

Abstract
We describe a new class of potent PD-L1/PD-1 inhibitors based on a terphenyl scaffold that is derived from the rigidified biphenyl-inspired structure. Using in silico docking, we designed and then experimentally demonstrated the effectiveness of the terphenyl-based scaffolds in inhibiting PD-1/PD-L1 complex formation using various biophysical and biochemical techniques. We also present a high-resolution structure of the complex of PD-L1 with one of our most potent inhibitors to identify key PD-L1/inhibitor interactions at the molecular level. In addition, we show the efficacy of our most potent inhibitors in activating the antitumor response using primary human immune cells from healthy donors.
Introduction
Cancer immunotherapy aims to stimulate the immune system’s ability to fight cancer as opposed to directly killing cancer cells with more traditional methods such as chemotherapy and radiation therapy.1−6 Inhibition of negative immune checkpoint regulators is now used in clinics around the world and was recognized with the 2018 Nobel Prize in Physiology or Medicine.7 One of the most important immune checkpoints (ICs) in cancer immunotherapy consists of programmed cell death protein 1 (PD-1, also known as CD279) with its ligand (PD-L1, known also as CD274 or B7-H1).4
The PD-1/PD-L1 axis is responsible for inhibiting excessive stimulation and normally aims at maintaining immune tolerance to self-antigens by negatively regulating the immune response.8−10 By blocking the PD-1/PD-L1 interaction, activation of T cells can be achieved for an improved antitumor response. Therefore, the immune checkpoint blockade (ICB) of PD-1 or PD-L1 at the tumor cell–T cell interface has become an attractive strategy for cancer immunotherapy.11,12
Traditionally, the inhibition of PD-1/PD-L1 interactions is achieved with monoclonal antibodies (mAbs) that target PD-1 (e.g., pembrolizumab, nivolumab, cemiplimab, etc.) or PD-L1 (e.g., atezolizumab, avelumab, and durvalumab).3 Despite their medical and commercial success, mAb-based immunotherapies suffer from several disadvantages, including high treatment costs, immune-related adverse events (irAE), and poor penetration of solid tumors.13,14 Small-molecule inhibitors (SMIs) are expected to overcome these limitations, with additional benefits such as oral bioavailability and lower production cost.15,16
Although the PD-1/PD-L1 interface is considered difficult to treat with SMIs due to its large, flat surface of interaction with no visible binding pockets, the number of patents and publications on anti-PD-L1 SMIs is constantly growing.17,18
A common feature of virtually all reported PD-1/PD-L1 SMI is that they target PD-L1 through a biphenyl-based scaffold originally developed by Bristol Myers Squibb (BMS).19−21 However, none of these molecules has progressed into clinical trials so far. As of now, only the peptide-derived compound CA-170 from Curis and Aurigene is undergoing clinical trials as a small molecule that directly targets the PD-L1, PD-L2, and the V-domain Ig suppressor of T cell activation (VISTA) IC. However, it turned out that CA-170 is not a classic PD-L1-blocker and alternative modes of its action have been postulated.22,23
Here, we present a new class of potent PD-L1 inhibitors based on a rigidified biphenyl structure. These compounds, which comprise the terphenyl scaffold, are effective in blocking PD-1/PD-L1 interactions in a variety of biophysical and cellular assays. We also provide a high-resolution structure of PD-L1 bound to the terphenyl small molecule to identify key PD-L1/ligand interactions on the PD-L1 interface.
Results and Discussion
Identification of Potent Terphenyl-Based PD-L1 Inhibitors
Rigidification of the scaffold is a well-known strategy for enhancing the selectivity and potency of SMIs that bind to protein targets.24−27 In our study, we focused on the effect of rigidifying the known PD-L1 inhibitors from the BMS family19,21 (Figure 1A). The core scaffold was extracted by comparison with several BMS molecules disclosed in patents19,21 (Figure 1A, BMS core). Rigidification was then implemented by removing the ether linker (highlighted in red), leading to the m-terphenyl derivative (Figure 1A). The rigidified terphenyl structure was tested in silico against the aromatic core of BMS molecules (Figure 1B) using AutoDock Vina28 integrated into PyRx software,29 with the dimeric structure of PD-L1 bound to BMS-1166 (PDB ID: 6R3K). Molecular docking shows the high potency of the designed, rigidified core, with a stronger PD-L1 binding affinity of 1.0 kcal/mol (−11.7 kcal/mol) than the corresponding fragment of BMS core (Figure 1A, −10.7 kcal/mol). Moreover, the calculations suggest that the deletion of the ether linker fragment allows for a better stabilization of the complex, mainly by a stronger π–π stacking with BTyr56 (Figure 1B). These promising results prompted us to structurally optimize the found m-terphenyl motif. All designed molecules were tested in a homogeneous time-resolved fluorescence (HTRF) assay to assess their activity as inhibitors of the PD-1/PD-L1 complex (Table S1).
Figure 1.
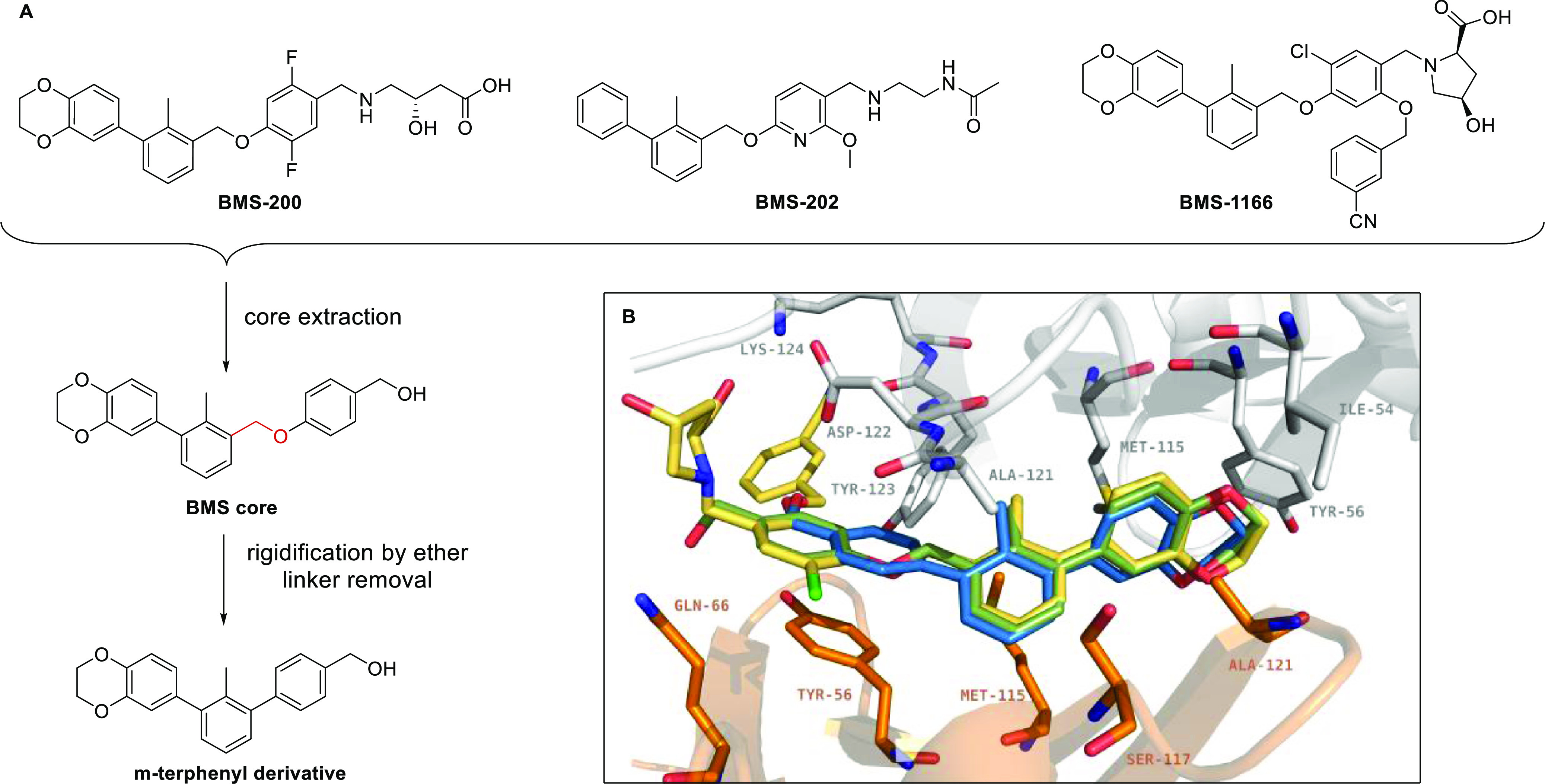
In silico comparison of the prototype m-terphenyl fragment and corresponding BMS core performed with AutoDock Vina integrated into PyRx software. (A) Structures used in molecular modeling: known PD-L1 inhibitors BMS-200, BMS-202, and BMS-1166 molecules; BMS core—fragment extracted by comparison of the structures of several BMS molecules; m-terphenyl derivative—rigidified motif obtained by the ether linker removal. (B) Imposition of the modeled complexes of found m-terphenyl derivative (blue), BMS core (green), and whole BMS-1166 structure (yellow) in complex with PD-L1 (subunit A—gray, subunit B—orange, PDB ID: 6R3K).
In the first step of the process, the impact of the type of the C-2′-attached substituent (R1), as well as the position of the hydroxymethyl group, for the binding affinity was tested (Scheme 1, Table 1).
Scheme 1. Synthesis Pathway Used in R1 and Hydroxymethyl Group Position Optimization of m-Terphenyls 4a–4h.

Reagents and conditions: (a) 1a–c, 4-benzodioxane-6-boronic acid, Pd(dppf)Cl2·DCM, K2CO3, dioxane/water 2:1, 80 °C, 3 h, 47–61%; (b) 2a–c, (hydroxymethyl)phenylboronic acid, Pd(dppf)Cl2·DCM, K2CO3, dioxane/water 2:1, 80 °C, 3 h, 32–56%; (c) for 4f: 4d (R1 = NH2), HBr, NaNO2, CuBr2, 70 °C, 2 h, 9%; for 4g: 4d (R1 = NH2), HCl, NaNO2, KI, RT, 16 h, 28%; for 4h: 4d (R1 = NH2), HCl, NaNO2, CuCN, KCN, RT, 4 h, 21%.
Table 1. Inhibitory Activities of the Optimized Terphenyls Obtained in the HTRF Assay at the First Stage of Optimization of the R1 Substituent.

| name | R1 | hydroxymethyl position | HTRF % of the undissociated complex at 5 μM | IC50 [μM] |
|---|---|---|---|---|
| 4a (m-terphenyl)a | Me | para | 42 | 5.52 ± 0.04 |
| 4b | Me | meta | 88 | |
| 4c | Me | ortho | 98 | |
| 4d | NH2 | para | 73 | |
| 4e | Cl | para | 1 | 0.51 ± 0.01 |
| 4f | Br | para | 15 | 0.95 ± 0.02 |
| 4g | I | para | 56 | |
| 4h | CN | para | 21 | 1.17 ± 0.12 |
To obtain the designed molecules, we applied a synthetic pathway based mainly on the Suzuki cross-coupling reaction. Appropriate 1,3-dibromoaryls 1a–c were coupled into their 3-benzodioxane derivatives 2a–c. Subsequently, biphenyls were taken into a second cross-coupling reaction with 2, 3, or 4-(hydroxymethyl)phenylboronic acid, leading to the corresponding m-terphenyl derivatives 4a–e. C-2′ nitrile and brominated and iodinated molecules (compounds 4f–h) were synthesized by the Sandmeyer reaction, followed by amine-based diazonium salt formation. All compounds generated in this step had their potency for the PD-1/PD-L1 complex dissociation tested using the HTRF assay in the scouting mode at 5 μM since IC50 determination would not be possible for some compounds due to their low solubility at this stage. In this assay, a lower percentage of the undissociated complex reported in Table 1 indicates higher potency of the tested compound. Full IC50s of several compounds were determined (Table 1) and compared with the corresponding percentages of the undissociated complex, indicating a correlation between the two measurements. This approach was used for the activity estimation of all molecules presented in this article. The HTRF assay was also validated on the BMS-1166 compound, yielding IC50 of 3.89 nM (Table 4) as compared to 1.40 nM reported in the literature.19,30 The results of the HTRF assay show clearly that the optimal substituent attached to C-2′ of the middle ring of terphenyl is chlorine with IC50 of approximately 500 nM (compound 4e); however, comparable results have been obtained for brominated and cyano derivatives (compounds 4f and 4h, respectively). Introduction of a polar fragment (compound 4d) in this region has a negative impact on the molecule affinity. Generally, the insertion of the halogen atom at the C-2′ position (the R1 substituent) is preferred and the activity of the tested fragments decreases with the growth of the halogen van der Waals radius (compounds 4e–g).
Table 4. Inhibitory Activities of the Optimized Terphenyls Obtained in HTRF Assay in the Second Step of Optimization of Hydrophilic Tail NR2.


Limit of quantification of the used assay was achieved.
In parallel, we assessed the influence of further molecular elongation on both the aromatic and aliphatic fragments with different hydrogen bond donor–acceptor properties (Scheme 2, Table 2). Molecules 5a–r were obtained following a nucleophilic substitution reaction of the previously prepared benzyl chloride derivatives. The corresponding alkyl chlorides were synthesized by the transformation of the 4-hydroxymethylterphenyl derivatives 4a–c with SOCl2 and a catalytic amount of anhydrous dimethylformamide (DMF). Then, they were treated with an appropriate nucleophile, yielding ethers or tertiary amines (compounds 5a–r and 6a–c). The double substitution of the amines was prevented by using an excess of the nucleophile. Furthermore, the bulkiness of the terphenyl fragment additionally precluded double substitution.
Scheme 2. Synthesis Routes Leading to Elongated m-Terphenyl Derivatives 5a–5r and 6a–6c.
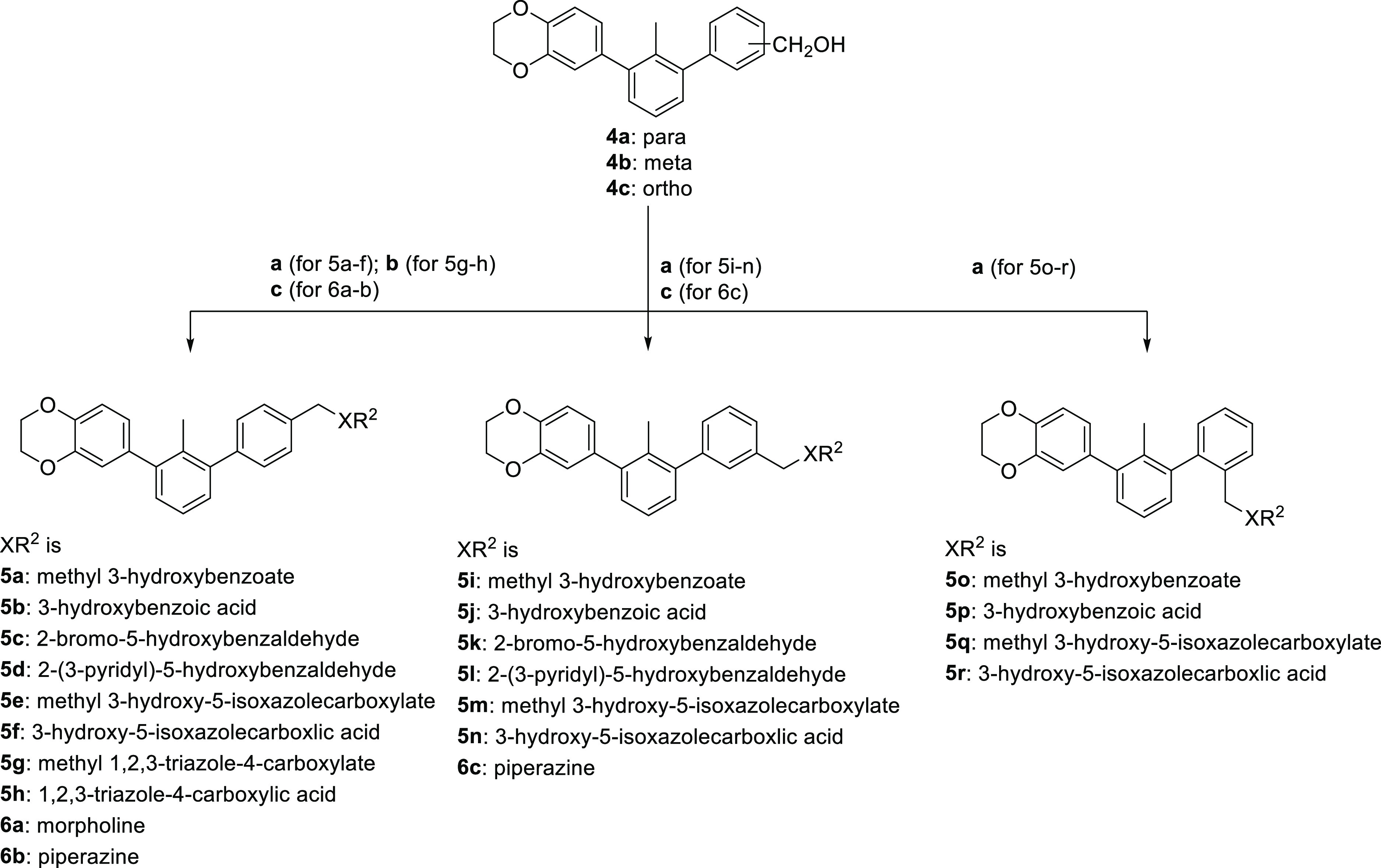
Reagents and conditions: (a) (i) 4a–4c, SOCl2, DCM, cat. DMF, RT, 2H; (ii) appropriate phenol, K2CO3/Cs2CO3, KI, DMF, 80 °C, 16 h, 42–85%; (iii) 5a/5e/5i/5m/5o/5q, LiOH, dioxane/H2O, 2:1, RT, 16 h, 45–89% or 5c/5k, 3-pyridinyl boronic acid, Pd(dppf)Cl2·DCM, K2CO3, dioxane/water 2:1, 80 °C, 5 h, 80–92%; (b) (i) 4a, MsCl, DCM, Et3N, RT, 2H (ii) NaN3, DMF, RT, 6 days; (iii) methyl propiolate, sodium ascorbate, CuSO4, RT, 24 h, 72%; (iv) 5g, LiOH, dioxane/H2O, 2:1, RT, 16 h, 61%; (c) (i): 4a or 4b, SOCl2, DCM, cat. DMF, RT, 2H (ii) appropriate amine, THF, RT, 48 h, 29–70%.
Table 2. Inhibitory Activities of the Optimized Terphenyls Obtained in HTRF Assay in the First Stage of Optimization of the XR2 Substituent and Position of Molecule Extension.


Percentage of the undissociated protein complex was determined at 50 μM concentration of the inhibitor.
The results obtained from these steps clearly indicated that only the para elongation, using amine as a building block is beneficial for the interaction (compound 6b, Table 2). Attempted meta extension of the motif causes a significant drop of the affinity from 5% of the undissociated complex for 6b to about 56% for 6c, tested at 5 μM concentrations. Furthermore, the introduction of an aromatic fragment in each position destabilizes the interaction.
In the next stage of the structure optimization, the type of the C3-attached ether substituent in the proximal phenyl ring of the terphenyl was considered. We synthesized a series of different alkoxy derivatives (Scheme 3, Table 3) implementing different-length alkyl and cycloalkyl substituents, as well as an acetonitrile fragment (compounds 6d–e and 7a–h).
Scheme 3. Synthetic Pathway Used during R3 Alkoxy Fragment and R2 Hydrophilic Tail Optimization of m-Terphenyls 6d–6e, 7a–7h, and 8a–8j.
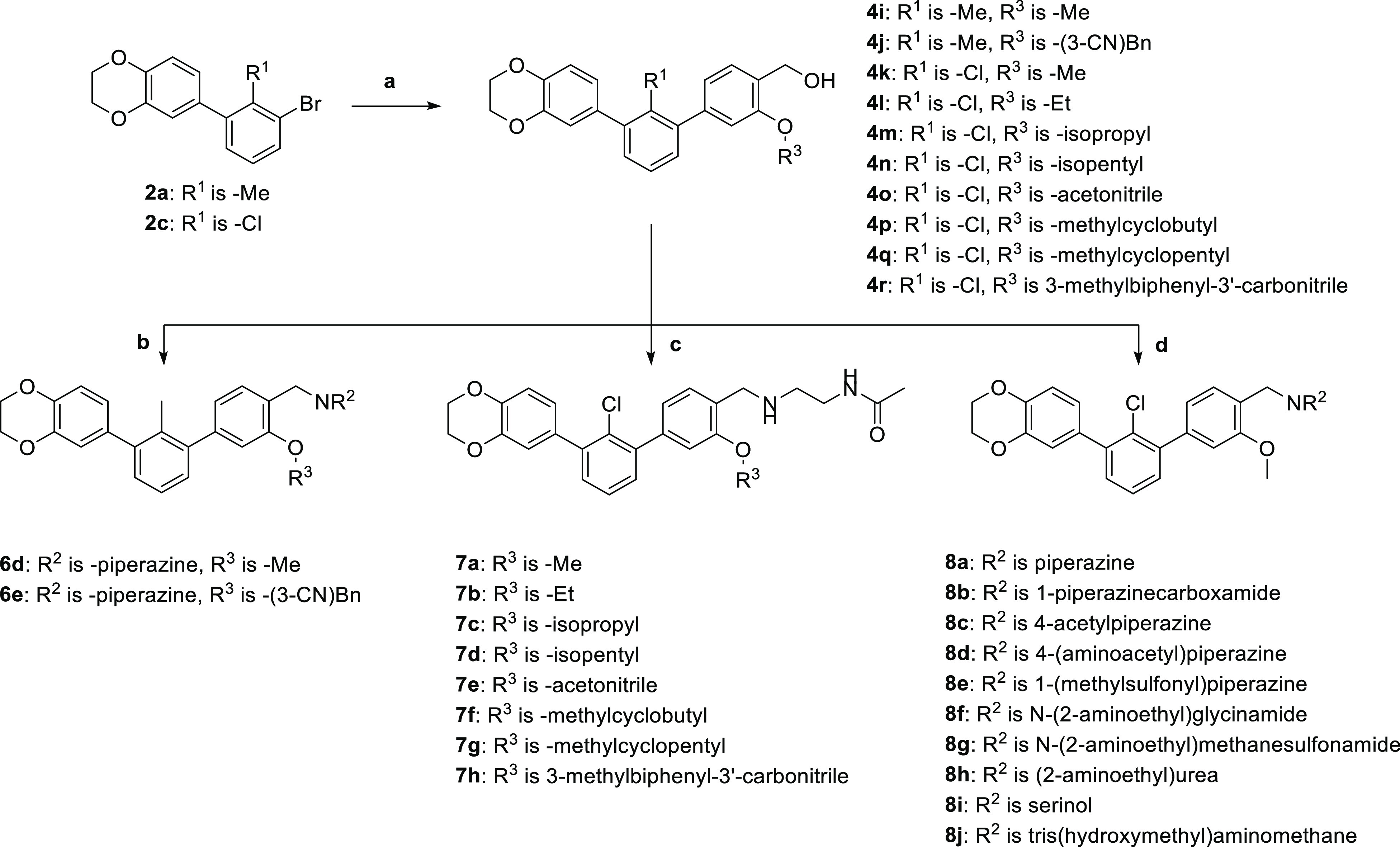
Reagents and conditions: (a) 2a or 2c, borane 3a–3i, Pd(dppf)Cl2·DCM, K2CO3, dioxane/water 2:1, 80 °C, 3 h, 29–89%; (b) (i) 4i or 4j, SOCl2, DCM, cat. DMF, RT, 2H; (ii) appropriate amine, THF, RT, 48 h, 20–38%; (c) (i) 4k–4r, SOCl2, DCM, cat. DMF, RT, 2H; (ii) appropriate amine, DIPEA, DMF, 80 °C, 16 h, 10–78% (d) (i) 4k, SOCl2, DCM, cat. DMF, RT, 2H (ii) appropriate amine, DIPEA, DMF, 80 °C, 16 h, 33–89%.
Table 3. Inhibitory Activities of the Optimized Terphenyls Obtained in HTRF Assay in the Second Step of Optimization of the R3 Alkoxyl Substituent.


To prepare the desired compounds, we carried out Williamson alkylation of the 4-bromo-2-hydroxybenzyl alcohol, leading to the appropriate alkoxy derivative. Then, the intermediates were transformed into the corresponding phenylboronic acid pinacol esters (compounds 3a–i, SI) and coupled with 3-bromobiphenyls 2a or 2c. Hydroxymethyl groups of terphenyls were then transformed into the corresponding chlorides, which were used as an amine alkylating agent. Initially, we tested the piperazine-extended derivatives of the ethers 6d–e, but we have observed precipitation during the HTRF assay. This forced us to change the amine to N-(2-aminoethyl)acetamide, significantly increasing the solubility in the aqueous buffer for compounds 7a–h. Almost all molecules obtained in this stage of the optimization were characterized by high affinity to the PD-L1 protein and caused complete dissociation of the complex at 5 μM. Therefore, to differentiate potencies of the tested molecules, the inhibitor concentration was set to 5 nM in further assays.
Introduction of the methoxy fragment in 7a has the greatest impact on binding to PD-L1, resulting in complete complex dissociation at 5 nM ligand concentration and a measured IC50 value of 0.82 nM. Further elongation of the ether substituent decreases the affinity to the target (compounds 7b–g). Addition of an extra biphenyl motif in compound 7h caused a drop in the inhibitory potency of the molecule. The acetonitrile derivative in molecule 7e shows the activity comparable with the O-methylated 7a molecule, causing 99% of the complex dissociation at 5 nM.
The last step of the SAR analysis was focused on the optimization of the hydrophilic tail (Scheme 3, Table 4). Maintaining all optimized molecule fragments, we synthesized and evaluated the inhibitory activities of the piperazine- (compounds 8a–e), ethylenediamine- (compounds 7a, 8f–h), and 2-aminoethanol-containing (compounds 8i–j) terphenyl derivatives.
The results showed that the type of solubilizer used has a great impact on the affinity to the targeted protein. The piperazine derivative family (compounds 8a–8e) was characterized by the lowest potencies in disrupting the PD-1/PD-L1 interaction, characterized by 88–5% undissociated complex at 5 nM ligand concentration. The best PD-L1 binder from this group is the glycine-containing 8d molecule with 5% of the remaining complex. For 8b–d, we could not precisely determine the values of the complex dissociation as they are beyond the quantification limits of the assay. Therefore, we assigned them the values below 1 nM, the level at which we can still confidently determine the assay results. The ethylenediamine derivatives (compounds 7a and 8f–h) were characterized by moderate to excellent potencies in the inhibition of the PD-1/PD-L1 interactions, with the percentage of the undissociated complex in 32–0% range at 5 nM inhibitor concentration. Determination of the half-maximal inhibitory concentration was successful only for 7a and 8g. The values of 0.82 and 2.07 nM, respectively, were obtained. For the rest of the tested molecules, the limit of the assay was exceeded. It has to be noted that HTRF results served only the purpose of the initial selection of “leads” for further cell-based assays and as such should not be considered as a sole determinant of the compound potency in the PD1-/PD-L1 complex formation inhibition.
Terphenyl-Based Inhibitors Induce Dimerization of PD-L1
Representative molecules from the optimized group were tested for the interaction with PD-L1 using a 1H NMR titration experiment and showed protein oligomerization upon addition of the ligand (Figure 2).
Figure 2.

1H NMR spectra of PD-L1 with terphenyl-based inhibitors. (A) 1H NMR spectra of the aliphatic part of human PD-L1 protein (blue) with compound 4a (red), 7a (green), 7e (purple), and 8j (orange) in the molar ratio 1:1. (B,C) Aliphatic region of PD-L1 (blue) with 8j (red): human PD-L1 (panel B) and mouse PD-L1 (panel C) in the molar ratio 1:1 and 1:10, respectively.
After adding compounds 4a, 7a, 7e, and 8j to human PD-L1 (hPD-L1), we observed the broadening of the hPD-L1 proton signals in 1D NMR spectra, characteristic of oligomerization of hPD-L1, previously observed by us with the BMS compounds.30 We additionally compared the spectra of the interaction of 8j with hPD-L1 (Figure 2B) and murine PD-L1 (mPD-L1) (Figure 2C). Only for human PD-L1, changes in NMR were observed upon addition of 8j to the protein. For murine PD-L1, no interaction was observed between mPD-L1 and the compound, even with the over-titration of mPD-L1 with 8j (1:10, molar ratio, respectively) as there was no change in the corresponding NMR spectra fingerprints (changes in the intensity of the peaks or their position compared to each other). The results are, therefore, consistent with our previous studies, in which we showed that the biphenyl moiety from anti-PD-L1 inhibitors binds to human PD-L1 and not to the murine analogue.31
Lead Compounds Activate Jurkat Effector Cells in the ICB Assay
After a successful optimization of the terphenyl compounds, the activity of the optimized molecules 7a and 8a–j in a cellular context was assessed. To this end, we first performed the PD-1/PD-L1 ICB assay. In this assay, T-cell receptor (TCR)-mediated activation of Jurkat effector cells (Jurkat-ECs) is reduced by the presence of PD-L1, exposed at the surface of CHO/TCRAct/PD-L1 cells (Figure 3A). Upon the addition of PD-1/PD-L1 blockers, full activation of Jurkat-ECs is restored, as presented for a therapeutic anti-PD-L1 antibody, atezolizumab (Figure 3B).
Figure 3.

Terphenyl compound bioactivity and toxicity in the ICB assay, (A) schematic representation of the ICB assay. (B) Dose–response curves of the reactivation of Jurkat-ECs with PD-L1-blocking agents: atezolizumab (left panel) and terphenyl compounds (right panel). Graphs show fold induction of the luminescence signal relative to either untreated (for atezolizumab) or dimethyl sulfoxide (DMSO)-treated (for compounds) cells. Data points represent mean ± SD values from 3 to 5 independent experiments. Gray data points were removed from curve-fitting analysis due to the toxicity of the compounds observed at the 10 μM concentration. (C) Long-term (48 h) cytotoxicity of the tested compounds toward Jurkat-ECs. The graph shows Jurkat-EC survival relative to DMSO-treated control cells. Data points represent mean ± SD values from three independent experiments. (D) Half-maximal effective concentrations (EC50) of the cytotoxicity of the compounds, calculated from (C). (E) EC50 values of the bioactivity of the compounds in the ICB assay, calculated from (B). (F) Selectivity of the bioactivity of the compounds in the ICB assay over the cytotoxicity effect toward Jurkat-ECs. Error bars are combined errors of EC50 (tox.) and EC50 (bio.). (G) Chemical structures of the three most potent terphenyl compounds.
Most of the compounds selected for the assay (molecules 7a, and 8f–8j) were able to increase the activation of Jurkat-ECs (Figure 3B). Only the piperazine-containing molecules do not show any changes after the treatment. The best activity, reflected by the lowest EC50 values (Figure 3E), was observed for 8j, followed by 8h, 8i, 7a, and 8f. These compounds provided higher activation of the Jurkat-ECs than the previously published BMS-1166 compound (Figure 3B). All active compounds were comparable in terms of toxicity toward Jurkat-ECs, with compounds 7a, 8f, and 8j slightly less cytotoxic than 8g, 8h, and 8i (Figure 3C,D). Importantly, none of the tested compounds was able to increase the activity of Jurkat-ECs where PD-L1 was absent (Figure S1). This provides evidence that the activity of the compounds is PD-L1-dependent and is not related to the unspecific activation of T cells. Compounds 8j, 8h, and 8i (Figure 3G) showed the highest selectivity of the desired PD-1/PD-L1-blocking activity versus cytotoxicity (Figure 3F). Compounds 8h and 8j, which, apart from their bioactivity, showed dissociation of the protein complex in HTRF at 5 nM, were directed for further study.
Lead Compound 8j Activates Primary T Cells in the T-Cell Activation Assay
A standard ICB assay relies on artificial Jurkat-ECs, which are surrogates of T-cells. To verify the potential of the compounds to reactivate the PD-L1-blocked primary T cells, a modified assay termed T-cell activation (TCA) was carried out.32 In this assay, human primary peripheral blood mononuclear cells (PBMCs) are allowed to make contact with CHO/TCRAct/PD-L1 cells alone or in the presence of PD-1/PD-L1 blockers, and the activation of CD4+ and CD8+ T cell is monitored by flow cytometry.
Following the addition of PBMCs to the CHO/TCRAct/PD-L1 cells, the activation of both CD4+ and CD8+ T cells was observed, as monitored by the expression of early (CD69), intermediate (CD25 and HLA-DR), and late (PD-1) activation/exhaustion markers (Figure 4A). This is due to the TCR-dependent activation of T-cells by CHO/TCRAct/PD-L1 cells. As reported before, the addition of PD-1/PD-L1-blockers in such a setup increases the expression of PD-1 on T-cells but rarely other markers.32 In the current experiment, this was confirmed for three therapeutic anti-PD-L1 antibodies, durvalumab, atezolizumab, and avelumab (Figure 4A). Also, for the tested compounds 8h and 8j, a significant increase in the numbers of the PD-1-positive cells was observed (Figure 4A). Surprisingly, for the antibodies, a decrease of CD69 and CD25 expression was observed for CD4+ cells, which has not been observed before in our studies. The addition of compound 8j, but neither 8h nor the therapeutic antibodies, increased the numbers of activated, CD25-positive, and HLA-DR-positive T-cells (Figure 4A). Altogether, the presented data clearly show the biological activity of 8j in a cellular context.
Figure 4.

Effect of 8j and 8h on the reactivation of primary T-cells preblocked by PD-L1-overexpressing cells. (A) Expression of early (CD69), intermediate (CD25, HLA-DR), and late (PD-1) T-cell activation/exhaustion markers was determined by flow cytometry. PBMCs from healthy donors were exposed to CHO/TCRAct/PD-L1 cells for 2 days in the presence of durvalumab (durva), atezolizumab (atezo), avelumab (avelu), or increasing concentrations of 8j and 8h (concentrations: 0.1, 0.5, or 2.5 μM). ø indicates untreated cells. The graphs show fractions of positive cells and represents cumulative data from four donors. Statistical significance was analyzed with one-way ANOVA, followed by Fisher’s posthoc test: *p < 0.05, **p < 0.01, ***p < 0.01. (B) Accumulation of the indicated cytokines in culture media from the TCA assay. The graphs show mean ± standard error of the mean values from four independent donors. For each donor, the data were normalized to untreated coculture of PBMCs and CHO/TCRAct cells as a determinant of donor-dependent cytokine level independent of the PD-L1/PD-1 axis. Statistical significance was analyzed with one-way ANOVA, followed by the Fisher posthoc test: *p < 0.05, **p < 0.01, ***p < 0.001.
In addition to the expression of surface markers, the accumulation of immune-related cytokines in the culture media collected in the TCA assay was monitored with the cytometric bead array (CBA) human Th1/Th2/Th17 kit. In the presence of atezolizumab, significantly increased levels of IL-17A, TNF-α, and IL-10 were observed (Figure 4B). Also, the levels of IL-6, IFN-γ, and IL-2 increased, but these increases were not statistically significant. Similarly, the treatment of PBMCs cocultured with the CHO/TCR-Act/PD-L1 cells with 8j also increased the accumulation of all six cytokines, with significant increases observed for IL-6, TNF-α, IFN-γ, IL-10, and IL-2 (Figure 4B).
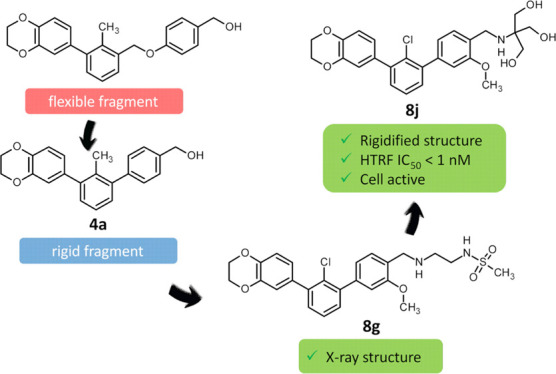
Distal Terphenyl Ring Provides Rigidification That Fits into the PD-L1 Cleft
We were able to crystalize and solve the structure of the 8g compound in complex with PD-L1 at the resolution of 2.3 Å that allowed us to decipher the molecular basis of the interactions between them (Figure 5 and Table S2). The asymmetric unit of the hPD-L1/8g complex contains six protein molecules organized into three dimers, with one inhibitor located at the center of the interface of each dimer. The electron density map describes each inhibitor molecule allowing the position of all moieties in the structure with the fully detailed density for the molecule located at the interface of the AB dimer. 8g is mostly buried in the deep and elongated tunnel, which is composed of two PD-L1 monomers. The 1,4-benzodioxane group creates the standard interaction with ATyr56 (the monomer molecules are annotated by subscripts A and B according to their chain arrangement in the crystal structure of the dimer), previously observed by us for the BMS-202, BMS-1001, and BMS-1166 compounds.30,33 The central terphenyl moiety is stabilized by plenty of hydrophobic contacts with BTyr56, AMet115, BMet115, AAla121, and BAla121. The chlorine substituent attached to this moiety forms an additional halogen bond with the backbone carbonyl of AAsp122, providing the rationale for the enhanced inhibitory effect of these halogenated inhibitors at the R1 position. The methoxy substituent at the meta position of the distal ring of the terphenyl is also additionally stabilized by hydrophobic interaction with BIle54 and BVal68, pointing out the importance of this substituent. At the other side of the tunnel, the sulfonamide polar extension forms a hydrogen bond with the carbonyl of the AAsp122 side chain and is largely water-exposed.
Figure 5.
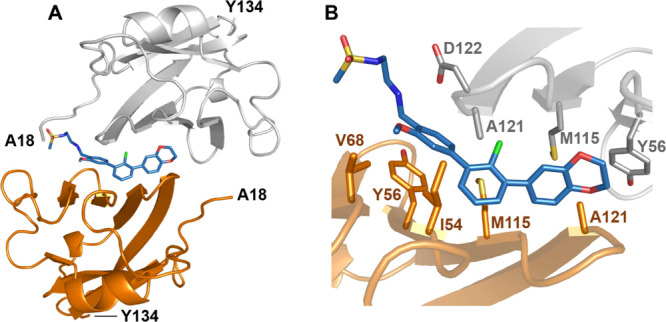
Cocrystal X-ray structure of 8g bound to the Ig-like V-type domain of human PD-L1 (PDB: 7NLD). (A) Arrangement of the molecules in the crystal structure—two PD-L1 molecules (gray and orange) form a single pocket accommodating 8g compound (blues). (B) Detailed interactions of 8g at the binding cleft of PD-L1. The inhibitor binds at a hydrophobic cavity formed upon PD-L1 dimerization. Color coding as in (A).
Our structural data confirm that the terphenyl compounds with the rigidified biphenyl unit bind to PD-L1 at the interaction site that has been seen in the compounds with the biphenyl scaffold. Comparison of the PD-L1/8g, PD-L1/BMS-1166, PD-L1/the C2-symmetric inhibitor, and PD-L1/ARB-272572 complexes shows that the crystallographic arrangement of the terphenyl core is close to the docking results (Figure S2).33−35
Conclusions
In this paper, we have demonstrated a new group of small-molecule anti-PD-L1 inhibitors based on the concept of rigidifying the biphenyl unit by removing the ether linker from the known PD-L1 inhibitors of the BMS family. Such terphenyl-based scaffolds exhibit high inhibitory activity against the PD-1/PD-L1 complex both in common biophysical assays, such as HTRF and NMR, as well as cell-based assays. Our most potent compounds activated Jurkat-ECs to levels comparable to those of the control antibodies, despite being considerably smaller. Similar to other anti-PD-L1 inhibitors reported in the literature, our compounds dimerize human PD-L1, as presented here by NMR and X-ray crystallography studies. They are specific for human PD-L1 and do not cross-react with the murine PD-L1 analogue. Our most potent compound, 8j, is characterized by an exceptionally high affinity to the molecular target (IC50 < 1 nM), as well as a proven potency in cell-based assays with an EC50 of ca. 1 μM. Most importantly, adding compound 8j to the coculture of PBMCs and CHO/TCRAct/PD-L1 cells not only induced PD-1 expression but also increased the numbers of CD25-positive and HLA-DR-positive T cells, which was not observed even in PD-L1 therapeutic antibodies. Also, the accumulation of immune-related cytokines was increased in the presence of either 8j or atezolizumab. This shows the potency of this compound toward primary human T cells and designates 8j as an outstanding candidate for further optimization in the anti-PD-L1 treatments of cancer. The presented crystal structure provides an explanation for the enhanced inhibitory effect of terphenyl-based inhibitors. The introduced phenyl ring replaces the benzyl linker of BMS-1166 and thus provides the rigidification of the structures in the conformation that fits perfectly into the PD-L1 cleft.
Experimental Procedures
Homogeneous Time-Resolved Fluorescence
HTRF was performed using the certified CisBio assay according to the manufacturer’s guidelines. The experiments were performed at 5 nM h-PD-L1 and 50 nM hPD-1 in the final formulation at 20 μL final volume in the well. To determine the half-maximal inhibitory concentration (IC50) of tested compounds, measurements were performed on two individual dilution series, unless states otherwise. After mixing all components according to the CisBio protocol, the plate was left for 2 h incubation at room temperature, followed by time-resolved fluorescence energy transfer (TR-FRET) measurement on Tecan Spark 20M. Collected data were background-subtracted on the negative control, normalized on the positive control, averaged, and fitted with normalized Hill’s equation to determine the IC50 value using Mathematica 12.
Protein Nuclear Magnetic Resonance
NMR spectra were recorded in PBS pH 7.4 containing 10% (v/v) of D2O added to the samples to provide the lock signal. Water suppression was carried out using the WATERGATE sequence. All the spectra were recorded at 300 K using a Bruker AVANCE 600 MHz spectrometer with the cryo-platform. The spectra were recorded at the ligand/protein ratio of 1:1, unless stated otherwise. The samples were prepared by adding small amounts of a 50 mM ligand stock solution in DMSO to the protein solution (0.20 mL) of PD-L1 fragment at a concentration of 0.2 mM. Spectra were visualized using TopSpin 4.0.2.
In Silico Molecular Docking
For molecular docking, we generated structures of BMS core and m-terphenyl derivative in the PDB format using Chem3D 17.1 and used AutoDock Vina1.1.2 integrated into PyRx with the dimeric structure of PD-L1 bound to BMS-1166 (PDB ID: 6R3K).
Protein Expression and Purification
Escherichia coli strain BL21 was transformed with a pET-21b plasmid carrying the PD-L1 gene (amino acids 18–134). The bacteria were cultured in LB at 37 °C until OD600nm of 0.6 when the recombinant protein production was induced with 1 mM IPTG. The protein production was continued overnight. Inclusion bodies were collected by centrifugation, washed twice with 50 mM Tris-HCl pH 8.0 containing 200 mM NaCl, 10 mM ethylenediaminetetraacetic acid, 10 mM 2-mercaptoethanol, and 0.5% Triton X-100, followed by a single wash with the same buffer but with no Triton X-100. The washed inclusion bodies were resuspended overnight in 50 mM Tris-HCl pH 8.0, 6 M GuHCl, 200 mM NaCl, and 10 mM 2-mercaptoethanol and clarified with centrifugation. Refolding of PD-L1 was performed by dropwise dilution into 0.1 M Tris-HCl pH 8.0 containing 1 M l-arginine hydrochloride, 0.25 mM oxidized glutathione, and 0.25 mM reduced glutathione. The refolded protein was dialyzed 3 times against 10 mM Tris-HCl pH 8.0 containing 20 mM NaCl and purified by size exclusion chromatography using Superdex 75 and dialysis buffer. The quality of the refolded protein was evaluated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis and NMR.
PD-L1 Cocrystallization, Determination, and Refinement
Purified PD-L1 in 10 mM Tris-HCl pH 8.0, 20 mM NaCl buffer was concentrated to 5 mg/mL, mixed with the 8g inhibitor in 1:3 molar ratio (protein/compound) and clarified. The supernatant was used for screening using commercially available buffer sets. Diffraction-quality crystals were obtained at room temperature from the condition containing 0.1 M sodium cocodylate pH 6.5, 25% PEG 4000. The crystal was flash-cooled in liquid nitrogen. The X-ray diffraction data were collected at the BL14.1 beamline operated by the Helmholtz-Zentrum Berlin (HZB) at the BESSY II (Berlin Adlershof, Germany).36 The data were indexed, integrated, and scaled using XDS, XSCALE, and Aimless.37−39 The initial phases were obtained by molecular replacement calculated in Phaser.40 The model building was performed in Coot and refinement was performed using Phenix or PDB-REDO server.41−43 Water molecules were added automatically and inspected manually. The coordinates and structure factors were deposited in the Protein Data Bank with accession number 7NLD.
Cell Culture
CHO-K1 cells, CHO-K1 cells overexpressing TCR activator (CHO/TCRAct), PD-L1-expressing CHO/TCRAct cells (CHO/TCRAct/PD-L1), and Jurkat-EC T-cells overexpressing PD-1 and carrying a luciferase reporter gene under the control of the nuclear factor of activated T-cell response element (NFAT-RE) were obtained from Promega and cultured in RPMI-1640 medium (Biowest) supplemented with 10% fetal bovine serum (FBS, Biowest) and 200 mM l-glutamine (Biowest). G418 (250 μg/mL, InvivoGen) and Hygromycin B Gold (50 μg/mL, InvivoGen) were also added to the culture medium as selection antibiotics. The overexpression of PD-L1 and TCR ligand in CHO/TCRAct/PD-L1 cells and overexpression of PD-1 in Jurkat-ECs were confirmed by flow cytometry and western blot analysis. PCR tests for Mycoplasma sp. contamination44 were routinely performed, which indicated negative results for both cell lines.
Cytotoxicity Assay
The cytotoxicity of compounds was verified toward Jurkat-ECs since these cells provide the readout in the ICB assay and present a much higher degree of susceptibility to the compounds’ toxicity than CHO-K1 cells (not shown). Jurkat-ECs were seeded on 96-well transparent plates in the presence of increasing concentrations of the compounds with DMSO-treated cells as a control. The concentration of DMSO was kept constant (0.1%). After 48 h of incubation, a tetrazolium reagent, Biolog redox dye MIX MB (Biolog), was added according to the manufacturer’s protocol, and the culture plates were incubated for an additional 2 h (37 °C, 5% CO2). The absorbance was measured using a Spark microplate reader (Tecan) at 590 nm with 750 nm as a reference. The data are presented as Jurkat-EC survival relative to DMSO-treated control cells. Data points represent mean ± SD values from three independent experiments. Half-maximal effective concentrations (EC50 values) were calculated from the dose–response curve using OriginPro 2020 (OriginLab) software.
PD-1/PD-L1 ICB Assay
The functional assay of the blockade of PD-1/PD-L1 interaction in vitro (PD-1/PD-L1 Blockade Bioassay, Promega) was performed according to the manufacturer’s protocol. CHO/TCRAct/PD-L1 cells or CHO/TCRAct or CHO-K1 cells were seeded on 96-well white plates at the density of 10,000 cells/well and the next day overlaid with Jurkat T cells (20,000 cells/well) in the presence of increasing concentrations of the compounds with DMSO only as a control (the concentration of DMSO was kept constant at 0.1%) or atezolizumab, as a positive control, anti-PD-L1 monoclonal antibody (Selleckchem). The 2.5-fold dilutions of atezolizumab were prepared in the assay buffer (RPMI 1640 + 1% FBS) on the day of the assay. Activation of the Jurkat T cells, reflected by luciferase activity, was monitored by luminescence measurements after 6 h of incubation (37 °C, 5% CO2), and 20 min of additional incubation with the Bio-Glo assay reagent (Promega) at room temperature. The luminescence was read on a Spark microplate reader (Tecan). The data are presented as fold induction of the luminescence signal relative to either untreated (for atezolizumab) or DMSO-treated (for compound) cells. Data points represent mean ± SD values from three to five independent experiments. Half-maximal effective concentrations (EC50 values) were calculated from the Hill curve fitting to the experimental data using Origin Pro 2020 software (OriginLab).
Isolation of PBMCs and TCA Assay
Anticoagulant citrate dextrose-A-treated blood from healthy donors was purchased from the Regional Center of Blood Donation and Blood Therapy in Krakow, Poland. PBMCs were isolated from whole blood by density gradient centrifugation using Pancoll human separating solution (PAN-Biotech GmbH, Aidenbach, Germany). The separated cells were washed and resuspended in RPMI 1640 medium (Biowest) containing 10% FBS (Biowest). PBMCs were added at an amount of 3 × 105 to CHO, CHO/TCRAct, or CHO/TCRAct/PD-L1 cells preseeded on 24-well plates in the presence of either anti-PD-L1 mAbs (at the final concentration 5 μg/mL): durvalumab (Selleckchem), atezolizumab (Selleckchem), or avelumab (MedChemExpress) or tested compounds at the concentrations 0.1, 0.5, and 2.5 μM. Untreated cells were used as controls for antibody treatment, while DMSO-treated cells [0.1% (v/v)] were used as controls for compound treatment. The cells were incubated for 48 h (37 °C, 5% CO2) and then detached from the plates with TrypLe Select Enzyme (Thermo Fisher). PBMCs were stained for 20 min at room temperature with the antibodies: anti-CD4-FITC, anti-CD8-BV510, anti-CD69-APC, anti-CD25-PE, anti-HLA-DR-PerCP, and anti-PD1-PECy7 (Becton Dickinson Biosciences, BD). Following two washing steps, the cells were analyzed with a FACSCanto II cytometer. Data analysis was carried out with FlowJo software, followed by statistical significance calculations done with Origin Pro 2020 software (OriginLab). Statistical significance was analyzed with one-way analysis of variance (ANOVA), followed by the Fisher’s posthoc test: *p < 0.05, **p < 0.01, ***p < 0.01.
Analysis of Cytokine Accumulation
The concentration of the cytokines (IL-2, IL-4, IL-6, IL-10, IL-17A, IFN-γ, and TNFα) in supernatants was determined using the CBA Human Th1/Th2/Th17 kit (BD Biosciences), with data acquisition using the FACSCanto flow cytometer. The CBA beads were discriminated in FL-4 and FL-5 channels, while the concentration of specified cytokines was determined by the intensity of FL-2 fluorescence, using the respective standard reference curve and FCAP Array software (BD Biosciences). IL-4 was below the detection level and is not presented in the manuscript. Statistical significance was analyzed with one-way ANOVA using the Origin Pro 2020 software, followed by the Fisher’s posthoc test: *p < 0.05, **p < 0.01, ***p < 0.01.
Syntheses
All syntheses were performed according to the procedures summarized in Schemes 1-3 and as described in the Supporting Information. Reagents were obtained from commercial suppliers (Sigma-Aldrich, ABCR, Acros Organics, Appollo Scientific, AK Scientific) and used without further purification unless otherwise noted. The anhydrous solvents were purchased from Sigma-Aldrich or Alfa Aesar. Nuclear magnetic resonance spectra were recorded at 300K on a Bruker AVANCE 600 spectrometer {1H NMR (600 MHz) and 13C NMR (151 MHz)}. Chemical shifts for 1H NMR and 13C NMR were reported in parts per million (δ) referenced to the appropriate deuterated solvent. Coupling constants were reported in hertz (Hz). The following abbreviations were used for spin multiplicity: s = singlet, sbroad = broad singlet, d = doublet, t = triplet, q = quartet, quin = quintet, dd = double of doublets, ddd = double doublet of doublets, and m = multiplet. IR absorption spectra were recorded on a Nicolet IR200 spectrometer using the attenuated total reflection (ATR) technique. Thin-layer chromatography (TLC) was performed on Sigma-Aldrich precoated silica gel plates (0.20 mm thick, particle size 25 mm). Spots were visualized by UV light at 254 and 365 nm. Column chromatography was performed using silica gel 60 (40–63 mm, Sigma-Aldrich). Flash chromatography was performed on a Reveleris X2 flash chromatography, using Grace Reveleris silica flash cartridges. The liquid chromatography–mass spectrometry (LC–MS) measurements were performed on an ultraperformance liquid chromatography (UPLC)–tandem mass spectrometry system consisting of a Waters ACQUITY UPLC (Waters Corporation, Milford, MA, USA) coupled to a Waters TQD mass spectrometer (electrospray ionization mode ESI-tandem quadrupole). Chromatographic separations were carried out using the Acquity UPLC BEH (bridged ethyl hybrid) C18 column, 2.1 × 100 mm and 1.7 μm particle size, equipped with an Acquity UPLC BEH C18 VanGuard precolumn; 2.1 × 5 mm and 1.7 μm particle size. The column was maintained at 40 °C and eluted under gradient conditions using 95–0% of eluent A over 10 min at a flow rate of 0.3 mL min–1. Eluent A: water/formic acid (0.1%, v/v); eluent B: acetonitrile/formic acid (0.1%, v/v). The purity of all final compounds determined using chromatographic LC–MS was >95%. HRMS was carried out by the Laboratory for Forensic Chemistry Faculty of Chemistry, Jagiellonian University, with a microOTOF-QII (Bruker) spectrometer using the ESI ionization technique.
General Procedure for Preparation of 3-Bromobiphenyl Intermediates (2a–2c)
A three-necked round-bottom flask was charged with appropriate halide 1a–1c, 1,4-benzodioxane-6-boronic acid, K2CO3, and dioxane/water mixture (2:1, 7/14 mL for 1 mmol) under an argon atmosphere. The mixture was deoxygenated by rinsing with argon for half an hour; then, the Pd(dppf)Cl2 dichloromethane complex was added. The reaction mixture was heated at 80 °C, using preheated bath, for 3 h and after this time, the progression of the reaction was controlled using TLC analysis (SiO2, hexane/ethyl acetate, 4:1). When the reaction was complete, water was added and the extraction with ethyl acetate followed. The organic phases were combined, dried over anhydrous MgSO4, and evaporated. The crude product was purified by flash chromatography (SiO2, hexane/ethyl acetate), giving the final products 2a–2c with 47–61% yield.
6-(3-Bromo-2-methylphenyl)-1,4-benzodioxane (2a)
2,6-Dibromotoluene (1a) (1.93 mL, 14.0 mmol, 1.4 equiv), 1,4-benzodioxane-6-boronic acid (1.79 g, 10.0 mmol, 1.0 equiv), K2CO3 (5.53 g, 40.0 mmol, 4.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.41 g, 0.5 mmol, 0.05 equiv). Product 2a was obtained as a colorless oil with 52.7% (1.61 g) yield. Compound 2a was previously described in patent application.45
Rf = 0.51 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.52 (dd, J = 8.0, 1.2 Hz, 1H), 7.14 (dd, J = 7.6, 1.1 Hz, 1H), 7.05 (t, J = 7.7 Hz, 1H), 6.90 (d, J = 8.2 Hz; 1H), 6.79 (d, J = 2.1 Hz, 1H), 6.74 (dd, J = 8.2, 2.2 Hz, 1H), 4.31 (s, 4H), 2.33 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.6, 143.2, 143.0, 135.7, 135.2, 131.5, 129.2, 126.8, 126.3, 122.5, 118.2, 117.1, 64.6, 21.2; IR (ATR) [cm–1]: 2927, 2874, 1582, 1505, 1457, 1303, 1276, 1240, 1066, 993, 897, 878, 820, 789, 780; LC–MS (DAD/ESI): tR = 9.03 compound did not ionize.
6-(2-Amino-3-bromophenyl)-1,4-benzodioxane (2b)
2,6-Dibromoaniline (1b) (5.27 g, 21.0 mmol, 1.4 equiv), 1,4-benzodioxane-6-boronic acid (2.70 g, 15.0 mmol, 1.0 equiv), K2CO3 (8.29 g, 60.0 mmol, 4.0 equiv), Pd(PPh3)4 (0.87 g, 0.75 mmol, 0.05 equiv). Product 2b was obtained as a colorless solid with 61.0% (2.80 g) yield. Product 2b was crystallized from cyclohexane.
Rf = 0.36 (SiO2, hexane/ethyl acetate 4:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.39 (dd, J = 8.0, 1.5 Hz, 1H), 7.02 (dd, J = 7.5, 1.4 Hz, 1H), 6.95–6.93 (m, 2H), 6.89 (dd, J = 8.3, 2.0 Hz, 1H), 6.65 (t, J = 7.3 Hz, 1H), 4.30 (s, 4H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.9, 143.3, 141.6, 132.4, 131.7, 129.6, 122.2, 119.0, 118.0, 117.9, 109.9, 64.6, 64.6; IR (ATR) [cm–1]: 3481,3389, 2932, 2876, 1603, 1504, 1462, 1308, 1239, 1055, 898, 881, 821, 780, 737; LC–MS (DAD/ESI): tr = 7.77 min, calcd for C14H12BrNO2 (m/z) [M + H]+ 306.01; found, [M + H]+ 306.10; HRMS (ESI): calcd for C14H12BrNO2 (m/z) [M + H]+ 306.0124; found, [M + H]+ 306.0123.
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c)
1,3-Dibromo-2-chlorobenzene (1c) (5.16 g, 19.09 mmol, 1.1 equiv), 1,4-benzodioxane-6-boronic acid (3.12 g, 17.4 mmol, 1.0 equiv), K2CO3 (9.62 g, 69.6 mmol, 4.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.71 g, 0.87 mmol, 0.05 equiv). Product 2c was obtained as a salmon solid with 46.8% (2.65 g) yield.
Rf = 0.38 (SiO2, hexane/ethyl acetate 4:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.59 (dd, J = 8.0, 1.6 Hz, 1H), 7.24 (dd, J = 7.6, 1.6 Hz, 1H), 7.13 (t, J = 7.8 Hz, 1H), 6.93–6.91 (m, 2H), 6.87 (dd, J = 8.3, 2.1 Hz, 1H), 4.30 (s, 4H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.5, 143.1, 142.3, 132.9, 132.9, 132.6, 130.2, 127.4, 123.8, 122.5, 118.3, 117.0, 64.5, 64.4; IR (ATR) [cm–1]: 2978, 2931, 2875, 1506, 1447, 1312; LC–MS (DAD/ESI+): tR = 8.68 compound did not ionize.
General Procedure for the Suzuki Reaction (4a–4e and 4i–4r)
A three-necked round-bottom flask was charged with appropriate halide 2a–2c, borane 3a–3i (for synthesis of compounds 3a–3i, see Supporting Information), K2CO3, and dioxane/water mixture (2:1, 5/10 mL for 1 mmol) under an argon atmosphere. The mixture was deoxygenated by rinsing with argon for half an hour, and then, the Pd(dppf)Cl2 dichloromethane complex was added. The reaction mixture was heated at 80 °C, using preheated bath, for 5 h. After this time, the progression of the reaction was controlled by TLC. When the reaction was complete, water was added and the extraction with ethyl acetate followed. The organic phases were combined, dried over anhydrous MgSO4, and evaporated. The crude product was purified by flash chromatography (SiO2, hexane/ethyl acetate), giving the final products 4a–e and 4i–r with 29–89% yield.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4a)
6-(3-Bromo-2-methylphenyl)-1,4-benzodioxane (2a) (2.98 g, 9.78 mmol, 1.0 equiv), 4-(hydroxymethyl)phenylboronic acid (2.03 g, 13.30 mmol, 1.2 equiv), K2CO3 (4.90 g, 35.56 mmol, 3.2 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.36 g, 0.45 mmol, 0.04 equiv). Product 4a was obtained as a colorless solid with 56.4% (1.62 g) yield.
Rf = 0.23 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.39 (d, J = 8.3 Hz, 2H), 7.33 (dt, J = 8.1, 1.8 Hz, 2H), 7.27 (t, J = 7.6 Hz, 1H), 7.17–7.15 (m, 2H), 6.92 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 2.1 Hz, 1H), 6.82 (dd, J = 8.2, 2.1 Hz, 1H), 5.22 (t, J = 5.8 Hz, 1H), 4.65 (d, J = 5.7 Hz, 2H), 4.28 (s, 4H), 2.07 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 143.4, 143.0, 142.8, 142.3, 141.7, 140.7, 135.4, 132.8, 129.4, 129.1, 129.0, 126.8, 126.0, 122.6, 118.2, 117.2, 64.6, 64.5, 63.2, 19.1; IR (ATR) [cm–1]: 3315, 2930, 1578, 1508, 1318; LC–MS (DAD/ESI): tR = 7.36 min, calcd for C22H20O3 [m/z] [M + H]+ 333.15, [M-H2O + H]+ 315.14; found, [M-H2O + H]+ 315.28; HRMS (ESI): calcd for C22H20O3 [m/z] [M + Na]+ 355.1305; found, [M + Na]+ 355.1301.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methanol (4b)
6-(3-Bromo-2-methylphenyl)-1,4-benzodioxane (2a) (2.98 g, 9.78 mmol, 1.0 equiv), 3-(hydroxymethyl)phenylboronic acid (2.03 g, 13.30 mmol, 1.2 equiv), K2CO3 (4.90 g, 35.56 mmol, 3.2 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.36 g, 0.45 mmol, 0.04 equiv). Product 4b was obtained as a colorless solid with 50.1% (1.48 g) yield.
Rf = 0.24 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.43–7.40 (m, 1H), 7.36 (m, 2H), 7.29 (dt, J = 7.5, 1.2 Hz, 1H), 7.26–7.24 (m, 1H), 7.22–7.19 (m, 2H), 6.91 (d, J = 8,2 Hz, 1H), 6.88 (d, J = 2.0, 1H), 6.83 (dd, J = 8.2, 2.1 Hz, 1H), 4.75 (s, 2H), 4.30 (s, 4H), 2.12 (s, 3H), 1.76 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.1, 142.8, 142.6, 142.6, 142.3, 140.7, 135.8, 133.1, 129.1, 128.8, 128.7, 128.3, 128.0, 125.4, 125.3, 122.6, 118.2, 116.9, 65.4, 64.4, 18.8; IR (ATR) [cm–1]: 3360, 2875, 1578, 1506, 1069; LC–MS (DAD/ESI): tR = 7.48 min, calcd for C22H20O3 [m/z] [M + H]+ 333.15, [M-H2O + H]+ 315.14; found, [M-H2O + H]+ 315.21; HRMS (ESI): calcd for C22H20O3 [m/z] [M + Na]+ 355.1305; found, [M + Na]+ 355.1304.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methanol (4c)
6-(3-Bromo-2-methylphenyl)-1,4-benzodioxane (2a) (2.98 g, 9.78 mmol, 1.0 equiv), 2-(hydroxymethyl)phenylboronic acid (2.03 g, 13.30 mmol, 1.2 equiv), K2CO3 (4.90 g, 35.56 mmol, 3.2 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.36 g, 0.45 mmol, 0.04 equiv). Product 4c was obtained as a colorless solid with 41.9% (1.24 g) yield.
Rf = 0.25 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.59 (dd, J = 7.6, 0.7 Hz, 1H), 7.43 (dt, J = 7.5, 1.4 Hz, 1H), 7.38 (dt, J = 7.5, 1.4 Hz, 1H), 7.28–7.25 (m, 2H), 7.22 (dd, J = 7.5, 1.3 Hz, 1H), 7.14 (dd, J = 6.6, 2.4 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.84 (dd, J = 8.2, 2.1 Hz, 1H), 4.49 (dd, J = 6.0, 1.2 Hz, 2H), 4.33 (s, 4H), 1.97 (s, 3H), 1.57 (t, J = 5.9 Hz, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.1, 142.6, 142.2, 140.9, 140.8, 138.5, 135.6, 133.5, 129.5, 129.3, 128.3, 127.7, 127.6, 127.5, 125.3, 122.5, 118.2, 116.9, 64.4, 63.2, 18.2; IR (ATR) [cm–1]: 3559, 3533, 2933, 1581, 1505, 1317, 1066; LC–MS (DAD/ESI): tR = 7.58 min, calcd for C22H20O3 [m/z] [M-H2O + H]+ 315.14; found, [M-H2O + H]+ 315.21; HRMS (ESI): calcd for C22H20O3: [M + Na]+ 355.1305; found, [M + Na]+ 355.1305.
(2′-Amino-3′-(benzo-1,4-dioxan-6-yl)-[1,1′-biphenyl]-4-yl)methanol (4d)
6-(2-Amino-3-bromophenyl)-1,4-benzodioxane (2b) (6.12 g, 20.00 mmol, 1.0 equiv), 4-(hydroxymethyl)phenylboronic acid (3.04 g, 20.00 mmol, 1.2 equiv), K2CO3 (9.22 g, 66.68 mmol, 4.2 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.68 g, 0.83 mmol, 0.04 equiv). Product 4d was obtained as a colorless solid with 32.0% (1.78 g) yield.
Rf = 0.19 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.53 (m, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.12 (m, 2H), 7.05 (d, J = 2.00 Hz, 1H), 7.00 (dd, J = 8.3, 2.0 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.88 (t, J = 7.5 Hz, 1H), 4.75 (s, 2H), 4.31 (s, 4H), 3.90 (sbroad, 2H), 2.23 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.7, 142.9, 141.0, 139.9, 139.2, 133.0, 129.8, 129.6, 129.5, 127.5, 127.5, 127.5, 122.4, 118.1, 117.6, 65.1, 64.5, 64.4; IR (ATR) [cm–1]: 3466, 3429, 3372, 3350, 3247, 1507, 1317, 1284; LC–MS (DAD/ESI): tR = 6.64 min, calcd for C21H19NO3 [m/z] [M + H]+ 334.14; found, [M + H]+ 334.22; HRMS (ESI): calcd for C21H19NO3 [m/z] [M + Na]+ 356.1257; found, [M + Na]+ 356.1259.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-[1,1′-biphenyl]-4-yl)methanol (4e)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.19 g, 0.59 mmol, 1.0 equiv), 4-(hydroxymethyl)phenylboronic acid (0.11 g, 0.71 mmol, 1.2 equiv), K2CO3 (0.24 g, 1.77 mmol, 4.2 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.048 g, 0.06 mmol, 0.1 equiv). Product 4e was obtained as a colorless solid with 32.6% (0.068 g) yield.
Rf = 0.50 (SiO2, hexane/ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.48 (m, 4H), 7.36–7.30 (m, 3H), 7.03 (d, J = 1.9 Hz, 1H), 6.98–6.95 (m, 2H), 4.79 (s, 2H), 4.33 (s, 4H), 1.75 (s, 1H);13C NMR (151 MHz, CDCl3): δ [ppm] 143.2, 143.0, 141.3, 141.1, 140.1, 139.6, 133.4, 131.0, 130.5, 130.2, 129.8, 126.7, 126.4, 122.8, 118.6, 116.9, 65.2, 64.5, 64.4; IR (ATR) [cm–1]: 3317, 1507, 1455, 1247, 795; LC–MS (DAD/ESI): tR = 7.29 min, calcd for C21H17ClO3 [m/z] [M + H]+ 353.09, [M-H2O + H]+ 335.08; found, [M-H2O + H]+ 335.15; HRMS (ESI): calcd for C21H17ClO3 [m/z] [M + Na]+ 375.0758; found, [M + Na]+ 375.0761.
(3′-(Benzo-1,4-dioxan-6-yl)-3-methoxy-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4i)
6-(3-Bromo-2-methylphenyl)-1,4-benzodioxane (2a) (1.50 g, 4.92 mmol, 1.0 equiv), (2-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (3a) (1.30 g, 4.92 mmol, 1.0 equiv), K2CO3 (2.72 g, 19.66 mmol, 4.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.20 g, 0.25 mmol, 0.05 equiv). Product 4i was obtained as a colorless solid with 68.7% (1.22 g) yield.
Rf = 0.38 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.34 (d, J = 7.5 Hz, 1H), 7.30–7.28 (m, 1H), 7.26–7.23 (m, 2H), 6.96 (dd, J = 7.5, 1.4 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.91 (m, 2H), 6.86 (dd, J = 8.2, 2.1 Hz, 1H), 4.77 (s, 2H), 4.33 (s, 4H), 3.91 (s, 3H), 2.20 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 157.1, 143.5, 143.1, 142.7, 142.6, 142.4, 135.7, 133.1, 129.2, 128.7, 128.5, 127.5, 125.3, 122.5, 121.6, 118.2, 116.9, 111.6, 64.5, 64.4, 62.0, 55.4, 18.8; IR (ATR) [cm–1]: 3547, 2936, 1505, 1302, 1229; LC–MS (DAD/ESI): tR = 7.58 min, calcd for C23H22O4 [m/z] [M + H]+ 363.15; found, [M + H]+ 363.26; HRMS (ESI): calcd for C23H22O4 [m/z] [M + Na]+ 385.1410; found, [M + Na]+ 385.1411.
3-(((3′-(Benzo-1,4-dioxan-6-yl)-4-(hydroxymethyl)-2′-methyl-[1,1′-biphenyl]-3-yl)oxy)methyl)benzonitrile (4j)
6-(3-Bromo-2-methylphenyl)-1,4-benzodioxane (2a) (0.60 g, 2.0 mmol, 1.0 equiv), 3-((2-(hydroxymethyl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)methyl)benzonitrile (3i) (0.86 g, 2.4 mmol, 1.2 equiv), K2CO3 (1.09 g, 7.9 mmol, 4.0 equiv), Pd(PPh3)4 (0.11 g, 0.1 mmol, 0.05 equiv). Product 4j was obtained as a bluish oil with 29.1% (0.266 g) yield.
Rf = 0.45 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.93 (s, 1H), 7.81 (t, J = 7.9 Hz, 2H), 7.62 (t, J = 7.8 Hz, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 7.15 (dd, J = 10.7, 3.5 Hz, 2H), 6.99 (d, J = 1.3 Hz, 1H), 6.95 (dd, J = 7.6, 1.3 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 6.84 (d, J = 2.1 Hz, 1H), 6.80 (dd, J = 8.2, 2.1 Hz, 1H), 5.24 (s, 2H), 5.07 (t, J = 5.6 Hz, 1H), 4.62 (d, J = 5.6 Hz, 1H), 4.28 (s, 4H), 1.99 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 154.3, 143.0, 142.5, 142.3, 141.8, 141.4, 139.2, 134.9, 132.4, 132.1, 131.5, 130.7, 129.7, 129.4, 128.7, 128.4, 127.2, 125.5, 122.1, 121.3, 118.8, 117.7, 116.8, 112.9, 111.4, 67.9, 64.1, 57.9, 18.6; IR (ATR) [cm–1]: 3390, 2924, 2873, 2230, 1574, 1505, 1459, 1402, 1301, 1278, 1221, 1066, 894, 791; LC–MS (DAD/ESI): tR = 8.16 min, calcd for C30H25NO4 (m/z) [M-H2O + H]+ 446.18; found, [M-H2O + H]+ 446.34; HRMS (ESI): calcd for C30H25NO4 (m/z) [M + Na]+ 486.1676; found, [M + Na]+ 486.1676.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.46 g, 1.41 mmol, 1.0 equiv), (2-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (3a) (0.37 g, 1.41 mmol, 1.00 equiv), K2CO3 (0.78 g, 5.63 mmol, 4.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.06 g, 0.07 mmol, 0.05 equiv). Product 4k was obtained as a colorless solid with 70.0% (0.377 g) yield.
Rf = 0.38 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.35 (d, J = 7.4 Hz, 1H), 7.33–7.28 (m, 3H), 7.02 (dd, J = 7.5, 1.5 Hz, 1H), 7.00 (m, 2H), 6.99–6.92 (m, 2H), 4.74 (d, J = 6.4 Hz, 2H), 4.31 (s, 4H), 3.89 (s, 3H), 2.30 (t, J = 6.5 Hz, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 156.9, 143.2, 143.0, 141.4, 141.1, 141.1, 133.3, 131.0, 130.6, 130.1, 128.4, 128.3, 126.4, 122.8, 121.8, 118.6, 116.9, 111.9, 64.5, 64.4, 62.0, 55.4; IR (ATR) [cm–1]: 3530, 2936, 1506, 1460, 1303, 1227; LC–MS (DAD/ESI): tR = 7.43 min, calcd for C22H19ClO4 [m/z] [M + H]+ 383.10, [M-H2O + H]+ 365.09; found, [M-H2O + H]+ 365.19; HRMS (ESI): calcd for C22H19ClO4 [m/z] [M + Na]+ 405.0864; found, [M + Na]+ 405.0863.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-ethoxy-[1,1′-biphenyl]-4-yl)methanol (4l)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.50 g, 1.53 mmol, 1.2 equiv), (2-ethoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (3b) (0.35 g, 1.27 mmol, 1.0 equiv), K2CO3 (0.70 g, 5.10 mmol, 4.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.05 g, 0.06 mmol, 0.05 equiv). Product 4l was obtained as a colorless solid with 37.9% (0.191 g) yield.
Rf = 0.40 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.38 (m, 1H), 7.35–7.31 (m, 3H), 7.05–7.03 (m, 2H), 7.02 (m, 1H), 6.99–6.95 (m, 2H), 4.79 (s, 2H), 4.33 (s, 4H), 4.16 (q, J = 7.0 Hz, 2H), 2.58 (sbroad, 1H), 1.48 (t, J = 7.0 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 156.3, 143.2, 143.0, 141.4, 141.1, 140.9, 133.3, 131.0, 130.5, 130.1, 128.4, 128.4, 126.4, 122.8, 121.7, 118.6, 116.9, 112.8, 64.5, 64.4, 63.7, 62.1, 14.9; IR (ATR) [cm–1]: 3219, 2972, 1509, 1399, 1279, 1070, 1052, 1044; LC–MS (DAD/ESI): tR = 7.92 min, calcd for C23H21ClO4 [m/z] [M + H]+ 397.12 [M-H2O + H]+ 379.11; found, [M-H2O + H]+ 379.21; HRMS (ESI): calcd for C23H21ClO4 [m/z] [M + Na]+ 419.1021; found, [M + Na]+ 419.1021.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-isopropoxy-[1,1′-biphenyl]-4-yl)methanol (4m)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.33 g, 1.03 mmol, 1.0 equiv), (2-izopropoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (3c) (0.36 g, 1.23 mmol, 1.2 equiv), K2CO3 (0.42 g, 3.08 mmol, 3.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.08 g, 0.10 mmol, 0.1 equiv). Product 4m was obtained as a colorless oil with 60.8% (0.256 g) yield.
Rf = 0.4 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.34–7.32 (m, 4H), 7.01–7.00 (m, 2H), 6.98 (dd, J = 7.6, 1.5 Hz, 1H), 6.96–6.92 (m, 2H), 4.72 (s, 2H), 4.66 (hept, J = 6.0 Hz, 1H), 4.31 (s, 4H), 1.39 (d, J = 6.0 Hz, 6H); 13C NMR (151 MHz, CDCl3): δ [ppm] 155.3, 143.3, 143.1, 141.5, 141.2, 140.9, 133.4, 131.1, 130.6, 130.2, 129.3, 128.7, 126.5, 122.9, 121.6, 118.7, 117.0, 114.3, 70.4, 64.6, 64.5, 62.6, 22.4; IR (ATR) [cm–1]: 3419, 3053, 2976, 2931, 2874, 1583, 1507, 1456, 1302, 1280, 1248, 1225, 1068, 797; LC–MS (DAD/ESI): tR = 8.23 min, calcd for C24H23ClNO4 (m/z) [M-H2O + H]+ 393.13; found, [M-H2O + H]+ 393.23; HRMS (ESI): calcd for C24H23ClO4 (m/z) [M + Na]+ 433.1177; found, [M + Na]+ 433.1177.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-isopentyloxy-[1,1′-biphenyl]-4-yl)methanol (4n)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.48 g, 1.47 mmol, 1.0 equiv), (2-izopentyloxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (3d) (0.66 g, 2.06 mmol, 1.4 equiv), K2CO3 (0.61 g, 4.41 mmol, 3.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.12 g, 0.15 mmol, 0.1 equiv). Product 4n was obtained as a colorless oil with 55.8% (0.361) yield.
Rf = 0.16 (SiO2, hexane/ethyl acetate 4:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.35 (m, 4H), 7.02–6.98 (m, 3H), 6.96–6.92 (m, 2H), 4.75 (s, 2H), 4.31 (s, 4H), 4.08 (t, J = 6.6 Hz, 2 H), 2.39 (sbroad, 1H), 1.89–1.79 (m, 1H), 1.73 (q, J = 6.6 Hz, 2H), 0.97 (d, J = 6.6 Hz, 6H); 13C NMR (151 MHz, CDCl3): δ [ppm] 156.6,143.3, 143.1, 141.6, 141.2, 141.1, 1335., 131.1, 130.7, 130.2, 128.5, 128.4, 126.5, 122.9, 121.8, 118.7, 117.0, 112.8, 66.7, 64.6, 64.6, 62.4, 38.2, 25.4, 227; IR (ATR) [cm–1]: 3376, 3055, 2955, 2930, 2871, 1583, 1507, 1456, 1303, 1280, 1248, 1225, 1068, 797; LC–MS (DAD/ESI): tR = 9.16 min, calcd for C26H27ClO4 (m/z) [M-H2O + H]+ 421.16; found, [M-H2O + H]+ 421.16; HRMS (ESI): calcd for C26H27ClO4 (m/z) [M + Na]+ 461.1490; found, [M + Na]+ 461.1490.
2-((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-4-(hydroxymethyl)-[1,1′-biphenyl]-3-yl)oxy)acetonitrile (4o)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.40 g, 1.24 mmol, 1.0 equiv), 2-(2-hydroxymethyl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)acetonitrile (3e) (0.50 g, 1.72 mmol, 1.4 equiv), K2CO3 (0.51 g, 3.72 mmol, 3.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.10 g, 0.12 mmol, 0.1 equiv). Product 4o was obtained as a colorless solid with 56.0% (0.283 g) yield.
Rf = 0.24 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.49 (d, J = 7.7 Hz, 1H), 7.36–7.32 (m, 2H), 7.29 (dd, J = 6.5, 2.8 Hz, 1H), 7.19 (dd, J = 7.7, 1.4 Hz, 1H), 7.06 (d, J = 1.3 Hz, 1H), 7.00–6.99 (m, 1H), 6.96–6.91 (m, 2H), 4.86 (s, 2H), 4.78 (s, 2H), 4.31 (s, 4H), 1.90 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 153.8, 143.4, 143.2, 141.4, 141.3, 133.2, 131.0, 130.2, 129.5, 129.2, 126.7, 124.5, 122.9, 118.7, 117.0, 115.0, 113.4, 64.6, 64.5, 60.8, 54.0; IR (ATR) [cm–1]: 3524, 3053, 2985, 2944, 2886, 1586, 1510, 1400, 1321, 1283, 1244, 1069, 1048, 890, 787; LC–MS (DAD/ESI): tR = 6.99 min, calcd for C23H18ClNO4 (m/z) [M-H2O + H]+ 390.09; found, [M-H2O + H]+ 390.18; HRMS (ESI): calcd for C23H28ClNO4 (m/z) [M + Na]+ 430.0816; found, [M + Na]+ 430.0817.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-cyclobutylmethoxy-[1,1′-biphenyl]-4-yl)methanol (4p)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.49 g, 1.50 mmol, 1.0 equiv), (2-(cyclobutylmethoxy)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (3f) (0.67 g, 2.10 mmol, 1.4 equiv), K2CO3 (0.62 g, 4.48 mmol, 3.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.12 g, 0.15 mmol, 0.1 equiv). Product 4p was obtained as a colorless solid with 64.8% (0.425 g) yield.
Rf = 0.16 (SiO2, hexane/ethyl acetate 4:1); IR (ATR) [cm–1]: 3414, 2976, 2935, 2869, 1583, 1507, 1456, 1303, 1280, 1249, 1226, 1069, 1046, 797, 746; 1H NMR (600 MHz, CDCl3): δ [ppm] 7.34–7.27 (m, 4H), 7.02–6.98 (m, 3H), 6.96–6.92 (m, 2H), 4.75 (s, 2H), 4.31 (s, 4H), 4.02 (d, J = 6.6. Hz, 2H), 2.8702.78 (m, 1H), 2.19–2.12 (m, 2H), 2.02–1.85 (m, 4H); 13C NMR (151 MHz, CDCl3): δ [ppm] 156.7, 143.3, 143.1, 141.5, 141.2, 141.1, 133.5, 131.1, 130.7, 130.2, 128.6, 128.4, 126.5, 1229., 121.8, 118.7, 117.0, 113.0, 72.2, 64.6, 64.6, 62.5, 34.7, 25.0, 18.8; LC–MS (DAD/ESI): tR = 9.00 min, calcd for C26H25ClNO4 (m/z) [M-H2O + H]+ 419.14; found, [M-H2O + H]+ 419.24; HRMS (ESI): calcd for C26H25ClO4 (m/z) [M + Na]+ 459.1334; found, [M + Na]+ 459.1333.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-cyclopentylmethoxy-[1,1′-biphenyl]-4-yl)methanol (4q)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.29 g, 0.89 mmol, 1.0 equiv), (2-(cyclobutylmethoxy)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (3g) (0.42 g, 1.26 mmol, 1.4 equiv), K2CO3 (0.37 g, 2.68 mmol, 3.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.07 g, 0.09 mmol, 0.1 equiv). Product 4q was obtained as a colorless solid with 62.4% (0.251 g) yield.
Rf = 0.16 (SiO2, hexane/ethyl acetate 4:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.35 (m, 4H), 7.02–6.97 (m, 3H), 6.96–6.91 (m, 2H), 4.75 (s, 2H), 4.31 (s, 4H), 3.93 (d, J = 6.9 Hz, 1H), 2.45–2.38 (m, 1H), 1.90–1.83 (m, 2H), 1.69–1.57 (m, 4H), 1.41–1.33 (m, 2H); 13C NMR (151 MHz, CDCl3): δ [ppm] 156.7, 143.3, 143.1, 141.6, 141.2, 141.1, 133.5, 131.1, 130.6, 128.5, 128.4, 126.5, 122.9, 121.7, 118.7, 117.0, 112.8, 72.4, 64.6, 64.6, 62.5, 39.2, 29.7, 25.6; IR (ATR) [cm–1]: 3390, 3054, 2952, 2868, 1583, 1507, 1456, 1303, 1280, 1249, 1225, 1069, 1040, 797, 747; LC–MS (DAD/ESI): tR = 9.38 min, calcd for C27H27ClO4 (m/z) [M-H2O + H]+ 433.16; found, [M-H2O + H]+ 433.25; HRMS (ESI): calcd for C27H27ClO4 (m/z) [M + Na]+ 473.1490; found, [M + Na]+ 473.1488.
3′-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-4-(hydroxymethyl)-[1,1′-biphenyl]-3-yl)oxy)methyl)-[1,1′-biphenyl]-3-carbonitrile (4r)
6-(3-Bromo-2-chlorophenyl)-1,4-benzodioxane (2c) (0.28 g, 0.86 mmol, 1.0 equiv), 3′-((2-(hydroxymethyl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)methyl)-[1,1′-biphenyl]-3-carbonitrile (3h) (0.38 g, 0.86 mmol, 1.0 equiv), K2CO3 (0.48 g, 3.45 mmol, 4.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.03 g, 0.04 mmol, 0.05 equiv). Product 4r was obtained as a colorless oil with 88.8% (0.427 g) yield.
Rf = 0.37 (SiO2, hexane/ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.86 (t, J = 1.6 Hz, 1H), 7.80 (m, 1H), 7.64 (dt, J = 7.7, 1.3 Hz, 1H), 7.62 (s, 1H), 7.56–7.48 (m, 4H), 7.41 (d, J = 7.6 Hz, 1H), 7.33–7.26 (m, 3H), 7.10 (d, J = 1.4 Hz, 1H), 7.06 (dd, J = 7.6, 1.5 Hz, 1H), 6.98 (t, J = 1.2 Hz, 1H), 6.92 (m, 2H), 5.22 (s, 2H), 4.81 (s, 2H), 4.30 (s, 4H), 2.27 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 155.8, 143.2, 143.0, 142.1, 141.2, 141.1, 141.0, 139.4, 137.7, 133.2, 131.6, 130.9, 130.8, 130.6, 130.1, 129.7, 129.6, 128.8, 128.6, 127.4, 126.9, 126.4, 126.2, 122.8, 122.4, 118.8, 118.6, 116.9, 113.4, 113.1, 70.0, 64.5, 64.4, 61.8; IR (ATR) [cm–1]: 3334, 3059, 2931, 2229, 1585, 1507, 1397, 1246, 781; LC–MS (DAD/ESI): tR = 8.83 min, calcd for C35H26ClNO4 [m/z] [M + H]+ 560.16 [M-H2O + H]+ 542.16; found, [M-H2O + H]+ 542.32; HRMS (ESI): calcd for C35H26ClNO4 (m/z) [M + Na]+ 582.1443; found, [M + Na]+ 582.1440.
Synthesis of C2′ Halogenated and C2′-Cyano Derivatives by the Sandmeyer Reaction (4f–h)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-bromo-[1,1′-biphenyl]-4-yl)methanol (4f)
In a three-necked round-bottom flask equipped with a thermometer, (2′-amino-3′-(benzo-1,4-dioxan-6-yl)-[1,1′-biphenyl]-4-yl)methanol (4d) (0.22 g, 0.63 mmol, 1 equiv) was added and dissolved in 1.2 mL of 48% HBr. The mixture was cooled in an ice bath and a solution of NaNO2 (0.09 g, 1.35 mmol, 2.0 equiv) in 2 mL of water was added dropwise, maintaining the temperature below 5 °C, and the mixture was stirred for 5 min and a solution of CuBr2 (0.37 g, 2.59 mmol, 4 equiv) in 1.8 mL of 48% HBr was added dropwise, maintaining the temperature below 5 °C. The mixture was heated in 70 °C for 2 h. After this time, the solution was transferred into a separatory funnel, 5 mL of water was added, and the aqueous phase was extracted with ethyl acetate (3 × 15 mL). The organic phases were collected, dried over anhydrous MgSO4, and the solvent was evaporated. The product was purified by flash chromatography (SiO2, hexane/ethyl acetate, 1:2), giving the final product 4f with 9.1% (0.02 g) yield.
Rf = 0.60 (SiO2, hexane/ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.37–2.28 (m, 5H), 7.21–7.16 (m, 2H), 6.88 (m, 1H), 6.86–6.80 (m, 2H), 4.69 (s, 2H), 4.23 (s, 4H), 1.55 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.5, 143.2, 143.1, 142.9, 141.6, 140.1, 135.4, 130.3, 123.0, 129.8, 126.9, 126.6, 123.3, 122.8, 118.5, 116.8, 65.2, 64.5, 64.4; IR (ATR) [cm–1]: 3291, 2927, 1505, 1454, 1251, 1070; LC–MS (DAD/ESI+): tR = 7.35 min, calcd for C21H17BrO3 [M-H2O + H]+ 379.03; found, [M-H2O + H]+ 379.14; HRMS (ESI+): calcd for C21H17BrO3 [m/z] [M + Na]+ 419.0253, [M + 2 + Na]+ 421.0233; found, [M + Na]+ 419.0251, [M + 2 + Na]+ 421.0233.
(3′-(Benzo-1,4-dioxan-6-yl)-2′-iodo-[1,1′-biphenyl]-4-yl)methanol (4g)
In a three-necked round-bottom flask equipped with a thermometer, (2′-amino-3′-(benzo-1,4-dioxan-6-yl)-[1,1′-biphenyl]-4-yl)methanol (4d) (0.21 g, 0.62 mmol, 1 equiv) was added and dissolved in 2 mL of dioxane/water (3/1, v/v) mixture. After dissolution, 2 mL of 10% HCl was added and the mixture was cooled in an ice bath; then, a solution of NaNO2 (0.05 g, 0.62 mmol, 1.0 equiv) in 2 mL of water was added dropwise, maintaining the temperature below 5 °C. The mixture was stirred for 1 h, and a solution of KI (0.42 g, 2.47 mmol, 4 equiv) in 4 mL of water was added dropwise, maintaining the temperature below 5 °C; then, the solution was stirred overnight at room temperature. After this time, the solution was transferred into a separatory funnel, 5 mL of water was added, and the aqueous phase was extracted with ethyl acetate (3 × 15 mL). The organic phases were collected, dried over anhydrous MgSO4, and the solvent was evaporated. The product was purified by flash chromatography (SiO2, hexane/ethyl acetate, 1:2), giving the final product 4g with 27.6% (0.08 g) yield.
Rf = 0.47 (SiO2, hexane/ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.44 (d, J = 8.2 Hz, 2H), 7.37–7.35 (m, 3H), 7.23 (dd, J = 7.6, 1.7 Hz, 1H), 7.21 (dd, J = 7.5, 1.7 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.89 (d, J = 2.1 Hz, 1H), 6.84 (dd, J = 8.3, 2.1 Hz, 1H), 4.77 (d, J = 2.6 Hz, 2H), 4.31 (s, 4H), 1.72 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 147.7, 147.5, 145.1, 143.1, 142.8, 140.2, 139.0, 129.7, 128.8, 128.5, 127.6, 126.5, 122.7, 118.4, 116.7, 104.0, 65.2, 64.5, 64.4; IR (ATR) [cm–1]: 3309, 2955, 1505, 1451, 1252, 1071; LC–MS (DAD/ESI+): tR = 7.47 min, calcd for C21H17IO3 [M-H2O + H]+ 427.02; found, [M-H2O + H]+ 427.13; HRMS (ESI+): calcd for C21H17IO3 [m/z] [M + Na]+ 467.0115; found, [M + Na]+ 467.0115.
3-(3′-(Benzo-1,4-dioxan-6-yl)-4′-(hydroxymethyl)-[1,1′-biphenyl]-2-carbonitrile (4h)
In a three-necked round-bottom flask equipped with a thermometer, (2′-amino-3′-(benzo-1,4-dioxan-6-yl)-[1,1′-biphenyl]-4-yl)methanol (4d) (0.21 g, 0.62 mmol, 1 equiv) was added and dissolved in 6 mL of dioxane/water (2/1, v/v) mixture. After dissolution, 0.2 mL of conc. HCl was added and the mixture was cooled in an ice bath; then, a solution of NaNO2 (0.07 g, 0.96 mmol, 1.5 equiv) in 2 mL of water was added dropwise, maintaining the temperature below 5 °C. The mixture was stirred for 5 min and then added to a previously prepared solution of CuCN (0.08 g, 0.86 mmol, 1.4 equiv) and KCN (0.16 g, 2.37 mmol, 3.8 equiv) in 4 mL of water. The solution was stirred for 4 h. After this time, 3 mL of 2 M NaOH was added and the mixture was transferred into a separatory funnel. The aqueous phase was extracted with ethyl acetate (3 × 15 mL). The organic phases were collected, dried over anhydrous MgSO4, and the solvent was evaporated. The product was purified by flash chromatography (SiO2, hexane/ethyl acetate, 1:2), giving the final product 4h with 20.6% (0.04 g) yield.
Rf = 0.44 (SiO2, hexane/ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.63 (t, J = 7.8 Hz, 1H), 7.58 (m, 2H), 7.50 (d, J = 8.2 Hz, 2H), 7.43–7.41 (m, 2H), 7.10 (d, J = 2.1 Hz, 1H), 7.08 (dd, J = 8.3, 2.2 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 4.77 (s, 2H), 4,31 (s, 4H), 1.82 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 146.6, 146.4, 144.2, 143.5, 141.4, 138.1, 132.3, 131.9, 129.3, 128.8, 128.5, 127.1, 122.3, 118.1, 118.0, 117.5, 110.3, 65.0, 64.5, 64.4; IR (ATR) [cm–1]: 3418, 2931, 2224, 1513, 1461, 1288; LC–MS (DAD/ESI+): tR = 6.60 min, calcd for C22H17NO3 [m/z] [M]+ 343.12; found, [M]+ 343.25; HRMS (ESI+): calcd for C22H17NO3 [m/z] [M + Na]+ 366.1101; found, [M + Na]+ 366.1100.
General Procedure for Williamson Ether Synthesis (5a, 5c, 5e, 5i, 5k, 5m, 5o, and 5q)
Appropriate alcohol 4a–c (1 equiv) was added in a round-bottom flask with anhydrous dichloromethane under an argon atmosphere and cooled in an ice bath; thionyl chloride (3 equiv) was added dropwise, and then, anhydrous DMF (0.2 mL per 2 mmol alcohol) was added. The reaction mixture was stirred approximately 2 h at room temperature and controlled by TLC (SiO2, hexane/ethyl acetate, 4/1). When all alcohol disappeared, the reaction mixture was poured onto saturated NaHCO3 and the aqueous phase extracted three times with dichloromethane. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The crude product was dissolved in anhydrous DMF; then, K2CO3 or Cs2CO3 (3 equiv), appropriate nucleophile (1.0–1.5 equiv), and KI (0.1 equiv) were added. The reaction mixture was heated at 80 °C overnight. After this time, the mixture was transferred into a separatory funnel with water and extracted three times with ethyl acetate. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The products were separated by column chromatography (SiO2, hexane/ethyl acetate, or CH2Cl2).
Methyl 3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)benzoate (5a)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4a) (0.30 g, 0.90 mmol, 1.0 equiv), SOCl2 (0.20 mL, 2.70 mmol, 3.0 equiv), K2CO3 (0.37 g, 2.70 mmol, 3.0 equiv), methyl 3-hydroxybenzoate (0.20 g, 1.35 mmol, 1.5 equiv), KI (0.015 g, 0.09 mmol, 0.1 equiv). Product 5a was obtained as a colorless solid with 77.7% (0.326 g) yield.
Rf = 0.50 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.62 (m, 1H), 7.58 (dt, J = 7.7, 1.0 Hz, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 7.28 (t, J = 7.9 Hz, 1H), 7.18–7.11 (m, 4H), 6.82 (d, J = 8.2 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 6.74 (dd, J = 8.2, 2.1 Hz, 1H), 5.06 (s, 2H), 4.20 (s, 4H), 3.83 (s, 3H), 2.06 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 167.0, 158.8, 143.1, 142.6, 142.4, 142.4, 135.8, 135.0, 133.1, 131.6, 129.7, 129.5, 129.2, 128.9, 127.5, 125.4, 122.6, 122.4, 120.3, 118.2, 116.9, 115.1, 70.1, 64.5, 64.5, 52.2, 18.8; IR (ATR) [cm–1]: 2952, 1712, 1301, 1278, 1234; LC–MS (DAD/ESI): tR = 9.80 min, calcd for C30H26O5 [m/z] [M + H]+ 467.19; found, [M + H]+ 467.41; HRMS (ESI): calcd for C30H26O5 [m/z] [M + Na]+ 489.1672; found, [M + Na]+ 489.1672.
2-Bromo-5-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)benzaldehydem (5c)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4a) (1.20 g, 3.62 mmol, 1.0 equiv), SOCl2 (0.79 mL, 10.86 mmol, 3.0 equiv), K2CO3 (1.50 g, 10.86 mmol, 3.0 equiv), 2-bromo-5-hydroxybenzaldehyde (1.09 g, 5.43 mmol, 1.5 equiv), KI (0.06 g, 0.36 mmol, 0.1 equiv). Product 5c was obtained as a colorless solid with 84.9% (1.58 g) yield.
Rf = 0.55 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 10.33 (s, 1H), 7.55 (m, 2H), 7.48 (d, J = 8.1 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 7.27–7.25 (m, 1H), 7.23–7.17 (m, 2H), 7.15 (dd, J = 8.8, 3.2 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.84 (dd, J = 8.2, 2.1 Hz, 1H), 5.14 (s, 2H), 4.31 (s, 4H), 2.13 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 191.8, 158.4, 143.0, 142.6, 142.6, 142.4, 142.2, 137.5, 134.7, 134.3, 134.0, 133.1, 129.7, 129.2, 128.8, 127.4, 125.3, 123.8, 122.5, 118.3, 118.2, 116.9, 113.8, 70.4, 64.4, 64.4, 18.8; IR (ATR) [cm–1]: 3052, 2953, 2874, 2754, 1688, 1508, 1305, 1167, 993; LC–MS (DAD/ESI): tR = 10.14 min, calcd for C29H23BrO4 [m/z] [M + H]+ 515.08; found, [M + H]+ 515.18; HRMS (ESI): calcd for C29H23BrO4 [m/z] [M + Na]+ 537.0672; found, [M + Na]+ 537.0679.
Methyl 3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)isoxazole-5-carboxylate (5e)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4a) (0.30 g, 0.90 mmol, 1.0 equiv), SOCl2 (0.20 mL, 2.70 mmol, 3.0 equiv), K2CO3 (0.37 g, 2.70 mmol, 3.0 equiv), methyl 3-hydroxyisoxazole-5-carboxylate (0.13 g, 0.90 mmol, 1.0 equiv), KI (0.015 g, 0.09 mmol, 0.1 equiv). Product 5e was obtained as a colorless solid with 69.2% (0.285 g) yield.
Rf = 0.34 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.54 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.1 Hz, 2H), 7.31–7.22 (m, 3H), 6.94 (d, J = 8.2 Hz, 1H), 6.92 (d, J = 2.0 Hz, 1H), 6.86 (dd, J = 8.2, 2.1 Hz, 1H), 6.63 (s, 1H), 5.41 (s, 2H), 4.33 (s, 4H), 3.99 (s, 3H), 2.17 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 171.4, 160.4, 157.1, 143.1, 142.6, 142.4, 142.2, 135.7, 133.7, 133.1, 129.7, 129.3, 128.8, 128.2, 125.4, 122.6, 118.2, 116.9, 101.0, 72.1, 64.5, 64.5, 52.9, 18.8; IR (ATR) [cm–1]: 3144, 1747, 1607, 1504, 1313; LC–MS (DAD/ESI): tR = 8.98 min, decomposition during ionization; HRMS (ESI): calcd for C27H23NO6 [m/z] [M + Na]+ 480.1418; found, [M + Na]+ 480.1418.
Methyl 3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzoate (5i)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methanol (4b) (0.30 g, 0.90 mmol, 1.0 equiv), SOCl2 (0.20 mL, 2.70 mmol, 3.0 equiv), K2CO3 (0.37 g, 2.70 mmol, 3.0 equiv), methyl 3-hydroxybenzoate (0.20 g, 1.35 mmol, 1.5 equiv), KI (0.015 g, 0.09 mmol, 0.1 equiv). Product 5i was obtained as colorless solid with 58.8% (0.247 g) yield.
Rf = 0.47 (SiO2, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.22 (m, 1H), 7.70 (dt, J = 7.7, 1.0 Hz, 1H), 7.49–7.46 (m, 3H), 7.40–7.37 (m, 2H), 7.32–7.25 (m, 3H), 7.23 (ddd, J = 8.2, 2.6, 0.9, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 2.0, 1H), 6.87 (dd, J = 8.2, 2.1 Hz, 1H), 5.20 (s, 2H), 4.33 (s, 4H), 3.95 (s, 3H), 2.17 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 166.9, 158.8, 143.1, 142.9, 142.6, 142.5, 142.4, 136.5, 135.8, 133.1, 131.5, 129.5, 129.2, 129.2, 128.8, 128.6, 128.4, 126.0, 125.4, 122.6, 122.3, 120.3, 118.2, 116.9, 115.2, 70.2, 64.5, 52.2, 18.8; IR (ATR) [cm–1]: 2950, 1722, 1507, 1279; LC–MS (DAD/ESI): tR = 9.81 min, calcd for C30H26O5 [m/z] [M + H]+ 467.19; found, [M + H]+ 467.28; HRMS (ESI): calcd for C30H26O5 [m/z] [M + Na]+ 489.1672; found, [M + Na]+ 489.1671.
2-Bromo-5-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzaldehyde (5k)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methanol (4b) (0.60 g, 1.81 mmol, 1.0 equiv), SOCl2 (0.40 mL, 5.43 mmol, 3.0 equiv), K2CO3 (0.75 g, 5.43 mmol, 3.0 equiv), 2-bromo-5-hydroxybenzaldehyde (0.55 g, 2.72 mmol, 1.5 equiv), KI (0.03 g, 0.18 mmol, 0.1 equiv). Product 5k was obtained as a colorless solid with 78.2% (0.73 g) yield.
Rf = 0.65 (SiO2, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ [ppm] 10.34 (s, 1H), 7.57–7.55 (m, 2H), 7.49–7.45 (m, 2H), 7.43 (m, 1H), 7.37 (dt, J = 7.5, 1.4 Hz, 1H), 7.31–7.28 (m, 1H), 7.26–7.23 (m, 2H), 7.14 (dd, J = 8.8, 3.2 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.86 (dd, J = 8.2, 2.1 Hz, 1H), 5.17 (s, 2H), 4.33 (s, 4H), 2.15 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 191.7, 158.4, 143.1, 143.0, 142.6, 142.4, 142.3, 135.8, 135.7, 134.7, 134.0, 133.1, 129.4, 129.3, 128.8, 128.5, 128.5, 125.9, 125.4, 123.8, 122.6, 118.3, 118.2, 116.9, 113.9, 70.5, 64.5, 18.8; IR (ATR) [cm–1]: 2976, 2928, 2873, 1692, 1507, 1467, 1278, 1229, 1069; LC–MS (DAD/ESI): tR = 10.13 min, calcd for C29H23BrO4 [m/z] [M + H]+ 515.08; found, [M + H]+ 515.22; HRMS (ESI): calcd for C29H23BrO4 [m/z] [M + Na]+ 537.0672; found, [M + Na]+ 537.0672.
Methyl 3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)isoxazole-5-carboxylate (5m)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methanol (4b) (0.30 g, 0.90 mmol, 1.0 equiv), SOCl2 (0.20 mL, 2.70 mmol, 3.0 equiv), K2CO3 (0.37 g, 2.70 mmol, 3.0 equiv), methyl 3-hydroxyisoxazole-5-carboxylate (0.13 g, 0.90 mmol, 1.0 equiv), KI (0.015 g, 0.09 mmol, 0.1 equiv). Product 5m was obtained as a colorless solid with 47.8% (0.197 g) yield.
Rf = 0.42 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.47–7.44 (m, 2H), 7.42 (m, 1H), 7.37 (dt, J = 7.4, 1.6 Hz, 1H), 7.28–7.25 (m, 1H), 7.23–7.20 (m, 2H), 6.91 (d, J = 8.2 Hz, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.83 (dd, J = 8.2, 2.1 Hz, 1H), 6.58 (s, 1H), 5.37 (s, 2H), 4.30 (s, 4H), 3.95 (s, 3H), 2.12 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 171.3, 160.4, 157.1, 143.1, 143.0, 142.6, 142.4, 142.2, 135.7, 135.1, 133.1, 129.7, 129.3, 129.3, 128.8, 128.4, 126.7, 125.4, 122.6, 118.2, 116.9, 101.0, 72.1, 64.5, 52.9, 18.8; IR (ATR) [cm–1]: 3140, 1743, 1506, 1303; LC–MS (DAD/ESI): tR = 9.07 min, calcd for C27H23NO6 [m/z] [M + H]+ 458.16; found, [M + H]+ 458.37; HRMS (ESI): calcd for C27H23NO6 [m/z] [M + Na]+ 480.1418; found, [M + Na]+ 480.1418.
Methyl 3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzoate (5o)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methanol (4c) (0.30 g, 0.90 mmol, 1.0 equiv), SOCl2 (0.20 mL, 2.70 mmol, 3.0 equiv), K2CO3 (0.37 g, 2.70 mmol, 3.0 equiv), methyl 2-hydroxybenzoate (0.20 g, 1.35 mmol, 1.5 equiv), KI (0.015 g, 0.09 mmol, 0.1 equiv). Product 5o was obtained as a colorless solid with 49.0% (0.206 g) yield.
Rf = 0.58 (SiO2, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.65 (m, 2H), 7.53 (m, 1H), 7.46–7.42 (m, 2H), 7.34–7.30 (m, 2H), 7.28–7.24 (m, 2H), 7.21 (dd, J = 7.0, 1.8 Hz, 1H), 7.04 (dd, J = 8.2, 2.6 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.81 (dd, J = 8.2, 2.0 Hz, 1H), 4.92 (s, 2H), 4.32 (s, 4H), 3.93 (s, 3H), 2.05 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 166.9, 158.7, 143.1, 142.6, 142.2, 141.7, 140.5, 135.6, 134.3, 133.6, 131.4, 123.0, 129.4, 129.4, 128.5, 128.3, 128.0, 127.6, 125.2, 122.5, 122.2, 120.1, 118.2, 116.9, 115.3, 68.3, 64.4, 52.2, 18.3; IR (ATR) [cm–1]: 2949, 1722, 1507, 1278; LC–MS (DAD/ESI): tR = 9.75 min, calcd for C30H26O5 [m/z] [M + H]+ 467.19, [M-OCH3]+ 435.16; found, [M-OCH3]+ 435.31; HRMS (ESI): calcd for C30H26O5 [m/z] [M + Na]+ 489.1672; found, [M + Na]+ 489.1672.
Methyl 3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methoxy)isoxazole-5-carboxylate (5q)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methanol (4c) (0.30 g, 0.90 mmol, 1.0 equiv), SOCl2 (0.20 mL, 2.70 mmol, 3.0 equiv), K2CO3 (0.37 g, 2.70 mmol, 3.0 equiv), methyl 3-hydroxyisoxazole-5-carboxylate (0.13 g, 0.90 mmol, 1.0 equiv), KI (0.015 g, 0.09 mmol, 0.1 equiv). Product 5q was obtained as a colorless solid with 42.1% (0.173 g) yield.
Rf = 0.35 (SiO2, hexane/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.50 (m, 1H), 7.35–7.31 (m, 2H), 7.19–7.17 (m, 1H), 7.13–7.10 (m, 2H), 7.02 (dd, J = 6.4, 2.6 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H), 6.75 (d, J = 2.0 Hz, 1H), 6.69 (dd, J = 8.2, 2.1 Hz, 1H), 6.38 (s, 1H), 5.05 (d, J = 11.7 Hz, 1H), 5.00 (d, J = 11.7 Hz, 1H), 4.20 (s, 4H), 3.84 (s, 3H), 1.86 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 171.2, 160.2, 157.1, 143.1, 142.6, 142.4, 142.2, 140.2, 135.5, 133.5, 133.1, 130.1, 129.5, 129.2, 128.7, 128.4, 127.6, 125.2, 122.5, 118.2, 116.8, 100.8, 70.3, 64.4, 52.8, 18.2; IR (ATR) [cm–1]: 3140, 2953, 1743, 1505, 1288; LC–MS (DAD/ESI): tR = 9.00 min, calcd for C27H23NO6 [m/z] [M + H]+ 458.16; found, [M + H]+ 458.30; HRMS (ESI): calcd for C27H23NO6 [m/z] [M + Na]+ 480.1418; found, [M + Na]+ 480.1418.
Methyl 1-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methyl)-1H-1,2,3-triazole-4-carboxylate (5g)
(3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4a) (0.33 g, 1.0, 1.0 equiv) was added in a round-bottom flask with anhydrous dichloromethane and triethylamine (0.21 mL, 1.5 mmol, 1.5 equiv) and cooled in an ice bath; then, mesyl chloride (0.12 mL, 1.5 mmol, 1.5 equiv) was added dropwise. The reaction mixture was stirred approximately 2 h at room temperature and controlled by TLC. When all substrate disappeared, the reaction mixture was poured onto water and extracted with dichloromethane. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The crude product was dissolved in anhydrous DMF, and NaN3 (0.097 g, 1.5 mmol, 1.5 equiv) was added. The reaction mixture was stirred at room temperature for 6 days. After this time, water was added and the reaction mixture was extracted with ethyl acetate. The organic layers were collected, washed with brine, dried over anhydrous MgSO4, and evaporated. Crude azide was roughly purified with flash chromatography on a silica pad (SiO2, hexane/ethyl acetate 4:1). The obtained azide (0.25 g, 0.69 mmol, 1.0 equiv) was added to a round-bottom flask, and then, methyl propiolate (0.041 mL, 0.69 mmol, 1.0 equiv) in a THF/water mixture (1:2), sodium ascorbate (0.068 g, 0.32 mmol, 0.5 equiv), and copper(II) sulfate pentahydrate (0.034 g, 0.14 mmol, 0.2 equiv) were added. The reaction mixture was stirred at room temperature for 24 h. After this time, water was added, and the reaction mixture was extracted with ethyl acetate. The organic layers were collected, washed with brine, dried over anhydrous MgSO4, and evaporated. The crude product was crystallized from the cyclohexane/ethyl acetate (4:1), giving the final product 5g with 72.1% (0.220 g)
Rf = 0.88 (SiO2, ethyl acetate/methanol 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 8.00 (s, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 7.19 (t, J = 7.5 Hz, 1H), 7.15 (dd, J = 7.6, 1.6 Hz, 1H), 7.09 (dd, J = 7.3, 1.6 Hz, 1H), 6.83 (d, J = 8.2 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H), 6.74 (dd, J = 8.2, 2.1 Hz, 1H), 5.56 (s, 2H), 4.23 (s, 4H), 3.87 (s, 3H), 2.03 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 161.3, 143.7, 143.2, 142.8, 142.6, 141.8, 140.5, 135.7, 133.1, 132.2, 130.4, 129.6, 128.8, 128.2, 127.6, 125.6, 122.6, 118.3, 117.0, 64.6, 64.6, 54.4, 52.4, 18.9; IR (ATR) [cm–1]: 3142, 2999, 1726, 1509, 1456, 1509, 1438, 1360, 1318, 1244, 1068, 1047, 901, 813, 780; LC–MS (DAD/ESI): tR = 7.78 min, calcd for C26H23N3O4 [m/z] [M + H]+ 442.18; found, [M + H]+ 442.30; HRMS (ESI): calcd for C26H23N3O4 (m/z) [M + Na]+ 464.1581; found, [M + Na]+ 464.1581.
General Procedure of Ester Saponification (5b, 5f, 5h, 5j, 5n, 5p, and 5r)
The appropriate ester was added in a one-necked round-bottom flask and dissolved in a dioxane/water (2:1, 10:5 mL per 1 mmol of ester) mixture. After complete dissolution, LiOH monohydrate was added and the mixture was stirred at room temperature overnight. The progression of the reaction was controlled using TLC analysis (SiO2, ethyl acetate/methanol, or dichloromethane (DCM)/methanol). After completion of the reaction, the mixture was transferred into a separatory funnel, 7 mL of water was added, and then extracted once with a small amount of ethyl acetate. The aqueous phase was acidified to pH ≈ 4 and extracted three times with ethyl acetate. The organic phases obtained from extraction after acidification were collected, washed with brine, dried over anhydrous MgSO4, and the solvent was evaporated. Further purification of the product was not required.
3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)benzoic Acid (5b)
Methyl 3-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)benzoate (5a) (0.093 g, 0.20 mmol, 1.0 equiv), LiOH·H2O (0.016 g, 0.40 mmol, 2.0 equiv). Product 5b was obtained as a colorless solid with 88.8% (0.080 g) yield.
Rf = 0.44 (SiO2, DCM/MeOH, 9:1); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.59–7.57 (m, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.43 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.30 (dd, J = 8.1, 2.4 Hz, 1H), 7.28–7.26 (m, 1H), 7.17 (d, J = 7.6 Hz, 2H), 6.91 (d, J = 8,2 Hz, 1H), 6.86 (d, J = 2.0, 1H), 6.81 (dd, J = 8.2, 2.0 Hz, 1H), 5.21 (s, 2H), 4.27 (s, 4H), 2.07 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 167.6, 158.9, 143.4, 143.0, 142.4, 142.3, 141.9, 135.9, 135.3, 132.8, 132.7, 130.3, 129.7, 129.3, 129.0, 128.1, 126.0, 122.6, 122.3, 120.2, 118.2, 117.2, 115.3, 69.7, 64.6, 64.6, 19.1; IR (ATR) [cm–1]: 2981, 2572, 1683, 1506, 1454, 1314, 1245; LC–MS (DAD/ESI): tR = 8.67 min, calcd for C29H24O5 [m/z] [M – H]− 451.16; found, [M – H]− 451.19; HRMS (ESI): calcd for C29H24O5 [m/z] [M + Na]+ 475.1516; found, [M + Na]+ 475.1517.
3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)isoxazole-5-carboxylic Acid (5f)
Methyl 3-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)isoxazole-5-carboxylate (5e) (0.090 g, 0.20 mmol, 1.0 equiv), LiOH·H2O (0.016 g, 0.40 mmol, 2.0 equiv). Product 5f was obtained as a colorless solid with 65.2% (0.058 g) yield.
Rf = 0.14 (SiO2, ethyl acetate/MeOH, 3:1); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.56 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 7.29 (t, J = 7.6 Hz, 1H), 7.18 (d, J = 7.7 Hz, 2H), 7.00 (s, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 2.0 Hz, 1H), 6.82 (dd, J = 8.2, 2.0 Hz, 1H), 2.17 (s, 2H), 4.28 (s, 4H), 2.07 (s, 3H) 13C NMR (151 MHz, DMSO-d6): δ [ppm] 171.8, 162.2, 158.0, 143.4, 143.0, 142.5, 142.4, 142.3, 135.3, 134.6, 132.8, 129.8, 129.4, 129.0, 128.9, 126.1, 122.6, 118.2, 117.3, 100.7, 71.9, 64.6, 64.6, 19.1; IR (ATR) [cm–1]: 2929, 2606, 1727, 1497, 1274; LC–MS (DAD/ESI): tR = 7.89 min, calcd for C26H21NO6 [m/z] [M – H]− 442.13; found, [M – H]− 442.15; HRMS (ESI): calcd for C26H21NO6 [m/z] [M + Na]+ 466.1261; found, [M + Na]+ 466.1263.
1-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methyl)-1H-1,2,3-triazole-4-carboxylic Acid (5h)
Methyl 1-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methyl)-1H-1,2,3-triazole-4-carboxylate (5g) (0.150 g, 0.34 mmol, 1.0 equiv), LiOH·H2O (0.042 g, 1.00 mmol, 3.0 equiv). Product 5h was obtained as a colorless solid with 61.4% (0.089 g) yield.
Rf = 0.39 (SiO2, ethyl acetate/methanol 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 8.85 (s, 1H), 7.42–7.37 (m, 4H), 7.26 (t, J = 7.6 Hz, 1H), 7.17–7.13 (m, 2H), 6.91 (d, J = 8.2 Hz, 1H), 6.84 (d, J = 2.0 Hz, 1H), 6.80 (dd, J = 8.2, 2.0 Hz, 1H), 5.71 (s, 2H), 4.27 (s, 4H), 2.04 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 162.1, 143.4, 134.0, 142.3, 142.2, 140.4, 135.2, 134.7, 132.8, 130.1, 129.6, 129.4, 129.0, 128.3, 126.1, 122.6, 118.2, 117.2, 64.6, 53.2, 19.1; IR (ATR) [cm–1]: 3115, 2977, 2874, 2559, 1687, 1506, 1439, 1229, 1071, 1053, 899, 783; LC–MS (DAD/ESI): tR = 6.99 min, calcd for C25H21N3O4 [m/z] [M + H]+ 428.16; found, [M + H]+ 428.28; HRMS (ESI): calcd for C25H21N3O4 (m/z) [M + Na]+ 450.1424; found, [M + Na]+ 450.1422.
3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzoic Acid (5j)
Methyl 3-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzoate (5i) (0.093 g, 0.20 mmol, 1.0 equiv), LiOH·H2O (0.016 g, 0.40 mmol, 2.0 equiv). Product 5j was obtained as a colorless solid with 58.8% (0.053 g) yield.
Rf = 0.58 (SiO2, DCM/MeOH, 9:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.66–7.64 (m, 2H), 7.38–7.34 (m, 3H), 7.30 (t, J = 8.2 Hz, 1H), 7.25 (dt, J = 6.9, 1.7 Hz, 1H), 7.19–7.13 (m, 4H), 6.82 (d, J = 8.2 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 6.75 (dd, J = 8.2, 2.0 Hz, 1H), 5.09 (s, 2H), 4.21 (s, 4H), 2.04 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 171.7, 158.8, 143.1, 142.9, 142.6, 142.5, 142.4, 136.4, 135.8, 133.1, 130.7, 129.6, 129.2, 129.2, 128.8, 128.6, 128.5, 126.0, 125.4, 123.0, 122.6, 121.3, 118.2, 116.9, 115.6, 70.2, 64.5, 18.8; IR (ATR) [cm–1]: 2922, 2565, 1679, 1506, 1455, 1302, 1247; LC–MS (DAD/ESI): tR = 8.68 min, calcd for C29H24O5 [m/z] [M – H]− 451.16; found, [M – H]− 451.12; HRMS (ESI): calcd for C29H24O5 [m/z] [M + Na]+ 475.1516; found, [M + Na]+ 475.1516.
3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)isoxazole-5-carboxylic Acid (5n)
Methyl 3-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)isoxazole-5-carboxylate (5m) (0.090 g, 0.20 mmol, 1.0 equiv), LiOH·H2O (0.016 g, 0.40 mmol, 2.0 equiv). Product 5n was obtained as a colorless solid with 45.1% (0.040 g) yield.
Rf = 0.31 (SiO2, ethyl acetate/MeOH, 3:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.47–7.45 (m, 2H), 7.43 (m, 1H), 7.37 (dt, J = 7.4, 1.4 Hz, 1H), 7.28–7.25 (m, 1H), 7.23–7.20 (m, 2H), 6.91 (d, J = 8.2 Hz, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.83 (dd, J = 8.2, 2.1 Hz, 1H), 6.68 (s, 1H), 5.38 (s, 2H), 4.31 (s, 4H), 2.12 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 171.4, 159.7, 159.3, 143.1, 143.0, 142.6, 142.4, 142.2, 135.7, 135.0, 133.1, 129.8, 129.3, 129.3, 128.8, 128.5, 126.7, 125.4, 122.6, 118.2, 116.9, 102.2, 72.3, 64.5, 64.5, 18.8; IR (ATR) [cm–1]: 3147, 2954, 2614, 360, 1736, 1505; LC–MS (DAD/ESI): tR = 7.79 min, calcd for C26H21NO6 [m/z] [M – H]− 442.13; found, [M – H]− 442.15; HRMS (ESI): calcd for C26H21NO6 [m/z] [M + Na]+ 466.1261; found, [M + Na]+ 466.1260.
3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzoic Acid (5p)
Methyl 3-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzoate (5o) (0.093 g, 0.20 mmol, 1.0 equiv), LiOH·H2O (0.016 g, 0.40 mmol, 2.0 equiv). Product 5p was obtained as a colorless solid with 58.8% (0.053 g) yield.
Rf = 0.70 (SiO2, DCM/MeOH, 9:1); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 12.96 (s, 1H), 7.61 (m, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.43 (m, 2H), 7.35 (t, J = 7.9 Hz, 1H), 7.31 (m, 1H), 7.27–7.21 (m, 2H), 7.14 (t, J = 7.5 Hz, 2H), 7.06 (dd, J = 8.2, 2.1 Hz, 1H), 6.88 (d, J = 8.2 Hz, 1H), 6.77 (d, J = 2.0 Hz, 1H), 6.72 (dd, J = 8.3, 2.0 Hz, 1H), 4.89 (d, J = 11.5 Hz, 1H), 4.85 (d, J = 11.5 Hz, 1H), 4.27 (s, 4H), 1.92 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 167.5, 158.7, 143.4, 142.9, 142.0, 142.0, 140.8, 135.2, 134.5, 133.4, 132.6, 130.2, 130.1, 129.5, 129.4, 128.6, 128.5, 128,00, 125.7, 122.5, 122.2, 119.9, 118.1, 117.2, 115.2, 68.3, 66.8, 64.6, 18.6; IR (ATR) [cm–1]: 2876, 2647, 1690, 1507, 1279, 1244; LC–MS (DAD/ESI): tR = 8.61 min, calcd for C29H24O5 [m/z] [M – H]− 451.16; found, [M – H]− 451.12 HRMS (ESI): calcd for C29H24O5 [m/z] [M + Na]+ 475.1516; found, [M + Na]+ 475.1516.
3-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methoxy)isoxazole-5-carboxylic Acid (5r)
Methyl 3-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-2-yl)methoxy)isoxazole-5-carboxylate (5q) (0.090 g, 0.20 mmol, 1.0 equiv), LiOH·H2O (0.016 g, 0.40 mmol, 2.0 equiv). Product 5r was obtained as a colorless solid with 55.3% (0.049 g) yield.
Rf = 0.45 (SiO2, ethyl acetate/MeOH, 1:3); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.21 (m, 1H), 7.46–7.41 (m, 2H), 7.26–7.23 (m, 2H), 7.16 (dd, J = 7.6, 0.8 Hz, 1H), 7.09 (dd, J = 7.4, 0.9 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.82 (d, J = 2.0 Hz, 1H), 6.76 (dd, J = 8.3, 2.0 Hz, 1H), 6.17 (s, 1H), 4.97 (d, J = 11.8 Hz, 1H), 4.93 (d, J = 11.8 Hz, 1H), 4.27 (s, 4H), 1.88 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 171.2, 170.6, 159.5, 143.4, 143.0, 142.0, 140.6, 135.1, 133.9, 133.3, 130.1, 129.5, 128.9, 128.6, 128.0, 125.7, 122.6, 118.1, 117.3, 95.6, 69.4, 64.6, 18.5; IR (ATR) [cm–1]: 3396, 2931, 1595, 1505, 1359; LC–MS (DAD/ESI): tR = 7.78 min, calcd for C26H21NO6 [m/z] [M – H]− 442.13; found, [M – H]− 442.15; HRMS (ESI): calcd for C26H21NO6 [M + Na]+ 466.1261; found, [M + Na]+ 466.1262.
General Procedure for 2-Pyridinebenzaldehyde Preparation (5d and 5l)
A three-neck round-bottom flask was charged with appropriate halide 5c or 5k, borane 3-pyridinylboronic acid, K2CO3, and a dioxane/water mixture (2:1, 5/10 mL for 1 mmol) under an argon atmosphere. The mixture was deoxygenated by rinsing with argon for half an hour; then, the Pd(dppf)Cl2 dichloromethane complex was added. The reaction mixture was heated at 80 °C, using preheated bath, for 5 h. After this time, the progression of the reaction was controlled by TLC. When the reaction was complete, water was added and the extraction with ethyl acetate followed. The organic phases were combined, dried over anhydrous MgSO4, and evaporated. The crude product was purified by flash chromatography (SiO2, hexane/ethyl acetate), giving the final products 5d or 5l with 93 and 80% yields, respectively.
5-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)-2-(pyridin-3-yl)benzaldehyde (5d)
2-Bromo-5-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methoxy)benzaldehyde (5c) (0.20 g, 0.40 mmol, 1.0 equiv), 3-pyridinylboronic acid (0.072 g, 0.60 mmol, 1.5 equiv), K2CO3 (0.16 g, 1.20 mmol, 3.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.016 g, 0.02 mmol, 0.05 equiv). Product 5d was obtained as a colorless solid with 92.6% (0.190 g) yield.
Rf = 0.37 (SiO2, ethyl acetate); 1H NMR (600 MHz, CDCl3): δ [ppm] 9.95 (s, 1H), 8.68 (dd, J = 4.9, 1.5 Hz, 1H), 8.66 (m, 1H), 7.71–7.69 (m, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.42–7.38 (m, 4H), 7.35 (dd, J = 8.4, 2.7 Hz, 1H), 7.27 (m, 1H), 7.24–7.21 (m, 2H), 6.91 (d, J = 8.2 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H), 6.83 (dd, J = 8.2, 2.0 Hz, 1H), 5.23 (s, 2H), 4.31 (s, 4H), 2.15 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 191.1, 159.1, 150.3, 149.1, 143.0, 142.6, 142.4, 142.3, 137.4, 135.7, 135.1, 134.8, 134.6, 133.4, 133.1, 132.4, 129.7, 129.2, 128.8, 127.4, 125.3, 123.2, 122.5, 122.2, 118.2, 116.9, 112.0, 70.3, 64.4, 64.4, 18.8; IR (ATR) [cm–1]: 3049, 2997, 2876, 1684, 1504, 1320, 1169; LC–MS (DAD/ESI): tR = 8.56 min, calcd for C34H27NO4 [m/z] [M + H]+ 514.19; found, [M + H]+ 514.33; HRMS (ESI): calcd for C34H27NO4: [M + Na]+ 536.1832; found, [M + Na]+ 536.1830.
5-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)-2-(yridine-3-yl)benzaldehyde (5l)
2-Bromo-5-((3′-(benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzaldehyde (5k) (0.20 g, 0.40 mmol, 1.0 equiv), 3-pyridinylboronic acid (0.072 g, 0.60 mmol, 1.5 equiv), K2CO3 (0.160 g, 1.20 mmol, 3.0 equiv), Pd(dppf)Cl2 complex with dichloromethane (0.016 g, 0.02 mmol, 0.05 equiv). Product 5l was obtained as a colorless solid with 79.9% (0.164 g) yield.
Rf = 0.51 (SiO2, ethyl acetate); 1H NMR (600 MHz, CDCl3): δ [ppm] 9.96 (s, 1H), 8.70 (dd, J = 4.9, 1.6 Hz, 1H), 8.67 (d, J = 1.7 Hz, 1H), 7.72–7.70 (m, 1H), 7.69 (d, J = 2.7 Hz, 1H), 7.51–7.46 (m, 3H), 7.43 (dd, J = 4.9, 0.7 Hz, 1H), 7.40–7.37 (m, 2H), 7.35 (dd, J = 8.5, 2.7 Hz, 1H), 7.31–7.28 (m, 1H), 7.26–7.24 (m, 2H), 6.94 (d, J = 8,2 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.86 (dd, J = 8.2, 2.1 Hz, 1H), 5.26 (s, 2H), 4.33 (s, 4H), 2.15 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 191.1, 159.0, 150.3, 149.0, 143.1, 143.0, 142.6, 142.4, 137.4, 136.0, 135.7, 135.0, 134.8, 133.5, 133.1, 132.4, 129.3, 129.3, 128.8, 128.6, 128.5, 126.0, 125.4, 123.2, 122.6, 122.2, 118.2, 116.9, 112.1, 70.4, 64.5, 64.5, 18.8; IR (ATR) [cm–1]: 2974, 2927, 2875, 1688, 1506, 1279, 1227, 1069; LC–MS (DAD/ESI): tR = 8.59 min, calcd for C34H27NO4 [m/z] [M + H]+ 514.19; found, [M + H]+ 514.33; HRMS (ESI): [M + Na]+ 536.1832; found, [M + Na]+ 536.1834.
General Procedure for the Amine Formation (6a–e, 7a–h, and 8a–j)
Appropriate alcohol 4a–h was added in a round-bottom flask with anhydrous dichloromethane and few drops of anhydrous DMF (0.2 mL for 2 mmol alcohol) and cooled in an ice bath; then, thionyl chloride (5 equiv) was added dropwise. The reaction mixture was stirred approximately 2 h at room temperature and controlled by TLC. When all substrates disappeared, the reaction mixture was poured onto saturated NaHCO3 and extracted with dichloromethane. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The crude product was used without further purification in the next step.
Method A. Crude alkyl chloride was dissolved in anhydrous acetonitrile or THF, and excess of appropriate amine dissolved in anhydrous acetonitrile or THF was added (if the hydrochloride of amine was used, an equimolar amount of triethylamine was added to the reaction mixture). The reaction mixture was stirred at room temperature for 48 h and controlled by TLC. After completion of the reaction, the mixture was poured into saturated NaHCO3 and extracted with ethyl acetate. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The crude products were purified by flash column chromatography (SiO2, hexane/ethyl acetate).
Method B. Crude alkyl chloride was dissolved in anhydrous THF, and excess of Boc-protected piperazine dissolved in anhydrous THF was added. The reaction mixture was stirred at room temperature 48 h and controlled by TLC. After the reaction was complete, the solvent was evaporated and the crude Boc-protected product was purified by flash chromatography (SiO2, hexane/ethyl acetate 1:1). The purified Boc-protected product was dissolved in anhydrous dioxane, and HCl in dioxane was added (10 equiv, 4 M). The reaction mixture was stirred at room temperature overnight and then poured into saturated NaHCO3 and extracted with ethyl acetate. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The products were purified by column chromatography (SiO2, pure methanol, or methanol/7 M NH3 in methanol).
Method C. Crude alkyl chloride was dissolved in anhydrous THF and an appropriate amine dissolved in anhydrous THF was added (if the hydrochloride of amine was used, an equimolar amount of triethylamine was added to the reaction mixture). The reaction mixture was stirred at 50 °C overnight. After completion of the reaction, it was poured into saturated NaHCO3 and extracted with ethyl acetate. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The crude products were purified by column chromatography (SiO2, DCM/methanol, methanol, or methanol/7 M NH3 in methanol).
Method D. Crude alkyl chloride was dissolved in anhydrous DMF, and an appropriate amine dissolved in anhydrous DMF was added (if the hydrochloride of the amine was used, equimolar amount of triethyl amine was added to the reaction mixture) together with diisopropylethylamine (DIPEA). The reaction mixture was stirred at 80 °C overnight. After completion of the reaction, it was poured into saturated NaHCO3 and extracted with ethyl acetate. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The crude products were purified by column chromatography (SiO2, ethyl acetate, DCM/methanol, or CHCl3/7 M NH3 in methanol or methanol/7 M NH3 in methanol).
Method E. Crude alkyl chloride was dissolved in anhydrous DMF, and an appropriate amine dissolved in anhydrous DMF was added (if the hydrochloride of the amine was used, an equimolar amount of triethylamine was added to the reaction mixture) together with DIPEA. The reaction mixture was stirred at 80 °C overnight. After completion of the reaction, it was poured into saturated NaHCO3, extracted three times with ethyl acetate, and the crude Boc-protected product was purified by flash chromatography (SiO2, hexane/ethyl acetate 1:1). The purified Boc-protected product was dissolved in anhydrous dioxane, and HCl in dioxane was added (10 equiv, 4 M). The reaction mixture was stirred at room temperature overnight, then poured into saturated NaHCO3, and extracted three times with ethyl acetate. The organic phases were collected, dried over anhydrous MgSO4, and evaporated. The products were purified by column chromatography (SiO2, DCM/methanol, or methanol/7 M NH3 in methanol).
4-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methyl)morpholine (6a)
Method A. (3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4a) (0.33 g, 1.0 mmol, 1.0 equiv), thionyl chloride (0.36 mL, 5.0 mmol, 5.0 equiv), morpholine (0.35 mL, 5.0 mmol, 5.0 equiv). The crude product was purified by flash chromatography (SiO2, hexane/ethyl acetate 2:1), giving the final compound 6a as a colorless oil with 70.1% (0.281 g) yield.
Rf = 0.36 (SiO2, ethyl acetate/hexane 1:1); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.37 (d, J = 8.1 Hz, 2H), 7.32 (d, J = 8.1 Hz, 2H), 7.27 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 7.6 Hz, 2H), 6.91 (d, J = 8.2 Hz, 1H), 6.85 (d, J = 2.1 Hz, 1H), 6.81 (dd, J = 8.2, 2.1 Hz, 1H), 4.28 (s, 4H), 3.62–3.57 (m, 4H), 3.51 (s, 2H), 2.43–2.32 (m, 4H), 2.06 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 142.9, 142.5, 142.1, 141.8, 140.5, 136.5, 134.9, 132.3, 29.0, 128.7, 128.5, 125.5, 122.1, 117.7, 116.8, 66.2, 64.1, 64.1, 62.2, 53.2, 18.7; IR (ATR) [cm–1]: 2957, 2923, 2842, 1582, 1506, 1455, 1317, 1292, 1242, 1228, 1114, 1070, 1006, 898, 881, 845, 791; LC–MS (DAD/ESI): tR = 5.65 min, calcd for C26H27NO3 (m/z) [M + H]+ 402.27; found, [M + H]+ 402.21; HRMS (ESI): calcd for C26H27NO3 (m/z) [M + H]+ 402.2064; found, [M + H]+ 402.2062.
4-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazine (6b)
Method B. (3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4a) (0.24 g, 0.72 mmol, 1.0 equiv), thionyl chloride (0.26 mL, 3.6 mmol, 5.0 equiv), 1-Boc-piperazine (0.67 g, 3.6 mmol, 5.0 equiv). Product 6b was obtained as a light-yellow oil with 66.3% (0.191 g) yield.
Rf = 0.09 (SiO2, methanol); IR (ATR) [cm–1]: 3298, 2933, 2811, 1578, 1507, 1463, 1414, 1317, 1279, 1245, 1123, 1069, 899, 790; 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.36 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.2 Hz, 2H), 7.27 (t, J = 7.4 Hz, 1H), 7.16 (dd, J = 7.6, 2.0 Hz, 2H), 6.92 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 2.1 Hz, 1H), 6.82 (dd, J = 8.2, 2.1 Hz, 1H), 4.28 (s, 4H), 3.47 (s, 2H), 3.17 (s, 1H), 2.71–2.66 (m, 4H), 2.35–2.27 (m, 4H), 2.07 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 142.9, 142.5, 142.2, 141.8, 140.3, 137.0, 134.9, 132.3, 128.9, 128.7, 128.6, 125.5, 122.1, 117.7, 116.8, 64.1, 62.6, 54.2, 45.6, 18.7; LC–MS (DAD/ESI): tR = 5.01 min, calcd for C26H28N2O2 [m/z] [M + H]+ 401.22; found, [M + H]+ 401.30; HRMS (ESI): calcd for C26H28N2O2 (m/z) [M + H]+ 401.2223; found, [M + H]+ 401.2226.
1-((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methyl)piperazine (6c)
Method B. (3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-[1,1′-biphenyl]-3-yl)methanol (4b) (0.20 g, 0.60 mmol, 1.0 equiv), SOCl2 (0.22 mL, 3.0 mmol, 5.0 equiv), 1-Boc-piperazine (0.56 g, 3.0 mmol, 5.0 equiv). Product 6c was obtained as a colorless solid with 29.1% (0.070 g) yield.
Rf = 0.11 (SiO2, MeOH); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.25 (t, J = 7.5 Hz, 1H), 7.23 (s, 1H), 7.19 (d, J = 7.6 Hz, 1H), 7.16–7.13 (m, 2H), 7.10 (m, 2H), 6.80 (d, J = 8.2 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H), 6.73 (dd, J = 8.2, 2.0 Hz, 1H), 4.17 (s, 4H), 3.45 (s, 2H), 2.80 (m, 5H), 2.36 (sbroad, 4H), 2.03 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 143.1, 142.8, 142.6, 142.3, 137.8, 135.9, 133.1, 130.3, 129.0, 128.8, 128.1, 128.0, 127.7, 125.3, 122.6, 118.2, 116.9, 64.5, 64.4, 63.6, 54.1, 45.8, 18.9; IR (ATR) [cm–1]: 3319, 2934, 1507, 1279, 1069; LC–MS (DAD/ESI): tR = 5.11 min, calcd for C26H28N2O2 [m/z] [M + H]+ 401.22; found, [M + H]+ 401.34; HRMS (ESI): calcd for C26H28N2O2 [m/z] [M + H]+ 401.2223; found, [M + H]+ 401.2222.
1-((3′-(Benzo-1,4-dioxan-6-yl)-3-methoxy-2′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazine (6d)
Method B. (3′-(Benzo-1,4-dioxan-6-yl)-3-methoxy-2′-methyl-[1,1′-biphenyl]-4-yl)methanol (4i) (0.20 g, 0.5 mmol, 1.0 equiv), thionyl chloride (0.19 mL, 2.7 mmol, 5.0 equiv), 1-Boc-piperazine (0.51 g, 2.7 mmol, 5.0 equiv). Product 6d was obtained as a light-yellow oil with 19.9% (0.047 g) yield.
Rf = 0.08 (SiO2, MeOH); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.35 (d, J = 7.6 Hz, 1H), 7.27 (t, J = 7.5 Hz, 1H), 7.19 (d, J = 6.6 Hz, 1H), 7.16 (dd, J = 7.5, 1.2 Hz, 1H), 6.95–6.89 (m, 3H), 6.88 (d, J = 2.1 Hz, 1H), 6.82 (dd, J = 8.2, 2.1 Hz, 1H), 4.28 (s, 4H), 3.80 (s, 3H), 3.45 (s, 2H), 3.17 (s, 1H), 2.74–2.60 (sbroad, 4H), 2.40–2.27 (sbroad, 4H), 2.09 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 157.4, 143.4, 143.0, 142.3, 142.0, 135.4, 132.9, 129.9, 129.1, 129.0, 125.9, 125.0, 122.6, 121.3, 118.2, 117.2, 112.3, 64.6, 56.6, 55.9, 54.9, 46.2, 19.2; IR (ATR) [cm–1]: 3322, 2934, 2829, 1577, 1507, 1463, 1399, 1301, 1280, 1227, 1068, 872, 792, 742; LC–MS (DAD/ESI): tR = 4.72 min, calcd for C27H30N2O3 [m/z] [M + H]+ 431.34; found, [M + H]+ 431.23; HRMS (ESI): calcd for C27H30N2O3 (m/z) [M + H]+ 431.2329; found, [M + H]+ 431.2329.
3-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-methyl-4-(piperazin-1-ylmethyl)-[1,1′-biphenyl]-3-yl)oxy)methyl)benzonitrile (6e)
Method B. 3-(((3′-(Benzo-1,4-dioxan-6-yl)-4-(hydroxymethyl)-2′-methyl-[1,1′-biphenyl]-3-yl)oxy)methyl)benzonitrile (4j) (0.24 g, 0.5 mmol, 1.0 equiv), thionyl chloride (0.14 mL, 2.0 mmol, 5.0 equiv), 1-Boc-piperazine (0.37 g, 2.0 mmol, 5.0 equiv). Product 6e was obtained as a colorless solid with 38.4% (0.104 g) yield.
Rf = 0.12 (SiO2, methanol1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.94 (s, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.26 (t, J = 7.3 Hz, 1H), 7.16 (d, J = 7.7 Hz, 2H), 7.02 (d, J = 1.4 Hz, 1H), 6.94–6.90 (m, 2H), 6.84 (d, J = 2.1 Hz, 1H), 6.81 (dd, J = 8.2, 2.1 Hz, 1H), 5.25 (s, 2H), 4.28 (s, 4H), 3.51 (s, 2H), 2.73–2.67 (m, 4H), 2.39–2.32 (m, 4H), 2.00 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 155.6, 143.0, 142.5, 142.2., 141.8, 141.7, 139.3, 134.9, 132.4, 132.0, 131.5, 130.6, 130.2, 129.7, 128.7, 128.4, 125.5, 125.5, 122.1, 121.3, 118.7, 117.7, 116.8, 113.6, 111.4, 68.0, 64.1, 56.7, 54.5, 45.7, 18.6); IR (ATR) [cm–1]: 2932, 2875, 2814, 2230, 1576, 1506, 1461, 1402, 1301, 1244, 1225, 1068, 1001, 792; LC–MS (DAD/ESI): tR = 5.93 min, calcd for C34H33N3O3 (m/z) [M + H]+ 532.26; found, [M + H]+ 532.41; HRMS (ESI): calcd for C34H33N3O3 (m/z) [M + H]+ 532.2595; found, [M + H]+ 532.2594.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7a)
Method C. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.1 mL, 1.95 mmol, 5.0 equiv) N-(2-aminoethyl)acetamide (0.19 mL, 1.95 mmol, 5.0 equiv). Product 7a was obtained as a brownish oil with 28.0% (0.051 g) yield.
Rf = 0.26 (SiO2, MeOH); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.33 (m, 4H), 7.00–6.98 (m, 3H), 6.95–6.91 (m, 2H), 6.33 (s, 1H), 4.29 (s, 4H), 3.86 (s, 3H), 3.81 (s, 2H), 3.36 (m, 2H), 2.78 (t, J = 5.8 Hz, 2H), 2.00 (sbroad, 1H), 1.98 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 170.3, 157.1, 143.2, 143.0, 141.4, 141.1, 140.5, 133.3, 130.9, 130.5, 130.1, 129.5, 127.2, 126.4, 122.8, 121.6, 118.6, 116.9, 112.0, 64.5, 64.4, 55.4, 48.7, 47.9, 39.1, 23.3; IR (ATR) [cm–1]: 3294, 2934, 1652, 1507, 1457, 1302, 1068, 732; LC–MS (DAD/ESI): tR = 5.43 min, calcd for C26H27ClN2O4 [m/z] [M + H]+ 467.17; found, [M + H]+ 467.28; HRMS (ESI): calcd for C26H27ClN2O4 [m/z] [M + H]+ 467.1732; found, [M + H]+ 467.1732.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-ethoxy-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7b)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-ethoxy-[1,1′-biphenyl]-4-yl)methanol (4l0.15 g, 0.27 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), N-(2-aminoethyl)acetamide (0.080 g, 0.78 mmol, 2.0 equiv), DIPEA (0.094 mL, 0.54 mmol, 2.0 equiv). Product 7b was obtained as a brownish oil with 48.7% (0.091 g) yield.
Rf = 0.29 (SiO2, MeOH); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.25–7.10 (m, 4H), 6.92–6.89 (m, 3H), 6.87–6.84 (m, 2H), 6.14 (sbroad, 1H), 4.22 (s, 4H), 4.02 (q, J = 7.0 Hz, 2H), 3.75 (s, 2H), 3.29 (m, 2H), 2.70 (m, 2H), 1.90 (s, 3H), 1.35 (t, J = 7.0 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 170.2, 156.4, 143.2, 143.0, 141.4, 141.1, 140.4, 133.3, 131.0, 130.5, 130.1, 129.5, 126.9, 126.4, 122.8, 121.5, 118.6, 116.9, 112.9, 64.5, 64.4, 63.6, 48.8, 47.8, 39.1, 23.4, 15.0; IR (ATR) [cm–1]: 3293, 3066, 2979, 2878, 1652, 1507, 1456, 1303, 1250, 1068; LC–MS (DAD/ESI): tR = 5.69 min, calcd for C27H29ClN2O4 [m/z] [M + H]+ 481.18; found, [M + H]+ 481.30; HRMS (ESI): calcd for C27H29ClN2O4 [m/z] [M + H]+ 481.1889; found, [M + H]+ 481.1889.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-isopropoxy-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7c)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-isopropoxy-[1,1′-biphenyl]-4-yl)methanol (4m) (0.21 g, 0.53 mmol, 1.0 equiv), SOCl2 (0.12 mL, 1.59 mmol, 3.0 equiv), N-(2-aminoethyl)acetamide (0.21 mL, 2.24 mmol, 4.0 equiv), DIPEA (0.20 mL, 1.12 mmol, 2.0 equiv). Product 7c was obtained as a colorless oil with 36.6% (0.096 g) yield.
Rf = 0.30 (SiO2, ethyl acetate/methanol 1:1;1H NMR (600 MHz, CDCl3): δ [ppm] 7.34–7.26 (m, 4H), 7.01–6.98 (m, 2H), 6.97–6.91 (m, 3H), 6.15 (sbroad, 1H), 4.65–4.58 (m, 1H), 4.31 (s, 4H), 3.81 (s, 2H), 3.37 (q, J = 5.8 Hz, 2H), 2.78 (t, J = 5.8 Hz, 2H), 1.99 (s, 3H), 1.36 (d, J = 6.0 Hz, 6H); 13C NMR (151 MHz, CDCl3): δ [ppm] 170.3, 155.5, 143.3, 143.1, 141.6, 141.2, 140.5, 139.5, 131.5, 130.6, 130.3, 129.9, 128.0, 126.5, 122.9, 121.4, 118.7, 117.0, 114.5, 70.2, 64.6, 64.6, 49.1, 47.8, 39.1, 23.5, 22.4); IR (ATR) [cm–1]: 3295, 3057, 2976, 2931, 2874, 1652, 1507, 1456, 1302, 1280, 1249, 1226, 1113, 1069, 796, 734; LC–MS (DAD/ESI): tR = 5.86 min, calcd for C28H31ClN2O4 (m/z) [M + H]+ 495.32; found, [M + H]+ 495.20; HRMS (ESI): calcd for C28H31ClN2O4 (m/z) [M + H]+ 495.2045; found, [M + H]+ 495.2045.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-isopentyloxy-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7d)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-isopentyloxy-[1,1′-biphenyl]-4-yl)methanol (4n) (0.15 g, 0.34 mmol, 1.0 equiv), SOCl2 (0.12 mL, 1.70 mmol, 5.0 equiv), N-(2-aminoethyl)acetamide (0.097 mL, 1.02 mmol, 3.0 equiv), DIPEA (0.12 mL, 0.68 mmol, 2.0 equiv). Product 7d was obtained as a colorless oil 73.7% (0.131 g) yield.
Rf = 0.30 (SiO2, ethyl acetate/methanol 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.34–7.26 (m, 4H), 7.00–6.97 (m, 3H), 6.95–6.91 (m, 2H), 6.23 (s, 1H), 4.31 (s, 1H), 4.04 (7, J = 6.6 Hz, 2H), 3.83 (s, 2H), 3.37 (q, J = 5.5 Hz, 2H), 2.79 (t, J = 5.9 Hz, 2H), 1.99 (s, 3H), 1.82 (sept, J = 6.7 Hz, 1H), 0.97 (d, J = 6.6 Hz, 6H); 13C NMR (151 MHz, CDCl3): δ [ppm] 170.4, 156.7, 143.3, 143.1, 141.5, 141.2, 140.7, 133.4, 131.1, 130.6, 130.2, 129.6, 126.9, 126.5, 122.9, 121.6, 118.7, 117.0,113.0, 66.6, 64.6, 64.5, 48.9, 47.9, 39.1, 38.2, 25.4, 23.5, 22.8; IR (ATR) [cm–1]: 3292, 3062, 2956, 2931, 2872, 1652, 1507, 1456, 1302, 1280, 1249, 1226, 1069, 796, 733; LC–MS (DAD/ESI): tR = 6.62 min, calcd for C30H35ClN2O4 (m/z) [M + H]+ 523.24; found, [M + H]+ 523.37; HRMS (ESI): calcd for C30H35ClN2O4 (m/z) [M + H]+ 523.2358; found, [M + H]+ 523.2358.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-(cyanomethoxy)-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7e)
Method D. 2-((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-4-(hydroxymethyl)-[1,1′-biphenyl]-3-yl)oxy)acetonitrile (4o) (0.22 g, 0.54 mmol, 1.0 equiv), SOCl2 (0.12 mL, 1.62 mmol, 3.0 equiv), N-(2-aminoethyl)acetamide (0.21 mL, 2.24 mmol, 4.0 equiv), DIPEA (0.20 mL, 1.12 mmol, 2.0 equiv). Product 7e was obtained as a colorless oil with 9.8% (0.026 g) yield.
Rf = 0.24 (SiO2, ethyl acetate/methanol 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.37 (d, 7.7 Hz, 1H), 7.35–7.31 (m, 2H), 7.28 (dd, J = 6.7, 2.7 Hz, 1H), 7.15 (dd, J = 7.6, 1.5 Hz, 1H), 7.05 (d, J = 1.4 Hz, 1H), 6.99–6.98 (m, 1H), 6.95–6.91 (m, 2H), 6.09 (sbroad, 1H), 4.86 (s, 2H), 4.30 (s, 4H), 3.85 (s, 2H), 3.38 (q, J = 5.5 Hz, 2H), 2.81 (t, J = 5.6 Hz, 2H), 1.99 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 170.3, 154.1, 143.3, 143.0, 141.2, 140.8, 140.5, 133.1, 130.9, 130.1, 128.5, 126.5, 124.3, 122.8, 118.5, 116.9, 115.1, 113.6, 64.5, 64.4, 53.9, 48.1, 48.0, 39.1, 23.4; IR (ATR) [cm–1]: 3292, 3059, 2932, 1652, 1582, 1508, 1457, 1319, 1303, 1281,1251, 1068, 798, 737; LC–MS (DAD/ESI): tR = 5.29 min, calcd for C27H26ClN3O4 (m/z) [M + H]+ 492.17; found, [M + H]+ 492.27; HRMS (ESI): calcd for C27H26ClN2O4 (m/z) [M + H]+ 492.1685; found, [M + H]+ 492.1683.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-(cyclobutylmethoxy)-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7f)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-cyclobutylmethoxy-[1,1′-biphenyl]-4-yl)methanol (4p) (0.36 g, 0.83 mmol, 1.0 equiv), SOCl2 (0.30 mL, 4.16 mmol, 5.0 equiv), N-(2-aminoethyl)acetamide (0.23 mL, 2.49 mmol, 3.0 equiv), DIPEA (0.29 mL, 1.66 mmol, 2.0 equiv). Product 7f was obtained as a colorless oil with 29.6% (0.128 g) yield.
Rf = 0.30 (SiO2, ethyl acetate/methanol 1:1); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.82 (t, J = 5.5 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.38–7.36 (m, 2H), 7.35 (dd, J = 7.5, 1.8 Hz, 1H), 7.00 (d, J = 1.5 Hz, 1H), 6.97 (dd, J = 7.6, 1.6 Hz, 1H), 6.95–6.93 (m, 2H), 6.90 (dd, J = 8.3, 2.1 Hz, 1H), 4.29 (s, 4H), 3.99 (d, J = 6.3 Hz, 2H), 3.72 (s, 2H), 3.15 (q, J = 6.3 Hz, 2H), 2.78–2.71 (m, 1H), 2.58 (t, J = 6.5 Hz, 2H), 2.11–2.03 (m, 2H), 1.94–1.85 (m, 4H), 1.79)s, 3H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 169.14, 156.1, 143.1, 142.8, 141.1, 140.4, 139.0, 132.5, 130.4, 130.3, 129.9, 128.5, 128.5, 126.9, 122.4, 121.0, 118.0, 116.7, 112.7, 79.2, 71.3, 64.1, 64.1, 48.5, 47.3, 34.1, 24.2, 22.6, 18.1; IR (ATR) [cm–1]: 3292, 3062, 2975, 2934, 2867, 1652, 1507, 1456, 1302, 1280, 1249, 1226, 1068, 796, 747; LC–MS (DAD/ESI): tR = 6.37 min, calcd for C30H33ClN2O4 (m/z) [M + H]+ 521.22; found, [M + H]+ 521.38; HRMS (ESI): calcd for C30H33ClN2O4 (m/z) [M + H]+ 521.2202; found, [M + H]+ 521.2202.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-(cyclopentylmethoxy)-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7g)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-cyclopentylmethoxy-[1,1′-biphenyl]-4-yl)methanol (4q) (0.21 g, 0.51 mmol, 1.0 equiv), SOCl2 (0.18 mL, 2.55 mmol, 5.0 equiv), N-(2-aminoethyl)acetamide (0.18 mL, 1.02 mmol, 3.0 equiv), DIPEA (0.15 mL, 1.53 mmol, 2.0 equiv). Product 7g was obtained as a colorless oil with 70.9% (0.193 g) yield.
Rf = 0.30 (SiO2, ethyl acetate/methanol 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.33–7.27 (m, 4H), 7.00–6.95 (m, 3H), 6.95–6.91 (m, 2H), 6.18 (s, 1H), 4.31 (s, 4H), 3.89 (d, J = 6.9 Hz, 2H0, 3.84 (s, 2H), 3.37 (q, J = 5.5 Hz, 2H), 2.79 (t, J = 5.8 Hz, 2H), 2.44–2.35 (m, 1H), 1.88–1.81 (m, 2H), 1.68–1.56 (m, 4H), 1.39–1.33 (m, 2H); 13C NMR (151 MHz, CDCl3): δ [ppm] 156.7, 143.2, 143.0, 141.4, 140.6, 133.3, 131.0, 130.5, 130.1, 129.6, 122.8, 121.4, 118.6, 116.9, 112.8, 72.2, 64.5, 64.4, 49.0, 47.8, 39.1, 39.0, 29.5, 25.5, 23.3; IR (ATR) [cm–1]: 3292, 3060, 2947, 2867, 1652, 1507, 1456, 1303, 1280, 1249, 1226, 1068, 796, 748; LC–MS (DAD/ESI): tR = 6.88 min, calcd for C31H35ClN2O4 (m/z) [M + H]+ 535.24; found, [M + H]+ 535.41; HRMS (ESI): calcd for C31H35ClN2O4 (m/z) [M + H]+ 535.2358; found, [M + H]+ 535.2358.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-((3′-cyano-[1,1′-biphenyl]-3-yl)methoxy)-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (7h)
Method D. 3′-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-4-(hydroxymethyl)-[1,1′-biphenyl]-3-yl)oxy)methyl)-[1,1′-biphenyl]-3-carbonitrile (4r) (0.15 g, 0.27 mmol, 1.0 equiv), SOCl2 (0.10 mL, 1.35 mmol, 5.0 equiv), N-(2-aminoethyl)acetamide (0.077 mL, 0.81 mmol, 3.0 equiv), DIPEA (0.094 mL, 0.54 mmol, 2.0 equiv). Product 7h was obtained as a brownish oil with 78.5% (0.136 g) yield.
Rf = 0.26 (SiO2, DCM/methanol, 9:1 1H NMR (600 MHz, CDCl3): δ [ppm] 7.77 (t, J = 1.5 Hz, 1H), 7.72 (dt, J = 7.9, 1.2 Hz, 1H), 7.54 (m, 2H), 7.47–7.39 (m, 4H), 7.29 (d, J = 7.7 Hz, 1H), 7.23–7.18 (m, 2H), 7.16–7.13 (m, 1H), 7.02 (d, J = 1.3 Hz, 1H), 6.93 (dd, J = 7.6, 1.4 Hz, 1H), 6.88 (m, 1H), 6.83–6.80 (m, 3H), 5.13 (s, 2H), 4.42 (sbroad, 1H), 4.20 (s, 4H), 3.92 (s, 2H), 3.30 (m, 2H), 2.82 (t, J = 5.5 Hz, 2H), 1.80 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 170.9, 156.2, 143.3, 143.0, 142.0, 141.6, 141.2, 140.9, 139.3, 137.7, 133.1, 131.6, 131.0, 130.8, 130.8, 130.7, 130.4, 130.0, 129.8, 129.6, 127.4, 126.9, 126.5, 126.2, 124.1, 122.7, 122.3, 118.8, 118.5, 116.9, 113.7, 113.0, 70.1, 64.5, 64.4, 47.9, 47.9, 38.0, 23.1); IR (ATR) [cm–1]: 3293, 3062, 2932, 2229, 1652, 1507, 1456, 1303, 1249, 1068; LC–MS (DAD/ESI): tR = 6.68 min, calcd for C39H34ClN3O4 (m/z) [M + H]+ 644.23; found, [M + H]+ 644.42; HRMS (ESI): calcd for C39H34ClN3O4 (m/z) [M + H]+ 644.2311; found, [M + H]+ 644.2311.
1-((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)piperazine (8a)
Method B. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), 1-Boc-piperazine (0.36 g, 1.95 mmol, 5.0 equiv). Product 8a was obtained as a colorless solid with 33.3% (0.058 g) yield.
Rf = 0.10 (SiO2, MeOH); 1H NMR (600 MHz, DMSO-d6): δ [ppm] 7.43 (m, 1H), 7.38 (m, 2H), 7.35 (dd, J = 7.5, 1.5 Hz, 1H), 7.02 (s, 1H), 6.99 (d, J = 7.6 Hz, 1H), 6.95–6.93 (m, 2H), 6.90 (dd, J = 8.3, 1.8 Hz, 1H), 4.29 (s, 4H), 3.80 (s, 3H), 3.46 (s, 2H), 2.70 (t, J = 4.3 Hz, 4H), 2.35 (m, 5H); 13C NMR (151 MHz, DMSO-d6): δ [ppm] 157.3, 143.6, 143.3, 141.5, 140.9, 139.6, 133.0, 130.9, 130.8, 130.4, 129.8, 127.4, 125.9, 122.9, 121.6, 118.5, 117.2, 112.5, 64.6, 64.6, 56.5, 55.9, 54.9, 46.2; IR (ATR) [cm–1]: 3306, 2931, 1507, 1457, 796; LC–MS (DAD/ESI): tR = 4.71 min, calcd for C26H27ClN2O3 [m/z] [M + H]+ 451.18; found, [M + H]+ 451.32; HRMS (ESI): calcd for C26H27ClN2O3 [M + H]+ 451.1783; found, [M + H]+ 451.1783.
4-((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)piperazine-1-carboxamide (8b)
Method C. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), piperazine-1-carboxamide hydrochloride (0.13 g, 0.78 mmol, 2.0 equiv), triethylamine (0.11 mL, 0.78 mmol, 2.00 equiv). Product 8b was obtained as a brownish solid with 36.3% (0.070 g) yield.
Rf = 0.38 (SiO2, DCM/MeOH, 9:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.31 (d, J = 7.7 Hz, 1H), 7.24–7.17 (m, 3H), 6.93 (dd, J = 7.7, 1.5 Hz, 1H), 6.91 (d, J = 1.9 Hz, 1H), 6.89 (d, J = 1.5 Hz, 1H), 6.86–6.83 (m, 2H), 4.71 (s, 2H), 4.20 (s, 4H), 3.74 (s, 3H), 3.54 (s, 2H), 3.34 (t, J = 5.0 Hz, 4H), 2.45 (t, J = 5.1 Hz, 4H); 13C NMR (151 MHz, CDCl3): δ [ppm] 158.3, 157.2, 143.2, 143.0, 141.5, 141.1, 140.2, 133.4, 130.9, 130.5, 130.2, 130.0, 126.4, 125.0, 122.8, 121.6, 118.6, 116.9, 112.1, 64.5, 64.4, 56.0, 55.6, 52.8, 44.1; IR (ATR) [cm–1]: 3352, 2937, 1586, 1507, 1459, 1301, 1228, 1068; LC–MS (DAD/ESI): tR = 5.22 min, calcd C27H28ClN3O4 [m/z] [M + H]+ 494.18; found, [M + H]+ 494.33; HRMS (ESI): calcd for C27H28ClN3O4 [m/z] [M + H]+ 494.1841; found, [M + H]+ 494.1841.
1-(4-((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)ethan-1-one (8c)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), 1-acetylpiperazine (0.10 g, 0.78 mmol, 2.0 equiv), DIPEA (0.14 mL, 0.78 mmol, 2.0 equiv). Product 8c was obtained as a brownish oil with 76.3% (0.147 g) yield.
Rf = 0.46 (SiO2, DCM/MeOH, 9:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.30 (d, J = 7.7 Hz, 1H), 7.23–7.17 (m, 3H), 6.92 (dd, J = 7.6, 1.5 Hz, 1H), 6.91 (d, J = 1.9 Hz, 1H), 6.88 (d, J = 1.5 Hz, 1H), 6.85–6.82 (m, 2H), 4.18 (s, 4H), 3.74 (s, 3H), 3.56 (t, J = 4.9 Hz, 2H), 3.54 (s, 2H), 3.39 (t, J = 4.8 Hz, 2H), 2.44 (m, 4H), 1.99 (s, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 169.0, 157.3, 143.2, 143.0, 141.5, 141.1, 140.3, 133.3, 130.9, 130.5, 130.2, 130.1, 126.4, 124.7, 122.8, 121.6, 118.6, 116.9, 112.1, 64.4, 64.4, 55.8, 55.5, 53.1, 52.7, 46.4, 41.5, 21.4; IR (ATR) [cm–1]: 3436, 2933, 1644, 1507, 1458, 1247, 1228; LC–MS (DAD/ESI): tR = 5.38 min, calcd for C28H29ClN2O4 [m/z] [M + H]+ 493.19; found, [M + H]+ 493.33; HRMS (ESI): calcd for C28H29ClN2O4 [M + H]+ 493.1888; found, [M + H]+ 493.1889.
2-Amino-1-(4-((3′-(benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)ethan-1-one (8d)
Method E. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), tert-butyl(2-oxo-2-(piperazin-1-yl)ethyl)carbamate (0.19 g, 0.78 mmol, 2.0 equiv), DIPEA (0.14 mL, 0.78 mmol, 2.0 equiv). Product 8d was obtained as a brownish solid with 57.3% (0.114 g) yield.
Rf = 0.26 (SiO2, methanol/7 M NH3 in methanol, 20:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.30 (d, J = 7.7 Hz, 1H), 7.25–7.19 (m, 3H), 6.94–6.92 (m, 2H), 6.89 (m, 1H), 6.87–6.83 (m, 2H), 4.22 (s, 4H), 3.75 (s, 3H), 3.59 (m, 2H), 3.54 (s, 2H), 3.48 (sbroad, 2H), 3,34 (m, 2H), 3.05 (sbroad, 2H), 2.45 (m, 4H); 13C NMR (151 MHz, CDCl3): δ [ppm] 170.2, 157.2, 143.2, 143.0, 141.5, 141.1, 140.3, 133.3, 130.9, 130.5, 130.2, 130.0, 126.3, 124.8, 122.8, 121.6, 118.6, 116.9, 112.1, 64.5, 64.4, 55.9, 55.6, 52.9, 52.8, 44.3, 42.7, 42.2; IR (ATR) [cm–1]: 3371, 2935, 1651, 1507, 1459, 1246, 1229, 732; LC–MS (DAD/ESI): tR = 4.50 min, calcd C28H30ClN3O4 [M + H]+ 508.20; found, [M + H]+ 508.35; HRMS (ESI+): calcd for C28H30ClN3O4 [M + H]+ 508.1998; found, [M + H]+ 508.1998.
1-((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)-4-(methylsulfonyl)piperazine (8e)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), 1-(methylsulfonyl)piperazine (0.13 g, 0.78 mmol, 2.0 equiv), DIPEA (0.14 mL, 0.78 mmol, 2.0 equiv). Product 8e was obtained as a brownish solid with 88.9% (0.183 g) yield.
Rf = 0.27 (SiO2, EtOAc); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.39 (d, J = 7.7 Hz, 1H), 7.36–7.31 (m, 3H), 7.03 (dd, J = 7.6, 1.5 Hz, 1H), 7.02 (d, J = 1.8 Hz, 1H), 7.00 (d, J = 1.4 Hz, 1H), 6.97–6.94 (m, 2H), 4.32 (s, 4H), 3.86 (s, 3H), 3.68 (s, 2H), 3.30 (m, 4H), 2.80 (s, 3H), 2.69 (t, J = 4.2 Hz, 4H); 13C NMR (151 MHz, CDCl3): δ [ppm] 157.3, 143.2, 143.0, 141.4, 141.1, 140.4, 133.3, 130.9, 130.5, 130.1, 130.0, 126.4, 124.7, 122.8, 121.6, 118.6, 116.9, 112.2, 64.5, 64.4, 55.7, 55.6, 52.3, 46.0, 34.1; IR (ATR) [cm–1]: 2929, 1507, 1323, 1160; LC–MS (DAD/ESI): tR = 5.81 min, calcd for C27H29ClN2O5S [m/z] [M + H]+ 529.16; found, [M + H]+ 529.29; HRMS (ESI): calcd for C27H29ClN2O5S [M + Na]+ 551.1378; found, [M + Na]+ 551.1378.
2-Amino-N-(2-(((3′-(benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)acetamide (8f)
Method E. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), tert-butyl (2-((2-aminoethyl)amino)-2-oxoethyl)carbamate (0.17 g, 0.78 mmol, 2.0 equiv), DIPEA (0.14 mL, 0.78 mmol, 2.0 equiv). Product 8f was obtained as a brownish solid with 43.3% (0.081 g) yield.
Rf = 0.29 (SiO2, methanol/7M NH3 in methanol, 20:1); IR (ATR) [cm–1]: 3306, 2934, 1650, 1581, 1507, 1459, 1068; 1H NMR (600 MHz, CDCl3): δ [ppm] 7.47 (s, 1H), 7.22 (m, 4H), 6.91–6.85 (m, 5H), 4.22 (s, 4H), 3.77 (s, 5H), 3.38–3.38 (m, 4H), 2.74 (s, 2H); 13C NMR (151 MHz, CDCl3): δ [ppm] 172.8, 157.1, 143.2, 143.0, 141.4, 141.1, 140.4, 133.3, 130.9, 130.5, 130.1, 129.5, 126.4, 122.8, 121.6, 118.6, 116.8, 112.0, 64.5, 64.4, 55.4, 48.5, 48.0, 44.9, 38.6; LC–MS (DAD/ESI): tR = 4.25 min, calcd C26H28ClN3O4 [m/z] [M + H]+ 482.18; found, [M + H]+ 482.30; HRMS (ESI): calcd for C26H28ClN3O4 [m/z] [M + H]+ 482.1841; found, [M + H]+ 482.1840.
N-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)methanesulfonamide (8g)
Method C. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), N-(2-aminoethyl)methanesulfonamide (0.27 g, 1.95 mmol, 5.0 equiv). Product 8g was obtained as a brownish oil with 36.2% (0.071 g) yield.
Rf = 0.28 (SiO2, DCM/methanol, 1:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.26–7.21 (m, 3H), 7.19 (dd, J = 6.6, 2.3 Hz, 1H), 6.94–6.91 (m, 3H), 6.87–6.84 (m, 2H), 4.85 (sbroad, 2H), 4.22 (s, 4H), 3.83 (s, 2H), 3.81 (s, 3H), 3.22 (t, J = 5.7 Hz, 2H), 2.87 (s, 3H), 2.84 (t, J = 5.6 Hz, 2H); 13C NMR (151 MHz, CDCl3): δ [ppm] 157.2, 143.2, 143.0, 141.4, 141.1, 141.1, 133.2, 130.9, 130.7, 130.1, 126.4, 124.5, 122.8, 121.8, 118.6, 116.9, 112.2, 64.5, 64.4, 55.6, 48.1, 47.6, 41.7, 40.0; IR (ATR) [cm–1]: 3320, 2932, 1507, 1457, 1318, 1303, 1151, 797; LC–MS (DAD/ESI): tR = 5.50 min, calcd C25H27ClN2O5S [m/z] [M + H]+ 503.14; found, [M + H]+ 503.30; HRMS (ESI): calcd for C25H27ClN2O5S [m/z] [M + H]+ 503.1402; found, [M + H]+ 503.1402.
1-(2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)amino)ethyl)urea (8h)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.142 mL, 1.95 mmol, 5.0 equiv), (2-aminoethyl)urea hydrochloride (0.11 g, 0.78 mmol, 2.0 equiv), triethylamine (0.054 mL, 0.39 mmol, 1.00 equiv), DIPEA (0.094 mL, 0.54 mmol, 2.0 equiv). Product 8h was obtained as a brownish oil with 54.9% (0.100 g) yield.
Rf = 0.18 (SiO2, CHCl3/7M NH3 in methanol, 10:1); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.33–7.27 (m, 3H), 7.24 (d, J = 7.5 Hz, 1H), 6.99–6.96 (m, 3H), 6.94–6.91 (m, 2H), 5.70 (sbroad, 1H), 4.93 (sbroad, 2H), 4.29 (s, 4H), 3.84 (s, 3H), 3.80 (s, 2H), 3.27 (m, 2H), 2.77 (t, J = 5.5 Hz, 2H), 2.19 (sbroad, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 159.7, 157.1, 143.2, 143.0, 141.4, 141.1, 140.6, 133.3, 130.9, 130.5, 130.1, 129.5, 126.9, 126.4, 122.8, 121.6, 118.6, 116.9, 112.0, 64.5, 64.4, 55.4, 48.9, 48.8, 40.3; IR (ATR) [cm–1]: 3335, 2933, 1658, 1582, 1507, 1457, 1303, 1168; LC–MS (DAD/ESI): tR = 5.23 min, calcd C25H26ClN3O4 [m/z] [M + H]+ 468.17; found, [M + H]+ 468.27; HRMS (ESI): calcd for C25H26ClN3O4 [m/z] [M + H]+ 468.1685; found, [M + H]+ 468.1687.
2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)amino)propane-1,3-diol (8i)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), serinol (0.071 g, 0.78 mmol, 2.0 equiv), DIPEA (0.094 mL, 0.54 mmol, 2.0 equiv). Product 8i was obtained as a brownish oil with 68.0% (0.121 g) yield.
Rf = 0.32 (SiO2, CHCl3/methanol, 1:2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.26 (d, J = 7.6 Hz, 1H), 7.23–7.21 (m, 2H), 7.18 (m, 1H), 6.93–6.90 (m, 3H), 6.86–6.83 (m, 2H), 4.84 (sbroad, 2H), 4.22 (s, 4H), 3.98 (s, 2H), 3.80 (s, 3H), 3.72 (m, 2H), 3.61 (m, 2H), 2.86 (m, 1H), 1.84 (s, 1H); 13C NMR (151 MHz, CDCl3): δ [ppm] 157.2, 143.2, 143.0, 141.8, 141.1, 141.1, 133.2, 130.9, 130.7, 130.3, 130.1, 126.4, 123.3, 122.8, 122.0, 118.6, 116.9, 112.2, 77.1, 64.5, 64.4, 60.7, 59.6, 55.6, 46.1, 24.0; IR (ATR) [cm–1]: 3304, 2935, 2246, 1507, 1458, 1303, 1246, 1069, 732; LC–MS (DAD/ESI): tR = 5.27 min, calcd C25H26ClNO5 [m/z] [M + H]+ 456.16; found, [M + H]+ 456.24; HRMS (ESI): calcd for C25H26ClNO5 [m/z] [M + H]+ 456.1572; found, [M + H]+ 456.1573.
2-(((3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methyl)amino)-2-(hydroxymethyl)propane-1,3-diol (8j)
Method D. (3′-(Benzo-1,4-dioxan-6-yl)-2′-chloro-3-methoxy-[1,1′-biphenyl]-4-yl)methanol (4k) (0.15 g, 0.39 mmol, 1.0 equiv), SOCl2 (0.14 mL, 1.95 mmol, 5.0 equiv), tris(hydroxymethyl)aminomethane (0.094 g, 0.78 mmol, 2.0 equiv), DIPEA (0.094 mL, 0.54 mmol, 2.0 equiv). Product 8j was obtained as a brownish oil with 77.7% (0.147 g) yield.
Rf = 0.26 (SiO2, CHCl3/methanol, 1:2); 1H NMR (600 MHz, CDCl3): δ [ppm] 7.23 (d, J = 7.8 Hz, 1H), 7.21–7.17 (m, 3H), 6.90 (m, 3H), 6.85–6.82 (m, 2H), 4.20 (s, 4H), 3.78 (s, 3H), 3.68 (s, 2H), 3.52 (s, 6H), 3.13 (sbroad, 3H); 13C NMR (151 MHz, CDCl3): δ [ppm] 156.9, 143.2, 143.0, 141.3, 141.1, 140.9, 133.3, 130.9, 130.6, 130.1, 129.8, 127.1, 126.4, 122.8, 122.0, 118.6, 116.9, 112.2, 64.5, 64.4, 62.9, 60.4, 55.6, 40.9; IR (ATR) [cm–1]: 3343, 2935, 1507, 1457, 1068, 1042, 796; LC–MS (DAD/ESI): tR = 5.18 min, calcd C26H28ClNO6 [m/z] [M + H]+ 486.17, found, [M + H]+ 486.29; HRMS (ESI): calcd for C26H28ClNO6 [m/z] [M + H]+ 486.1678; found, [M + H]+ 486.1.
Acknowledgments
This research was funded by Preludium grant no. UMO-2020/37/N/ST4/02691 from the National Science Centre, Poland (to D.M.), by the project POIR.04.04.00-00-420F/17-00, which is carried out within the TEAM program of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund (to T.A.H.), by Preludium grant no. UMO-2019/35/N/NZ5/04435 from the National Science Centre, Poland (to J.K.-K.). Further funding was received to A.D. through an NIH Grant (2R01GM097082-05). We acknowledge the MCB Structural Biology Core Facility (supported by the TEAM TECH CORE FACILITY/2017-4/6 grant from the Foundation for Polish Science) for valuable support. J.K.-K. acknowledges the fellowship with project no. POWR.03.02.00-00-I013/16 from the National Center for Research and Development. The research was carried out with the equipment purchased, thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08). We would like to thank Louis Gadina, Patryk Kuczynski, and Marta Stefańska for the excellent support in organic syntheses.
Glossary
Abbreviations
- DMSO
dimethyl sulfoxide
- EC50
half-maximal effective concentration
- FBS
fetal bovine serum
- GuHCl
guanidinium chloride
- HTRF
homogeneous time-resolved fluorescence
- ICB
immune checkpoint blockade
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- irAE
immune-related adverse events
- Jurkat ECs
Jurkat effector cells
- LB
Luria–Bertani broth Miller
- mAbs
monoclonal antibodies
- NFAT-RE
nuclear factor of activated T-cell response element
- PBMCs
peripheral blood mononuclear cells
- PD-1
programmed cell death protein 1
- PD-L1
programmed cell death ligand 1 m-murine, h-human
- SMI
small-molecule inhibitors
- TCA
T-cell activation
- TCR
T-cell receptor
- TCRAct
TCR activator
- TPSA
topological polar surface area
- TR-FRET
time-resolved fluorescence energy transfer
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00957.
Experimental procedures for substrate preparation (S1, S2a–i, and 3a–3i); results of the HTRF assay; data collection and refinement statistics (molecular replacement) for the PD-L1 cocrystal structure with 8j; results of the ICB assay; comparison of the terphenyl-based with biphenyl-based inhibitor structures in complex with PD-L1; and spectral data and purity of target compounds (PDF)
Molecular formula strings (CSV)
Accession Codes
7NLD; authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
Supplementary Material
References
- Khalil D. N.; Smith E. L.; Brentjens R. J.; Wolchok J. D. The Future of Cancer Treatment: Immunomodulation, CARs and Combination Immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney K. M.; Rennert P. D.; Freeman G. J. Combination Cancer Immunotherapy and New Immunomodulatory Targets. Nat. Rev. Drug Discovery 2015, 14, 561–584. 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- Ribas A.; Wolchok J. D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Allison J. P. Dissecting the Mechanisms of Immune Checkpoint Therapy. Nat. Rev. Immunol. 2020, 20, 75–76. 10.1038/s41577-020-0275-8. [DOI] [PubMed] [Google Scholar]
- Sharma P.; Allison J. P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Shin D. S.; Ribas A. The Evolution of Checkpoint Blockade as a Cancer Therapy: What’s Here, What’s Next?. Curr. Opin. Immunol. 2015, 33, 23–35. 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Ledford H.; Else H.; Warren M. Cancer Immunologists Scoop Medicine Nobel Prize. Nature 2018, 562, 20–21. 10.1038/d41586-018-06751-0. [DOI] [PubMed] [Google Scholar]
- O’Shea J. J.; Paul W. E. Mechanisms Underlying Lineage Commitment and Plasticity of Helper CD4+T Cells. Science 2010, 327, 1098–1102. 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Chong M. M. W.; Littman D. R. Plasticity of CD4+ T Cell Lineage Differentiation. Immunity 2009, 30, 646–655. 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Chen L.; Flies D. B. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K.; Apetoh L.; Sullivan J. M.; Blazar B. R.; Kuchroo V. K.; Anderson A. C. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J. Exp. Med. 2010, 207, 2187–2194. 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoos A. Development of Immuno-Oncology Drugs-from CTLA4 to PD1 to the next Generations. Nat. Rev. Drug Discovery 2016, 15, 235–247. 10.1038/nrd.2015.35. [DOI] [PubMed] [Google Scholar]
- Baldo B. Adverse Events to Monoclonal Antibodies Used for Cancer Therapy: Focus on Hypersensitivity Responses. Oncoimmunology 2013, 2, e26333 10.4161/onci.26333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid S. S. Process Economics of Industrial Monoclonal Antibody Manufacture. J. Chromatogr. B 2007, 848, 8–18. 10.1016/j.jchromb.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big Opportunities for Small Molecules in Immuno-Oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Huck B. R.; Kötzner L.; Urbahns K. Small Molecules Drive Big Improvements in Immuno-Oncology Therapies. Angew. Chem., Int. Ed. 2018, 57, 4412–4428. 10.1002/anie.201707816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak K. M.; Grudnik P.; Guzik K.; Zieba B. J.; Musielak B.; Dömling A.; Dubin G.; Holak T. A. Structural Basis for Small Molecule Targeting of the Programmed Death Ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. 10.18632/oncotarget.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak K. M.; Kitel R.; Przetocka S.; Golik P.; Guzik K.; Musielak B.; Dömling A.; Dubin G.; Holak T. A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. 10.1016/j.str.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chupak L.; Ding M.; Martin S.; Zheng X.; Hewawasam P.; Connoly T.; Xu N.; Yeung K.; Zhu J.; Langley D.; Tenney D.; Scola P.. Compounds Useful as Immunomodulators. WO 2015160641 A2, Bristol-Myers Squibb, 2015.
- Guzik K.; Tomala M.; Muszak D.; Konieczny M.; Hec A.; Błaszkiewicz U.; Pustuła M.; Butera R.; Dömling A.; Holak T. A. Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—a Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. 10.3390/molecules24112071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chupak L. S.; Zheng X.. Compounds Useful as Immunomodulators. WO 2015034820 A1, Bristol-Myers Squibb Company, 2015.
- Blevins D. J.; Hanley R.; Bolduc T.; Powell D. A.; Gignac M.; Walker K.; Carr M. D.; Hof F.; Wulff J. E. In Vitro Assessment of Putative PD-1/PD-L1 Inhibitors: Suggestions of an Alternative Mode of Action. ACS Med. Chem. Lett. 2019, 10, 1187–1192. 10.1021/acsmedchemlett.9b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musielak B.; Kocik J.; Skalniak L.; Magiera-Mularz K.; Sala D.; Czub M.; Stec M.; Siedlar M.; Holak T. A.; Plewka J. CA-170 - A Potent Small-Molecule PD-L1 Inhibitor or Not?. Molecules 2019, 24, 2804. 10.3390/molecules24152804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Qian D.-Q.; Lu L.; Lajkiewicz N.; Konkol L. C.; Li Z.; Zhang F.; Li J.; Wang H.; Xu M.; Xiao K.; Ywao W.. Heterocyclic Compounds as Immunomodulators. U.S. Patent 20,180,177,784 A1, Incyte Corporation, 2018.
- Assadieskandar A.; Yu C.; Maisonneuve P.; Kurinov I.; Sicheri F.; Zhang C. Rigidification Dramatically Improves Inhibitor Selectivity for RAF Kinases. ACS Med. Chem. Lett. 2019, 10, 1074–1080. 10.1021/acsmedchemlett.9b00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson W. M.; Domek G. J.; Horvath M. P.; Goldenberg D. P. Rigidification of a Flexible Protease Inhibitor Variant upon Binding to Trypsin. J. Mol. Biol. 2007, 366, 230–243. 10.1016/j.jmb.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalut J.; Payan H.; Davis A.; Lecoutey C.; Legay R.; Sopkova-de Oliveira Santos J.; Claeysen S.; Dallemagne P.; Rochais C. Rational Design of Novel Benzisoxazole Derivatives with Acetylcholinesterase Inhibitory and Serotoninergic 5-HT4 Receptors Activities for the Treatment of Alzheimer’s Disease. Sci. Rep. 2020, 10, 3014. 10.1038/s41598-020-59805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf L. K. 122 ’Verpleeg- En Verzorgingshuizen Vaak Veel Te Warm’. Zorg en Financiering 2009, 8, 87. 10.1007/bf03097791. [DOI] [Google Scholar]
- Guzik K.; Zak K. M.; Grudnik P.; Magiera K.; Musielak B.; Törner R.; Skalniak L.; Dömling A.; Dubin G.; Holak T. A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. 10.1021/acs.jmedchem.7b00293. [DOI] [PubMed] [Google Scholar]
- Magiera-Mularz K.; Kocik J.; Musielak B.; Plewka J.; Sala D.; Machula M.; Grudnik P.; Hajduk M.; Czepiel M.; Siedlar M.; Holak T. A.; Skalniak L. Human and Mouse PD-L1: Similar Molecular Structure, but Different Druggability Profiles. iScience 2021, 24, 101960. 10.1016/j.isci.2020.101960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera-Mularz K.; Kuska K.; Skalniak L.; Grudnik P.; Musielak B.; Plewka J.; Kocik J.; Stec M.; Weglarczyk K.; Sala D.; Wladyka B.; Siedlar M.; Holak T. A.; Dubin G. Macrocyclic Peptide Inhibitor of PD-1/PD-L1 Immune Checkpoint. Adv. Ther. 2021, 4, 2000195. 10.1002/adtp.202000195. [DOI] [Google Scholar]
- Skalniak L.; Zak K. M.; Guzik K.; Magiera K.; Musielak B.; Pachota M.; Szelazek B.; Kocik J.; Grudnik P.; Tomala M.; Krzanik S.; Pyrc K.; Dömling A.; Dubin G.; Holak T. A. Small-Molecule Inhibitors of PD-1/PD-L1 Immune Checkpoint Alleviate the PD-L1-Induced Exhaustion of T-Cells. Oncotarget 2017, 8, 72167–72181. 10.18632/oncotarget.20050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S.; Yang J.; Xu B.; Magiera-Mularz K.; Skalniak L.; Musielak B.; Kholodovych V.; Holak T. A.; Hu L. Design, Synthesis, Evaluation, and Structural Studies of C2-Symmetric Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein-Protein Interaction. J. Med. Chem. 2019, 62, 7250–7263. 10.1021/acs.jmedchem.9b00795. [DOI] [PubMed] [Google Scholar]
- Park J.-J.; Thi E. P.; Carpio V. H.; Bi Y.; Cole A. G.; Dorsey B. D.; Fan K.; Harasym T.; Iott C. L.; Kadhim S.; Kim J. H.; Lee A. C. H.; Nguyen D.; Paratala B. S.; Qiu R.; White A.; Lakshminarasimhan D.; Leo C.; Suto R. K.; Rijnbrand R.; Tang S.; Sofia M. J.; Moore C. B. Checkpoint Inhibition through Small Molecule-Induced Internalization of Programmed Death-Ligand 1. Nat. Commun. 2021, 12, 1222. 10.1038/s41467-021-21410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller U.; Darowski N.; Fuchs M. R.; Förster R.; Hellmig M.; Paithankar K. S.; Pühringer S.; Steffien M.; Zocher G.; Weiss M. S. Facilities for Macromolecular Crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 2012, 19, 442–449. 10.1107/s0909049512006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How Good Are My Data and What Is the Resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/s0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug M.; Weiss M. S.; Heinemann U.; Mueller U. XDSAPP: A Graphical User Interface for the Convenient Processing of Diffraction Data Using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. 10.1107/s0021889812011715. [DOI] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Joosten R. P.; Long F.; Murshudov G. N.; Perrakis A. The PDB-REDO Server for Macromolecular Structure Model Optimization. IUCrJ 2014, 1, 213–220. 10.1107/s2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kuppeveld F. J.; Van der Logt J. T.; Angulo A. F.; Van Zoest M. J.; Quint W. G.; Niesters H. G.; Galama J. M.; Melchers W. J. Genus- and Species-Specific Identification of Mycoplasmas by 16S RRNA Amplification. Appl. Environ. Microbiol. 1992, 58, 2606–2615. 10.1128/aem.58.8.2606-2615.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M.; Wang Y.; Wu T.; Xu Z.; Zhang N.. Aromatic Acetylene or Aromatic Ethylene Compound Intermediate, Preparation Method, Pharmaceutical Composition and Use Thereof. EP 3483142 A1, Guangzhou Maxinovel Pharmaceuticals Co. LTD., 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.