

Abstract
The continuing rise of antibiotic resistance, particularly among Gram-negative pathogens, threatens to undermine many aspects of modern medical practice. To address this threat, novel antibiotics that utilize unexploited bacterial targets are urgently needed. Over the past decade, a number of studies have highlighted the antibacterial potential of macrocyclic peptides that target Gram-negative outer membrane proteins (OMPs). Recently, it was reported that the antibacterial activities of OMP-targeting macrocyclic peptidomimetics of the antimicrobial peptide protegrin-1 are dramatically enhanced upon linking to polymyxin E nonapeptide (PMEN). In this study, we describe a convergent, chemoenzymatic route for the convenient preparation of such conjugates. Specifically, we investigated the use of both amide bond formation and azide-alkyne ligation for connecting an OMP-targeting macrocyclic peptide to a PMEN building block (obtained by enzymatic degradation of polymyxin E). The conjugates obtained via both approaches display potent antibacterial activity against a range of Gram-negative pathogens including multi-drug-resistant isolates.
Introduction
While the rate of multi- and pan-drug-resistant bacterial infections increases worldwide, the antibiotic development pipeline offers little reason for optimism.1 Most notable in this regard is the lack of effective antibiotic therapies available for the treatment of infections due to Gram-negative pathogens.2,3 To address this shortfall, antibiotics that operate via unique and unexploited mechanisms are desperately needed. While, in recent years, there has been some progress made in the development of novel treatments for Gram-positive infections, the same cannot be said for anti-Gram-negative agents with the last major advancement coming nearly four decades ago with the clinical introduction of the carbapenems.2,4,5 The development of antibiotics for Gram-negative bacteria is particularly challenging given the presence of their outer membrane (OM), which is composed of an inner phospholipid leaflet and a lipopolysaccharide (LPS)-decorated outer leaflet.6 This OM provides an effective barrier, shielding the bacterial cell from many antibiotics. Notably, the polymyxin antibiotics, typified by the clinically used polymyxin E, more commonly known as colistin 1 (Figure 1A), are cyclic lipopeptides that act specifically against Gram-negative bacteria via a mode of action involving binding to lipid A, the lipid membrane anchor of LPS.7−9 Despite colistin’s inherent (nephro)toxicity, its use has been revived in recent years due to the lack of effective alternative treatments.10,11 Interestingly, polymyxin derivatives that lack the lipophilic fatty acyl chain are devoid of antimicrobial activity but can, in some cases, potentiate the action of other antibiotics by permeabilizing the Gram-negative OM.12−14 Best studied in this regard are the cyclic nonapeptide fragments of polymyxin B and polymyxin E (PMBN and PMEN, respectively), which have also been investigated as leads for the development of antibiotic adjuvants.12,15−17
Figure 1.
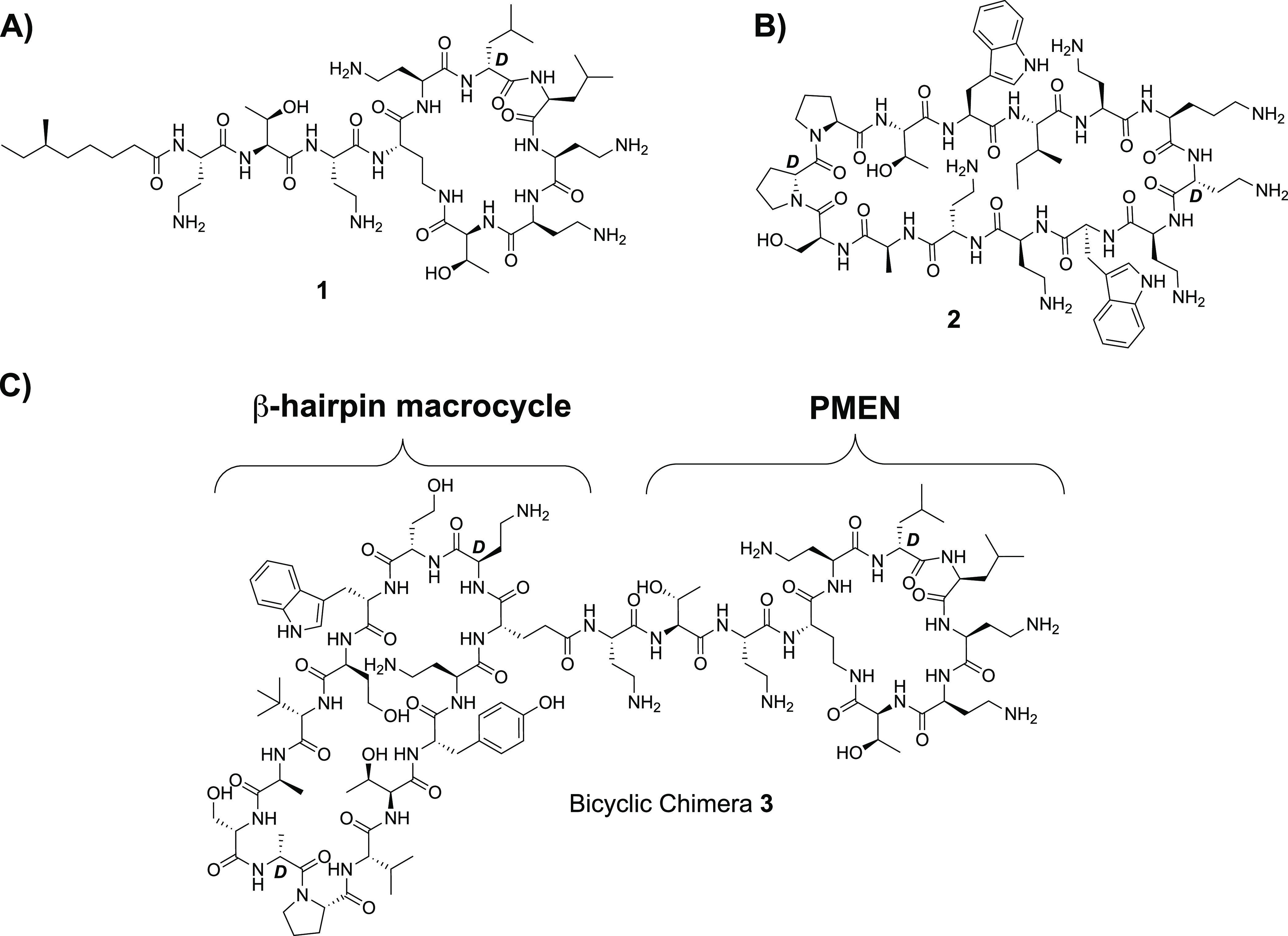
Macrocyclic peptide antibiotics targeting the Gram-negative OM. (A) Structure of clinically used colistin; (B) structure of the protegrin I β-hairpin peptidomimetic murepavadin; (C) representative structure of recently reported chimeric antibiotics consisting of a β-hairpin mimetic ligated to the nonapeptide derived from colistin.
The Gram-negative OM contains a select number of surface-exposed β-barrel outer membrane proteins (OMPs) that are essential for bacterial viability. In recent years, increasing efforts have been made to identify novel antibiotics that operate by targeting Gram-negative OMPs. In principle, such antibiotics would circumvent two of the main impediments presented by Gram-negative bacterial cells, namely, OM penetration and efflux pump-mediated export. Most notable among OMP-targeting antibiotics is the β-hairpin peptidomimetic murepavadin 2 (Figure 1B) first reported by Robinson and co-workers and subsequently taken into clinical development by Polyphor for the treatment of Pseudomonas aeruginosa infections.18 Murepavadin specifically targets the OMP LptD and, in doing so, interferes with OM biogenesis, leading to selective killing of Pseudomonas spp.18,19 In a recent study, the same group described a new series of β-hairpin peptidomimetics of the antimicrobial peptide protegrin I that demonstrated a moderate but broader Gram-negative specific antibacterial effect.20 More notable, however, was the finding that when covalently linked to the PMEN, the antibacterial activity of these macrocyclic peptides is enhanced by many orders of magnitude.20,21 Interestingly, mechanistic studies revealed that these bicyclic conjugates, including compound 3 illustrated in Figure 1C, function by targeting another OMP, namely, BamA, a key component of the β-barrel assembly machine (BAM) that plays an essential role in OMP folding in Gram-negative bacteria.22−25 Importantly, these new BamA-targeting macrocyclic peptide conjugates were found to retain their potent activity in vivo(20) and represent a promising new class of antimicrobials for further development.
The synthesis reported for the preparation of the PMEN-conjugated BamA-targeting macrocyclic peptides relies completely upon a linear SPPS approach (Scheme 1). The PMEN macrocycle is first assembled on resin after which the remaining 17 amino acids are sequentially added. Following selective allyl ester removal and cleavage from the resin, the BamA-targeting macrocycle is subsequently formed in solution followed by global deprotection to yield the desired bicyclic conjugate 3. Given our interest in macrocyclic peptide antibiotics and semisynthesis,26−29 we were drawn to explore alternative strategies for the preparation of these constructs. Specifically, rather than synthesizing the PMEN motif “from scratch”, we considered a chemoenzymatic approach involving degradation of readily available colistin followed by coupling to the BamA-targeting macrocycle. In addition, as a means of further simplifying both the synthesis of the BamA-targeting macrocycle and subsequent assembly of the bicyclic constructs, we also explored the application of the well-established azide-alkyne ligation.30,31 We here report the applicability of these alternative synthetic approaches to provide convenient access to both the original class of BamA-targeting conjugates and new triazole-linked variants with varying spacer lengths. The antibacterial activity of these constructs was evaluated against a panel of Gram-negative bacteria, including several drug-resistant isolates, confirming the activity of the parent compound as well as revealing new analogues of similar potency.
Scheme 1. Published Synthesis of Bicyclic Chimera 3.
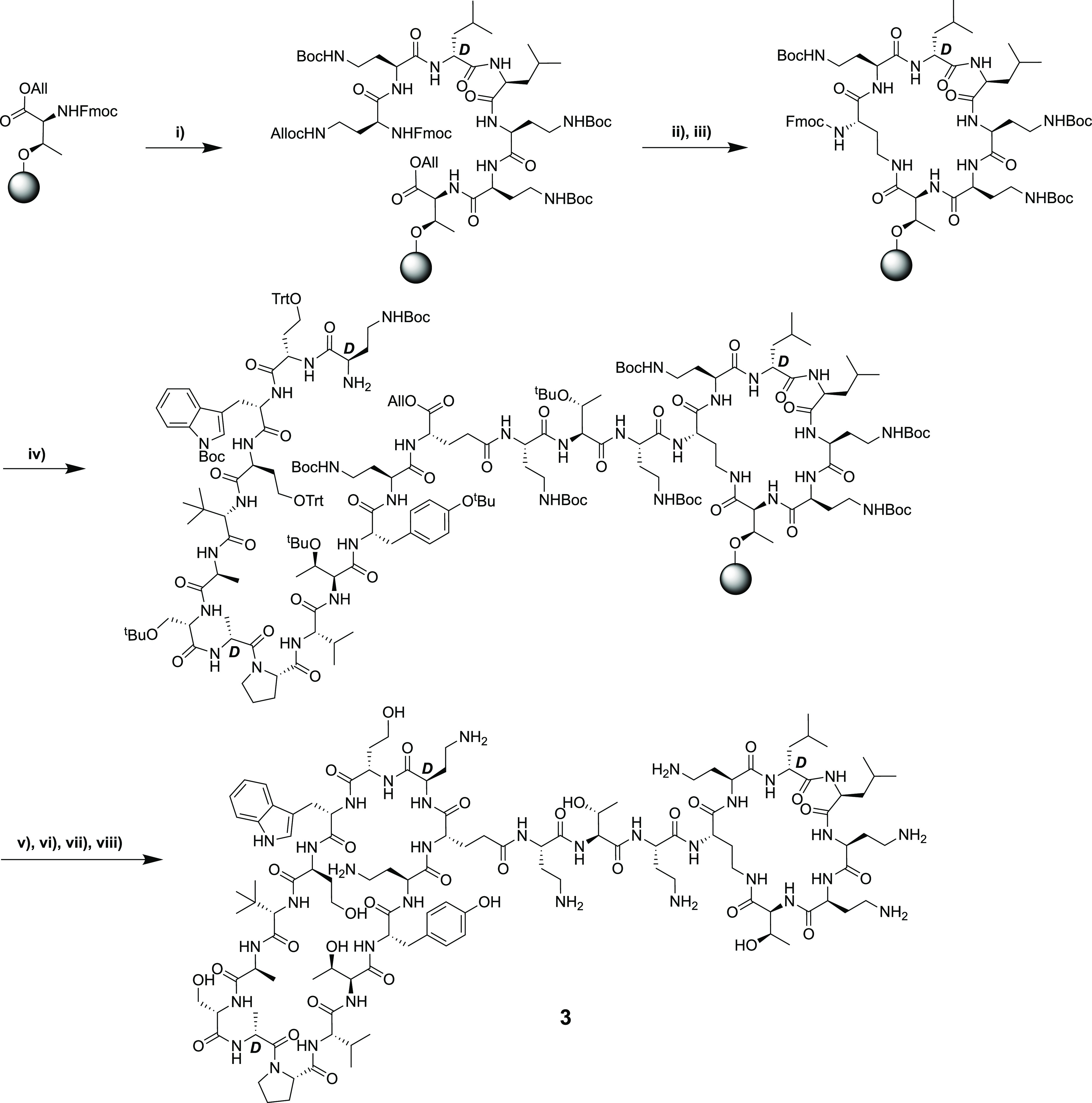
(i) Fmoc SPPS on CTC resin up to and including Fmoc-Dab (Alloc)-OH; (ii) Alloc/Allyl deprotection; (iii) ring closing on resin; (iv) Fmoc SPPS (with incorporation of Fmoc-Glu-OAllyl via side-chain carboxyl) up to and including final Fmoc-d-Dab(Boc)-OH, including removal of the final Fmoc group; (v) Allyl deprotection; (vi) mild acid cleavage from resin; (vii) solution-phase cyclization; (vii) TFA deprotection and RP-HPLC purification.
Results and Discussion
As illustrated in Scheme 1, the published synthesis of the bicyclic peptides was accomplished completely by the use of SPPS. As an alternative, we were interested in examining whether the PMEN fragment could instead be sourced by degradation of the readily available parent colistin (polymyxin E) and then coupled to the second macrocycle (Scheme 2). As a proof of concept for this approach, we selected bicyclic chimera 3, previously identified as a lead compound by the Polyphor group.20 To begin, PMEN was obtained by following a previously described protocol of enzymatic degradation of the parent polymyxin using the readily available ficin.32,33 The crude PMEN thus obtained was then selectively protected at the four Dab side chains by treating with 2-(Boc-oxyimino)-2-phenylacetonitrile (Boc-ON) in water/dioxane/NEt3 (1:1:1) to yield PMEN-Boc4 (4a).33 In parallel, the protected β-hairpin macrocyclic peptide (5) was obtained by SPPS starting from Fmoc-Dab(Boc)-OH loaded onto CTC resin. The next amino acid to be added was the orthogonally protected Fmoc-Glu(OH)-OAll, which was coupled via its free side-chain carboxylate. The remaining residues were added in a linear fashion followed by allyl ester cleavage using Pd(0). Subsequent on-resin cyclization was found to proceed cleanly after which the protected peptide macrocycle was detached from the resin using mild acid conditions to yield the protected intermediate 5. Analysis of crude 5 indicated that the desired product was the major species in good purity, and it was therefore used directly in the subsequent coupling with PMEN-Boc4 (4a). The solution-phase coupling of fragments 4a and 5 was accomplished by stirring overnight in DCM with activation using BOP/DIPEA. The following morning, LC–MS analysis indicated the clean formation of the bicyclic conjugate after which TFA deprotection and HPLC purification provided compound 3a. Using the same approach, the corresponding conjugate incorporating the PMBN macrocycle, compound 3b, was also successfully prepared by starting from PMBN-Boc44b. In addition, the 14-mer monocyclic β-hairpin peptidomimetic (6) was prepared to serve as a reference compound. Briefly, Rink amide resin was loaded with the free side chain of Fmoc-Glu(OH)-OAll followed by coupling of the next 13 amino acids (essentially following the same steps as in Scheme 2). Subsequent allyl ester removal and on-resin cyclization followed by strong acid cleavage/deprotection then provided compound 6, terminated as an amide at the glutamic acid side-chain position (Scheme 3).
Scheme 2. Convergent, Chemoenzymatic Synthesis of Bicyclic Chimera 3.
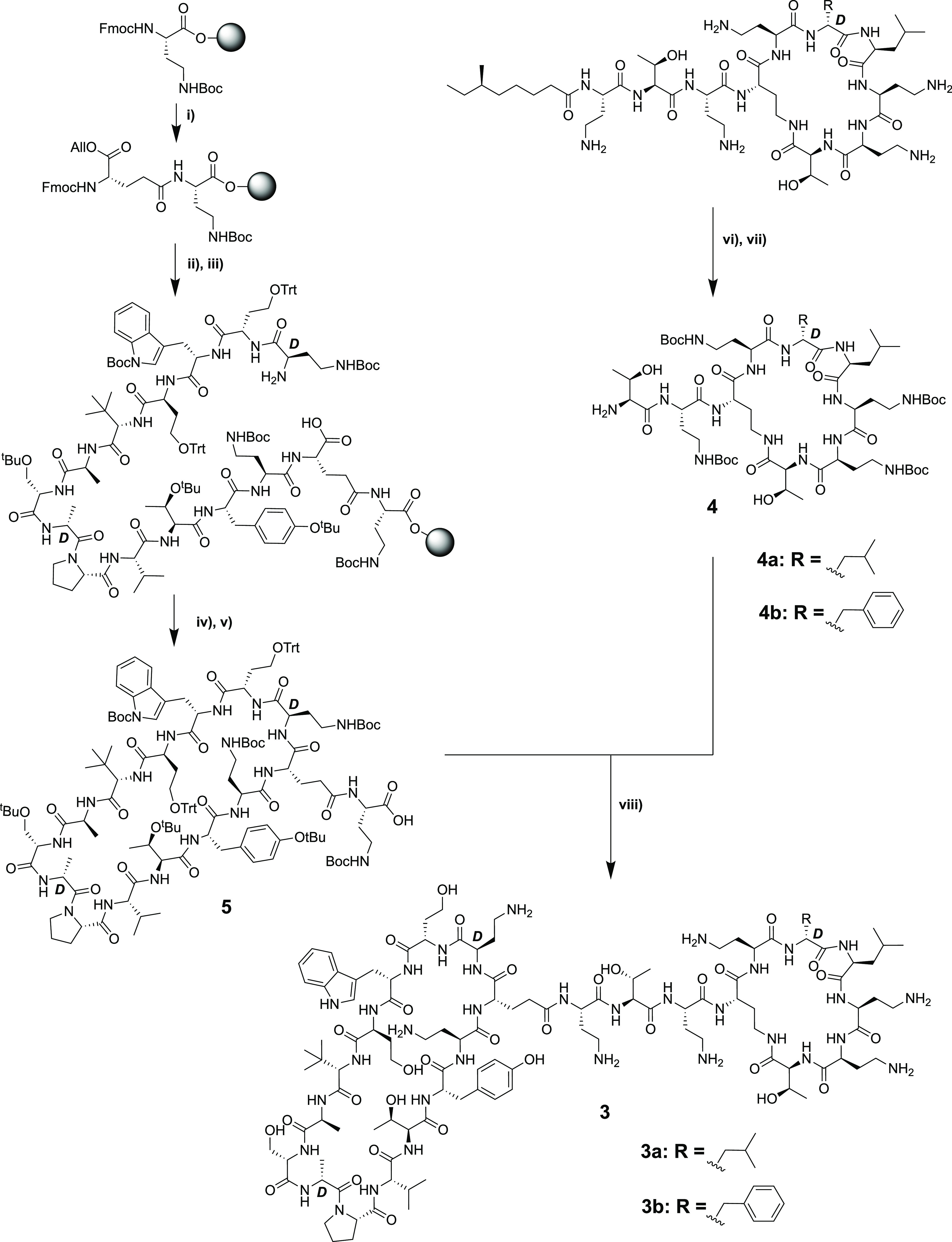
(i) Fmoc SPPS starting on CTC resin and coupling to the free side chain of Fmoc-Glu-OAllyl (BOP/DIPEA); (ii) SPPS till d-Dab(Boc) + removal of final Fmoc; (iii) Allyl deprotection with Pd; (iv) on-resin cyclization (BOP/DIPEA); (v) resin cleavage (HFIP/DCM); (vi) enzymatic digestion by ficin; (vii) site-selective bocylation by Boc-ON; (viii) solution-phase coupling using BOP/DIPEA and HPLC purification.
Scheme 3. Synthesis of the Monocyclic Peptide 6.
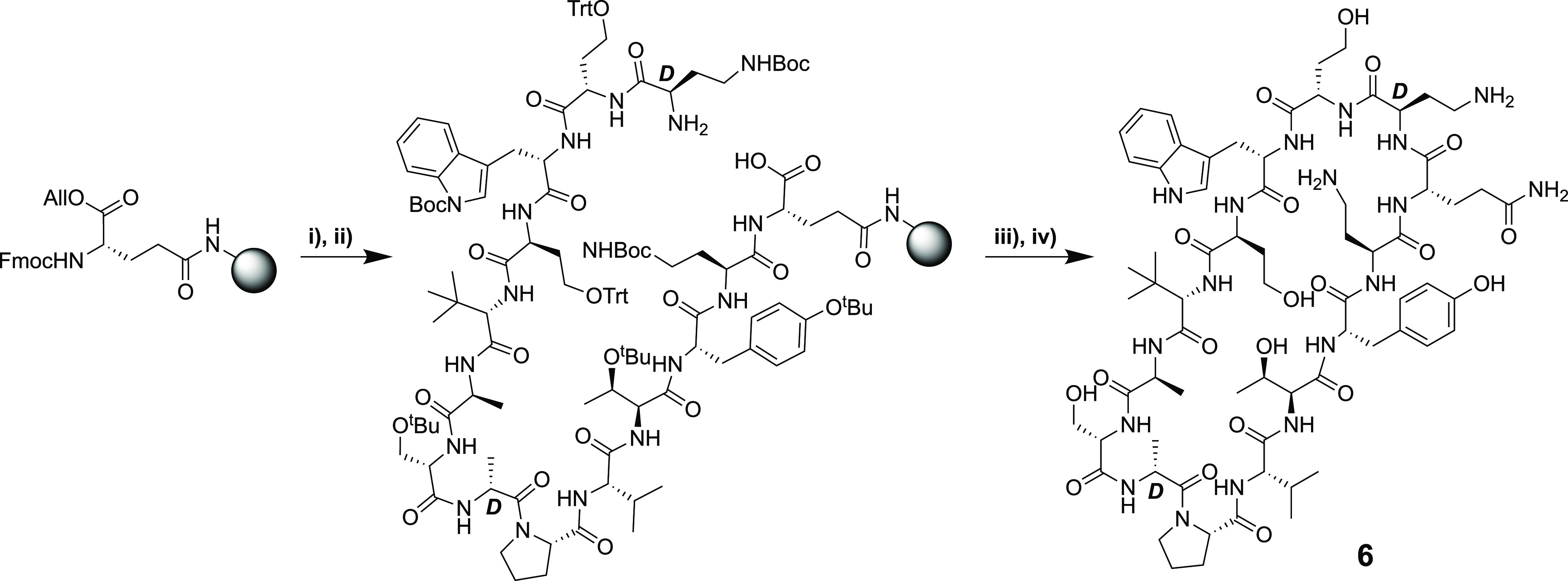
(i) Fmoc SPPS starting from Rink amide resin loaded with Fmoc-Glu-OAll via the carboxyl side chain; (ii) Allyl ester deprotection with Pd; (iii) on-resin cyclization (BOP/DIPEA); (iv) TFA deprotection + resin cleavage followed by RP-HPLC purification.
The antibacterial activity of bicyclic constructs 3a and 3b was assessed against a range of Gram-negative pathogens including several drug-resistant isolates (Table 1). For comparison, colistin, PMEN, and the monocyclic β-hairpin mimetic 6 were also tested. As reported in the literature, 3a was found to exhibit potent antibacterial activity, on par or superior to that of colistin, among the strains tested. In addition, the activity of the PMBN conjugate 3b was found to largely mirror that of the PMEN conjugate 3a. Also of note was the confirmation that the activity of bicyclic constructs was not diminished against colistin-resistant strains possessing mcr-type plasmids that encode for phosphoethanolamine transferases that modify the structure of lipid A. Both PMEN and the monocyclic β-hairpin mimetic 6 were also assessed, neither of which demonstrated appreciable antibacterial activity. Furthermore, none of the compounds tested showed any activity against a representative Gram-positive strain (MRSA USA300).
Table 1. Minimum Inhibitory Concentration Values for Analogues 3a, 3b, 6, Colistin, and PMENa.
| 3a | 3b | colistin | β-hairpin 6 | PMEN | |
|---|---|---|---|---|---|
| Acinetobacter baumannii ATCC 9955 | 0.2 | 0.2 | 0.4 | >64 | >64 |
| A. baumannii ATCC 17978 | 0.4 | 0.4 | 0.4 | >64 | >64 |
| A. baumannii NDM2 (NRZ 00687) | 0.1 | 0.2 | 0.2 | >64 | >64 |
| A. baumannii MDR | 0.2 | 0.4 | 0.4 | >64 | >64 |
| Klebsiella pneumoniae ATCC 11228 | 0.2 | 0.2 | 0.1 | >64 | >64 |
| K. pneumoniae NDM1 | 0.8 | 0.4 | 1.6 | >64 | >64 |
| P. aeruginosa ATCC 27853 | 0.8 | 1.6 | 1.6 | >64 | >64 |
| P. aeruginosa NDM1 (NRZ 08418) | 0.8 | 0.8 | 0.8 | >64 | >64 |
| P. aeruginosa NDM1 | 0.4 | 0.8 | 0.4 | >64 | >64 |
| P. aeruginosa M-120 | 1.6 | 1.6 | 0.8 | >64 | 32 |
| Escherichia coli ATCC 35218 | 0.2 | 0.1 | 0.1 | >64 | >64 |
| E. coli NDM1 | 0.1 | 0.1 | 0.1 | >64 | >64 |
| E. coli MCR1 | 0.2 | 0.4 | 3.2 | >64 | >64 |
| E. coli MCR2 | 0.2 | 0.4 | >6.4 | >64 | >64 |
| E. coli MCR3 | 0.2 | 0.4 | 3.2 | >64 | >64 |
| MRSA USA300 | >6.4 | >6.4 | >6.4 | >64 | >64 |
Minimum inhibitory concentration reported in μg/mL. All compounds were tested in triplicates on two separate days.
Upon confirming the potent anti-Gram-negative activity of conjugates 3a and 3b, we next considered other options for the preparation of similar bicyclic constructs. The use of the well-established copper-catalyzed azide-alkyne cycloaddition (“click chemistry”) was deemed particularly attractive.34 To this end, we envisioned a convenient approach whereby the monocyclic β-hairpin mimetic, modified to bear an appropriately placed alkyne moiety, could be ligated to the PMEN moiety containing a complementary azide group. The preparation of each fragment was achieved by extension of the methods used to access the building blocks employed in the synthesis of compounds 3a and 3b. The alkyne-modified peptidomimetic β-hairpin 7 was conveniently synthesized completely on resin (Scheme 4A). To begin, Rink amide resin was loaded with Fmoc-propargyl-glycine followed by addition of the orthogonally protected Fmoc-Glu(OH)-OAll via its free side-chain carboxylate. The remaining 13 amino acids were then added and, following final Fmoc removal and Pd(0)-catalyzed allyl ester removal, the macrocycle was cleanly formed on resin. Strong acid cleavage/deprotection then provided compound 7. Notably, the use of microwave-assisted SPPS enabled the convenient preparation of 7 in as little as 4 h including cyclization and cleavage/deprotection. When it came to preparing the azide-modified PMEN fragment, we elected to incorporate azido-carboxylic acids of varying lengths to also assess how the distance between the two peptide macrocycles might impact antibacterial activity. To this end, we generated azido PMEN analogues 8a–8c containing a short spacer based on azido glycine, a medium-length C5 spacer, and a longer PEG3-based spacer (Scheme 4B). With these azido-PMEN building blocks in hand, ligation with alkyne 7 was accomplished by treatment with CuSO4 and ascorbic acid to yield the expected triazole-linked conjugates 9a–9c, in yields ranging from 55 to 71% after RP-HPLC (Scheme 4C).
Scheme 4. Synthesis of Triazole-Linked Bicyclic Conjugates 9a–9c.
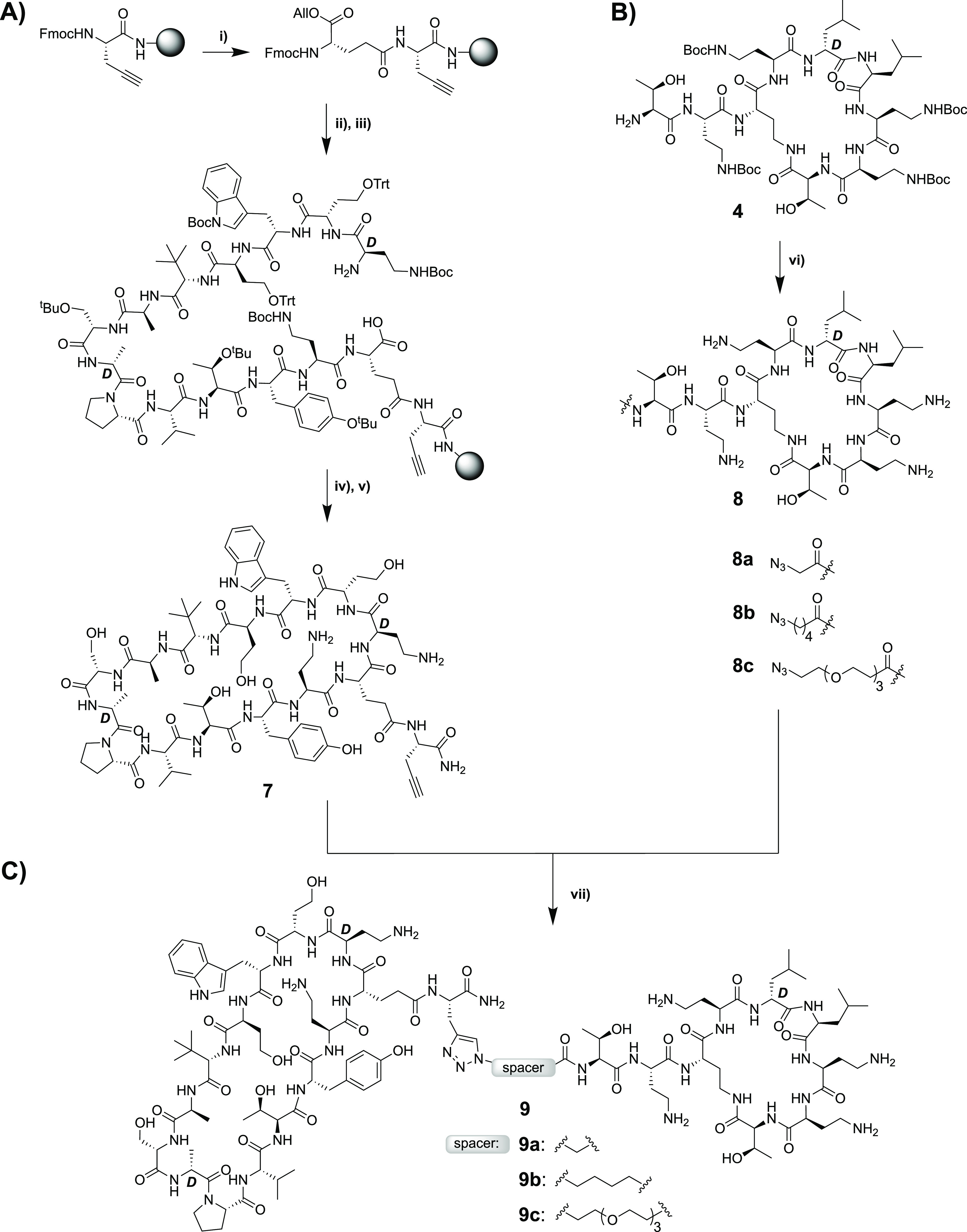
(i) Starting from Fmoc-propargyl-Gly-loaded Rink resin, Fmoc-Glu-OAll was coupled via its carboxyl side chain; (ii) Fmoc SPPS till the final Fmoc-d-Dab(Boc)-OH + removal of final Fmoc; (iii) Glu-OAll deprotection with Pd; (iv) on-resin cyclization (BOP/DIPEA); (v) TFA deprotection + resin cleavage followed by RP-HPLC purification; (vi) coupling with azido carboxylic acids (BOP/DIPEA); (vii) triazole formation with CuSO4 and sodium ascorbate followed by RP-HPLC purification.
The antibacterial activities of triazole conjugates 9a–9c were assessed against the same panel of Gram-negative bacteria, revealing a clear trend (Table 2). Conjugate 9a containing the shortest linker consistently demonstrated potent antibacterial activity with MIC values ranging from 0.25 to 2.0 μg/mL. By comparison, 9b and 9c, containing the C5 and PEG3 spacers, respectively, exhibited progressively weaker activities. These findings indicate that the proximity of the two monocyclic peptides to each other is an important factor in achieving maximal activity.
Table 2. Minimum Inhibitory Concentration Values for Analogues 9a, 9b, 9c, 12, and Colistina.
| 9a | 9b | 9c | 12 | colistin | |
|---|---|---|---|---|---|
| A. baumannii ATCC 9955 | 2 | 2 | 8 | 64 | 0.4 |
| A. baumannii ATCC 17978 | 0.5 | 1 | 4 | 64 | 0.4 |
| A. baumannii NDM2 (NRZ 00687) | 0.5 | 1 | 2 | 64 | 0.2 |
| A. baumannii MDR | 1 | 2 | 4 | 32 | 0.4 |
| K. pneumoniae ATCC 11228 | 1 | 2 | 8 | 16 | 0.1 |
| K. pneumoniae NDM1 | 2 | 4 | 16 | 32 | 1.6 |
| P. aeruginosa ATCC 27853 | 1 | 2 | 4 | 64 | 1.6 |
| P. aeruginosa NDM1 (NRZ 08418) | 2 | 4 | 8 | 64 | 0.8 |
| P. aeruginosa NDM1 | 2 | 2 | 4 | 32 | 0.4 |
| P. aeruginosa M-120 | 4 | 4 | 8 | 64 | 0.8 |
| E. coli ATCC 35218 | 0.5 | 1 | 2 | 8 | 0.1 |
| E. coli NDM1 | 0.25 | 0.5 | 2 | 2 | 0.1 |
| E. coli MCR1 | 2 | 4 | 16 | 16 | 3.2 |
| E. coli MCR2 | 2 | 4 | 16 | 32 | >6.4 |
| E. coli MCR3 | 4 | 8 | 32 | 64 | 3.2 |
| MRSA USA300 | >6.4 | >64 | >64 | >64 | >6.4 |
Minimum inhibitory concentration reported in μg/mL. All compounds were tested in triplicates on two separate days.
Based on the trend revealed by conjugates 9a–9c, we were also curious to know how bringing the two macrocycles even closer together might impact activity. To address this question, we employed the polymyxin E heptapeptide (PMEH) building block 10, a more truncated fragment of colistin lacking the exocyclic amino acids present in the corresponding PMEN. It is known that polymyxin-derived heptapeptides can also bind LPS35 and permeabilize the Gram-negative OM in a manner similar to polymyxin nonapepides.12 In addition, the polymyxin B heptapeptide has also been shown to potentiate various antibiotics against Gram-negative bacteria, albeit at 3-fold higher concentrations than required for the corresponding nonapeptide.12 The preparation of 10 (Scheme 5) followed a literature protocol whereby colistin was first converted to the penta-Boc-protected species followed by enzymatic degradation with Savinase to yield the tri-Boc-protected polymyxin E heptapeptide (PMEH-Boc3) 10.36 Attempts at directly coupling the free N-terminal amine of 10 to the free carboxylic acid of the protected peptide 5, as in the synthesis of conjugates 3a/b, in this case, failed to yield any of the expected bicyclic conjugate, presumably due to increased steric demands introduced by bringing the large macrocycles closer together. As an alternative, we then coupled azido glycine to 10, which, following deprotection, provided the N-terminally azide-modified PMEH species 11. As for azido-PMEN analogues 8a–8c, the ligation of 11 with alkyne-modified 7 proceeded smoothly upon treatment with CuSO4 and ascorbic acid to yield conjugate 12. Interestingly, the antibacterial activity of 12 was found to be much lower than those of the PMEN conjugates 9a–9c containing the longer linkers (Table 2). This finding indicates that there is an optimal spacer length for the conjugation of BamA-targeting β-hairpin peptidomimetics to the cyclic polymyxin fragment. Additionally, the reduced activity of compound 12 may point to a role for the exocyclic amino acids l-Thr and l-Dab (missing in 12 but present in 3a, 3b, and 9a–9c) for achieving potent antibacterial activity.
Scheme 5. Synthesis of Triazole-Linked Bicyclic Conjugates 12.
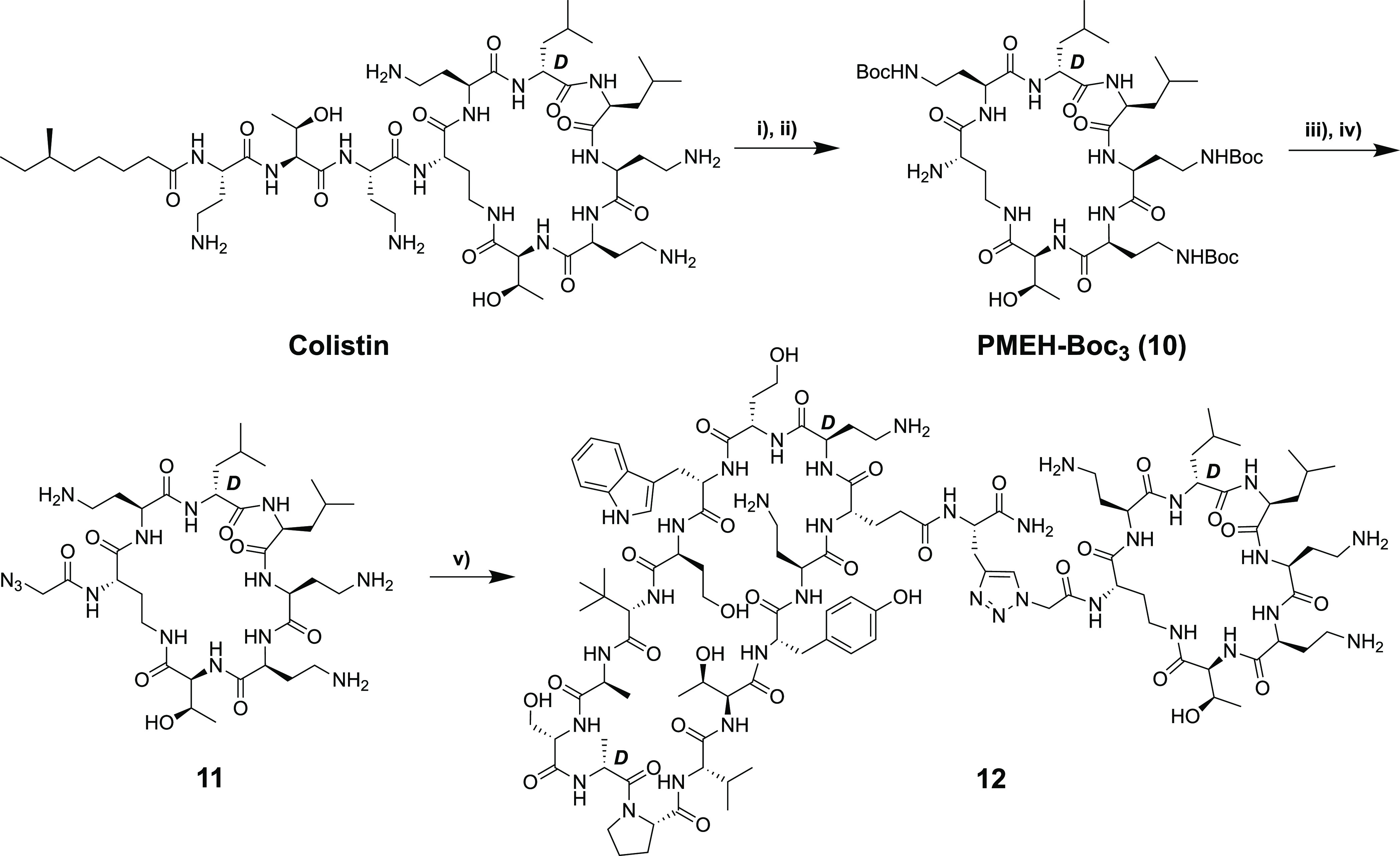
(i) (Boc)2O, Et3N; (ii) Savinase; (iii) 2-azidoacetic acid, HCTU, HOBt, 2,4,6-trimethylpyridine; (iv) TFA deprotection and HPLC purification; (v) alkyne-containing peptide macrocycle 7, CuSO4, and sodium ascorbate, followed by RP-HPLC purification.
Conclusions
While peptide-based therapeutics often face specific hurdles in drug development, their success in the field of antibacterials demonstrates that such compounds have potential. Specially, peptide antibiotics that operate by exploiting targets on the bacterial cell surface offer much promise. In this regard, a new class of Gram-negative-specific macrocyclic peptide antibiotics that target the OMP BamA have recently gained attention.18−20 Essential to the activity of these BamA-targeting antibiotics is ligation to the polymyxin nonapeptide, which is known to bind the lipid A component of LPS. In this study, we developed a convenient convergent, chemoenzymatic synthesis of these chimeric bicyclic peptide antibiotics. The antibacterial activities of the compounds prepared by our route compare well with MIC values previously reported. Notably, we also confirmed the potent activity of these compounds against colistin-resistant mcr-positive strains of E. coli. Furthermore, a new series of chimeric peptides were readily assembled using copper-catalyzed azide-alkyne cycloaddition (“click chemistry”) via the ligation of various PMEN building blocks bearing an N-terminal azide ranging in size from a simple azido glycine spacer to a C5 spacer and a (PEG)3 spacer. The β-hairpin macrocycle containing propargyl-glycine was readily prepared entirely on solid support and cleanly conjugated to the different azides containing PMEN units. The resulting triazole-linked bicyclic peptides demonstrated a range of antibacterial activities with the azido-glycine-linked conjugate demonstrating the most potent activity. Interestingly, conjugates containing either longer or shorter linkers between the β-hairpin macrocycle and the PMEN moiety were significantly less active. These findings point to the importance of identifying the optimal linker properties to achieve potent antibacterial activity. In summary, the chemoenzymatic routes here reported for the preparation of BamA-targeting bicyclic peptide antibiotics simplify the synthetic effort required to do so and enable the convenient preparation of new analogues to further evaluate the potential of this exciting new class of anti-Gram-negative agents.
Experimental Section
General Procedures
All reagents employed were of American Chemical Society (ACS) grade or finer and were used without further purification, unless otherwise stated. For compound characterization, HRMS analysis was performed on a Shimadzu Nexera X2 UHPLC system with a Waters Acquity HSS C18 column (2.1 × 100 mm, 1.8 μm) at 30 °C and equipped with a diode array detector. The following solvent system, at a flow rate of 0.5 mL/min, was used: solvent A, 0.1% formic acid in water; solvent B, 0.1% formic acid in acetonitrile. Gradient elution was as follows: 95:5 (A/B) for 1 min, 95:5 to 15:85 (A/B) over 6 min, 15:85 to 0:100 (A/B) over 1 min, 0:100 (A/B) for 3 min, then reversion back to 95:5 (A/B) for 3 min. This system was connected to a Shimadzu 9030 QTOF mass spectrometer (ESI ionization) calibrated internally with Agilent’s API-TOF reference mass solution kit (5.0 mM purine, 100.0 mM ammonium trifluoroacetate, and 2.5 mM hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazine) diluted to achieve a mass count of 10,000.
The purity of the peptides was confirmed to be ≥95% by analytical RP-HPLC using a Shimadzu Prominence-i LC-2030 system with a Dr. Maisch ReproSil Gold 120 C18 column (4.6 × 250 mm, 5 μm) at 30 °C and equipped with a UV detector monitoring at 214 nm. The following solvent system, at a flow rate of 1 mL/min, was used: solvent A, 0.1% TFA in water/acetonitrile (95/5); solvent B, 0.1% TFA in water/acetonitrile (5/95). Gradient elution was as follows: 95:5 (A/B) for 2 min, 95:5 to 0:100 (A/B) over 13 min, 0:100 (A/B) for 2 min, then reversion back to 95:5 (A/B) over 1 min, 95:5 (A/B) for 2 min.
The compounds were purified via preparative HPLC using a BESTA-Technik system with a Dr. Maisch Reprosil Gold 120 C18 column (25 × 250 mm, 10 μm) and equipped with a ECOM Flash UV detector monitoring at 214 nm. The following solvent system, at a flow rate of 12 mL/min, was used: solvent A, 0.1% TFA in water/acetonitrile (95/5); solvent B, 0.1% TFA in water/acetonitrile (5/95). Gradient elution was as follows: 95:5 (A/B) for 2 min, 95:5 to 0:100 (A/B) over 45 min, 0:100 (A/B) for 2 min, then reversion back to 95:5 (A/B) over 1 min, 95:5 (A/B) for 2 min.
Peptide Synthesis
Synthesis of the Protected Peptide 5
Chlorotrityl (CTC) resin (5.0 g, 1.60 mmol g–1) was loaded with Fmoc-Dab(Boc)-OH. Resin loading was determined to be 0.477 mmol g–1. Linear peptides encompassing AA-1 to AA-15 were assembled manually via standard Fmoc solid-phase peptide synthesis (SPPS) (resin-bound AA:Fmoc-AA:BOP:DIPEA, 1:4:4:8 molar equiv) on a 0.1 mmol scale. DMF was used as a solvent and Fmoc deprotections were carried out with piperidine:DMF (1:4, v:v). Amino acid side chains were protected as follows: tBu for Tyr, Allyl for Glu C-terminus, Trt for homoserine (Hse), and Boc for Trp/Dab. Following coupling and Fmoc deprotection of the final Fmoc-d-Dab(Boc)-OH, the resin-bound, Allyl-protected intermediate was next washed with CH2Cl2 and treated with Pd(PPh3)4 (30 mg, 0.03 mmol) and PhSiH3 (0.30 mL, 3.0 mmol) in CH2Cl2 (ca. 7 mL) under argon for 1 h. The resin was subsequently washed with CH2Cl2 (5 × 10 mL), followed by a solution of diethyldithiocarbamic acid trihydrate sodium salt (5 mg mL–1 in DMF, 5 × 10 mL), and DMF (5 × 10 mL). The linear side chain-protected peptide with free C- and N-terminus resin was subjected to on-resin cyclization conditions for 1 h (BOP/DIPEA, 4:8 molar equiv). The cyclic peptide still on resin was treated with (CF3)2CHOH:CH2Cl2 (1:4, 10 mL) for 1 h and rinsed with additional (CF3)2CHOH:CH2Cl2 and CH2Cl2. The combined washings were then evaporated to yield the side chain-protected cyclic peptide 5 with a free C-terminus for use in subsequent couplings.
Coupling of Protected Peptide 5 and Tetra-Boc-Protected PMEN
The protected cyclic peptide 5 and PMEN-Boc4 were dissolved in CH2Cl2 (150 mL) and treated with BOP (0.22 g, 0.5 mmol) and DIPEA (0.17 mL, 1.0 mmol), and the solution was stirred overnight. The reaction mixture was concentrated and directly treated with TFA:TIS:H2O (95:2.5:2.5, 10 mL) for 90 min. The reaction mixture was added to cold MTBE:hexanes (1:1) and spun down at 4500 RPM for 5 min, and the resulting precipitate was washed once more with MTBE:hexanes (1:1). The crude peptide was lyophilized from tBuOH:H2O (1:1) and purified with reversed-phase HPLC. Pure fractions were pooled and lyophilized to yield the desired cyclic lipopeptide products in >95% purity as white powders in 22–62 mg quantities (8–23% yield based on the resin loading used in the preparation of peptide 5).
Synthesis of the Monocyclic Peptide 6
Rink amide resin (150 mg, 0.684 mmol g–1) was loaded into a CEM Liberty Blue μwave peptide synthesizer at a 0.1 mmol scale. Linear peptides encompassing AA-1 to AA-15 were assembled using microwave irradiation at 90 °C (resin-bound AA:Fmoc-AA:DIC:Oxyma, 1:5:5:5 molar equiv). DMF was used as a solvent and Fmoc deprotections were carried out with piperidine:DMF (1:4, v:v). Amino acid side chains were protected as follows: tBu for Tyr, Allyl for Glu C-terminus, Trt for homoserine (Hse), and Boc for Trp/Dab. Following coupling and Fmoc deprotection of the final Fmoc-d-Dab(Boc)-OH, the linear peptide was removed from the synthesizer and washed with DCM, and the Allyl-protected intermediate was treated with Pd(PPh3)4 (30 mg, 0.03 mmol) and PhSiH3 (0.30 mL, 3.0 mmol) in CH2Cl2 (ca. 7 mL) under argon for 1 h. The resin was subsequently washed with CH2Cl2 (5 × 10 mL), followed by a solution of diethyldithiocarbamic acid trihydrate sodium salt (5 mg mL–1 in DMF, 5 × 10 mL) and DMF (5 × 10 mL). The protected, resin-bound peptide was next subjected to on-resin cyclization conditions for 1 h (BOP/DIPEA, 4:8 molar equiv), washed with DCM, and directly treated with TFA:TIS:H2O (95:2.5:2.5, 10 mL) for 90 min. The reaction mixture was added to cold MTBE:hexanes (1:1) and spun down at 4500 RPM for 5 min, and the resulting precipitate was washed once more with MTBE:hexanes (1:1). The crude cyclic peptide was lyophilized from tBuOH:H2O (1:1) and purified with reversed-phase HPLC. Pure fractions were pooled and lyophilized to yield the desired cyclic lipopeptide products in >95% purity as white powders, typically in 30–70 mg quantities (11–26% yield based on resin loading).
Antibacterial Assays
Minimum inhibitory concentrations (MICs) were determined by broth microdilution according to CLSI guidelines.37 Blood agar plates were inoculated with glycerol stocks of the chosen bacteria strains, followed by incubation for 16 h at 37 °C. Cation-adjusted Mueller-Hinton broth (CAMHB) containing 10 mg L–1 Mg2+ and 25 mg L–1 Ca2+ was inoculated with individual colonies of the chosen bacteria. The peptides were dissolved in CAMHB, supplemented with polysorbate-80 (P-80 or Tween-80, sterile-filtered) at a 0.002% (v/v) final concentration, and serially diluted on polypropylene microtiter plates with a volume of 50 μL per well. Inoculated CAMHB (5 × 105 CFU mL–1) was added to reach a total volume of 100 μL per well. The microtiter plates were sealed with an adhesive membrane and after 16 h of incubation at 37 °C the wells were visually inspected for bacterial growth. All reported MIC values resulted from three or more measurements performed on multiple days.
Glossary
Abbreviations
- Alloc
allyloxycarbonyl
- BAM
β-barrel assembly machine
- Boc
tert-butyloxycarbonyl
- Boc-ON
2-(Boc-oxyimino)-2-phenylacetonitrile
- BOP
benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate
- CAMHB
cation-adjusted Mueller-Hinton broth
- CTC
2-chlorotrityl chloride
- Dab
2,4-diaminobutyric acid
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- Fmoc
fluorenylmethoxycarbonyl
- Glu
glutamic acid
- HCTU
O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HFIP
hexafluoroisopropanol
- HOBt
hydroxybenzotriazole
- LC–MS
liquid chromatography mass spectrometry
- LPS
lipopolysaccharide
- LptD
LPS-assembly protein
- MCR
mobilized colistin resistance
- MIC
minimum inhibitory concentration
- MRSA
methicillin-resistant Staphylococcus aureus
- MTBE
methyl tert-butyl ether
- OM
outer membrane
- OMP
outer membrane protein
- PEG
polyethylene glycol
- PMBN
polymyxin B nonapeptide
- PMEN
polymyxin E nonapeptide
- RP-HPLC
reversed-phase high-performance liquid chromatography
- SPPS
solid-phase peptide synthesis
- TIF
tri-isopropyl silane
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00176.
Author Contributions
§ T.M.W. and C.J.S. contributed equally to this work.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work received financial support provided by the European Research Council (ERC consolidator grant to N.I.M.; grant agreement no. 725523).
The authors declare no competing financial interest.
Supplementary Material
References
- Roope L. S. J.; Smith R. D.; Pouwels K. B.; Buchanan J.; Abel L.; Eibich P.; Butler C. C.; Tan P. S.; Sarah Walker A.; Robotham J. V.; et al. The challenge of antimicrobial resistance: what economics can contribute. Science 2019, 364, 6435. 10.1126/science.aau4679. [DOI] [PubMed] [Google Scholar]
- MacNair C. R.; Tsai C. N.; Brown E. D. Creative targeting of the gram-negative outer membrane in antibiotic discovery. Ann. N. Y. Acad. Sci. 2020, 1459, 69–85. 10.1111/nyas.14280. [DOI] [PubMed] [Google Scholar]
- Årdal C.; Balasegaram M.; Laxminarayan R.; McAdams D.; Outterson K.; Rex J. H.; Sumpradit N. Antibiotic Development — Economic, regulatory and societal challenges. Nat. Rev. Microbiol. 2020, 18, 267–274. 10.1038/s41579-019-0293-3. [DOI] [PubMed] [Google Scholar]
- Codjoe F. S.; Donkor E. S. Carbapenem resistance: a review. Med. Sci. 2018, 6, 1. 10.3390/medsci6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri A. M.; Doi Y.; Potoski B. A.; Harris P. N. A.; Paterson D. L.; Righi E. Antimicrobial treatment challenges in the era of carbapenem resistance. Diagn. Microbiol. Infect. Dis. 2019, 94, 413–425. 10.1016/j.diagmicrobio.2019.01.020. [DOI] [PubMed] [Google Scholar]
- Henderson J. C.; Zimmerman S. M.; Crofts A. A.; Boll J. M.; Kuhns L. G.; Herrera C. M.; Trent M. S. The power of asymmetry: architecture and assembly of the gram-negative outer membrane lipid bilayer. Annu. Rev. Microbiol. 2016, 70, 255–278. 10.1146/annurev-micro-102215-095308. [DOI] [PubMed] [Google Scholar]
- Stojanoski V.; Sankaran B.; Prasad B. V.; Poirel L.; Nordmann P.; Palzkill T. Structure of the catalytic domain of the colistin resistance enzyme MCR-1. BMC Biol. 2016, 14, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubery H.; Ofek I.; Cohen S.; Fridkin M. The functional association of polymyxin B with bacterial lipopolysaccharide is stereospecific: studies on polymyxin B nonapeptide. Biochemistry 2000, 39, 11837–11844. 10.1021/bi000386q. [DOI] [PubMed] [Google Scholar]
- Moore R. A.; Bates N. C.; Hancock R. E. Interaction of polycationic antibiotics with pseudomonas aeruginosa lipopolysaccharide and lipid a studied by using dansyl-polymyxin. Antimicrob. Agents Chemother. 1986, 29, 496–500. 10.1128/AAC.29.3.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas M. E.; Kasiakou S. K.; Saravolatz L. D. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. 10.1086/429323. [DOI] [PubMed] [Google Scholar]
- Velkov T.; Thompson P. E.; Nation R. L.; Li J. Structure—activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y.; Matsunaga H.; Vaara M. Polymyxin B octapeptide and polymyxin b heptapeptide are potent outer membrane permeability-increasing agents. J. Antibiot. (Tokyo) 1992, 45, 742–749. 10.7164/antibiotics.45.742. [DOI] [PubMed] [Google Scholar]
- Zabawa T. P.; Pucci M. J.; Parr T. R. Jr.; Lister T. Treatment of gram-negative bacterial infections by potentiation of antibiotics. Curr. Opin. Microbiol. 2016, 33, 7–12. 10.1016/j.mib.2016.05.005. [DOI] [PubMed] [Google Scholar]
- French S.; Farha M.; Ellis M. J.; Sameer Z.; Côté J. P.; Cotroneo N.; Lister T.; Rubio A.; Brown E. D. Potentiation of antibiotics against gram-negative bacteria by polymyxin B analogue SPR741 from unique perturbation of the outer membrane. ACS Infect. Dis. 2020, 6, 1405–1412. 10.1021/acsinfecdis.9b00159. [DOI] [PubMed] [Google Scholar]
- Ofek I.; Cohen S.; Rahmani R.; Kabha K.; Tamarkin D.; Herzig Y.; Rubinstein E. Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob. Agents Chemother. 1994, 38, 374–377. 10.1128/AAC.38.2.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaara M.; Siikanen O.; Apajalahti J.; Fox J.; Frimodt-Møller N.; He H.; Poudyal A.; Li J.; Nation R. L.; Vaara T. A novel polymyxin derivative that lacks the fatty acid tail and carries only three positive charges has strong synergism with agents excluded by the intact outer membrane. Antimicrob. Agents Chemother. 2010, 54, 3341–3346. 10.1128/AAC.01439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Espada R.; Sánchez-Gómez S.; Pitts B.; Stewart P. S.; Martínez-de-Tejada G. Permeability enhancers sensitize β-lactamase-expressing enterobacteriaceae and pseudomonas aeruginosa to β-lactamase inhibitors, thereby restoring their β-lactam susceptibility. Int. J. Antimicrob. Agents 2020, 56, 105986. 10.1016/j.ijantimicag.2020.105986. [DOI] [PubMed] [Google Scholar]
- Srinivas N.; Jetter P.; Ueberbacher B. J.; Werneburg M.; Zerbe K.; Steinmann J.; Van der Meijden B.; Bernardini F.; Lederer A.; Dias R. L. A.; Misson P. E.; Henze H.; Zumbrunn J.; Gombert F. O.; Obrecht D.; Hunziker P.; Schauer S.; Ziegler U.; Kach A.; Eberl L.; Riedel K.; DeMarco S. J.; Robinson J. A. Peptidomimetic antibiotics target outer-membrane biogenesis in pseudomonas aeruginosa. Science 2010, 327, 1010–1013. 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- Martin-Loeches I.; Dale G. E.; Torres A. Murepavadin: a new antibiotic class in the pipeline. Expert Rev. Anti-infect. Ther. 2018, 16, 259–268. 10.1080/14787210.2018.1441024. [DOI] [PubMed] [Google Scholar]
- Luther A.; Urfer M.; Zahn M.; Müller M.; Wang S. Y.; Mondal M.; Vitale A.; Hartmann J. B.; Sharpe T.; Monte F.; Kocherla H. Chimeric peptidomimetic antibiotics against gram-negative bacteria. Nature 2019, 576, 452–458. 10.1038/s41586-019-1665-6. [DOI] [PubMed] [Google Scholar]
- McLaughlin M. I.; Van Der Donk W. A. The fellowship of the rings: macrocyclic antibiotic peptides reveal an anti-gram-negative target. Biochemistry 2020, 59, 343–345. 10.1021/acs.biochem.9b01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan C. L.; Wzorek J. S.; Kahne D. Inhibition of the β-barrel assembly machine by a peptide that binds BamD. Proc. Natl. Acad. Sci. 2015, 112, 2011–2016. 10.1073/pnas.1415955112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan C. L.; Silhavy T. J.; Kahne D. Β-barrel membrane protein assembly by the Bam complex. Annu. Rev. Biochem. 2011, 80, 189–210. 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- Tomasek D.; Rawson S.; Lee J.; Wzorek J. S.; Harrison S. C.; Li Z.; Kahne D. Structure of a nascent membrane protein as it folds on the Bam complex. Nature 2020, 583, 473–478. 10.1038/s41586-020-2370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J. A. Folded synthetic peptides and other molecules targeting outer membrane protein complexes in gram-negative bacteria. Front. Chem. 2019, 7, 45. 10.3389/fchem.2019.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans T.; Wood T. M.; ‘t Hart P.; Kleijn L. H.; Hendrickx A. P.; Willems R. J.; Breukink E.; Martin N. I. Semisynthetic lipopeptides derived from nisin display antibacterial activity and lipid ii binding on par with that of the parent compound. J. Am. Chem. Soc. 2015, 137, 9382–9389. 10.1021/jacs.5b04501. [DOI] [PubMed] [Google Scholar]
- ‘t Hart P.; Wood T. M.; Tehrani K. H. M. E.; Van Harten R. M.; Śleszyńska M.; Rebollo I. R.; Hendrickx A. P.; Willems R. J.; Breukink E.; Martin N. I. De novo identification of lipid II binding lipopeptides with antibacterial activity against vancomycin-resistant bacteria. Chem. Sci. 2017, 8, 7991–7997. 10.1039/C7SC03413J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijn L. H.; Oppedijk S. F.; ’t Hart P.; Van Harten R. M.; Martin-Visscher L. A.; Kemmink J.; Breukink E.; Martin N. I. Total synthesis of laspartomycin C and characterization of its antibacterial mechanism of action. J. Med. Chem. 2016, 59, 3569–3574. 10.1021/acs.jmedchem.6b00219. [DOI] [PubMed] [Google Scholar]
- Wood T. M.; Bertheussen K.; Martin N. I. The contribution of achiral residues in the laspartomycin family of calcium-dependent lipopeptide antibiotics. Org. Biomol. Chem. 2020, 18, 514–517. 10.1039/C9OB02534K. [DOI] [PubMed] [Google Scholar]
- Silverman S. M.; Moses J. E.; Sharpless K. B. Reengineering antibiotics to combat bacterial resistance: click chemistry [1,2,3]-triazole vancomycin dimers with potent activity against mrsa and vre. Chem. - Eur. J. 2017, 23, 79–83. 10.1002/chem.201604765. [DOI] [PubMed] [Google Scholar]
- Meyer J. P.; Adumeau P.; Lewis J. S.; Zeglis B. M. Click chemistry and radiochemistry: the first 10 years. Bioconjugate Chem. 2016, 27, 2791–2807. 10.1021/acs.bioconjchem.6b00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danner R. L.; Joiner K. A.; Rubin M.; Patterson W. H.; Johnson N.; Ayers K. M.; Parillo J. E. Purification, toxicity, and antiendotoxin activity of polymyxin B nonapeptide. Antimicrob. Agents Chemother. 1989, 33, 1428–1434. 10.1128/AAC.33.9.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dowd H.; Kim B.; Margolis P.; Wang W.; Wu C.; Lopez S. L.; Blais J. Preparation of tetra-boc-protected polymyxin B nonapeptide. Tetrahedron Lett. 2007, 48, 2003–2005. 10.1016/j.tetlet.2007.01.071. [DOI] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Thomas C. J.; Surolia A. Kinetics of the interaction of endotoxin with polymyxin B and its analogs: a surface plasmon resonance analysis. FEBS Lett. 1999, 445, 420–424. 10.1016/S0014-5793(99)00150-7. [DOI] [PubMed] [Google Scholar]
- Li B.; Akin A.; Magee T. V.; Martinez C.; Szeliga J.; Vuong D. V. Syntheses of dap-3 polymyxin analogues via a tris-boc-protected polymyxin B heptapeptide. Synthesis 2015, 47, 2088–2092. 10.1055/s-0034-1380549. [DOI] [Google Scholar]
- Abmm D.; Tamma D.; Kirn J.; Cullen S. K.. Clinical and Laboratory Standards Institute (CLSI). In Performance Standards for Antimicrobial Susceptibility Testing. 30th Ed. CLSI Supplement M100: Clinical and Laboratory Standards Institute, 2020, 30. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.