

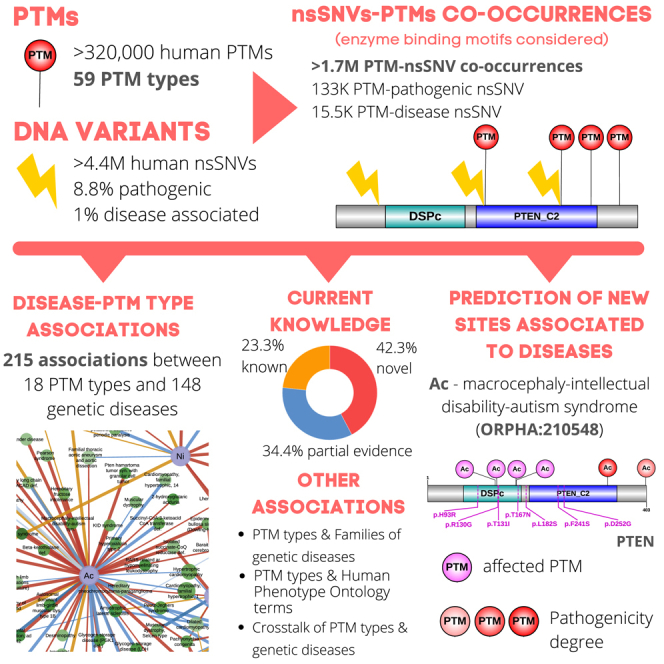
Summary
There are >200 types of protein posttranslational modifications (PTMs) described in eukaryotes, each with unique proteome coverage and functions. We hypothesized that some genetic diseases may be caused by the removal of a specific type of PTMs by genomic variants and the consequent deregulation of particular functions. We collected >320,000 human PTMs representing 59 types and crossed them with >4M nonsynonymous DNA variants annotated with predicted pathogenicity and disease associations. We report >1.74M PTM-variant co-occurrences that an enrichment analysis distributed into 215 pairwise associations between 18 PTM types and 148 genetic diseases. Of them, 42% were not previously described. Removal of lysine acetylation exerts the most pronounced effect, and less studied PTM types such as S-glutathionylation or S-nitrosylation show relevance. Using pathogenicity predictions, we identified PTM sites that may produce particular diseases if prevented. Our results provide evidence of a substantial impact of PTM-specific removal on the pathogenesis of genetic diseases and phenotypes.
Subject areas: Omics, Proteomics, Systems Biology
Graphical abstract
Highlights
-
•
There is an enrichment of disease-associated nsSNVs preventing certain types of PTMs
-
•
We report 215 pairwise associations between 18 PTM types and 148 genetic diseases
-
•
The removal of lysine acetylation exerts the most pronounced effect
-
•
We report a set of PTM sites that may produce particular diseases if prevented
Omics, Proteomics, Systems Biology
Introduction
Posttranslational modifications (PTMs) are an essential source of protein regulation. They are mostly reversible additions of small moieties or proteins that are capable of rapidly reprogramming protein function. There are several hundred PTM types, each with characteristic chemical and functional features (Buuh et al., 2018; Larsen et al., 2006).
Sources of lack of occupancy of PTMs in modifiable residues include the regulation and failure of writer and eraser enzymes, differential isoform transcription (De La Fuente et al., 2020), or genomic variation. In particular, nonsynonymous single-nucleotide variations (nsSNVs) in codons coding for a modifiable residue or within the enzyme recognized motif permanently impede modification. This effect has been widely studied in the context of somatic genomic variation in cancers (Narayan et al., 2016; Radivojac et al., 2008; Reimand et al., 2013). Regarding germline DNA variation, a large-scale survey described enrichment of rare and disease-annotated variants in PTM regions (Reimand et al., 2015), suggesting that functions provided by PTMs are linked to disease phenotypes and this effect is extended to the PTM site flanking region. Several public resources provide mappings between nsSNVs and PTMs (Hornbeck et al., 2015; Huang et al., 2019; Krassowski et al., 2018; Xu et al., 2018; Yang et al., 2019) and tools for their usage for genetic diagnosis (Peng et al., 2020; Wagih et al., 2018) or algorithms to predict alteration in kinase-binding motifs (Wagih et al., 2015). Not surprisingly, the main features of PTM sites, functionality, exposure, and conservation are also the main input for most variant pathogenicity/tolerance predictors (Adzhubei et al., 2013; Kumar et al., 2009).
The functional landscape of every type of PTM is still being decoded (Barber and Rinehart, 2018), and their combination seems to provide conserved functions (Minguez et al., 2012; Prabakaran et al., 2012). Meanwhile, their dysfunction has been associated with hundreds of diseases already. Major attention has been given to phosphorylation, which has been associated with cancers (Radivojac et al., 2008) and other disorders such as Alzheimer (Perluigi et al., 2016) and cardiovascular diseases (Wieland and Attwood, 2015). Target residues of acetylation, methylation, glycosylation, and S-nitrosylation are reported to be dysfunctional in cardiovascular and neurological disorders (Brosnan et al., 2004; Dudman et al., 1996; Lu et al., 2009; Mattson, 2010; Ngoh et al., 2010). Glycation was found to be deregulated in diabetes mellitus (Méndez et al., 2010), and N- and O-glycosylation deregulation has been reported in a number of congenital genetic disorders (Jaeken, 2006). The ubiquitination writer system is found to be key in many rare congenital disorders such as Angelman or von Hippel-Lindau syndromes (Jiang and Beaudet, 2004).
In this work, we hypothesized that the loss of specific protein functions defined by particular types of PTMs may map onto similar disease phenotypes. Thus, we cross-referenced protein modification sites and DNA variation positions to detect PTMs matched by nsSNVs. The PTMs were classified by types and the variants with disease annotation and deleterious predictions. By means of an enrichment analysis, we found a large number of associations between diseases and the prevention of specific PTM types, some of them with a very less number of instances described in the public databases. We report the first systematic global survey of associations between genetic diseases and the lack of specific PTM types. These results will bring attention to particular PTM types in the study of genetic disorders and provide a significant volume of new hypotheses in the research of rare diseases.
Results
Mapping PTMs and genomic variants positions to extract modifications prevented by nsSNVs
We collected 321,643 human PTMs pertaining to 59 types (Bateman, 2019; Chen et al., 2014, 2015; Gnad et al., 2011; Gupta et al., 1999; Hornbeck et al., 2015; Lu et al., 2013; Minguez et al., 2015; Sadowski et al., 2013; Sun et al., 2012) (Figure 1A) and 4,464,412 nsSNVs (Bateman, 2019; Koch, 2020; Landrum et al., 2014; Piñero et al., 2017; Sayers et al., 2011), 392,185 (8.8%) of which are classified as pathogenic as predicted deleterious by at least 3 out of 5 pathogenicity predictors: PolyPhen2 (Adzhubei et al., 2013), SIFT (Ng and Henikoff, 2001), PROVEAN (Choi and Chan, 2015), FATHMM (Shihab et al., 2014), and MutationTaster (Schwarz et al., 2014), and 44,969 (1%) with annotated disease associations obtained from Humsavar (Bateman, 2019), ClinVar (Landrum et al., 2014), and DisGeNET (Piñero et al., 2017) databases with the disease annotated as OMIM (Hamosh et al., 2002) and Orphanet (Pavan et al., 2017) (Figures 1B, Tables S1–S3). All PTMs and nsSNV variants were mapped into the same reference sequences (Huerta-Cepas et al., 2016) to identify residues with reported modifications that are prevented by certain nsSNVs. We annotated a PTM as affected and identified a PTM-nsSNV co-occurrence, when a nonsynonymous variant mutates: 1) the codon coding the modifiable amino acid or 2) the codons coding a window of ±5 amino acids from the modifiable residue, for PTM types with motifs reported (see STAR Methods, Figure S1). We identified a total of 1,743,207 PTM-nsSNVs co-occurrences (Table S4, Data S1), 15,607 with nsSNVs involved in diseases (Table S5), and 133,292 with nsSNVs predicted to be pathogenic (Table S6). Ordered and disordered protein regions were calculated using MobiDB-lite (Necci et al., 2017), and the PTM-nsSNV co-occurrences mapped into them. For each of these protein regions, we next obtained the percentage of the compiled PTMs of each type to be impacted by i) any type of DNA variant, ii) predicted pathogenic variants, and iii) disease-associated variants. Thus, the first observation is that different PTM types have different percentages of co-occurrence with nsSNVs, pathogenic nsSNVs, and disease-associated nsSNVs in both ordered and disordered protein regions (Figure 1C). Furthermore, we also annotated variations involved in the regulation of protein-protein interactions extracted from the IntAct database (Orchard et al., 2014) that can provide additional functional insights into the prevention of the PTMs by nsSNVs (Data S1 and Table S7).
Figure 1.

Human PTMs and nsSNVs compiled
(A) Number of PTMs classified by type.
(B) Number of nsSNVs classified in Orphanet families. Donut charts represent the percentage of variants predicted to be pathogenic and involved in diseases.
(C) Mapping between PTMs and nsSNVs per PTM type. Horizontal colored lines represent the average percentage of all PTM types to match all pathogenic and disease-associated nsSNVs.
(D) Enrichment of pathogenic and disease-causing variants affecting PTM types. PTM types are abbreviated as follows: phosphorylation (Ph), ubiquitination (Ub), acetylation (Ac), SUMOylation (SUMO), methylation (Me), N-linked glycosylation (NG), O-GalNAc glycosylation (OGa), S-glutathionylation (Glu), S-nitrosylation (Ni), O-linked glycosylation (OG), proteolytic cleavage (PC), hydroxylation (Hy), O-GlcNAc glycosylation (OGl), caspase cleavage aspartic acid (CCAsp), citrullination (Ci), palmitoylation (Pal), intra-polypeptide disulfide bond formation (DisB), ADP-ribosylation (ADP-r), sulfation (Sul), carboxylation (Ca), neddylation (Nedd), malonylation (Ma), oxidation (Oxi), amidation (Ami), pyrrolidone carboxylic acid formation (PyCB), nitration (Nit), myristoylation (My), protein-pyridoxal-5-phosphate linkage (P5PI), C-linked glycosylation (CG), and prenylation (Pre).
In order to assess whether there are PTM types that, when removed, are more deleterious than others, we determined whether pathogenic and disease-associated nsSNVs impact recurrently, not just PTMs but affecting certain PTM types (Figure 1D, Tables S8 and S9). As PTM functionality might depend on the protein region where the PTM is located, we independently tested ordered and disordered regions. Our first observation was that PTM types are, in general, more enriched in predicted pathogenic nsSNVs than in disease-associated nsSNVs. Phosphorylation, ubiquitination, and methylation show a functionality that seems significantly impacted by nsSNVs in both ordered and disordered regions. Seven additional PTM types showed enrichments only in one sort of protein region, three in disordered, and four in ordered regions. ADP-ribosylation, carboxylation, and myristoylation are the PTM types with the least number of instances (123, 85, and 78, respectively) that are hit significantly by disease-related or pathogenic DNA variants.
A global network of associations between PTM types and inherited diseases
To extract significant pairwise associations between PTM types and genetic diseases, for every pair of a genetic disease and a PTM type, we performed a Fisher’s exact test to determine differences in the proportions of 1) the number of disease nsSNVs hitting a PTM of that type compared with 2) the number of disease nsSNVs hitting in other PTM types and 3) the number of nsSNVs causing other diseases affecting the PTM type compared with 4) the number of nsSNVs causing other diseases affecting other PTM types. In this analysis, we distinguished between N-terminal and lysine acetylations (Nt-Ac, K-Ac), between serine/threonine and tyrosine phosphorylations (S/T-Ph, Y-Ph), and between arginine and lysine methylations (R-Me, K-Me). We found a total of 222 significant associations (FDR<0.05) between 18 PTM general types and 148 inheritance diseases (Table S10), where one disease is associated to both K-Ac and Nt-Ac and 6 to S/T-Ph and Y-Ph, so 215 if those are merged into general PTM types. If recognition motifs are not considered, we report 42 associations (Table S11). We annotated the full set of significant associations as i) known, ii) with partial evidence, or iii) novel according to a literature review. In the category “having partial evidence,” we grouped several heterogeneous examples that indirectly suggest a link between the disease and the PTM type. These include i) reports of one variant case, ii) associations of the PTM type with a related or similar disease, and iii) regulatory evidence when the PTM type is involved in the deregulation causing the disease. Summing up, our strategy led to the documentation of 50 known associations, generalized 74, and evidenced 91 novel links (Figure 2A) which are unequally distributed across PTM types (Figure 2B). Figure 2C displays all PTM types and disease significant associations in a network format. Thus, acetylation stands out as the PTM with the most associations, 58 associations with K-Ac and 2 with Nt-Ac, one of the latest also caught by K-Ac, so 59 in total. Of them, 34 were not previously reported, and only 6 of which were known. Next, ubiquitination and N-linked glycosylation were associated with 29 and 22 pathologies, respectively, and the removal of another 6 PTM types (phosphorylation, methylation, S-glutathionylation, S-nitrosylation, SUMOylation, and protein cleavage) are proposed to be the cause of 10 or more inheritance pathologies. Regarding phosphorylation, S/T-Ph is found to be associated with 18 diseases, while Y-Ph with 7 diseases, 6 of them shared between both, so 19 unique associations. The distinct role of K-Me and R-Me in genetic diseases seems to be more clear; we detect 3 diseases associated to K-Me and 11 diseases associated to R-Me, none of them in common. Importantly, even infrequent PTM types such as malonylation or neddylation, with only 45 and 40 modified instances each, show associations. In terms of novelty, besides acetylation, S-glutathionylation, S-nitrosylation, O-GalNAc glycosylation, O-GlcNAc glycosylation, ADP-ribosylation, and hydroxylation are the most underestimated types, with ≥50% of previously undescribed associations, although the latter are linked only to 4, 3, and 1 diseases, respectively. Proportionally, phosphorylation is the PTM with more previous evidence, with only 3 novel associations of the 19 extracted (16%). Interestingly, a number of diseases were associated with several PTM types, with Li-Fraumeni syndrome and Pachyonychia congenita at the top, with 5 associations, and followed by 7 diseases with 4 associations: amyotrophic lateral sclerosis, hemophilia B, Hutchinson-Gilford progeria syndrome, lissencephaly due to TUBA1A mutation, lissencephaly type 3, hyperinsulinism-hyperammonemia syndrome, and dilated cardiomyopathy. Nevertheless, 74% of the diseases studies were found to have a unique PTM type involved as a molecular cause.
Figure 2.

Predicted associations between PTM types and genetic diseases
(A) Classification of the associations based on the current knowledge obtained by means of a literature review.
(B) Per each PTM type, we show the number of diseases for which associations have been predicted, classified using current knowledge.
(C) Network providing all predicted associations between PTM types and genetic diseases. Two types of nodes are present: PTM types and diseases, and links represent a predicted association that is colored according to its current knowledge status. PTM types are abbreviated as follows: phosphorylation (Ph), ubiquitination (Ub), acetylation (Ac), SUMOylation (SUMO), methylation (Me), N-linked glycosylation (NG), O-GalNAc glycosylation (OGa), S-glutathionylation (Glu), S-nitrosylation (Ni), O-linked glycosylation (OG), proteolytic cleavage (PC), hydroxylation (Hy), O-GlcNAc glycosylation (OGl), ADP-ribosylation (ADP-r), sulfation (Sul), carboxylation (Ca), neddylation (Nedd), and malonylation (Ma).
We also checked the degree to which the signal of the associations may come from the particular location of the PTMs into the proteins, as it has been described for cancer somatic mutations (Woodsmith et al., 2013), or from a saturation of modifications so any location would give a match with the disease causal variants. Thus, for each significant association between a PTM type and a pathology, we randomly permuted the location of the modifications, maintaining the ratios of PTMs in ordered and disordered regions and only in residues modifiable by the PTM type. This exercise was performed 100 times, and for each, a Fisher's exact test was performed. We refer to the reallocation score (RAS) as the number of times an association has been found to be significant when the PTMs are randomly located (Figure 3A). The maximum value for the RAS is 100, meaning that PTMs of a given type are affected by variants associated with a particular disease even if the modifications are randomly located. In contrast, the smallest possible value is 0, indicating that the actual location of the PTMs is, almost, the unique possible location affected by the nsSNVs. Thus, 95 of our associations (42.8%) had from very low to medium values of RAS and 125 (56.3%) high or very high RASs (Figures 3B and S2). Interestingly, we observed that associations not previously reported displayed more robust RASs (RASmean = 55) than known associations and those with partial evidence (RASmean = 62), still with no significant p value (0.154). The novel associations also have a lower numbers of PTMs and nsSNVs than known associations and those with partial evidence (p values = 0.001) (Figures 3C and 3D). In addition, RAS levels were unequally distributed over associations in terms of the PTM types involved, with phosphorylation showing the highest level of RASs and protein cleavage the smallest, considering PTM types with ≥10 associations (Figure S3).
Figure 3.

Assessment of the PTM type-disease associations in terms of PTM density
(A) Scheme for the calculation of the reallocation score (RAS).
(B) Classification of the associations based on their RASs.
(C) Boxplots for RAS distributions in associations annotated as known, having partial evidence, and novel.
(D) Boxplots with the distribution of the sum of the number of nsSNVs associated to the disease and the PTMs of the type being studied co-occurring in the proteins annotated as involved in the disease. For the comparisons of these distribution at (C and D), known and partial evidence was merged and compared with novel associations using Wilcoxon rank-sum tests.
Associations between the removal of PTM types, disease families, and phenotypic features
We set out to describe patterns of PTM types for which removal affected similar pathologies. Thus, we used the Orphanet disease ontology to classify diseases into families (Table S12) and merge our associations between PTM types and diseases. We calculated the Jaccard index for every pair of PTMs measuring their similarities in the connections to disease families (Figure S4) and used it to define clusters of PTM types (Figure 4A). Three main clusters are observed. First, a core of 8 PTM types (cluster 1) formed by phosphorylation, ubiquitination, acetylation, SUMOylation, methylation, N-linked glycosylation, S-nitrosylation, and S-glutathionylation, which are proposed to be behind the molecular basis of 71% of the associations in the network of associations between PTM types and disease families (124 links out of a total of 175). This core is suggested to be responsible for the deregulation of important functions producing 88 diseases grouped into 17 families, of which skin and cardiac diseases make up the largest groups; second, a cluster of 5 PTM types (cluster 2) formed by O-GalNAc glycosylation, O-linked glycosylation, proteolytic cleavage, O-GlcNAc glycosylation, and ADP-ribosylation, which are connected to 11 disease families, being neurologic diseases the largest family (72 diseases) followed by developmental defect during embryogenesis, neoplastic, endocrine, hematologic, and eye diseases. Cluster 2 shows an exclusive link between proteolytic cleavage and allergic diseases. The third cluster of PTM types (cluster 3) comprises hydroxylation, sulfation, and carboxylation; removal of these types is suggested to affect hematologic diseases only. Finally, neddylation and malonylation affected 2 disease families, neurologic and systemic or rheumatologic, and inborn errors of metabolism and endocrine, respectively.
Figure 4.

Associations between PTM types and disease families and phenotypes
(A) The PTM type-disease predicted associations were merged into PTM type-disease family associations and represented in a network. Nodes are PTM types and disease families, and an edge represents the number of predicted associations between the PTM type and disease from a particular family. PTM types are distributed according to the Jaccard index calculated with the families they share. Connections based on the Jaccard index between PTM types are not represented. PTM types are arranged in clusters based on the disease families they share, and disease families are colored according to their connections to clusters.
(B) Network providing all predicted associations between PTM types and human phenotypes using Human Ontology Phenotype (HPO) terms. Two types of nodes are present: PTM types and HPO terms, and links represent a predicted association that is colored according to its reallocation score. PTM types are abbreviated as follows: phosphorylation (Ph), ubiquitination (Ub), acetylation (Ac), SUMOylation (SUMO), methylation (Me), N-linked glycosylation (NG), O-GalNAc glycosylation (OGa), S-glutathionylation (Glu), S-nitrosylation (Ni), O-linked glycosylation (OG), proteolytic cleavage (PC), hydroxylation (Hy), O-GlcNAc glycosylation (OGl), ADP-ribosylation (ADP-r), sulfation (Sul), carboxylation (Ca), neddylation (Nedd), malonylation (Ma).
Next, we extracted associations between PTM types and phenotypic features taken from the Human Phenotype Ontology (HPO) (Köhler et al., 2019) using the same methods (Table S13). One hundred thirty-three significant associations were found between 11 PTM types and 101 HPO terms, 37 parent and 64 child terms (Table S14 and Figure S5). An overall network is presented in Figure 4B with only HPO parent terms for clarity. Here, the removal of acetylated and phosphorylated sites shows the greatest number of associations, both with 15 HPOs associated, 3 of them shared (muscular hypotonia and 2 leukemia-related phenotypes). Phosphorylation shares 3 other cancer-related terms, 2 with ubiquitination (non-Hodgkin lymphoma and non-small cell lung carcinoma) and one with methylation (transitional cell carcinoma of the bladder). The link between phosphorylation and cardiopathies is seen here again, although most of the terms are indeed related to cancer, same happening with ubiquitination. Finally, acetylation seems to be crucial to phenotypes related to development and growth.
Associations between the removal of cross talking PTM types and genetic diseases
We were also interested in detecting genetic diseases which phenotype might be caused by nsSNVs affecting residues that can be modified by several PTM types as a cross talk mechanism. Thus, we compiled i) serines and threonines modified with any type of O-linked glycosylation and a phosphorylation (OG-Ph) and ii) lysines modified by at least two PTM types (acetylation, methylation, SUMOylation, or ubiquitination). With this collection of sites grouped by pairs of PTM types, we performed a similar enrichment analysis as with the PTM types and diseases (Table S15). Thus, OG/Ph sites are found enriched in nsSNVs causing four genetic diseases: ORPHA:276152 (Multiple endocrine neoplasia type 4), ORPHA:603 (Distal myopathy, Welander type), ORPHA:93,616 (Hemoglobin H disease), and ORPHA:740 (Hutchinson-Gilford progeria syndrome) (Figure 5A). For any of the six possible combinations of PTM types modifying lysines, we found associations with 34 genetic diseases; for many of them, we report a suggested role of three or more PTM types. An interesting case is Pachyonychia congenita (ORPHA:2309) whose causing nsSNVs seem to prevent the modification of sites regulated by all the possible PTM types (Figure 5A). Also to highlight is the full overlap in the associations for the PTM combinations Ub-Me and Me-SUMO (Figure 5B).
Figure 5.

Associations between genetic diseases and pairs of PTM types modifying the same residue as a cross talk mechanism
(A) Network with the significant associations (FDR<0.05) of pairs of cross talking PTM types that modify the same residue and gene diseases. Cross talking PTM types represented by blue nodes: acetylation-methylation (Ac-Me), acetylation-ubiquitination (Ac-Ub), acetylation-SUMOylation (Ac-SUMO), ubiquitination-methylation (Ub-Me), ubiquitination-SUMOylation (Ub-SUMO), methylation-SUMOylation (Me-SUMO), and phosphorylation-O-GlcNAc glycosylation (Ph-OGl).
(B) Overlap of diseases predicted to be associated with the pairs of cross talking PTMs.
Prediction of potential disease-associated mutation sites
If there is a global association between a PTM type and a genetic disease, other modifications of the same type in the proteins involved in the disease may become candidate sites for eventual nsSNVs to produce the same phenotype. Here, sites refer to nucleotides in codons coding for the modified amino acids. For each of our detected associations, we identified the sites of the same PTM type that are not affected by an nsSNV causing the disease (candidate PTMs). For those, we calculated the pathogenicity, measured using Polyphen2 (Adzhubei et al., 2013), for in silico randomly generated nsSNVs that prevent the PTM. In parallel, for each PTM type, we define a background distribution of Polyphen2 scores of in silico random mutations in PTMs of the same type in proteins not involved in any disease detected as associated to that PTM type (background PTMs). This background distribution tries to represent the Polyphen2 score for regular PTMs of a type. As a proof of concept, we compared both distributions of Polyphen2 scores at candidate and background PTMs. In Figure 6A, we show the general scheme for this comparison. In total, 37 associations between PTM types and diseases showed a significantly higher pathogenicity in PTMs not affected by a disease variant (Table 1).
Figure 6.

Prediction of pathogenicity for PTM sites in the context of associations between PTM types and genetic diseases
(A) General schema of the comparison between 1) the potential pathogenicity of selected PTM sites not affected by disease nsSNVs when the disease and its type are predicted to be associated (colored in blue) and 2) reference distribution of the potential pathogenicity scores of PTM sites of the type but not linked to diseases (colored in orange).
(B) Barplot with the number of PTM sites with pathogenicity predictions per PTM type and scheme of the rps calculation. (C-F) Four examples of proteins involved in predicted associations between PTM types and genetic diseases. PTM types are abbreviated as follows: methylation (Me), SUMOylation (SUMO), acetylation (Ac), S-glutathionylation (Glu).
Table 1.
Associations between PTM types and genetic diseases where there is significant higher pathogenicity in the alteration of modified sites of the same type but not affected by the disease variants and other PTM sites
| PTM type | Disease name | Disease ID | Proteins | PTMs (rps>95) |
|---|---|---|---|---|
| Acetylation | Dilated cardiomyopathy | ORPHA:217604 | KIAA1652,LMNA,MYH6,MYH7 | 20 |
| Acetylation | Cardiomyopathy, familial hypertrophic, 1 | OMIM:192600 | MYH6,MYH7,RYR2,TPM1 | 17 |
| Acetylation | Rubinstein-Taybi syndrome | ORPHA:783 | CREBBP,EP300 | 26 |
| Acetylation | Rubinstein-Taybi syndrome due to CREBBP mutations | ORPHA:353277 | CREBBP,EP300 | 26 |
| Acetylation | Muscular dystrophy | ORPHA:98,473 | KIAA0723,LMNA | 4 |
| Acetylation | Pten hamartoma tumor syndrome with granular cell tumor, included | OMIM:601728 | PTEN | 1 |
| Acetylation | Syndrome with limb malformations as a major feature | ORPHA:109009 | CREBBP,EP300 | 32 |
| Acetylation | Hypertrophic cardiomyopathy | ORPHA:217569 | MYH6,MYH7 | 17 |
| Acetylation | Distal myopathy, Welander type | ORPHA:603 | MYH7 | 8 |
| Acetylation | Cardiomyopathy, familial hypertrophic, 14 | OMIM:613251 | MYH6 | 4 |
| Acetylation | Alexander disease | ORPHA:58 | GFAP | 1 |
| Acetylation | Macrocephaly-intellectual disability-autism syndrome | ORPHA:210548 | PTEN | 1 |
| Acetylation | Desminopathy | ORPHA:98,909 | DES | 1 |
| Carboxylation | Congenital factor X deficiency | ORPHA:328 | F10 | 2 |
| S-glutathionylation | Cornelia de Lange syndrome | ORPHA:199 | SMC1A | 1 |
| Methylation | Pachyonychia congenita | ORPHA:2309 | KRT6B | 1 |
| Methylation | Inclusion body myopathy with Paget disease of bone and frontotemporal dementia | ORPHA:52,430 | VCP | 2 |
| Methylation | Hyperinsulinism-hyperammonemia syndrome | ORPHA:35,878 | GLUD1 | 1 |
| S-nitrosylation | Lethal encephalopathy due to mitochondrial and peroxisomal fission defect | ORPHA:330050 | DNM1L | 0 |
| O-GlcNAc glycosylation | Multiple endocrine neoplasia type 4 | ORPHA:276152 | CDKN1B | 1 |
| O-GlcNAc glycosylation | Multiple endocrine neoplasia | ORPHA:276161 | CDKN1B | 1 |
| O-linked glycosylation | Glanzmann thrombasthenia | ORPHA:849 | ITGA2B | 1 |
| Proteolytic cleavage | Hyperproinsulinemia | OMIM:616214 | INS | 2 |
| Proteolytic cleavage | Permanent neonatal diabetes mellitus | ORPHA:99,885 | ISN | 0 |
| Proteolytic cleavage | Hereditary thrombophilia due to congenital protein S deficiency | ORPHA:743 | PROS1 | 1 |
| Phosphorylation | Turcot syndrome with polyposis | ORPHA:99,818 | APC | 6 |
| Phosphorylation | Hereditary breast cancer | ORPHA:227535 | BACH1,ESR1 | 5 |
| Phosphorylation | Baraitser-Winter cerebrofrontofacial syndrome | ORPHA:2995 | ACTB, ACTG1 | 8 |
| Phosphorylation | Hypomyelination with atrophy of basal ganglia and cerebellum | ORPHA:139441 | TUBB3 | 10 |
| SUMOylation | Familial dilated cardiomyopathy with conduction defect due to LMNA mutation | ORPHA:300751 | LMNA | 1 |
| SUMOylation | Dilated cardiomyopathy | ORPHA:217604 | LMNA | 1 |
| SUMOylation | Roberts syndrome | ORPHA:3103 | ESCO2 | 3 |
| Ubiquitination | lung cancer | OMIM:211980 | EGFR, ERBB2,MAP2K1 | 8 |
| Ubiquitination | Fanconi anemia | ORPHA:84 | BRCA2,FANCI,SLX4 | 10 |
| Ubiquitination | Cardiofaciocutaneous syndrome | ORPHA:1340 | BRAF,MAP2K1,MAP2K2 | 4 |
| Ubiquitination | Aicardi-Goutieres syndrome | ORPHA:51 | ADAR,NPAS1,RNASEH2A,RNASEH2B | 15 |
| Ubiquitination | X-linked intellectual disability-hypotonia-movement disorder syndrome | ORPHA:457260 | DDX3X | 3 |
Proteins and sites predicted to be potentially involved in the disease if a genetic alteration occurs are given.
In order to predict new pathogenic sites associated to a disease, we introduced the relative pathogenicity score (rps). The rps is calculated in the context of PTM type-disease associations for PTM sites of the same type but not affected by a disease variant, such as the percentile of the pathogenicity estimation of the site in a reference distribution (Figure 6B, Material and Methods); rps>95 is set as significant. Thus, we evaluated the potential pathogenicity of the deletion of 2,205 PTMs sites in 153 proteins, of which 156 sites (7.6%) had a significant rps (Table S16). Figure 6B shows the number of sites and their rps per PTM type. Unsurprisingly, we see that phosphorylation is the type with the highest number of sites evaluated, still with a below-average percentage of significant pathogenic sites (5.3%). Although carboxylation and O-GlcNAc glycosylation had the highest percentage of significant pathogenic sites (57.1% and 33.3%, respectively), acetylation again deserves special attention: with a large number of PTM sites evaluated (292), it is an essential type for which deletion may produce disease-related phenotypes, with 18.9% of sites showing significant pathogenicity. See Table S17 for all sites and their evaluation including complementary cancer associations from the BioMuta database (Dingerdissen et al., 2017).
Finally, we will discuss a few examples that illustrate some of the findings of this work. Amyotrophic lateral sclerosis (ALS, ORPHA:803), the most common genetic disorder due to the degeneration of motor neurons, is a neurodegenerative disease producing muscular paralysis. Several genes have been reported to carry mutations that lead to this phenotype, including FUS, an RNA-binding protein with genomic variations leading to the formation of stress granules. Methylation of FUS is required to bind the autophagy receptor p62, which, in association with other proteins, can control the aberrant accumulation of granules (Chitiprolu et al., 2018). Other studies have reported methylation as an important modification type in the accumulation and elimination of these toxic granules (Simandi et al., 2018), thus supporting our predicted association with ALS. Figure 6C shows FUS with methylation sites affected and not affected by ALS mutations; the former is likely to be more pathogenic than regular methylation sites, measured using rps.
The second example (Figure 6D) shows protein ESCO2 and acetyltransferase with genomic variants causing Roberts syndrome (RBS, ORPHA:3103), an ultrarare disorder causing limb and facial abnormalities and slow down prenatal and postnatal growth. In RBS, the dysfunction of ESCO2 produces a lack of acetylation in the cohesin complex, which regulates a proper sister chromatid cohesion in the DNA replication process (Xu et al., 2014). We predict an association between the lack of SUMO sites and RBS. Figure 6D shows 4 SUMO sites in ESCO2 affected by RBS variants and 7 more that might be candidates for the same type of regulation, 3 with a significant rps. A sister chromatid cohesion impairment has already been observed owing to a lack of SUMOylation directly in the cohesin complex (Almedawar et al., 2012) as well as in a knockout of SUMO protease SENP1 in chicken cell lines (Era et al., 2012), suggesting an important role played by this PTM type in this process, although still with no reports linking it to RBS. We annotated the association as constituting partial evidence.
Next, we briefly describe 2 examples of associations with no previous evidence. Starting with the PTM type with the highest number of associations, acetylation, we show the phosphatase PTEN (Figure 6E) with 7 mutations linked to the macrocephaly intellectual disability—autism syndrome disorder (ORPHA:210548), a rare neurological disorder with macrocephaly and facial deformity as notable anatomical features and psychomotor delay, intellectual disability, and autism as the primary disease-associated disabilities. PTEN acts at the phosphoinositide 3-kinase pathway and is described to be intensively regulated by acetylation (Ikenoue et al., 2008; Meng et al., 2016). Figure 6E shows PTEN protein with 4 acetylation sites affected by variants and K332 predicted to be a functionally associated site to the mentioned phenotype. In a second novel case, we focus on S-glutathionylation, the eighth most abundant PTM type in our collection, although nearly 80% of our predictions have not been previously reported in the scientific literature (11 out of 14). One of these new predicted associations is with Darier disease (DD, ORPHA:218), a rare skin disorder characterized by the formation of keratotic papules during puberty that are frequently subject to infection. Causative mutations have been described only in the gene ATP2A2, which codes for protein SERCA Ca2+-ATPase, responsible for ER Ca2+ uptake. Malfunctions in this protein alter Ca2+ homeostasis in the cytoplasm, which alters keratonocyte functions downstream (Savignac et al., 2011). Figure 6F shows ATP2A2 with DD variants and S-glutathionylation sites mapped, 2 of these residues are directly removed by the variants, and we calculated a proxy score to determine the probability that other glutathionylated residues will be key sites in the disease. Although S-glutathionylation has been described to be involved in the regulation of Ca2+ ER pumps, for instance, by activating SERCA in arterial relaxation (Adachi et al., 2004) or promoting its optimal oscillation by diamide in bovine aortic endothelial cells (Lock et al., 2011), to the best of our knowledge, this is the first report suggesting an association with the disorder. Table S18 has details about the PTM sites of the four examples.
Discussion
There are 2 motivations to study PTMs in the context of genetic diseases, the first and most obvious of which is to deepen knowledge of the genetic basis of these pathologies. Systematic and global approaches are of special interest in rare diseases, where current resources cover only a small percentage of these diseases owing to their low incidence individually, although together they affect around 7% of the population (Dharssi et al., 2017). Here we have studied >3,200 genetic diseases and have obtained significant results for 148, including new hypotheses linking the loss of specific functions to diseases and phenotypes as well as describing general patterns of association. The second motivation is to add functional information to individual PTMs (Beltrao et al., 2013; Minguez et al., 2012; Naegle et al., 2010), frequently extracted from high-throughput experiments and most with succinct annotation (Gnad et al., 2011). The annotation of PTMs and the study of the types as functional entities and their interactions is essential to decode the regulation of proteins and establish how cells respond to a changing environment, named the PTM code (Hunter, 2007). Although several previous resources have cross-referenced genomic variation and PTM data (Hornbeck et al., 2015; Huang et al., 2019; Xu et al., 2018), for the first time, we add a global interpretation of PTM types in the context of genetic diseases as well as prediction capacities, even to the less studied PTM types.
We believe ours is the largest study to cross-reference human genomic variation and protein modifications, comprising 4,464,412 germline nsSNVs (44,969 involved in diseases and 392,185 with predicted pathogenicity) and 321,643 of 59 types that revealed 1,743,207 PTM-nsSNVs co-occurrences in 16,541 proteins. Of those PTMs and nsSNVs co-occurrences, 15,607 involved diseases and 133,292 conferred pathogenicity. Previous research by Reimand et al (Reimand et al., 2015) focused on 4 PTM types (130,000 sites), reporting 77,829 matches between PTM regions and nsSNVs. Kim et al. (Kim et al., 2015) extended the number of PTM types to 20, extracting 179,325 PTM-SNVs associations using 517,466 missense and stop gain SNVs and 599,387 PTMs, most of which were predicted (404,501) and generally reported with low accuracy (Piovesan et al., 2020). More recently, the AWESOME database (Yang et al., 2019) also provides a large data set with 1,043,668 nsSNVs and experimentally verified and predicted PTMs of 6 types totaling 481,557 matches. Similar to our study, other approaches have also focused on disease associations, but compiling curated associations from the scientific literature: PTMD (Xu et al., 2018) and PhosphoSitePlus (Hornbeck et al., 2015), for instance, report 1,950 and 1,230 PTMs-SNVs associations, respectively. The topic is clearly of interest (Pascovici et al., 2019).
Our framework proposed a total of 215 cases where the nsSNVs causing a particular disease show a preference to prevent a particular type of PTM in the proteins causing a disease. Thus, we defined associations between the loss of a function provided by a PTM type and a genetic disease and represented them as pairs of PTM types and genetic diseases. Although other types of genomic variations are reported to affect functions led by PTMs (Gonçalves et al., 2017), we concentrated on reported causal nsSNVs for each of the disorders. Because the overall diagnostic rate of these disorders is about 50%, and the number of predicted pathogenic variants substantially surpasses the numbers for annotated disease-causing variants (Figure 1B), it can be stated that the scenario described here is underestimated. In addition, other sources of implications of PTMs in harmful phenotypes include, for instance, the possibility of genetic variants to produce new modifiable sites or PTM motifs (Krassowski et al., 2018; Yang et al., 2019) or the alteration of the enzymatic cascades that attach the PTMs, which we have applied as confirmatory rule (Figures 2A and 2C).
This wealth of information has been scanned in 2 different manners. First, we report an extensive literature review where we searched for previous evidence for all the proposed associations between PTM types and diseases. Thus, we found that 23% of the total cases we extracted were already reported and 34% had evidence not directly linking the PTM type to the disease but showing related functional proofs. Altogether they represent 57% of our associations supported by external evidence. Besides, we provide new insights for about 165 associations, 91 completely novel. We are aware that with a literature review of such a magnitude, associations may go overlooked, although we believe these would be cases of partial evidence as they are less obvious links. Additionally, we checked if the associations are extracted owing to a high number of modifications using a permutation approach, as done by others (Woodsmith et al., 2013). Our RAS measures this possibility (Figure 3B) showing that up to 43.6% of the pairwise proposed associations have PTMs displayed in proteins with a medium to high chance of being the unique conformation providing the association (Figure 3B). Importantly, the RAS is lower in the associations not previously described and higher for associations involving PTM types such as phosphorylation, ubiquitination, or SUMOylation, which may indicate that the discovery of types of PTMs and diseases depends at least partially on the level of knowledge accumulated on PTM types and proteins/diseases. Thus, potential associations involving less studied proteins or PTM types with less instances described in the databases seem to be more probable to be missed unless systematic studies like this one are performed. As an example of this behavior, we found that phosphorylation, by far the most studied type of PTM, represented a great proportion of the predicted associations with some kind of evidence, with only 3 completely novel. Thus, our conclusion is that the RAS cannot be used as a confidence estimator for the association but as an additional descriptor.
Summing up, we highlight lysine acetylation with not only histone functions (Narita et al., 2019) predicted to be involved in up to 58 diseases, ∼60% of them no previously reported, and linked to several phenotypes. Its implication seems to be relevant in several disease families, with neurologic and inborn errors of metabolism comprising 19% and 14% of its associations, respectively, but also being connected to urogenital, hepatic, and immune diseases almost uniquely. Other PTMs seem to be of special interest, such as ubiquitination due to its role in protein degradation, which has been described widely associated with human pathologies (Jiang and Beaudet, 2004), but also S-glutathionylation, S-nitrosylation, or the different glycosylations, that are normally not included in the systematic studies but with a wealth of predictions here. Displayed in a network format and merging diseases in families, we were able to extract general patterns that provide clues for researchers to explore the implication of PTM types in diseases that do not have enough genetic information to be explored. Similarly, a network of links between PTMs and phenotypes is also produced. In this sense, it is expected that disorders with similar phenotypes are caused by similar deregulation processes (Xue et al., 2019). Last about network analysis capacities, it is feasible to think that the convergence of several PTM types implicated in similar diseases may be a signal of functional cross talk (Beltrao et al., 2012; Minguez et al., 2012). In this sense, we have found several diseases with nsSNVs affecting residues with two different types of PTMs with significant associations, so reinforcing the important regulatory role of these molecular switches, in here linked to genetic diseases.
Finally, if our strategy up to this point has been to establish general patterns from individual events (PTM-nsSNV co-occurrences), we wanted to add value to the predictions by scaling down the functional implications and providing a bunch of PTM sites whose removal by genomic variations is predicted to produce specific disorders. The score we built for this task, the rps, is quite conservative in the sense of making validated pathogenicity scores (Adzhubei et al., 2013), being referenced to functional sites that are expected to be pathogenic themselves (Holehouse and Naegle, 2015). Besides the 4 examples discussed in detail, we add as Table S17 all the PTM sites predicted to be functionally linked to the diseases, with detailed Polyphen2 scores and rps values. This, together with all the PTM-nsSNV co-occurrences available as Data S1 (>1.7M), represents a valuable and unique source of information to be included in bioinformatics pipelines of DNASeq analysis to annotate genomic variants. Because the filtering and prioritization of variants is an essential task in genetic diagnosis based on deep sequencing (Roy et al., 2018), we believe the resources reported in this work may contribute to new hypotheses on specific variants that could lead to more conclusive diagnosis of patients.
Limitations of the study
As any study that manages genome- or proteome-wide annotations, our work is mainly limited by the status of the current knowledge on those fields. In this case, the repertoires of DNA variants-disease associations and the human PTMs are not complete. On the top of this, long-term studied PTM types and genes are obviously more annotated than others, so they, genes and PTM types, represent an important part of the final signal. Thus, the associations provided have to be understood as a subset of the real implication of PTMs in genetic diseases where, apart from the lack of annotation, other types of variants and deregulations could be involved.
For the PTM types that were reported to have recognition motifs, we established a unique length of the PTM site flanking region were an nsSNV could be considered to disrupt the modification. Here, there might be motifs with a longer extension as well as mutations that do not alter the capacity of the enzyme to recognize the site. Because previous reports have seen PTM regions significantly enriched in rare substitutions (Reimand et al., 2015), and our analysis aims for global associations based on enrichments, we believe there is no bias in the mapping of PTMs and nsSNVs that would favor the detection of associations between PTM types and diseases.
Regarding our classification of the PTM type-disease associations into known, partial evidence, and novel, we are aware that with a literature review of such a magnitude, associations may go overlooked, although we believe these would be cases of partial evidence as they are less obvious links.
Regarding PTM functionality, as in every systematic and computational study, we are aware of the cell state dependence of their occupancy (Wagih et al., 2018).
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and algorithms should be directed to and will be fulfilled by the lead contact, Pablo Minguez (pablo.minguez@quironsalud.es).
Materials availability
This study did not generate new materials.
Data and code availability
-
•
The data sets supporting the conclusions of this article are included within the article and its additional files. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the data sets are listed in the key resources table.
-
•
This paper does not report original code.
Method details
Compilation of nsSNVs and annotation as disease-associated and pathogenic
We compiled human nsSNVs from the following databases: dbSNP (Sherry et al., 2001), gnoMAD v2 (Collins et al., 2019; Koch, 2020), Humsavar (UniProt) (Bateman, 2019), ClinVar (Landrum et al., 2014), and DisGeNET (Piñero et al., 2017). Protein IDs were mapped to STRING v10 (Szklarczyk et al., 2015) dictionary, and sites were mapped onto the eggNOG v4.5 (Huerta-Cepas et al., 2016) set of human sequences confirming the wild-type amino acid.
The disease annotation of nsSNVs was retrieved from Humsavar (Bateman, 2019), ClinVar (Landrum et al., 2014), and DisGeNET (Piñero et al., 2017) databases as OMIM (Hamosh et al., 2002) and Orphanet (Pavan et al., 2017) diseases. Phenotype information was retrieved from ClinVar as HPO terms (Köhler et al., 2017). OMIM and Orphanet IDs for the same diseases were merged into Orphanet IDs.
To assess the pathogenicity of the nsSNVs, we used pathogenicity predictions from 5 algorithms: PolyPhen2 (Adzhubei et al., 2013), SIFT (Ng and Henikoff, 2001), PROVEAN (Choi and Chan, 2015), FATHMM (Shihab et al., 2014), and MutationTaster (Schwarz et al., 2014), all calculated using the Variant Effect Predictor pipeline (McLaren et al., 2016). An nsSNV is classified as “pathogenic” if >50% of the predictions are deleterious.
Compilation of PTMs
Human experimentally verified, not predicted, PTM sites were obtained from dbPTM3 (Huang et al., 2016), PhosphoSitePlus (Hornbeck et al., 2012), PhosphoELM (Dinkel et al., 2011), PHOSIDA (Gnad et al., 2011), PhosphoGrid (Stark et al., 2010), OGlycoBase (Gupta et al., 1999), UniProt (Bateman, 2019), HPRD (Keshava Prasad et al., 2009), RedoxDB (Sun et al., 2012), dbGSH (Chen et al., 2014), and dbSNO (Chen et al., 2015), and several data sets from scientific papers (Nousianen et al., 2006; Malik et al., 2009; Olsen et al., 2010; Wang et al., 2010; Matic et al., 2010; Choudhary et al., 2009; Wagner et al., 2011; Danielsen et al., 2011; Blasius et al., 2011; Phanstiel et al., 2011; Hendriks et al., 2018). Protein IDs were mapped to STRING v10 (Szklarczyk et al., 2015) dictionary, and PTM sites were mapped onto eggNOG v4.5 (Huerta-Cepas et al., 2016) set of human sequences. In order to avoid unspecific mapping, we accept PTMs for a particular source in a specific protein if all of them match the correct amino acid. PTM types are annotated for all accepted modifications, and the same PTM types in the same residues from different sources are merged as in the study by Minguez et al., 2015.
Co-occurrences between PTMs and nsSNVs
We identified nsSNVs and PTMs matching same protein positions. In addition, we identified the PTM types with enzymatic motifs in the target proteins phosphorylation, ubiquitination, acetylation, SUMOylation, methylation, N-linked glycosylation, S-glutathionylation, S-nitrosylation, O-GalNAc glycosylation, O-linked glycosylation, O-GlcNAc glycosylation, neddylation, and malonylation (Aicart-Ramos et al., 2011; Hamby and Hirst, 2008; Lee et al., 2011; 2014; Lu et al., 2014; Marino and Gladyshev, 2010; Rust and Thompson, 2011; Sigrist et al., 2002; Su et al., 2017). A consensus of window of ±5 amino acids from the PTM was agreed to consider the nsSNV matching motifs.
Enrichment of disease-associated and pathogenic nsSNVs affecting PTMs in PTM types
We extracted ordered and disordered regions in every protein of our reference set of human sequences (Huerta-Cepas et al., 2016), using the consensus predictions from MobiDB-lite v1.0 (Necci et al., 2017). PTMs-nsSNVs co-occurrences were assigned to ordered or disordered regions.
We measured whether the set of nsSNVs affecting specific PTM types are enriched in disease-causing nsSNVs. Thus, we counted disease-nsSNVs matching PTM sites, disease-nsSNVs not matching PTM sites, nondisease-nsSNVs matching PTM sites, and nondisease-nsSNVs not matching PTM sites, applied a Fisher’s exact test, and p-values adjusted by Benjamini-Hochberg false discovery rate (FDR). FDR < 0.05 was considered significant. The same analysis was performed for predicted pathogenic nsSNVs.
Extraction of associations between PTM types and diseases or phenotypes
We measured for associations between specific PTM types and specific genetic diseases. For each PTM type and genetic disease in our compilation, we counted the following: nsSNVs causing the disease affecting the PTM type, nsSNVs causing other diseases affecting the PTM type, nsSNVs causing the disease not affecting the PTM type, and nsSNVs causing other diseases not affecting the PTM type. In the case of phosphosites, we did not consider a modification affected by an nsSNV if the change produces an aspartic or glutamic acid. We applied Fisher’s exact test and p-values adjusted by FDR. FDR < 0.05 was considered significant. The same analysis was performed for nsSNVs affecting phenotypes annotated as HPO terms.
A literature review to classify the predicted PTM type and disease associations based on current knowledge
We performed a literature review to establish current knowledge for each of the 215 predicted associations between PTM types and genetic diseases. Owing to the large number of the possible combinations between PTM types and genetic diseases (>190K), it was not possible to calculate false negatives. The searches were performed with the 2 terms (PTM type and disease) in PubMed and Google considering derived and related terms if appropriate. The associations were classified as i) known, if at least a scientific article described the implication of the loss of the PTM type in the disease; ii) having partial evidence; or iii) novel, if no information was found linking both terms. Cases of partial evidence include i) reports of one variant case, where a single nsSNV, if more than 2 are described, is reported to prevent the modification of the residue; ii) associations of the PTM type with a related or similar disease; and iii) regulatory evidence, where at least one scientific publication reports that the disease is produced by a deregulation involving the PTM type within the regulatory neighborhood of the proteins with the deleterious genomic variations. PubMed IDs are provided for all the cases with some kind of evidence; if multiple are found, we provide a selection.
Reallocation score calculation
To evaluate whether the predicted associations were due to a high density of PTMs of a type in the proteins with variants annotated with the disease, we used the reallocation score (RAS). The RAS was calculated for each predicted association as follows: 1) same number of PTMs of the selected type is randomly located in modifiable residues of the PTM type, maintaining their ratio of ordered and disordered regions; 2) new co-occurrences of PTMs and nsSNVs associated with the disease are extracted; 3) same Fisher’s exact test described earlier is performed and p-values adjusted; 4) steps 1, 2, and 3 are performed 100 times; 5) the RAS is assigned to the association as the number of times the association is found significant. The comparisons between RAS distributions were performed by means of Wilcoxon rank sum tests.
The same methodology was applied to calculate RAS for phenotype-PTM type associations.
Classification of diseases in families
We used Orphanet disease families to classify the 150 disorders that had a significant association with at least one PTM type. OMIM diseases that did not have a correspondence with Orphanet diseases were manually mapped to the families.
Jaccard index calculation
We merged the associations of PTM types and diseases into supra association between PTM types and disease families. We calculated the Jaccard index (Bass et al., 2013) of every pair of the 18 PTM types with at least a significant association. We built a network with only PTM types as nodes and edges annotated with their Jaccard index, which was uploaded to Cytoscape (Shannon et al., 2003) and Prefuse Force Directed layout applied on the Jaccard index. We uploaded into Cytoscape the network between PTM types and disease families maintaining the conformation of PTM types produced by their similarity. Clusters of PTM types were created according to their sharing disease families.
Enrichment analysis to extract associations between the removal of cross talking PTM types and genetic diseases
We compiled i) serines and threonines modified with any type of O-linked glycosylation and a phosphorylation (OG-Ph) and ii) lysines modified by at least two PTM types (acetylation, methylation, SUMOylation, or ubiquitination). For every set of sites grouped by PTM cross talk types, we counted the following: nsSNVs causing the disease affecting the cross talk type, nsSNVs causing other diseases affecting the cross talk type, nsSNVs causing the disease not affecting the cross talk type, and nsSNVs causing other diseases not affecting the cross talk type. We applied Fisher’s exact test and p-values adjusted by FDR. FDR < 0.05 was considered significant.
Pathogenicity comparison of PTMs of a type in proteins causing the disease but not affected by variants and PTMs of the same type in other proteins
For each of the predicted associations between PTM types and genetic diseases, we extracted the PTMs of the selected type that are not affected by the nsSNVs causing the disease. For them, we randomly produced 5 nonsynonymous amino acid in silico changes (mimicking the effect of nsSNVs), measured their pathogenicity using Polyphen2 (Adzhubei et al., 2013), and calculated their mean. The distribution of these means was compared with a reference distribution composed of the same values for PTMs of the same type in proteins not associated with any disease where the PTM type is predicted to be involved. The comparison was made using Wilcoxon rank-sum tests and p-values adjusted by FDR. FDR<0.05 was taken as significant.
rps calculation
For a PTM site with suspicion to be associated with a disease, an rps can be calculated as follows: 1) produce 5 random nonsynonymous amino acid in silico changes; 2) obtain a Polyphen2 score for them; 3) calculate the mean; and 4) calculate the percentile of the mean in the reference distribution (see the previous section). Thus, the rps ranges from 0 to 100. An rps >95 was considered significant. Variant associations with cancer were extracted from the BioMuta database v4 (Dingerdissen et al., 2017).
Quantification and statistical analysis
We applied a Fisher's exact test in the results section "mapping PTMs and genomic variants positions to extract modifications prevented by nsSNVs" to perform the enrichment of pathogenic and disease-causing variants affecting PTM types. p-Values were adjusted by FDR, and FDR<0.05 was taken as significant. Tables S8 and S9 have all the details on this analysis. We applied Fisher's exact test in the results section "a global network of associations between PTM types and inherited diseases" to extract pairwise associations between PTM types and genetic diseases. p-Values were adjusted by FDR, and FDR<0.05 was taken as significant. Tables S10 and S11 have all the details on this analysis. In the same section, we used the Wilcoxon rank-sum test to compare our reallocation score between novel, with partial evidence and known associations, p-values are within the text and Figure 3C. We also applied the Wilcoxon rank-sum test to compare the number of nsSNVs and PTMs involved in the associations classified as novel, with partial evidence and known, p-values are within the text and Figure 3C. In the results section "associations between the removal of PTM types, disease families, and phenotypic features" we applied a Fisher's exact test to extract associations between phenotypes and genetic diseases. p-Values were adjusted by FDR, and FDR<0.05 were taken as significant. Tables S13 and S14 have all the details on this analysis. Again, Fisher's exact test with p-value adjustment by FDR was applied in the section "associations between the removal of cross talking PTM types and genetic diseases" to get associations between pairs of PTM types modifying the same site and genetic diseases. FDR<0.05 was taken as significant; details can be found in Table S15. In the results section "prediction of potential disease-associated mutation sites", we compared the relative pathogenicity score of candidate PTMs and the reference distribution using a Wilcoxon rank-sum test. p-Values were corrected by FDR, and FDR<0.05 was taken as significant; details are in Table 1.
Acknowledgments
This work was supported by the Instituto de Salud Carlos III (ISCIII)-Fondos FEDER by means of the Miguel Servet Program (CP16/00116) and the PI18/00579 project and by the Ramón Areces Foundation (CIVP18A3862). PM is supported by the Miguel Servet Program from the ISCIII (CP16/00116). PV was supported by ISCIII (CP16/00116) and the Ramón Areces Foundation (CIVP18A3862). We wish to thank members of the Bioinformatics Unit and the Genetics Department of the IIS-FJD for their comments and critical review of the work. We also thank IIS-FJD and ISCIII for their support and the RAREGenomics network for providing a forum of discussion. We also thank Oliver Shaw (IIS-FJD) for editorial assistance.
Author contributions
PV collected the data, designed and performed the analyses, interpreted the data, and reviewed the manuscript. PM conceived of the project, designed and performed analyses, interpreted the data, and wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102917.
Supplemental information
Together with p-values and FDR, reallocation scores (RAS) are also shown. Next to last columns holds the category related to the previous knowledge of the association, three classes: known previous evidence divided into one variant case study (OVCS), evidence in related diseases (ERD), and regulatory evidence (RE), and novel. The last column shows the PubMed IDs of the scientific papers supporting the classification.
For every predicted association between PTM types and genetic diseases, we show the p-values and FDRs for the tests comparing pathogenicity predictions of in silico nsSNVs in PTMs of the same type but not affected by a disease-related nsSNV and pathogenicity predictions of in silico nsSNVs affecting PTMs of the same type not predicted to be involved in diseases. We add the number of PTM sites predicted to be involved in the disease and those that are not, based on relative pathogenicity scores (rps).
Grouped by PTM type and genetic disease associated, we show the PTM sites (protein and position), the Polyphen2 score as the mean of the scores of 5 random in silico nsSNVs, and its relative pathogenicity score (rps), which represents the percentile within the reference distribution of Polyphen2 scores built by 5 random nsSNVs in same type of PTM types outside the proteins involved in the disease. Last column shows entries, separated by semicolon, of the variants in the database BioMuta that collects nsSNVs associated with cancer.
References
- Adachi T., Weisbrod R.M., Pimentel D.R., Ying J., Sharov V.S., Schöneich C., Cohen R.A. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- Adzhubei I., Jordan D.M., Sunyaev S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013;7:Unit7.20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicart-Ramos C., Valero R.A., Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta. 2011;1808:2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- Almedawar S., Colomina N., Bermúdez-López M., Pociño-Merino I., Torres-Rosell J. A SUMO-dependent step during establishment of sister chromatid cohesion. Curr. Biol. 2012;22:1576–1581. doi: 10.1016/j.cub.2012.06.046. [DOI] [PubMed] [Google Scholar]
- Barber K.W., Rinehart J. The ABCs of PTMs. Nat. Chem. Biol. 2018;14:188–192. doi: 10.1038/nchembio.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass J.I.F., Diallo A., Nelson J., Soto J.M., Myers C.L., Walhout A.J.M. Using networks to measure similarity between genes: association index selection. Nat. Methods. 2013;10:1169–1176. doi: 10.1038/nmeth.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P., Albanèse V., Kenner L.R., Swaney D.L., Burlingame A., Villén J., Lim W.A., Fraser J.S., Frydman J., Krogan N.J. Systematic functional prioritization of protein posttranslational modifications. Cell. 2012;150:413–425. doi: 10.1016/j.cell.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P., Bork P., Krogan N.J., van Noort V. Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 2013;9:714. doi: 10.1002/msb.201304521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius M., Forment J.V., Thakkar N., Wagner S.A., Choudhary C., Jackson S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011;12:R78. doi: 10.1186/gb-2011-12-8-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosnan J.T., Jacobs R.L., Stead L.M., Brosnan M.E. Methylation demand: a key determinant of homocysteine metabolism. Acta Biochim. Pol. 2004;51:405–413. [PubMed] [Google Scholar]
- Buuh Z.Y., Lyu Z., Wang R.E. Interrogating the roles of post-translational modifications of non-histone proteins. J. Med. Chem. 2018;61:3239–3252. doi: 10.1021/acs.jmedchem.6b01817. [DOI] [PubMed] [Google Scholar]
- Chen Y.-J., Lu C.-T., Lee T.-Y., Chen Y.-J. dbGSH: a database of S-glutathionylation. Bioinformatics. 2014;30:2386–2388. doi: 10.1093/bioinformatics/btu301. [DOI] [PubMed] [Google Scholar]
- Chen Y.-J., Lu C.-T., Su M.-G., Huang K.-Y., Ching W.-C., Yang H.-H., Liao Y.-C., Chen Y.-J., Lee T.-Y. dbSNO 2.0: a resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation. Nucleic Acids Res. 2015;43:D503–D511. doi: 10.1093/nar/gku1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitiprolu M., Jagow C., Tremblay V., Bondy-Chorney E., Paris G., Savard A., Palidwor G., Barry F.A., Zinman L., Keith J. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 2018;9:2794. doi: 10.1038/s41467-018-05273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y., Chan A.P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Collins R., Brand H., Karczewski K., Zhao X., Alföldi J., Francioli L., Khera A., Lowther C., Gauthier L., Wang H. An open resource of structural variation for medical and population genetics. bioRxiv. 2019 doi: 10.1101/578674. [DOI] [Google Scholar]
- Danielsen J.M., Sylvestersen K.B., Bekker-Jensen S., Szklarczyk D., Poulsen J.W., Horn H., Jensen L.J., Mailand N., Nielsen M.L. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.003590. M110.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharssi S., Wong-Rieger D., Harold M., Terry S. Review of 11 national policies for rare diseases in the context of key patient needs. Orphanet J. Rare Dis. 2017;12:63. doi: 10.1186/s13023-017-0618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingerdissen H.M., Torcivia-Rodriguez J., Hu Y., Chang T.C., Mazumder R., Kahsay R. BioMuta and BioXpress: mutation and expression knowledgebases for cancer biomarker discovery. Nucleic Acids Res. 2017;46:D1128–D1136. doi: 10.1093/nar/gkx907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel H., Chica C., Via A., Gould C.M., Jensen L.J., Gibson T.J., Diella F. Phospho.ELM: a database of phosphorylation sites--update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudman N.P., Guo X.W., Gordon R.B., Dawson P.A., Wilcken D.E. Human homocysteine catabolism: three major pathways and their relevance to development of arterial occlusive disease. J. Nutr. 1996;126:1295S–1300S. doi: 10.1093/jn/126.suppl_4.1295S. [DOI] [PubMed] [Google Scholar]
- Era S., Abe T., Arakawa H., Kobayashi S., Szakal B., Yoshikawa Y., Motegi A., Takeda S., Branzei D. The SUMO protease SENP1 is required for cohesion maintenance and mitotic arrest following spindle poison treatment. Biochem. Biophys. Res. Commun. 2012;426:310–316. doi: 10.1016/j.bbrc.2012.08.066. [DOI] [PubMed] [Google Scholar]
- Gnad F., Gunawardena J., Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011;39:D253–D260. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves E., Fragoulis A., Garcia-Alonso L., Cramer T., Saez-Rodriguez J., Beltrao P. Widespread post-transcriptional Attenuation of genomic copy-number variation in cancer. Cell Syst. 2017;5:386–398.e4. doi: 10.1016/j.cels.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R., Birch H., Rapacki K., Brunak S., Hansen J.E. O-GLYCBASE version 4.0: a revised database of O-glycosylated proteins. Nucleic Acids Res. 1999;27:370–372. doi: 10.1093/nar/27.1.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby S.E., Hirst J.D. Prediction of glycosylation sites using random forests. BMC Bioinformatics. 2008;9:500. doi: 10.1186/1471-2105-9-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A., Scott A.F., Amberger J., Bocchini C., Valle D., McKusick V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002;30:52–55. doi: 10.1093/nar/30.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks I.A., Lyon D., Su D., Skotte N.H., Daniel J.A., Jensen L.J., Nielsen M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018;9:2456. doi: 10.1038/s41467-018-04957-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holehouse A.S., Naegle K.M. Reproducible analysis of post-translational modifications in proteomes—Application to human mutations. PLoS One. 2015;10:e0144692. doi: 10.1371/journal.pone.0144692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.-Y., Su M.-G., Kao H.-J., Hsieh Y.-C., Jhong J.-H., Cheng K.-H., Huang H.-D., Lee T.-Y. dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 2016;44:D435–D446. doi: 10.1093/nar/gkv1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.Y., Lee T.Y., Kao H.J., Ma C.T., Lee C.C., Lin T.H., Chang W.C., Huang H.D. DbPTM in 2019: exploring disease association and cross-Talk of post-Translational modifications. Nucleic Acids Res. 2019;47:D298–D308. doi: 10.1093/nar/gky1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J., Szklarczyk D., Forslund K., Cook H., Heller D., Walter M.C., Rattei T., Mende D.R., Sunagawa S., Kuhn M. Eggnog 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Ikenoue T., Inoki K., Zhao B., Guan K.-L.L. PTEN acetylation modulates its interaction with PDZ domain. Cancer Res. 2008;68:6908–6912. doi: 10.1158/0008-5472.CAN-08-1107. [DOI] [PubMed] [Google Scholar]
- Jaeken J. Congenital disorders of glycosylation. In: Balu N., Hoffmann G.F.F., Leonard J.V., Clarke J.T.R., editors. Physician’s Guide to the Treatment and Follow-Up of Metabolic Diseases. Springer Berlin Heidelberg; 2006. pp. 217–220. [Google Scholar]
- Jiang Y.H., Beaudet A.L. Human disorders of ubiquitination and proteasomal degradation. Curr. Opin. Pediatr. 2004;16:419–426. doi: 10.1097/01.mop.0000133634.79661.cd. [DOI] [PubMed] [Google Scholar]
- Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A. Human protein reference database - 2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Kang C., Min B., Yi G.S. Detection and analysis of disease-associated single nucleotide polymorphism influencing post-translational modification. BMC Med. Genomics. 2015;8:S7. doi: 10.1186/1755-8794-8-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch L. Exploring human genomic diversity with gnomAD. Nat. Rev. Genet. 2020;21:448. doi: 10.1038/s41576-020-0255-7. [DOI] [PubMed] [Google Scholar]
- Köhler S., Vasilevsky N.A., Engelstad M., Foster E., McMurry J., Aymé S., Baynam G., Bello S.M., Boerkoel C.F., Boycott K.M. The human phenotype ontology in 2017. Nucleic Acids Res. 2017;45:D865–D876. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S., Carmody L., Vasilevsky N., Jacobsen J.O.B., Danis D., Gourdine J.-P., Gargano M., Harris N.L., Matentzoglu N., McMurry J.A. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019;47:D1018–D1027. doi: 10.1093/nar/gky1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krassowski M., Paczkowska M., Cullion K., Huang T., Dzneladze I., Ouellette B.F.F., Yamada J.T., Fradet-Turcotte A., Reimand J. ActiveDriverDB: human disease mutations and genome variation in post-translational modification sites of proteins. Nucleic Acids Res. 2018;46:D901–D910. doi: 10.1093/nar/gkx973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- De La Fuente L., Arzalluz-Luque Á., Tardáguila M., Del Risco H., Martí C., Tarazona S., Salguero P., Scott R., Lerma A., Alastrue-Agudo A. TappAS: a comprehensive computational framework for the analysis of the functional impact of differential splicing. Genome Biol. 2020;21:119. doi: 10.1186/s13059-020-02028-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M.R., Trelle M.B., Thingholm T.E., Jensen O.N. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Biotechniques. 2006;40:790–798. doi: 10.2144/000112201. [DOI] [PubMed] [Google Scholar]
- Lee T.Y., Chen Y.J., Lu T.C., Huang H.D., Chen Y.J. Snosite: exploiting maximal dependence decomposition to identify cysteine S-Nitrosylation with substrate site specificity. PLoS One. 2011;6:e21849. doi: 10.1371/journal.pone.0021849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.Y., Chang C.W., Lu C.T., Cheng T.H., Chang T.H. Identification and characterization of lysine-methylated sites on histones and non-histone proteins. Comput. Biol. Chem. 2014;50:11–18. doi: 10.1016/j.compbiolchem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Lock J.T., Sinkins W.G., Schilling W.P. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am. J. Physiol. 2011;300:H493. doi: 10.1152/ajpheart.01073.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.-T., Huang K.-Y., Su M.-G., Lee T.-Y., Bretaña N.A., Chang W.-C., Chen Y.-J.Y.-J., Chen Y.-J.Y.-J., Huang H.-D. dbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2013;41:D295–D305. doi: 10.1093/nar/gks1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.T., Lee T.Y., Chen Y.J., Chen Y.J. An intelligent system for identifying acetylated lysine on histones and nonhistone proteins. Biomed. Res. Int. 2014;2014:528650. doi: 10.1155/2014/528650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Scott I., Webster B.R., Sack M.N. The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ. Res. 2009;105:830–841. doi: 10.1161/CIRCRESAHA.109.204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R., Lenobel R., Santamaria A., Ries A., Nigg E.A., Körner R. Quantitative analysis of the human spindle phosphoproteome at distinct mitotic stages. J. Proteome Res. 2009;10:4553–4563. doi: 10.1021/pr9003773. [DOI] [PubMed] [Google Scholar]
- Marino S.M., Gladyshev V.N. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 2010;395:844–859. doi: 10.1016/j.jmb.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic I., Schimmel J., Hendriks I.A., van Santen M.A., van de Rijke F., van Dam H., Gnad F., Mann M., Vertegaal A.C. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol. Cell. 2010;39:641–652. doi: 10.1016/j.molcel.2010.07.026. [DOI] [PubMed] [Google Scholar]
- Mattson M.P. Acetylation unleashes protein demons of dementia. Neuron. 2010;67:900–902. doi: 10.1016/j.neuron.2010.09.010. [DOI] [PubMed] [Google Scholar]
- McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez J.D., Xie J., Aguilar-Hernández M., Méndez-Valenzuela V. Molecular susceptibility to glycation and its implication in diabetes mellitus and related diseases. Mol. Cell. Biochem. 2010;344:185–193. doi: 10.1007/s11010-010-0541-3. [DOI] [PubMed] [Google Scholar]
- Meng Z., Jia L.F., Gan Y.H. PTEN activation through K163 acetylation by inhibiting HDAC6 contributes to tumour inhibition. Oncogene. 2016;35:2333–2344. doi: 10.1038/onc.2015.293. [DOI] [PubMed] [Google Scholar]
- Minguez P., Parca L., Diella F., Mende D., Kumar R., Helmer-Citterich M., Gavin A., van Noort V., Bork P. Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 2012;8:599. doi: 10.1038/msb.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguez P., Letunic I., Parca L., Garcia-Alonso L., Dopazo J., Huerta-Cepas J., Bork P. PTMcode v2: a resource for functional associations of post-translational modifications within and between proteins. Nucleic Acids Res. 2015;43:D494–D502. doi: 10.1093/nar/gku1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naegle K.M., Gymrek M., Joughin B.A., Wagner J.P., Welsch R.E., Yaffe M.B., Lauffenburger D.A., White F.M. PTMScout, a Web resource for analysis of high throughput post-translational proteomics studies. Mol. Cell. Proteomics. 2010;9:2558–2570. doi: 10.1074/mcp.M110.001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan S., Bader G.D., Reimand J. Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Med. 2016;8:55. doi: 10.1186/s13073-016-0311-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita T., Weinert B.T., Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019;20:156–174. doi: 10.1038/s41580-018-0081-3. [DOI] [PubMed] [Google Scholar]
- Necci M., Piovesan D., Dosztanyi Z., Tosatto S.C.E. MobiDB-lite: Fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics. 2017;33(9):1402–1404. doi: 10.1093/bioinformatics/btx015. [DOI] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoh G.A., Facundo H.T., Zafir A., Jones S.P. O-GlcNAc signaling in the cardiovascular system. Circ. Res. 2010;107:171–185. doi: 10.1161/CIRCRESAHA.110.224675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousiainen M., Silljé H.H., Sauer G., Nigg E.A., Körner R. Phosphoproteome analysis of the human mitotic spindle. Proc. Natl. Acad. Sci. 2006;103(14):5391–5396. doi: 10.1073/pnas.0507066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J.V., Vermeulen M., Santamaria A., Kumar C., Miller M.L., Jensen L.J., Gnad F., Cox J., Jensen T.S., Nigg E.A. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascovici D., Wu J.X., McKay M.J., Joseph C., Noor Z., Kamath K., Wu Y., Ranganathan S., Gupta V., Mirzaei M. Clinically relevant post-translational modification analyses—maturing workflows and bioinformatics tools. Int. J. Mol. Sci. 2019;20:16. doi: 10.3390/ijms20010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavan S., Rommel K., Mateo Marquina M.E., Höhn S., Lanneau V., Rath A. Clinical practice guidelines for rare diseases: the Orphanet database. PLoS One. 2017;12:e0170365. doi: 10.1371/journal.pone.0170365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D., Li H., Hu B., Zhang H., Chen L., Lin S., Zuo Z., Xue Y., Ren J., Xie Y. PTMsnp: a web server for the identification of driver mutations that affect protein post-translational modification. Front. Cell Dev. Biol. 2020;8:1330. doi: 10.3389/fcell.2020.593661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M., Barone E., Di Domenico F., Butterfield D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta. 2016;1862:1871–1882. doi: 10.1016/j.bbadis.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Phanstiel D.H., Brumbaugh J., Wenger C.D., Tian S., Probasco M.D., Bailey D.J., Swaney D.L., Tervo M.A., Bolin J.M., Ruotti V. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods. 2011;8:821–827. doi: 10.1038/nmeth.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero J., Bravo A., Queralt-Rosinach N., Gutierrez-Sacristan A., Deu-Pons J., Centeno E., Garcia-Garcia J., Sanz F., Furlong L.I. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45:D833–D839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan D., Hatos A., Minervini G., Quaglia F., Monzon A.M., Tosatto S.C.E. Assessing predictors for new post translational modification sites: a case study on hydroxylation. PLoS Comput. Biol. 2020;16:e1007967. doi: 10.1371/journal.pcbi.1007967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran S., Lippens G., Steen H., Gunawardena J. Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012;4:565–583. doi: 10.1002/wsbm.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P., Baenziger P.H., Kann M.G., Mort M.E., Hahn M.W., Mooney S.D. Gain and loss of phosphorylation sites in human cancer. Bioinformatics. 2008;24:i241–i247. doi: 10.1093/bioinformatics/btn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Wagih O., Bader G.D. The mutational landscape of phosphorylation signaling in cancer. Sci. Rep. 2013;3:2651. doi: 10.1038/srep02651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Wagih O., Bader G.D. Evolutionary constraint and disease associations of post-translational modification sites in human genomes. PLoS Genet. 2015;11:e1004919. doi: 10.1371/journal.pgen.1004919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Coldren C., Karunamurthy A., Kip N.S., Klee E.W., Lincoln S.E., Leon A., Pullambhatla M., Temple-Smolkin R.L., Voelkerding K.V. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: a joint recommendation of the association for molecular pathology and the college of American pathologists. J. Mol. Diagn. 2018;20:4–27. doi: 10.1016/j.jmoldx.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Rust H.L., Thompson P.R. Kinase consensus sequences: a breeding ground for crosstalk. ACS Chem. Biol. 2011;6:881–892. doi: 10.1021/cb200171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I., Breitkreutz B.-J., Stark C., Su T.-C., Dahabieh M., Raithatha S., Bernhard W., Oughtred R., Dolinski K., Barreto K. The PhosphoGRID saccharomyces cerevisiae protein phosphorylation site database: version 2.0 update. Database. 2013;2013:bat026. doi: 10.1093/database/bat026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savignac M., Edir A., Simon M., Hovnanian A. Darier disease: a disease model of impaired calcium homeostasis in the skin. Biochim. Biophys. Acta. 2011;1813:1111–1117. doi: 10.1016/j.bbamcr.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Sayers E.W., Barrett T., Benson D.A., Bolton E., Bryant S.H., Canese K., Chetvernin V., Church D.M., DiCuccio M., Federhen S. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2011;39:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz J.M., Cooper D.N., Schuelke M., Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nat. Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab H.A., Gough J., Mort M., Cooper D.N., Day I.N.M., Gaunt T.R. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genomics. 2014;8:11. doi: 10.1186/1479-7364-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C.J.A., Cerutti L., Hulo N., Gattiker A., Falquet L., Pagni M., Bairoch A., Bucher P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 2002;3:265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- Simandi Z., Pajer K., Karolyi K., Sieler T., Jiang L.L., Kolostyak Z., Sari Z., Fekecs Z., Pap A., Patsalos A. Arginine methyltransferase PRMT8 provides cellular stress tolerance in aging motoneurons. J. Neurosci. 2018;38:7683–7700. doi: 10.1523/JNEUROSCI.3389-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C., Su T.C., Breitkreutz A., Lourenco P., Dahabieh M., Breitkreutz B.J., Tyers M., Sadowski I. PhosphoGRID: a database of experimentally verified in vivo protein phosphorylation sites from the budding yeast saccharomyces cerevisiae. Database. 2010;2010:bap026. doi: 10.1093/database/bap026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M.-G., Weng J.T.-Y., Hsu J.B.-K., Huang K.-Y., Chi Y.-H., Lee T.-Y. Investigation and identification of functional post-translational modification sites associated with drug binding and protein-protein interactions. BMC Syst. Biol. 2017;11:132. doi: 10.1186/s12918-017-0506-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M.-a., Wang Y., Cheng H., Zhang Q., Ge W., Guo D. RedoxDB--a curated database for experimentally verified protein oxidative modification. Bioinformatics. 2012;28:2551–2552. doi: 10.1093/bioinformatics/bts468. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O., Reimand J., Bader G.D. MIMP: predicting the impact of mutations on kinase-substrate phosphorylation. Nat. Methods. 2015;12:531–533. doi: 10.1038/nmeth.3396. [DOI] [PubMed] [Google Scholar]
- Wagih O., Galardini M., Busby B.P., Memon D., Typas A., Beltrao P. A resource of variant effect predictions of single nucleotide variants in model organisms. Mol. Syst. Biol. 2018;14:e8430. doi: 10.15252/msb.20188430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S.A., Beli P., Weinert B.T., Nielsen M.L., Cox J., Mann M., Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.013284. M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Udeshi N.D., Slawson C., Compton P.D., Sakabe K., Cheung W.D., Shabanowitz J., Hunt D.F., Hart G.W. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010;3:ra2. doi: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland T., Attwood P.V. Alterations in reversible protein histidine phosphorylation as intracellular signals in cardiovascular disease. Front. Pharmacol. 2015;6:173. doi: 10.3389/fphar.2015.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodsmith J., Kamburov A., Stelzl U. Dual coordination of post translational modifications in human protein networks. PLoS Comput. Biol. 2013;9:e1002933. doi: 10.1371/journal.pcbi.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B., Lu S., Gerton J.L. Roberts syndrome. Rare Dis. 2014;2:e27743. doi: 10.4161/rdis.27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Wang Y., Lin S., Deng W., Peng D., Cui Q., Xue Y. PTMD: a database of human disease-associated post-translational modifications. Genomics Proteomics Bioinformatics. 2018;16:244–251. doi: 10.1016/j.gpb.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H., Peng J., Shang X. Predicting disease-related phenotypes using an integrated phenotype similarity measurement based on HPO. BMC Syst. Biol. 2019;13:34. doi: 10.1186/s12918-019-0697-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Peng X., Ying P., Tian J., Li J., Ke J., Zhu Y., Gong Y., Zou D., Yang N. AWESOME: a database of SNPs that affect protein post-translational modifications. Nucleic Acids Res. 2019;47:D874–D880. doi: 10.1093/nar/gky821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Together with p-values and FDR, reallocation scores (RAS) are also shown. Next to last columns holds the category related to the previous knowledge of the association, three classes: known previous evidence divided into one variant case study (OVCS), evidence in related diseases (ERD), and regulatory evidence (RE), and novel. The last column shows the PubMed IDs of the scientific papers supporting the classification.
For every predicted association between PTM types and genetic diseases, we show the p-values and FDRs for the tests comparing pathogenicity predictions of in silico nsSNVs in PTMs of the same type but not affected by a disease-related nsSNV and pathogenicity predictions of in silico nsSNVs affecting PTMs of the same type not predicted to be involved in diseases. We add the number of PTM sites predicted to be involved in the disease and those that are not, based on relative pathogenicity scores (rps).
Grouped by PTM type and genetic disease associated, we show the PTM sites (protein and position), the Polyphen2 score as the mean of the scores of 5 random in silico nsSNVs, and its relative pathogenicity score (rps), which represents the percentile within the reference distribution of Polyphen2 scores built by 5 random nsSNVs in same type of PTM types outside the proteins involved in the disease. Last column shows entries, separated by semicolon, of the variants in the database BioMuta that collects nsSNVs associated with cancer.
Data Availability Statement
-
•
The data sets supporting the conclusions of this article are included within the article and its additional files. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the data sets are listed in the key resources table.
-
•
This paper does not report original code.