Hoffmann et al, 2020b; Ou et al, 2020; Wang et al, 2020). SARS‐CoV‐2 and other CoVs have apparently found a way to use diverse entry mechanisms to infect target cells and spread throughout the host.
In this study, we developed reliable and accurate assays for the investigation of SARS‐CoV‐2 infection of lung, intestine, and kidney epithelial cells, from proteolytic activation to membrane fusion. In agreement with other reports (Bojkova et al, 2020; Hoffmann et al, 2020b), our results showed that SARS‐CoV‐2 infection was sensitive to inhibitors of TMPRSS2 and cathepsin L. We further found that blocking TMPRSS2 abrogated infection even when the cells were expressing cathepsin L, indicating that the virus does not reach endolysosomal cathepsins when TMPRSS2 is present. Others have shown that infection by MERS pseudoviruses was suppressed by trypsin‐like protease inhibitors in the presence of tetraspanin CD9, while entry was unaffected, but further infection progression was blocked by cathepsin inhibitors in the absence of CD9 (Earnest et al, 2017). These authors proposed that tetraspanins concentrate CoV entry factors into localized positions on or near the cell surface, allowing rapid and efficient activation of viral fusion (Hantak et al, 2019).
We observed that SARS‐CoV‐2 used two distinct routes to enter cells, one fast (˜10 min), corresponding to the timing of TMPRSS2 activation, and one slower (40–50 min), corresponding to cathepsin L priming. Although other cellular factors are likely necessary, our results support the view that TMPRSS2 is a major determinant of the SARS‐CoV‐2 fast entry mechanism. Similar observations have been made for human CoV 229E (hCoV‐229E), which prefers cell surface TMPRSS2 to endosomal cathepsins for cell entry (Bertram et al, 2013; Shirato et al, 2017; Shirato et al, 2018).
It is clear from our data that, in the presence of TMPRSS2, SARS‐CoV‐2 did not rely on endosomal acidification and late endosomal maturation for infectious penetration. Concanamycin B, which specifically inhibits vATPases and elevates endosomal pH, affected UUKV, an enveloped virus that penetrates host cells by acid‐activated membrane fusion (Lozach et al, 2010), but did not affect SARS‐CoV‐2 infection. MG‐132, known to divert IAV and UUKV away from LEs (Khor et al, 2003; Lozach et al, 2010), was no longer able to impede SARS‐CoV‐2 infection when TMPRSS2 was overexpressed. This was consistent with reports that TMPRSS2 processes CoV S and other substrates at or near the plasma membrane (Kleine‐Weber et al, 2018; Wang & Xiang, 2020), i.e., at neutral pH. Using aprotinin, we found that half of the bound viral particles had completed the TMPRSS2‐dependent step within 5–10 min. We cannot completely exclude the possibility that aprotinin was not instantaneously effective when it was added to the infected cells. In this case, the timing of the TMPRSS2‐requiring step was therefore faster. SARS‐CoV‐2 activation and penetration would then likely take place at the plasma membrane following proteolytic activation, as proposed for hCoV‐229E and MERS‐CoV (Qian et al, 2013; Shirato et al, 2017).
An alternative scenario is that SARS‐CoV‐2 is sorted into the endocytic machinery regardless of TMPRSS2 expression. The time course of the TMPRSS2‐requiring step resembled that of cargo sorting into EEs, approximately 5–10 min (Huotari & Helenius, 2011). Another observation supporting this hypothesis was that colcemid partially hampered infection. This drug perturbs LE maturation by disrupting the microtubule network and in turn causes EE accumulation and dysfunction (Lozach et al, 2010). Such a strategy has been proposed for reoviruses, which use a similar uptake pathway but different trafficking pathways depending on whether viral particles are activated or not (Boulant et al, 2013). As is the case for other CoVs (Wang & Xiang, 2020), additional functional investigations are required to determine exactly where, whether from the plasma membrane or from EEs, SARS‐CoV‐2 enters the cytosol of TMRPSS2+ cells, as well as whether the processing of the S protein is followed by transport of the virus to downstream organelles for penetration.
In the absence of TMPRSS2, it was evident that SARS‐CoV‐2 depended on endocytosis and transport through the late endosomal system for infectious penetration. Infectious entry was inhibited by endosomal pH neutralizing drugs. Impairing LE maturation, either by colcemid treatment or by the expression of Rab7a T22N or Q67L, affected SARS‐CoV‐2 infection. The sensitivity to MG‐132 mirrored those observed for UUKV, IAV, and murine CoVs, which accumulated in cytosolic vesicles and failed to infect the cells (Khor et al, 2003; Yu & Lai, 2005; Lozach et al, 2010). Other researchers have reported that SARS‐CoV‐2 depends on PIKfyve for the infection of 293T cells, a line devoid of TMPRSS2 (Ou et al, 2020). PIKfyve is a phosphoinositide kinase involved in the first stages of LE maturation. Collectively, our results indicate that SARS‐CoV‐2, like other CoVs (Simmons et al, 2005; Kawase et al, 2009; Qian et al, 2013), is dependent on functional endolysosomes and cathepsins for infectious penetration when the viral particles are not activated at or near the cell surface.
Our results suggested that the proteolytic activation of the spike S protein was sufficient and necessary for SARS‐CoV‐2 fusion. The Vero cells used in our virus‐mediated cell–cell fusion assay did not express TMPRSS2 on the cell surface, or at least at a detectable level. In this assay, exogenous furin failed to promote syncytia formation, indicating that furin cleavage was either inefficient or not sufficient to achieve the full activation of the S protein at the plasma membrane. The S1/S2 site of SARS‐CoV‐2 S protein exhibits an RRAR motif instead of the typical RX(R/K)R furin motif, and a recent structural study indicates that cleavage by furin at this site in S trimers is rather low, approximately 30% (Bestle et al, 2020; Cai et al, 2020; Wrobel et al, 2020). However, we found that, unlike furin, trypsin prompted the formation of syncytia, which supports the proposed involvement of the proteases within target cells, such as TMPRSS2 and cathepsin L, in completing the proteolytic processing of the S protein. Others have shown that SARS‐CoV‐2 and MERS‐CoV mediate cell–cell fusion at neutral pH without any further proteolytic treatment when target cells express TMPRSS2 (Shirato et al, 2013; Buchrieser et al, 2020). It is tempting to postulate that the more protease is expressed in the target cells, the more molecules of spike are cleaved and activated, and the more fusogenic the virus is. Additionally, work evaluating SARS‐CoV‐2 infection of primary human intestinal epithelial cells showed that TMPRSS2 expression was the best indicator of cell tropism (Triana et al, 2021).
It is also apparent from our results that the SARS‐CoV‐2 progeny were not fully processed and activated. Trypsin pretreatment increased virus infectivity. More work is required to decipher the SARS‐CoV‐2 fusion mechanism. The list of the involved host cell proteases is most likely not restricted to furin, TMPRSS2, and cathepsin L, as suggested by recent biochemical studies (Jaimes et al, 2020; Tang et al, 2021). S proteolytic activation might involve the cleavage of sites other than S1/S2 and S2′, as was found for the MERS‐CoV S protein (Kleine‐Weber et al, 2018). Consistent with the results from others, our own data indicated that the cleavage to S1 and S2 is incomplete on SARS‐CoV‐2 particles, most likely with only one of the three S1/S2 sites within S trimers cut by furin in producer cells. As recently proposed, cutting at all the S1/S2 sites would be achieved in this model with the aid of proteases within target cells, such as TMPRSS2 and cathepsin L (Ou et al, 2021; Tang et al, 2021). The fusogenic conformational change would then occur and be completed by the cleavage of the S2′ sites to unmask the fusogenic units. The amino acid sequence of the S1/S2 site differs significantly among CoVs (Bestle et al, 2020), and it is highly likely that the sequence influences the overall viral fusion process.
We found that the level of viral mRNA and amount of infectious viral progeny released in the outer media were lower in the absence of TMPRSS2. The TMPRSS2‐dependent entry mechanisms occurred more rapidly than the cathepsin L‐activated mechanisms, and it might be that the early route results in a more productive infection than the late‐penetrating process. Separate studies support the view that early entry results in productive infection, while late penetration would be an alternative backup route, at least for some CoV strains including HCoV‐229E (Shirato et al, 2017; Shirato et al, 2018; Hantak et al, 2019). Other works on candidate therapeutics have linked host cell proteases to CoV spread. Inhibitors of TMPRSS2, but not of cathepsins, effectively prevent the pathogenesis of SARS‐CoV in mice, suggesting that SARS‐CoV mainly uses cell surface proteases rather than endosomal cathepsins in vivo (Zhou et al, 2015). The identification of all host cell proteases involved in SARS‐CoV‐2 and other CoV infections, as well as the tissues and organs that express them, remains an important objective for better understanding viral propagation and virus‐induced diseases.
Intriguingly, SARS‐CoV‐2 showed a strong resistance to acidic buffers. Exposure to pH ˜5.0 only marginally inactivated the virus, and infectivity was rescued or even enhanced by proteolytic treatment. In addition, trypsin activation appeared to protect the virus from acid inactivation, which could explain how it infects the gastrointestinal tract in vivo. SARS‐CoV‐2 has evidently developed a remarkable ability to adapt to an acidic environment. Interestingly, low pH has been shown to alter the positioning of the receptor‐binding domain in the SARS‐CoV‐2 S trimers, which could help the virus escape the immune system (Zhou et al, 2020). Overall, this property certainly endows the virus with the ability to sustain high infectivity by entering host cells not only from endosomes but also from the extracellular space, especially during the spread of the virus throughout the host.
Reports on the cell entry mechanisms of SARS‐CoV‐2 and other CoVs often describe only one cell model system, and the literature in this field remains unclear in general. Our study recapitulates the SARS‐CoV‐2 entry process within a single investigation and provides an overview of the cellular mechanisms used by SARS‐CoV‐2 to penetrate and infect target cells. Although it remains to be confirmed under physiological conditions, we propose that SARS‐CoV‐2 can enter cells through two distinct and mutually exclusive pathways. When target cells express TMPRSS2, the virus is activated at or close to the cell surface and penetrates early in a pH‐independent manner. When target cells lack TMPRSS2, SARS‐CoV‐2 is endocytosed and sorted into endolysosomes, from which the virus is activated in a pH‐dependent manner and penetrates the cytosol at a late timepoint. With the ability to utilize diverse cell entry routes, SARS‐CoV‐2 has likely found a way to expand its number of target tissues and organs, which certainly contributes to the broad tropism of the virus in vivo.
Materials and Methods
Cells
African green monkey Vero kidney epithelial cells (ATCC CRL 1586), human Caco‐2 colorectal adenocarcinoma (ATCC HTB‐37), human Calu‐3 lung adenocarcinoma (ATCC HTB‐55), and A549 human epithelial lung cells stably expressing ACE2 or ACE2 and TMPRSS2 (A549* and TMPRSS2+ A549*, respectively) were all maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 µg/ml streptomycin. A549 cell lines were a kind gift from Prof. Ralf Bartenschlager (Steuten et al, 2021). Baby hamster kidney cells (BHK‐21) were grown in Glasgow’s minimal essential medium containing 10% tryptose phosphate broth, 5% FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin. All cell lines were grown in an atmosphere of 5% CO2 in air at 37°C. All products used for cell culture were obtained from Thermo Fisher Scientific and Sigma‐Aldrich.
Viruses
SARS‐CoV‐2 (strain BavPat1) was obtained from Prof. Christian Drosten at the Charité in Berlin, Germany, and provided via the European Virology Archive. The virus was amplified in Vero cells in the presence of 2% serum, and working stocks were used after three passages. Uukuniemi (UUKV) and Semliki forest (SFV) viruses were previously described and amplified in BHK‐21 cells (Helenius et al, 1980; Mazelier et al, 2016). For titration of SARS‐CoV‐2, confluent monolayers of Vero cells were infected with 10‐fold dilutions of virus in serum‐free medium and then grown in the presence of complete medium containing 2% serum and 0.05% agarose to prevent virus spread. Plaques were stained by crystal violet 3 days post‐infection. The MOI was assigned for SARS‐CoV‐2 according to the titer determined on Vero cells. The MOI for SFV and UUKV was given based on the titers determined on BHK‐21 cells as previously described (Lozach et al, 2010).
Antibodies
The mouse mAb against SARS‐CoV‐2 nucleoprotein NP (40143‐MM05) was purchased from Sino Biologicals and used at dilutions of 1:500 for flow cytometry analysis and 1:1,000 for titration in TCID50 assays. The rabbit polyclonal antibody against SARS‐CoV‐2 spike protein was obtained from Thermo Fisher Scientific (PA1‐41165). The rabbit polyclonal antibody U2 targets all the UUKV structural proteins and was used at a dilution of 1:4,000 for immunohistochemistry in foci‐forming unit assays (Lozach et al, 2011b). The mouse mAb 8B11A3 against the UUKV nucleoprotein N was a kind gift from Ludwig Institute for Cancer Research (Stockholm, Sweden) (Persson & Pettersson, 1991). The mouse mAb against SFV glycoprotein E2 was kindly provided by Prof. Margaret Kielian (Albert Einstein College of Medicine, USA). mAb 8B11A3 and mAb against SFV E2 were used at a dilution of 1:400 for flow cytometry analysis. Rabbit antibodies against TMPRSS2 (ab92323) and actin (A2066) were obtained from Abcam and Sigma, respectively. Mouse mAb against cathepsin L (BMS1032) and α‐tubulin (T5158) were purchased from Thermo Fisher Scientific and Sigma, respectively. The goat polyclonal antibody against the elongation factor 2 (EF2, SC‐13004) was obtained from Santa Cruz. Anti‐mouse secondary antibodies were conjugated to Alexa Fluor (AF) 405 (Molecular Probes), AF488 (Molecular Probes), IRDye 700 (LI‐COR), IRDye 800CW (LI‐COR), and horseradish peroxidase (HRP; Vector Laboratories). Anti‐rabbit and anti‐goat secondary antibodies conjugated to IRDye 800CW were purchased from LI‐COR.
Reagents and plasmids
Aprotinin (Cayman Chemical), camostat mesylate (Sigma), chloroquine diphosphate (Sigma), and NH4Cl (Sigma) stocks were dissolved in water. Bafilomycin A1 (BioViotica), colcemid (Cayman Chemical), concanamycin B (BioViotica), MG‐132 (Selleck Chemicals), SB412515 (Cayman Chemical), and DL‐threo‐1‐phenyl‐2‐palmitoylamino‐3‐morpholino‐1‐propanol (PPMP, Cayman Chemical) were all dissolved in DMSO. All drugs were assessed for cytotoxicity at the indicated concentrations using the CytoTox96 Non‐Radioactive Cytotoxicity colorimetric assay (Promega) according to the provider’s recommendations. Furin was purchased from R&D, and thermolysin and trypsin were purchased from Sigma. Plasmids encoding EGFP‐tagged Rab7a, Rab7a T22N, and Rab7a Q67L have been described elsewhere (Lozach et al, 2010).
Protein analysis
Cells were lysed with phosphate‐buffered saline (PBS) containing 0.1% Triton X‐100 (Merck Millipore) according to a standard procedure (Lozach et al, 2011b). Cell lysates were then diluted in LDS sample buffer (Thermo Fisher Scientific) and analyzed by SDS–PAGE (Nu‐PAGE Novex 10% Bis‐Tris gels; Thermo Fisher Scientific). Proteins were subsequently transferred to polyvinylidene difluoride membranes (iBlot transfer stacks; Thermo Fisher Scientific). The membranes were first blocked with intercept blocking buffer (LI‐COR) and then incubated with primary antibodies against the SARS‐CoV‐2 spike, TMPRSS2, cathepsin L, EF2, actin, and α‐tubulin, all diluted in Tris‐buffered saline containing 0.1% Tween and intercept blocking buffer (1:1,000, 1:1,000, 1:400, 1:1,000, 1:5,000, and 1:2,000, respectively). After extensive washing, the membranes were incubated with the corresponding secondary antibodies conjugated to IRDye 700 or 800CW (both at 1:10,000) or HRP (1:1,000). Proteins were analyzed with a LI‐COR Odyssey CLx scanner, or alternatively, detected with SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific) and an Intas ChemoStar ECL analyzer.
Virus infection
Cells were exposed to viruses at the indicated MOIs in the presence of 2% FBS for 1 h at 37°C. Viral input was then replaced with complete culture medium, and the infected cells were incubated for 8 h before fixation. For virus‐mediated cell–cell fusion, cells were infected for 24 h. Cells that transiently expressed EGFP‐Rab7a and related mutants were infected 18 h post‐transfection. For pH inactivation, citric acid, 2‐(N‐morpholino)‐ethanesulfonic acid (MES), and 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) were used at 100 mM as buffers with pH values of pH < 5.5, 5.5 < pH < 6.5, and 6.5 < pH, respectively. Viral inputs were exposed to buffers at the indicated pH for 10 min at 37°C and then to buffers at neutral pH prior to infection. For furin, trypsin, or thermolysin activation, SARS‐CoV‐2 was pretreated with furin (1 µg/ml), trypsin (100 µg/ml), or thermolysin (1 mg/ml), respectively, for 15 min at 37°C and then allowed to infect cells. For inhibition assays, cells were pretreated with drugs for 30 min at 37°C or 3 h on ice for colcemid pretreatment and then exposed to viruses in the continuous presence of the inhibitors. For inhibitor add‐in time courses, virus binding to cells was synchronized on ice for 90 min. Cells were then rapidly warmed to 37°C, and SB412515 (10 µM), aprotinin (30 µM), NH4Cl (at indicated concentrations), concanamycin B (50 nM), and MG‐132 (at indicated concentrations) were added at the indicated times. Cells were subsequently incubated at 37°C and harvested 8 h after the initial warm shift. Infection was monitored by flow cytometry, fluorescence microscopy, or qRT–PCR. For analysis of infection by microscopy, cells were seeded on Lab‐Tek glass‐bottom 8‐well chamber slides.
DNA transfection
As previously described (Meier et al, 2014), Vero cells in 24‐well plates were transfected with 750 ng of plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations and washed 5 h later.
Immunofluorescence microscopy
Fluorescence microscopy was performed as extensively described in Ref. Leger et al (2020). Briefly, infected cells were rinsed with PBS, permeabilized with 0.5% Triton X‐100 (Sigma) for 15 min at room temperature (RT), and stained with primary antibodies diluted in PBS for 1 h at RT. Subsequently, the cells were extensively washed and incubated with secondary antibodies for 1 h at RT. Samples were then stained with Hoechst 33258 (0.5 µg/ml, Thermo Fisher Scientific) and imaged with a Leica TCS SP8 confocal microscope.
Flow cytometry
The flow cytometry‐based infection assay has been described previously (Mazelier et al, 2016). Briefly, infected cells were fixed with 4% formaldehyde for 30 min at RT and then permeabilized with 0.1% saponin (SERVA). Cells were then exposed to primary antibody at RT for 1 h, washed, and subsequently incubated with secondary anti‐mouse antibodies at RT for another 1 h. Infected cells were quantified with a FACSCelesta cytometer (Becton Dickinson) and FlowJo software (TreeStar).
Viral RNA quantification
As previously reported (Woelfl et al, 2020), RNA was harvested from cells using the NucleoSpin RNA extraction kit (Macherey‐Nagel) according to the manufacturer’s instructions. cDNA was synthesized using iSCRIPT reverse transcriptase (Bio‐Rad) from 250 ng of total RNA as per supplier recommendations. qPCR was performed using iTaq SYBR green (Bio‐Rad) following the manufacturer’s instructions for the SARS‐CoV‐2 genome using the forward primer, GCCTCTTCTCGTTCC, and the reverse primer, AGCAGCATCACCGCC. HPRT1 was used as a housekeeping gene and amplified using the forward primer CCTGGCGTCGTGATTAGTGAT and reverse primer AGACGTTCAGTCCTGTCCATAA.
TCID50 assay
Confluent monolayers of Vero and Caco‐2 cells in 96‐well plates were infected with 10‐fold serial dilutions of SARS‐CoV‐2. Infected cells were fixed at 24 hpi and subjected to immunostaining using mouse mAb anti‐SARS‐CoV‐2 NP as the primary antibody and then anti‐mouse antibody 800CW (1:10,000) as the secondary antibody. Samples were finally scanned on LI‐COR.
Cell–cell fusion
Infected cells and fresh cells, not infected but prestained with 2.5 µM of CellTracker Green CMFDA dye (Thermo Fisher Scientific) in serum‐free medium for 30 min at 37°C, were detached with 0.5 mM ethylenediaminetetraacetic acid (EDTA, Thermo Fisher Scientific) and cocultured at a 3:1 ratio in complete medium for 5 h at 37°C. Cells were then washed in PBS, exposed to furin (1 µg/ml) and trypsin (100 µg/ml) for 5 min at 37°C, and left to incubate for an additional hour at 37°C prior fixation. Alternatively, cells were not fixed but treated with DMEM containing 0.2% bovine serum albumin (Gibco) buffered at pH 7.4, 6.0, or 5.0 with 30 mM HEPES, MES, or citric acid, respectively, for 5 min at 37°C. Subsequently, the cells were washed and incubated in complete medium for an additional hour at 37°C prior fixation. After fixation, cells were subjected to immunofluorescence staining against SARS‐CoV‐2 NP, and nuclei stained with Hoechst 33258 (0.5 µg/ml). The formation of syncytia was evaluated by fluorescence microscopy by counting the number of nuclei and cells positive for CMFDA present in a microscope field. A fusion index (f) was calculated according to the equation f = (1 – [c/n]), where c is the number of cells in a field after fusion and n is the number of nuclei. An average field contained 30–60 nuclei.
Statistical analysis
Graph plotting of numerical values, as well as the statistics, was achieved with Prism v9.1.1 (GraphPad Software). The sample sizes (n), reproducibility information, and statistical methods, including parameters and P values, are indicated in the figure legends when appropriate.
Author contributions
JK, ZMU, and P‐YL designed research; JK, ZMU, PD, and MS performed research; JK, ZMU, MS, SB, and P‐YL analyzed data; P‐YL wrote the original draft; and JK, ZMU, MS, SB, and P‐YL reviewed and edited the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
This work was supported by grants from CellNetworks Research Group funds, Heidelberg, and from the Deutsche Forschungsgemeinschaft (DFG) project numbers LO‐2338/1‐1 and LO‐2338/3‐1 to PYL. This work was also supported by INRAE starter funds, IDEX‐Impulsion 2020 (University of Lyon), and FINOVI (Fondation pour l’Université de Lyon), all to PYL. SB received support from DFG project numbers 415089553 (Heisenberg program), 240245660 (SFB1129), 278001972 (TRR186), and 272983813 (TRR179) as well as the state Baden Wuerttemberg (AZ: 33.7533.‐6‐21/5/1) and the Bundesministerium Bildung und Forschung (BMBF) (01KI20198A). MS was supported by DFG project 416072091. We acknowledge funding from the German Academic Exchange Service (DAAD, Research Grant 57440921) to PD. We also acknowledge Vibor Laketa and the Imaging Platform at the Center for Integrative Infectious Disease Research, Heidelberg. We thank Felix Rey and Ari Helenius for fruitful discussions. Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2021) 40: e107821.
Contributor Information
Megan Stanifer, Email: m.stanifer@dkfz-heidelberg.de.
Steeve Boulant, Email: s.boulant@dkfz-heidelberg.de.
Pierre‐Yves Lozach, Email: pierre-yves.lozach@med.uni-heidelberg.de.
Data availability
This study includes no data deposited in external repositories.
References
- Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann‐Winkler Het al (2013) TMPRSS2 activates the human coronavirus 229E for cathepsin‐independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87: 6150–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestle D, Heindl MR, Limburg H, van Lam Van T, Pilgram O, Moulton H, Stein DA, Hardes K, Eickmann M, Dolnik Oet al (2020) TMPRSS2 and furin are both essential for proteolytic activation of SARS‐CoV‐2 in human airway cells. Life Sci Alliance 3: e202000786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojkova D, Bechtel M, McLaughlin K‐M, McGreig JE, Klann K, Bellinghausen C, Rohde G, Jonigk D, Braubach P, Ciesek Set al (2020) Aprotinin inhibits SARS‐CoV‐2 replication. Cells 9: 2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Stanifer M, Kural C, Cureton DK, Massol R, Nibert ML, Kirchhausen T (2013) Similar uptake but different trafficking and escape routes of reovirus virions and infectious subvirion particles imaged in polarized Madin‐Darby canine kidney cells. Mol Biol Cell 24: 1196–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratt MA, Gallaher WR (1969) Preliminary analysis of the requirements for fusion from within and fusion from without by Newcastle disease virus. Proc Natl Acad Sci USA 64: 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser J, Dufloo J, Hubert M, Monel B, Planas D, Rajah MM, Planchais C, Porrot F, Guivel‐Benhassine F, Van der Werf Set al (2020) Syncytia formation by SARS‐CoV‐2‐infected cells. EMBO J 39: e106267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Zhang J, Xiao T, Peng H, Sterling SM, Walsh RM Jr, Rawson S, Rits‐Volloch S, Chen B (2020) Distinct conformational states of SARS‐CoV‐2 spike protein. Science 369: 1586–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly L, Druce J, Roberts J, Bond K, Tran T, Kostecki R, Yoga Y, Naughton W, Taiaroa G, Seemann Tet al (2020) Isolation and rapid sharing of the 2019 novel coronavirus (SARS‐CoV‐2) from the first patient diagnosed with COVID‐19 in Australia. Med J Aust 212: 459–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y‐W, Lee M‐S, Lucht A, Chou F‐P, Huang W, Havighurst TC, Kim KM, Wang J‐K, Antalis TM, Johnson MDet al (2010) TMPRSS2, a serine protease expressed in the prostate on the apical surface of luminal epithelial cells and released into semen in prostasomes, is misregulated in prostate cancer cells. Am J Pathol 176: 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Bertram S, Glowacka I, Park YW, Pohlmann S (2009) Type II transmembrane serine proteases in cancer and viral infections. Trends Mol Med 15: 303–312 [DOI] [PubMed] [Google Scholar]
- Clausen TM, Sandoval DR, Spliid CB, Pihl J, Painter CD, Thacker BE, Glass CA, Narayanan A, Majowicz SA, Zhang Yet al (2020) SARS‐CoV‐2 infection depends on cellular heparan sulfate and ACE2. Cell 183: 1043–1057.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard B, Valle C, de Lamballerie X , Canard B, Seidah NG, Decroly E (2020) The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Res 176: 104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Anton‐Plagaro C, Shoemark DK, Simon‐Gracia L, Bauer M, Hollandi Ret al (2020) Neuropilin‐1 is a host factor for SARS‐CoV‐2 infection. Science 370: 861–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnest JT, Hantak MP, Li K, McCray PB Jr, Perlman S, Gallagher T (2017) The tetraspanin CD9 facilitates MERS‐coronavirus entry by scaffolding host cell receptors and proteases. PLoS Pathog 13: e1006546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantak MP, Qing E, Earnest JT, Gallagher T (2019) Tetraspanins: architects of viral entry and exit platforms. J Virol 93: e01429‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SC (2015) Viral membrane fusion. Virology 479–480: 498–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenian E, Nandakumar D, Lari A, Ly M, Tucker JM, Glaunsinger BA (2020) The molecular virology of coronaviruses. J Biol Chem 295: 12910–12934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A (2013) Virus entry: what has pH got to do with it? Nat Cell Biol 15: 125 [DOI] [PubMed] [Google Scholar]
- Helenius A, Kartenbeck J, Simons K, Fries E (1980) On the entry of Semliki forest virus into BHK‐21 cells. J Cell Biol 84: 404–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Pohlmann S (2020a) A multibasic cleavage site in the spike protein of sars‐cov‐2 is essential for infection of human lung cells. Mol Cell 78: 779–784.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N‐H, Nitsche Aet al (2020b) SARS‐CoV‐2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J, Helenius A (2011) Endosome maturation. EMBO J 30: 3481–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaimes JA, André NM, Millet JK, Whittaker GR (2020) Phylogenetic analysis and structural modeling of SARS‐CoV‐2 spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. J Mol Biol 432: 3309–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase M, Shirato K, Matsuyama S, Taguchi F (2009) Protease‐mediated entry via the endosome of human coronavirus 229E. J Virol 83: 712–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z, Oton J, Qu K, Cortese M, Zila V, McKeane L, Nakane T, Zivanov J, Neufeldt CJ, Cerikan Bet al (2020) Structures and distributions of SARS‐CoV‐2 spike proteins on intact virions. Nature 588: 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor R, McElroy LJ, Whittaker GR (2003) The ubiquitin‐vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 4: 857–868 [DOI] [PubMed] [Google Scholar]
- Kleine‐Weber H, Elzayat MT, Hoffmann M, Pohlmann S (2018) Functional analysis of potential cleavage sites in the MERS‐coronavirus spike protein. Sci Rep 8: 16597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AL, Millet JK, Daniel S, Freed JH, Whittaker GR (2017) The SARS‐CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium‐dependent manner. J Mol Biol 429: 3875–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léger P, Nachman E, Richter K, Tamietti C, Koch J, Burk R, Kummer S, Xin Q, Stanifer M, Bouloy Met al (2020) NSs amyloid formation is associated with the virulence of Rift Valley fever virus in mice. Nat Commun 11: 3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Luo S, Libby P, Shi GP (2020) Cathepsin L‐selective inhibitors: a potentially promising treatment for COVID‐19 patients. Pharmacol Ther 213: 107587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozach PY, Huotari J, Helenius A (2011a) Late‐penetrating viruses. Curr Opin Virol 1: 35–43 [DOI] [PubMed] [Google Scholar]
- Lozach PY, Kuhbacher A, Meier R, Mancini R, Bitto D, Bouloy M, Helenius A (2011b) DC‐SIGN as a receptor for phleboviruses. Cell Host Microbe 10: 75–88 [DOI] [PubMed] [Google Scholar]
- Lozach PY, Mancini R, Bitto D, Meier R, Oestereich L, Overby AK, Pettersson RF, Helenius A (2010) Entry of bunyaviruses into mammalian cells. Cell Host Microbe 7: 488–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, Nagata N, Sekizuka T, Katoh H, Kato Fet al (2020) Enhanced isolation of SARS‐CoV‐2 by TMPRSS2‐expressing cells. Proc Natl Acad Sci USA 117: 7001–7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazelier M, Rouxel RN, Zumstein M, Mancini R, Bell‐Sakyi L, Lozach PY (2016) Uukuniemi virus as a tick‐borne virus model. J Virol 90: 6784–6798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier R, Franceschini A, Horvath P, Tetard M, Mancini R, von Mering C , Helenius A, Lozach PY (2014) Genome‐wide small interfering RNA screens reveal VAMP3 as a novel host factor required for Uukuniemi virus late penetration. J Virol 88: 8565–8578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrow S, Falke D, Truyen U, Schätzl H (2013) Viruses with single‐stranded, positive‐sense RNA genomes. In Molecular virology, Modrow S, Falke D, Truyen U, Schätzl H (eds.), pp 185–349. Berlin, Heidelberg: Springer; [Google Scholar]
- Mohamed MM, Sloane BF (2006) Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 6: 764–775 [DOI] [PubMed] [Google Scholar]
- Ohkuma S, Poole B (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci USA 75: 3327–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou T, Mou H, Zhang L, Ojha A, Choe H, Farzan M (2021) Hydroxychloroquine‐mediated inhibition of SARS‐CoV‐2 entry is attenuated by TMPRSS2. PLoS Pathog 17: e1009212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo Li, Guo R, Chen T, Hu Jet al (2020) Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun 11: 1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa G, Mallery DL, Albecka A, Welch LG, Cattin‐Ortola J, Luptak J, Paul D, McMahon HT, Goodfellow IG, Carter Aet al (2021) Furin cleavage of SARS‐CoV‐2 Spike promotes but is not essential for infection and cell‐cell fusion. PLoS Pathog 17: e1009246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules CI, Marston HD, Fauci AS (2020) Coronavirus infections‐more than just the common cold. JAMA 323: 707–708 [DOI] [PubMed] [Google Scholar]
- Persson R, Pettersson RF (1991) Formation and intracellular transport of a heterodimeric viral spike protein complex. J Cell Biol 112: 257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Dominguez SR, Holmes KV (2013) Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS‐CoV) in virus entry and syncytia formation. PLoS One 8: e76469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirin K, Eschli B, Scheu I, Poort L, Kartenbeck J, Helenius A (2008) Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 378: 21–33 [DOI] [PubMed] [Google Scholar]
- Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J (2006) Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol 80: 4174–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F (2020) Cell entry mechanisms of SARS‐CoV‐2. Proc Natl Acad Sci USA 117: 11727–11734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K, Kanou K, Kawase M, Matsuyama S (2017) Clinical isolates of human coronavirus 229E bypass the endosome for cell entry. J Virol 91: e01387‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K, Kawase M, Matsuyama S (2013) Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87: 12552–12561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K, Kawase M, Matsuyama S (2018) Wild‐type human coronaviruses prefer cell‐surface TMPRSS2 to endosomal cathepsins for cell entry. Virology 517: 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P (2005) Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA 102: 11876–11881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuten K, Kim H, Widen JC, Babin BM, Onguka O, Lovell S, Bolgi O, Cerikan B, Neufeldt CJ, Cortese Met al (2021) Challenges for targeting SARS‐CoV‐2 proteases as a therapeutic strategy for COVID‐19. ACS Infect Dis 7: 1457–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T, Jaimes JA, Bidon MK, Straus MR, Daniel S, Whittaker GR (2021) Proteolytic activation of SARS‐CoV‐2 spike at the S1/S2 boundary: potential role of proteases beyond Furin. ACS Infect Dis 7: 264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triana S, Metz‐Zumaran C, Ramirez C, Kee C, Doldan P, Shahraz M, Schraivogel D, Gschwind AR, Sharma AK, Steinmetz LMet al (2021) Single‐cell analyses reveal SARS‐CoV‐2 interference with intrinsic immune response in the human gut. Mol Syst Biol 17: e10232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B, Sikora M, Schürmann C, Hagen WJH, Welsch S, Blanc FEC, von Bülow S, Gecht M, Bagola K, Hörner Cet al (2020) In situ structural analysis of SARS‐CoV‐2 spike reveals flexibility mediated by three hinges. Science 370: 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the sars‐cov‐2 spike glycoprotein. Cell 181: 281–292.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xiang Y (2020) Spike glycoprotein‐mediated entry of SARS coronaviruses. Viruses 12: 1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res 30: 269–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, Matlin K, Helenius A (1981) Cell fusion by Semliki forest, influenza, and vesicular stomatitis viruses. J Cell Biol 89: 674–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelfl F, Leger P, Oreshkova N, Pahmeier F, Windhaber S, Koch J, Stanifer M, Roman Sosa G, Uckeley ZM, Rey FAet al (2020) Novel toscana virus reverse genetics system establishes nss as an antagonist of type I interferon responses. Viruses 12: 400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS (2020) Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science 367: 1260–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, Skehel JJ, Gamblin SJ (2020) SARS‐CoV‐2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin‐cleavage effects. Nat Struct Mol Biol 27: 763–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GY, Lai MM (2005) The ubiquitin‐proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J Virol 79: 644–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecha J, Lee C‐Y, Bayer FP, Meng C, Grass V, Zerweck J, Schnatbaum K, Michler T, Pichlmair A, Ludwig Cet al (2020) Data, reagents, assays and merits of proteomics for SARS‐CoV‐2 research and testing. Mol Cell Proteomics 19: 1503–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Tsybovsky Y, Gorman J, Rapp M, Cerutti G, Chuang G‐Y, Katsamba PS, Sampson JM, Schön A, Bimela Jet al (2020) Cryo‐EM structures of SARS‐CoV‐2 spike without and with ACE2 reveal a pH‐dependent switch to mediate endosomal positioning of receptor‐binding domains. Cell Host Microbe 28: 867–879.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R, Nunneley JW, Barnard D, Pöhlmann S, McKerrow JH, Renslo ARet al (2015) Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 116: 76–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Data Availability Statement
This study includes no data deposited in external repositories.