

Abstract

CAMKK2 is a serine/threonine kinase and an activator of AMPK whose dysregulation is linked with multiple diseases. Unfortunately, STO-609, the tool inhibitor commonly used to probe CAMKK2 signaling, has limitations. To identify promising scaffolds as starting points for the development of high-quality CAMKK2 chemical probes, we utilized a hinge-binding scaffold hopping strategy to design new CAMKK2 inhibitors. Starting from the potent but promiscuous disubstituted 7-azaindole GSK650934, a total of 32 compounds, composed of single-ring, 5,6-, and 6,6-fused heteroaromatic cores, were synthesized. The compound set was specifically designed to probe interactions with the kinase hinge-binding residues. Compared to GSK650394 and STO-609, 13 compounds displayed similar or better CAMKK2 inhibitory potency in vitro, while compounds 13g and 45 had improved selectivity for CAMKK2 across the kinome. Our systematic survey of hinge-binding chemotypes identified several potent and selective inhibitors of CAMKK2 to serve as starting points for medicinal chemistry programs.
Introduction
Calcium (Ca2+)/calmodulin-dependent protein kinase kinase 2 (CAMKK2) is a serine/threonine kinase that is one of the calmodulin (CaM)-binding proteins of the CaMK family.1−3 Ca2+ is an important second messenger that aids in the regulation of a wide range of signaling events in part through binding to its intracellular receptor CaM.4 Ca2+-bound CaM modulates various cellular responses via activation of an array of downstream proteins including CAMKK2.5 Upon activation, CAMKK2 phosphorylates and activates its substrates including CAMK1, CAMK4, AMP-activated protein kinase, (AMPK) and in some cases AKT. This signal transduction results in the regulation of many important physiological and pathological processes.6−16
Due to CAMKK2’s important role in cell signaling, dysregulation of CAMKK2 has been implicated in several diseases. Aberrant activation and overexpression of CAMKK2 have been linked to multiple cancer types including prostate, breast, ovarian, gastric, and hepatic cancers.8−14,17,18 Knockdown of CAMKK2 via siRNA or pharmacological inhibition of CAMKK2 reduced cell proliferation, migration, and invasion as well as induced apoptosis and cell cycle arrest in numerous cancer cell lines and tumor models.8,12−14,17,19−21 Mechanistically, CAMKK2 is an important regulator of metabolic and inflammatory processes, which are contributory factors to its impact on cancer growth.9,14
In addition, therapeutic interventions targeting CAMKK2 may be beneficial beyond oncology. For example, hepatocellular carcinoma (HCC) is often preceded by non-alcoholic fatty liver disease (NAFLD), which is driven by several risk factors including obesity and type 2 diabetes.22−24 Inhibition of CAMKK2 reduced food intake in mice, and Camkk2-null mice are leaner than wild-type mice when fed a high-fat diet.25,26 Consistent with these findings, CaMKK2 was recently shown to be inhibited by liraglutide, a glucagon-like peptide-1 receptor agonist that decreases food intake and promotes weight loss.27 In relation to skeletal disease, CAMKK2 is expressed in osteoblasts and osteoclasts, which play an essential role in bone tissue maintenance.28 Inhibition of CAMKK2 stimulated bone formation and reversed age-associated decline in bone health by promoting osteoblast differentiation and inhibiting osteoclast differentiation.29 Taken together, these findings suggest that inhibition of CAMKK2 may be effective for the treatment of a variety of diseases including various types of cancers, metabolic disorders, and bone diseases like osteoporosis. While CAMKK2 has emerged as an attractive therapeutic target, there remains a shortage of high-quality CAMKK2 inhibitors, which has impaired progress in the field.
At present, almost all CAMKK2-related pharmacological studies rely on the use of the ATP-competitive inhibitor STO-609 to study CAMKK2 signaling events (Figure 1).8,11,17,28−32 However, STO-609 is not an ideal chemical tool.33 It binds to multiple kinases beyond CAMKK2. When screened against a panel of over 400 wild-type human kinases (KINOMEscan, Eurofins DiscoverX) at 1 μM, STO-609 bound with strong affinity to 14 kinases (<20% activity remaining).33 Among these kinases were CDKL2, GRK3, and CK2, which are all overexpressed in several cancer types.34−36 In addition to these collateral kinase targets, STO-609 potently activates the aryl hydrocarbon receptor.37 STO-609 is a planar molecule with poor aqueous solubility, yet it is routinely used at high doses in cell culture (>10 μM) and in vivo.24 Due to the liabilities of STO-609, the field would benefit from the discovery and development of potent and highly selective small-molecule inhibitors of CAMKK2 as high-quality probes.
Figure 1.

Structures of known CAMKK2 inhibitors: STO-609 and GSK650394.
We recently conducted an extensive literature search for CAMKK2 inhibitors to identify starting points for medicinal chemistry optimization.33 A promising series of potent CAMKK2 inhibitors were recently disclosed by GlaxoSmithKline (GSK).25 The 7-azaindole GSK650394 (also called a pyrrolopyridine) (Figure 1) was a potent CAMKK2 inhibitor (IC50 = 0.63 nM). The co-crystal structure of GSK650394 with CAMKK2 (PDB 6BKU) revealed the critical role of the carboxylic acid and cyclopentyl moiety (Figure 2a), which was consistent with the reported structure–activity relationship (SAR) studies.25 The co-crystal structure reveals that the nitrogen atoms of the pyridine and pyrrole rings act as hydrogen bond acceptor and donor pairs, respectively, and form interactions with the protein backbone of the ATP-binding site. The acid functionality contributes to critical hydrogen bonding interactions with both the protonated amine of Lys194 and the carboxylate group of Glu236 in a water-mediated manner.38 Additionally, CH−π interactions (aromatic edge–face interactions) between the aromatic ring of gatekeeper Phe267 with both the CH in the 2-position of the 7-azaindole and the methine group in the ortho-position of the 3-aryl moiety of the inhibitor stabilize GSK650394 in this orientation. The pendant phenyl group in the 5-position of the 7-azaindole scaffold occupies a hydrophobic pocket which can potentially be exploited to gain selectivity and potency and modulate physical properties to optimize pharmacokinetic parameters. Although a crystal structure of CAMKK2 co-crystallized with ATP is not available, the cyclopentyl group appears to mimic at least the position of the ribosyl moiety of ATP based on the proximity of the cyclopentyl group to the P-loop of CAMKK2. The 7-azaindole pharmacophore in GSK650394 is commonly found in kinase inhibitors and is considered a powerful but sometimes promiscuous hinge binder.39−43 When screened against a panel of 334 kinases (Reaction Biology Corporation), GSK650394 inhibited 29 kinases by more than 90%, limiting its utility as a tool compound for studying CAMKK2 or any other kinase.33
Figure 2.

(a) Binding mode of the co-crystallized ligand GSK650394 in the ATP-binding site of CAMKK2 (PDB-ID: 6BKU). The image was generated with PyMOL. The protein is colored in gray. Blue-dashed lines indicate H-bond interactions; green-dashed lines display CH−π interactions. GSK650394 is shown as purple sticks and the water molecule as a red sphere. Oxygen and nitrogen atoms are colored in red and blue, respectively. (b) Structure of GSK650394 highlighting important sites for design of new CAMKK2 inhibitors.
To improve the kinase selectivity of GSK650394, we targeted replacement of the 7-azaindole with other heterocycles that would modulate interactions with the hinge-binding residues and in some cases reduce interactions with the hinge-binding residues. Our compound design strategy is depicted in Figure 2b. Our central hypothesis was based on the belief that the 3-position aryl ring, decorated with the carboxylic acid and cyclopentyl groups, plays a dominant role in CAMKK2 recognition and that inhibitors with enhanced selectivity would arise from compounds with the binding contribution from the hinge binder diminished. This led to the synthesis and evaluation of structurally diverse novel chemotypes as potential inhibitors of CAMKK2 (Figure 3).
Figure 3.

New molecules based on scaffold hopping from GSK650394 designed to interrogate several aspects of hinge binding interactions between CAMKK2 and the proposed inhibitors. Top row: Inhibitors retaining the topology of GSK650394 (5,6-fused ring systems with the cyclopentyl-substituted benzoic acid moiety attached to the 5-membered ring). Middle row: 5,6-fused systems with the cyclopentyl-substituted benzoic acid moiety appended to the 6-membered ring. Bottom row: Inhibitors with ring expansion (6,6-fused ring system) or ring deletion (single 6-membered ring as the hinge-binding core).
Thus, we performed a scaffold-hopping exercise wherein the hinge-binding 7-azaindole core of GSK650394 was substituted with structurally diverse alternate hinge-binding moieties. We retained the ortho-cyclopentyl benzoic acid moiety hypothesizing that the interactions observed in the crystal structure would bias the new set toward CAMKK2 affinity.
Results
Chemistry
To develop analogues of GSK650394 that could be potent and selective CAMKK2 inhibitor scaffolds, all our synthesized compounds incorporated the critical pharmacophore, ortho-cyclopentyl benzoic acid. In parallel, to investigate the importance of the pendant phenyl ring, we synthesized matched pairs with and without this group. We theorized that removal of the phenyl ring could significantly lower the cLog P and increase ligand efficiency (LE) if it was dispensable without being detrimental to potency. To this end, we first synthesized pinacol borate esters 3 and 4 to enable attachment to the various hinge-binding pharmacophores via Suzuki coupling reactions (Scheme 1). 4-Bromo-2-fluorobenzoic acid 1 underwent a Grignard reaction with cyclopentylmagnesium bromide to afford the ortho-cyclopentyl-substituted benzoic acid 2, which was then converted into the corresponding pinacol boronate ester using Miyaura borylation conditions yielding 3.44 The analogous methyl ester 4 was made in a similar fashion following esterification of 2 in methanol in the presence of thionyl chloride.
Scheme 1. Synthesis of Pinacol Boronate Esters 3 and 4.
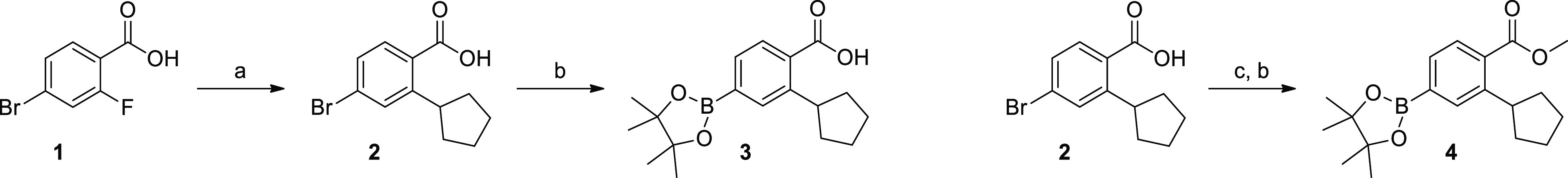
Reagents and conditions: (a) cyclopentylmagnesium bromide, THF, −10 to 25 °C, 5 h, then aq HCl; (b) PdCl2(dppf)·CH2Cl2, KOAc, bis(pinacolato)diboron, 100 °C, 2 h; (c) SOCl2, MeOH, 16 h, 75 °C.
3- and 3,5-Substituted Fused 6,5-Ring Systems
Initial analogues were fused 5,6-ring systems with 3- and 3,5- substitution patterns that retained the topology of GSK650394 but modify the nature and/or location of heteroatoms in the ring system and thus can alter the compound’s ability to form H-bond interactions with the hinge region. GSK650394 is commercially available, but its matched pair 7, truncated at the 5-position, required synthesis (Scheme 2). 3-Bromo-7-azaindole 5 was converted to the silylethoxymethyl (SEM)-protected azaindole and then subjected to Suzuki–Miyaura conditions to afford cyclopentyl analogue 6. SEM deprotection, followed by base-mediated saponification, afforded the target azaindole 7.
Scheme 2. Synthesis of Azaindole 7, the Matched Pair for GSK650394.

Reagents and conditions: (a) SEM-Cl, NaH, DMF, 0–21 °C, 2 h; (b) 4, Pd(OAc)2, K3PO4, P(Cy)3, PhMe/H2O, 80 °C, 16 h; (c) CF3COOH, CH2Cl2, then NaOAc, EtOH, 21 °C, 24 h; (d) aq NaOH, MeOH, 75 °C, 1 h, then aq HCl.
The N-methyl analogues of GSK650394 and 7 were synthesized as depicted in Scheme 3. To access 10, 5-chloro-3-iodo azaindole 8b was N-alkylated.45 Consecutive Suzuki reactions with the ortho-cyclopentyl methyl benzoate 4 and then phenyl boronic acid placed the key pharmacophore in the 3-position and a phenyl ring in the 5-position. Saponification of the methyl ester proceeded smoothly affording azaindole 10. Coupling of boronate ester 4 with 3-bromo-N-methyl azaindole, followed by saponification of the methyl ester, afforded target molecule 11.
Scheme 3. Synthesis of N-Methyl Azaindole Hinge Binders 10 and 11.
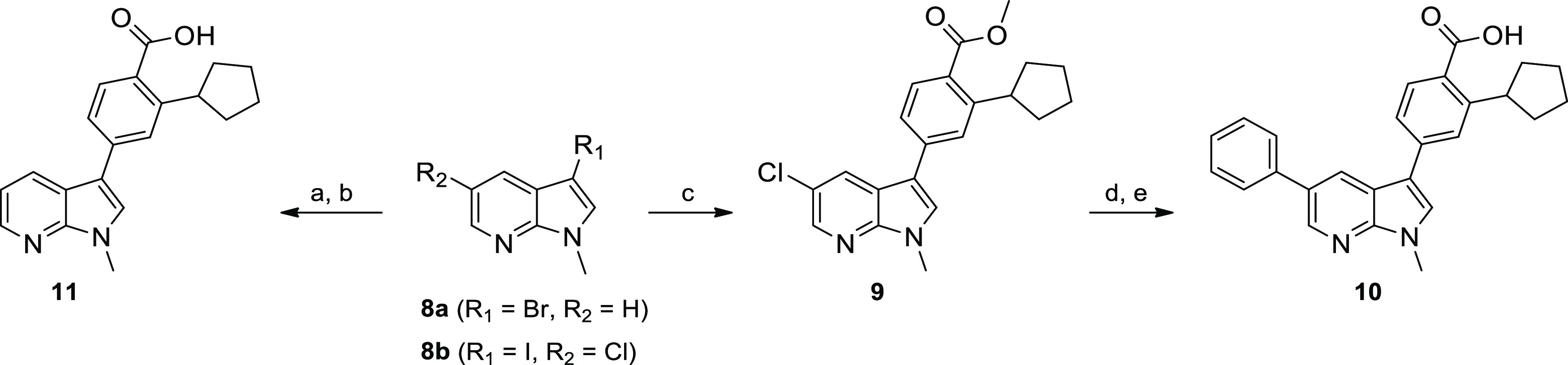
Reagents and conditions: (a) 3, Pd2(dba)3, XPhos, Cs2CO3, dioxane, H2O, 120 °C, 16 h; (b) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl; (c) 4, PdCl2(dppf)·CH2Cl2, Cs2CO3, dioxane, H2O, rt, 16 h; (d) PhB(OH)2, Pd2(dba)3, XPhos, Cs2CO3, dioxane, H2O, 120 °C, 16 h; (e) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl.
The synthesis of the structurally similar furopyridine and thienopyridine cores is described in Scheme 4. Nicotinic acid derivatives 12a and 13a were converted into ethyl esters 12–14b and then treated with ethyl glycolate or ethyl thioglycolate in the presence of sodium hydride (NaH) to give the respective furopyridines or thienopyridines 12–14c.46 A one-pot saponification–decarboxylation of the β-keto esters afforded the 2-unsubstituted heterocycles 12–14d. These aryl alcohols were converted to the corresponding triflates 12–14d, which were able to undergo Suzuki–Miyaura reactions to yield analogues 12f, 13g, and 14g.4712g was obtained directly from the commercially available 3-bromothieno[2,3-b]pyridine 15e using Suzuki–Miyaura reaction.
Scheme 4. General Route to the Furo- and Thienopyridines.
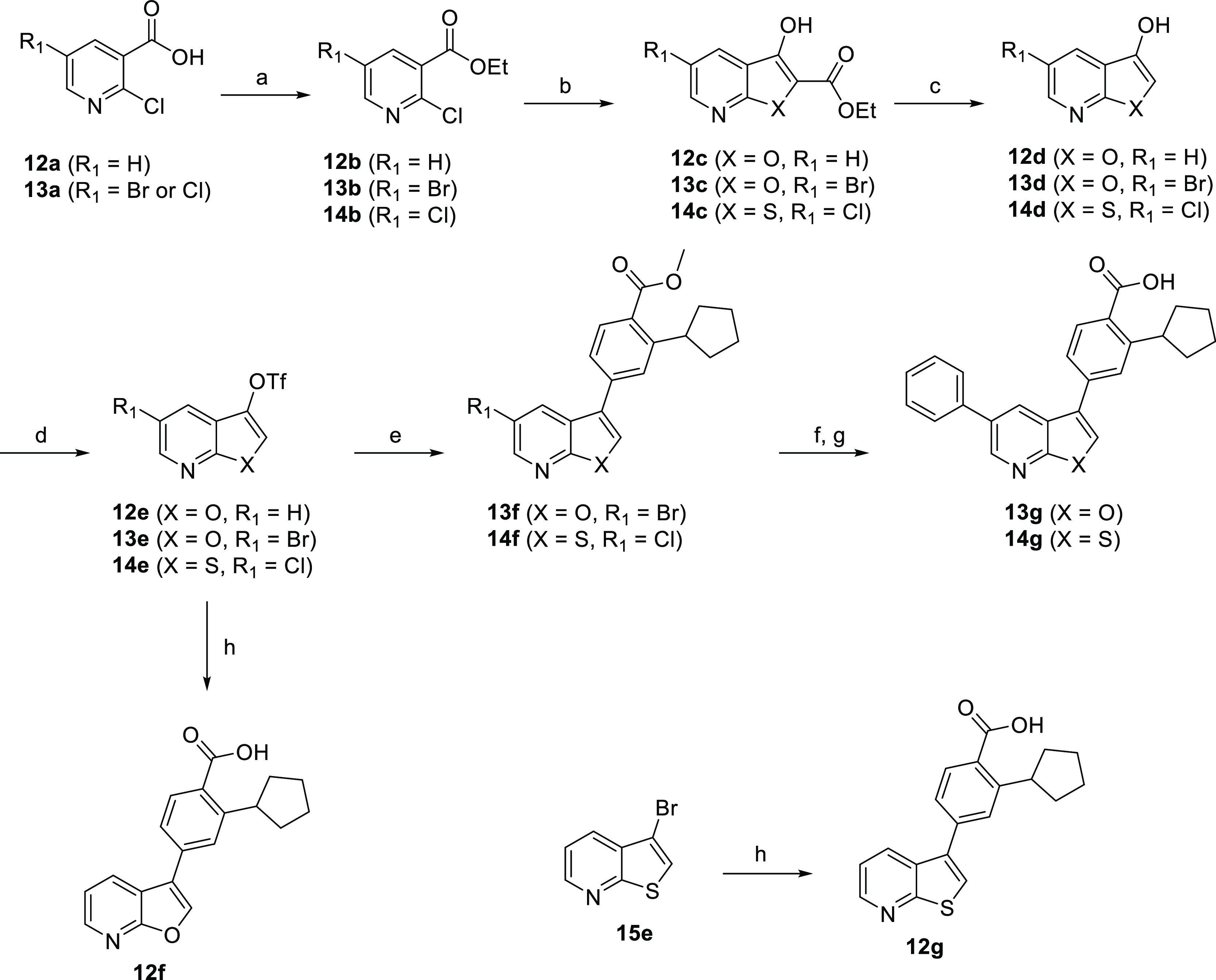
Reagents and conditions: (a) (EtO)3CH, PhMe, 100 °C, 2 h or H2SO4, EtOH, reflux, 24 h; (b) ethyl glycolate or ethylthioglycolate, NaH, DME, 0–75 °C, 2.5 h; (c) aq NaOH, EtOH then aq HCl, 100 °C, 2 h; (d) Tf2O, DIPEA, CH2Cl2, −10 to 25 °C, 3 h; (e) 4, Pd(PPh3)4, Na2CO3, MeOH/CH2Cl2, 90 °C, 1 h; (f) PhB(OH)2, Pd(PPh3)4, Na2CO3, MeOH/CH2Cl2, 90 °C, 1 h or PhB(OH)2, Pd2(dba)3, XPhos, Cs2CO3, dioxane/H2O 90 °C, 16 h; (g) aq NaOH, MeOH then aq HCl, 1 h; (h) 3, Pd(PPh3)4, Na2CO3, MeOH/CH2Cl2, 90 °C, 1 h.
The subsequent set of fused 5,6-ring derivatives synthesized in Scheme 5 (pyrazolopyridines and pyrazolopyrimidines) dramatically altered the placement of the heteroatoms based on the parent compound. Analogues 19 and 20 were obtained by stepwise Suzuki couplings, first with 4 and then with phenylboronic acid, followed by methyl ester hydrolysis. The monosubstituted complementary matched pairs were synthesized from commercially available 3-bromopyrazolo[1,5-a]pyridine or -pyrimidine via Suzuki couplings with 4 to afford 21 and 22. The same set of reaction conditions was used to afford the triazolopyridazine analogues 25 and 26 (Scheme 6).
Scheme 5. Synthesis of Pyrazolopyridine and Pyrazolopyrimidine Analogues.
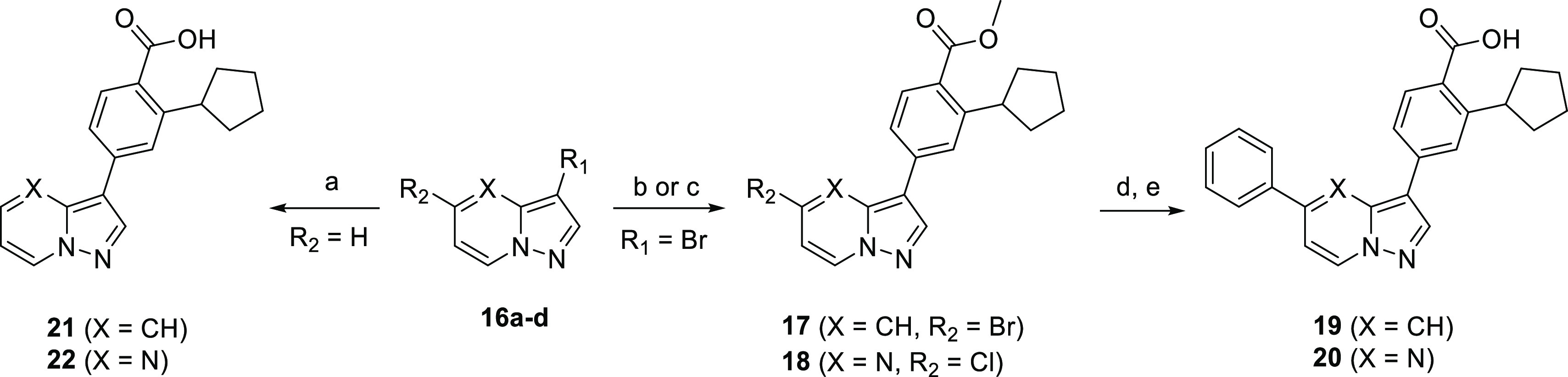
Reagents and conditions: (a) 3, Pd2(dba)3, XPhos, Cs2CO3, dioxane, H2O, 120 °C, 16 h; 17via (b) 4, Pd2(dba)3, XPhos, Cs2CO3, dioxane, H2O, 120 °C, 16 h; 18via (c) PdCl2(dppf)·CH2Cl2, Cs2CO3, dioxane, H2O, 80 °C, 16 h; (d) PhB(OH)2, Pd(PPh3)4, Cs2CO3, dioxane, H2O, 120 °C, 30 min, μW; (e) aq LiOH, dioxane, 100 °C, 16 h then aq HCl.
Scheme 6. Synthesis of the Triazolopyridazine Analogues 25 and 26.
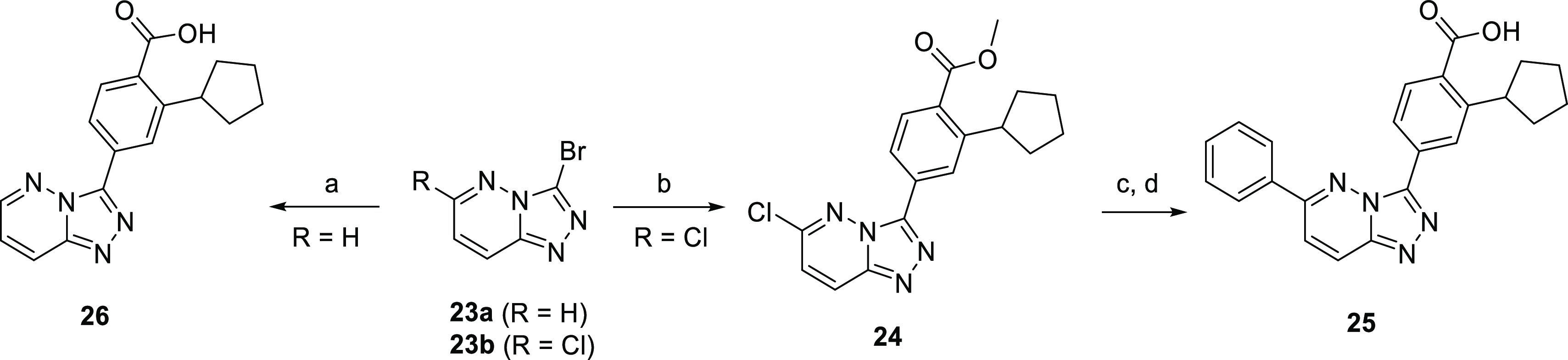
Reagents and conditions: (a) 3, Pd2(dba)3, XPhos, Cs2CO3, dioxane/H2O, 90 °C, 16 h; (b) 4, Pd(PPh3)4, K2CO3, dioxane/H2O, 90 °C, 16 h; (c) Pd2(dba)3, XPhos, Cs2CO3, dioxane/H2O, 90 °C, 16 h; (d) aq NaOH, MeOH 75 °C, 1 h, then aq HCl.
2- and 2,4-Substituted Fused 5,6-Ring Systems
The first scaffolds in this category were N-methyl azaindoles and are described in Scheme 7. The disubstituted azaindole 29 was obtained from coupling phenylboronic acid at the more reactive 2-position of 2-iodo-4-chloro-N-methyl azaindole, followed by coupling with intermediate 4 at the 4-position. Saponification of the methyl ester afforded acid 29. The monosubstituted compound 30 was synthesized from commercially available 4-chloro-N-methyl-7-azaindole with a Suzuki coupling reaction, followed by saponification of the methyl ester.
Scheme 7. Synthesis of 2,4-Substituted N-Methyl Azaindoles.

Reagents and conditions: (a) 4, Pd(PPh3)4, Cs2CO3, dioxane/H2O, 120 °C, 30 min; (b) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl; (c) PhB(OH)2, Pd(PPh3)4, Cs2CO3, dioxane/H2O, 120 °C, 30 min; (d) 4, Pd(PPh3)4, Cs2CO3, dioxane/H2O, 120 °C, 30 min; (e) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl.
Imidazopyridine analogues were obtained from initial coupling of 2,3-diamino-4-chloropyridine with 4 to afford the aryl-substituted pyridine 32 (Scheme 8). Imidazopyridine 33 was synthesized by condensation of diamine 32 with trimethyl orthoformate in methanol, followed by saponification of the ester to afford the desired carboxylic acid. The 2-aryl analogue 34 was obtained by converting benzoic acid into an activated ester with 1,1′-carbonyldiimidazole (CDI) and addition of diamine 32 to form a putative amide intermediate that underwent a ring closure under thermal conditions resulting in the imidazopyridine intermediate. Ester hydrolysis yielded the desired imidazopyridine 34.
Scheme 8. Synthesis of the Imidazopyridine Hinge Binders.
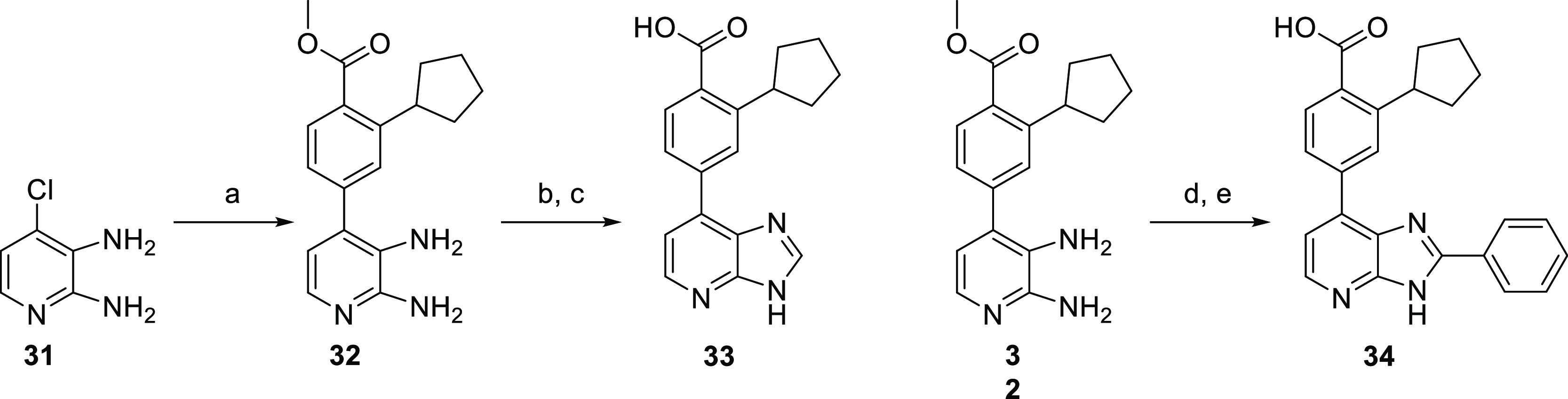
Reagents and conditions: (a) 4, Pd2(dba)3, XPhos, Cs2CO3, dioxane, H2O, 100 °C, 16 h; (b) CH(OMe)3, H3NSO3, MeOH, rt, 1 h; (c) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl; (d) benzoic acid, CDI, DMF, 0 °C, 30 min, then 32, 200 °C, 10 min; (e) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl.
Mono- and disubstituted thienopyrimidine analogues of the [3,2-d] and [2,3-d] core scaffolds were synthesized from commercially available starting materials (Scheme 9). The monosubstituted analogues 38 and 42 were easily accessed in one step via Suzuki coupling using commercially available 35a and 39a, respectively. 37 was synthesized by first coupling 35b with phenylboronic acid to afford 2-aryl-substituted intermediate 36, which was then coupled with 3 to yield the disubstituted thieno[3,2-d]pyrimidine. The reaction sequence was reversed in the synthesis of the disubstituted thieno[2,3-d]pyrimidine 41 since the chlorine substituent in 39b was found to be more reactive than the thiophene bromine. Intermediate 4 was coupled to the 4-position of 39b affording 40. Subsequent Suzuki coupling with phenylboronic acid, followed by saponification of the ester, yielded disubstituted analogue 41.
Scheme 9. Synthesis of Thieno[3,2-d]pyrimidine and Thieno[2,3-d]pyrimidine Hinge Binders.

Reagents and conditions: (a) 3, Pd(PPh3)4, Cs2CO3, dioxane/water, 125 °C, μW, 15 min; (b) PhB(OH)2, NaHCO3, CsF, Pd(PPh3)4, dioxane/H2O, 95 °C, 3 h; (c) 3, Pd(PPh3)4, Cs2CO3, dioxane/water, 125 °C, 15 min; (d) 3, Pd(PPh3)4, Cs2CO3, dioxane/water, 125 °C, μW, 15 min; (e) 4, Pd(PPh3)4, Cs2CO3, dioxane/water, 145 °C, μW, 15 min; (f) PhB(OH)2, NaHCO3, CsF, Pd(PPh3)4, dioxane/H2O, 80 °C, 4 h; (g) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl.
Synthesis of Substituted Fused 6,6-Ring Hinge Binders
This set of hinge binders moves from 5,6-fused ring systems to 6,6-fused systems. The disubstituted quinoline analogue 45 was synthesized in a straightforward manner from commercially available 4,6-dichloroquinoline 43bvia sequential Suzuki couplings with 4 and phenylboronic acid, followed by saponification of the methyl ester. The monosubstituted quinoline 46 was synthesized in one step via Suzuki coupling between the commercially available 43a and 3.
The quinazoline hinge binder derivatives were obtained via similar routes utilized in the synthesis of the quinolines (Scheme 10) with the exception of compound 49. The disubstituted quinazoline 49 was prepared from 4-chloroquinazolin-6-ol 47b with initial installation of boronate 4 and subsequent conversion of the hydroxy group of 48 into a triflate which readily underwent Suzuki coupling with phenylboronic acid. Saponification then afforded the target compound. 4-Aryl-2-methylquinoline 52 and 1,6-naphthyridine 54 analogues were synthesized in one step via Suzuki coupling with 3 (Scheme 10c).
Scheme 10. Synthesis of Quinoline and Quinazoline Hinge Binders.

Reagents and conditions: (a) 3, Pd2(dba)3, XPhos, Cs2CO3, dioxane/H2O, 90 °C, 16 h; (b) 4, Pd2(dba)3, XPhos, Cs2CO3, rt, 16 h; (c) PhB(OH)2, Pd2(dba)3, XPhos, Cs2CO3, 120 °C, 16 h; (d) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl; (e) 4, Pd(PPh3)4, K2CO3, dioxane/H2O, 120 °C, 16 h; (f) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl; (g) 4, Pd(PPh3)4, K2CO3, dioxane/H2O, 90 °C, 16 h; (h) (i) DIPEA, Tf2O, CH2Cl2, 0–21 °C, 2 h, (ii) PhB(OH)2, Pd(PPh3)4, K2CO3, CH2Cl2/MeOH, 90 °C, 1 h; (i) aq NaOH, MeOH, 75 °C, 1 h, then aq HCl; (j) 4, Pd2(dba)3, XPhos, Cs2CO3, dioxane/H2O, 90 °C, 16 h.
Synthesis of Pyrimidine Hinge Binders
The final set of hinge binders we explored consisted of substituted pyrimidines. The monosubstituted pyrimidine 56 was obtained via Suzuki coupling of 55 with 3 (Scheme 11a). 2-Phenyl-4-aryl pyrimidine 58 was synthesized from 2-phenyl-4-chloropyrimidine 57 by Suzuki coupling with 3. The synthesis of aminopyrimidines 61 and 62 is described in Scheme 11b. Starting from commercially available 4-chloropyrimidin-2-amine 59, Suzuki coupling with 4 afforded 60. Saponification of the resulting methyl ester afforded 61. 60 was utilized in a Buchwald–Hartwig coupling with bromobenzene, and subsequent ester hydrolysis yielded 2-phenylaminopyrimidine 62.
Scheme 11. Synthesis of Pyrimidine and Aminopyrimidine Hinge Binders.
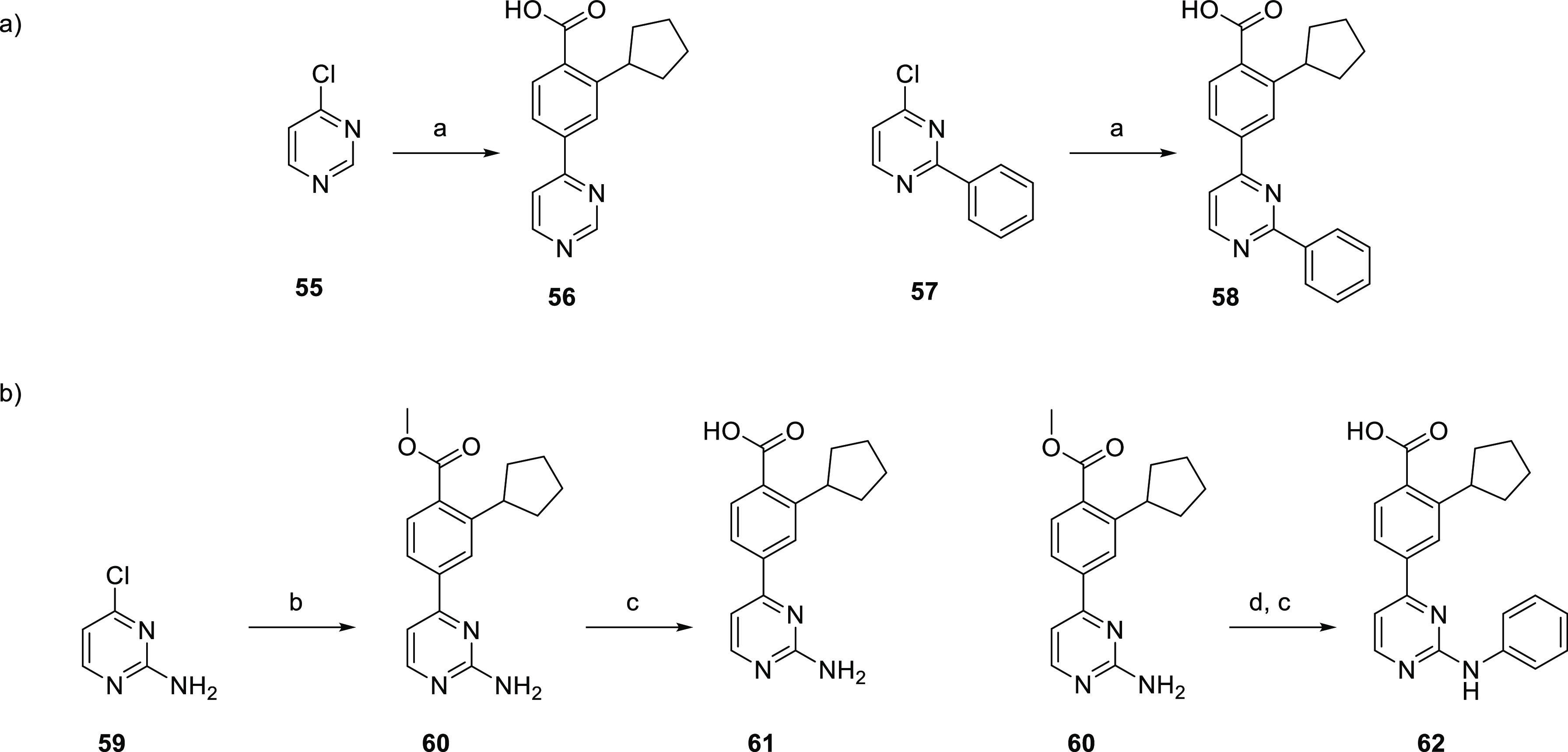
Reagents and conditions: (a) 3, Pd(PPh3)4, Cs2CO3, 1,2-DME, H2O, 120 °C, 0.5 h. (b) 4, Pd(PPh3)4, Cs2CO3, 1,2-DME, H2O, 120 °C, 0.5 h; (c) aq LiOH, dioxane, 100 °C, 16 h, then aq HCl; (d) PhBr, Pd2(dba)3, XantPhos, NaOtBu, PhMe, 80 °C, 19 h.
Assays Used for In Vitro Compound Affinity Evaluation
DSF Assay
We used a thermal-shift assay (differential scanning fluorimetry, DSF) to detect protein–ligand interactions.48 For many proteins, including kinases, this is a useful high-throughput method to identify compounds that bind to a protein of interest, and in many cases, the melting temperature correlates with binding affinity. CAMKK1 and CAMKK2 are ∼60% identical at the amino acid sequence level.49 We measured DSF ΔTm for all analogues against both CAMKK2 and CAMKK1 using the purified kinase domains of these two proteins. As in the end we did not utilize the DSF data to drive the project, all DSF data are reported in the Supporting Information.
In Vitro CAMKK1 and CAMKK2 Enzyme Inhibition Assays
We used purified recombinant CAMKK1 and CAMKK2 in an assay that measured the transfer of radiolabeled phosphate from [γ-32P]-ATP to a synthetic CAMKK2 substrate (CAMKKtide). Initially, we evaluated CAMKK2 inhibitory activity of all synthesized compounds at three different concentrations (10, 100, and 1000 nM) to rank compounds and provide preliminary dose response information. In the percent of control (PoC) experiments, the assay with no inhibitor present serves as the control. Half-maximal inhibitory concentrations (IC50) were then determined for analogues with PoC values <10 at the 1 μM screening concentration. For a subset of CAMKK2 inhibitors, we also determined half-maximal inhibitory concentration (IC50) values. STO-609 and GSK650394 were routinely used as reference compounds in the assay to ensure that the assay performed correctly and CAMKK2 inhibition was observed.
In Vitro Compound Screening Results
The initial set of compounds evaluated was focused on those with similar topology to GSK650394. Data in Table 1 depict compounds, the enzyme CAMKK2 inhibition data (PoC) at three concentrations, and CAMKK2 enzyme inhibition IC50 data for selected compounds. Although we collected thermal shift (DSF) data for our inhibitors with both CAMKK1 and CAMKK2, we found that they did not provide enough information to drive chemistry decisions and did not always correlate with the enzymatic activity. The DSF data are provided in the Supporting Information for reference. We thus relied on the CAMKK2 enzyme inhibition data as our primary assay.
Table 1. CAMKK2 Enzyme Inhibition Data for Compounds with 3- and 3,5-Substituted Fused 5,6-Ring Systemsa.

R = ortho-cyclopentyl benzoic acid moiety; PoC (percent of control = percent of enzyme activity remaining, when compared with the control); IC50 (half-maximal inhibitory concentration); NG (not generated); IC50 values were generated in an 8-point full dose response assay.
Removal of the phenyl group from the 5-position of GSK650394 led to a roughly 3-fold decrease in enzyme inhibitory activity (IC50 = 26 vs 3 nM), implying that the phenyl group or an appropriately adorned phenyl group may be useful to enhance CAMKK2 enzyme affinity and/or activity. Similar pairs of analogues for the different hinge binders allowed for direct comparison (Table 1).
The goal of this project was to discover alternate scaffolds for CAMKK2 with sufficient potency and suitable kinome selectivity that can be used as starting points for medicinal chemistry optimization programs. We hypothesized that carefully modulating the hydrogen bonding interactions at the hinge-binding region via a variety of heterocyclic scaffolds could identify a new series that maintained CAMKK2 affinity and improved selectivity profiles. Kinase inhibitors with multiple hinge-binding interactions can suffer from off-target effects owing to their potential for poor selectivity across the kinome.39,41,50 However, careful modification of even promiscuous starting points can in some cases lead to desired levels of selectivity. There are examples demonstrating that modifications to the hydrogen bonding interactions between a kinase inhibitor and the hinge region of the kinase can improve selectivity without severely impacting potency.41N-Methyl azaindoles 10 and 11 (Table 1) offer a particularly drastic change in hinge binding by blocking the H-bonding donor property of the pyrrole ring. This resulted in a complete loss in enzyme inhibition potency in vitro. This suggested that the steric bulk of the N-methyl group is not accommodated within the binding pocket of CAMKK2 when the cyclopentyl benzoic acid moiety is attached at the 3-position of the azaindole scaffold.
Further structural modifications to the hinge-binding moiety that maintained the fused 5,6-ring system are outlined here. Compounds 13g and 12f replaced the pyrrole ring with a furan ring. Compounds 14g and 12g replaced the pyrrole ring of GSK650394 with a thiophene ring. The pyridine nitrogen that makes a key hydrogen bond with the hinge is maintained, but the NH hydrogen bonding opportunity has been removed. Compounds 19, 20, 21, 22, 25, and 26 all maintain the 5,6-fused core but have key differences from GSK650394. They do not have a nitrogen in a position analogous to the nitrogen in the 7-position of GSK650394, thus necessitating different hinge-binding interactions. They also incorporate nitrogen atoms in other locations within the 5,6-ring system.
Nearly all the phenyl-substituted analogues of active CAMKK2 inhibitor scaffolds showed greater enzyme potency than their corresponding truncated analogues. An exception was compound 19 (phenyl version), which had an IC50 = 145 nM compared to an IC50 = 44 nM for compound 21 (truncated version). Replacement of the pyrrole ring in the azaindole with either a furan or thiophene ring retained potency in the phenyl-substituted analogues (13g IC50 = 65 nM, 14g IC50 = 5 nM), but their corresponding truncated analogues (12f, 12g) were relatively inactive. We would like to point out that an analogue of compound 13g is listed as a CAMKK2 chemical probe (https://www.thesgc.org/chemical-probes/SGC-CAMKK2-1) with the name SGC-CAMKK2-1. The kinome-wide selectivity profiles and cellular potencies of these two close analogues are comparable (vide infra and the SGC probe link above). In addition, SGC-CAMKK2-1 is currently commercially available from Sigma-Aldrich. The results depicted in Table 1 demonstrate that the choice of a hinge binder plays a role in the type of substitutions that are tolerated. Both versions of the triazolopyridazine hinge binder (25, 26) showed no activity. We hypothesize that this is due to repulsion between the lone pair on the nitrogen in the 2-position and a backbone protein carbonyl (carbonyl from E268 in CAMKK2). In many kinase/inhibitor co-crystal structures, this carbonyl is engaged in hydrogen bonding with a NH from the inhibitor or at least pointing toward a polarized CH on the inhibitor. In summary, four scaffolds (furopyridine 13g, thienopyridine 14g, pyrazolopyridine 19 and 21, and pyrazolopyrimidine 20) in this set exhibited robust CAMKK2 enzyme inhibition (IC50 values < 150 nM), with 14g (IC50 = 5 nM) exhibiting comparable enzymatic inhibitory activity to GSK650394 (IC50 = 3 nM).
To further evaluate fused 5,6-ring structures as CAMKK2 inhibitors, we switched from 3,5- to 2,4-ring substitutions as depicted in Table 2. N-methyl azaindoles 29 and 30 were the first pair of analogues synthesized. Interestingly, these two N-methyl-substituted azaindole analogues were well tolerated, displaying good CAMKK2 enzyme inhibitory activity (29 IC50 = 120 nM, 30 IC50 = 56 nM). This result is in stark contrast to 3,5-N-methyl azaindoles 10 and 11 in Table 1. The truncated analogue 30 was significantly more potent than its corresponding phenyl analogue 29.
Table 2. CAMKK2 Enzyme Inhibition Data for Compounds with 2- and 2,4-Substituted Fused 6,5-Ring Systemsa.

R = ortho-cyclopentyl benzoic acid moiety; PoC (percent of control = percent of enzyme activity remaining, when compared with control); IC50 (half-maximal inhibitory concentration); IC50 values were generated in an 8-point full dose response assay.
The 2,4-substituted analogues of Table 2 are all inhibitors of CAMKK2. Incorporation of an extra nitrogen into the pyrrole ring to create imidazopyridine analogues 33 and 34 was well tolerated, with the phenyl-substituted analogue demonstrating higher potency (34 IC50 = 23 nM) than the corresponding non-phenyl version (33 IC50 = 193 nM). Similar to N-methyl azaindole 30, non-phenyl-substituted thienopyrimidines (38 IC50 = 24 nM, 42 IC50 = 31 nM) were more potent than their corresponding phenyl-substituted analogues (37 IC50 = 169 nM, 41 IC50 = 232 nM). Both [3,2-d] analogues 37 and 38 (with the sulfur oriented away from the hinge-binding region) and [2,3-d] analogues 41 and 42 (with the sulfur oriented toward the hinge) inhibit CAMKK2. For both these isomeric thienopyrimidines, removal of the pendant phenyl actually increases CAMKK2 potency (37 with phenyl IC50 = 169 nM 38 without phenyl IC50 = 24 nM; 41 with phenyl IC50 = 232 nM 42 without phenyl IC50 = 31 nM).
Next, we explored replacement of the 5,6-bicyclic cores with 6,6-bicyclic structures. Quinazolines and quinolines are frequently used as kinase hinge-binding heterocycles and were identified as active scaffolds for CAMKK2 inhibition (Table 3). Both quinoline 45, with a phenyl in the 6-position, and 46, unsubstituted in the 6-position, inhibited CAMKK2 (45 IC50 = 137 nM, IC5046 = 13 nM). The potency of the low-molecular-weight compound 46 suggests that further exploration may be fruitful. Similarly, the two quinazolines 49 (IC50 = 12 nM) and 50 (IC50 = 96 nM) were also potent CAMKK2 inhibitors.
Table 3. CAMKK2 Enzyme Inhibition Data for Compounds with Fused 6,6-Ring Systemsa.

R = ortho-cyclopentyl benzoic acid moiety; PoC (percent of control = percent of enzyme activity remaining, when compared with the control); IC50 (half-maximal inhibitory concentration); NG (not generated); IC50 values were generated in an 8-point full dose response assay.
As expected, introduction of a methyl group at the 2-position of 52 resulted in a complete loss of CAMKK2 enzyme activity. The steric bulk of the 2′ methyl group is not tolerated and likely hinders the ability of the quinoline nitrogen to effectively participate in hydrogen bonding with the hinge region. Naphthyridine 54 showed poor CAMKK2 enzymatic activity.
Finally, we evaluated a small set of pyrimidines, well-known kinase ATP-competitive inhibitor scaffolds, for their CAMKK2 inhibitory activity (Table 4). Pyrimidine 56, with a phenyl in the 2-position, had a CAMKK2 IC50 = 21 nM. 2-Anilino-pyrimidine 62 was also potent with CAMKK2 IC50 = 51 nM. Pyrimidine 58, unsubstituted in the 2-position, lost considerable activity relative to 56. 2-Amino-pyrimidine 61 retained CAMKK2 potency (IC50 = 108 nM).
Table 4. CAMKK2 Enzyme Inhibition Data for Substituted Pyrimidines and 4-Aminopyrimidinesa.

R = ortho-cyclopentyl benzoic acid moiety; PoC (percent of control = percent of enzyme activity remaining, when compared with the control); IC50 (half-maximal inhibitory concentration); NG (not generated); IC50 values were generated in an 8-point full dose response assay.
X-ray Crystallography and In Silico Docking Studies
In order to more fully understand the binding modes and to plan optimization studies on these scaffolds, we turned to X-ray crystallography and in silico docking analysis of key molecules. We previously reported the crystal structure of 7-azaindole GSK650394 bound to CAMKK2 (PDB ID 6BKU).51 A crystal structure of the closely related 7-azaindole GSK650393 is also available (PDB ID 6CMJ).25 These two structures clearly demonstrate a hydrogen bond interaction between the NH of the pyrrole in the 7-azaindole to the carbonyl group of Glu268 at the CAMKK2 hinge region. The nitrogen atom at the 7-position forms an interaction with the hinge via the hydrogen bond with the NH of Val270. We were able to obtain a crystal structure of furopyridine 13g (PDB ID 5UY6). Data collection statistics and crystallization conditions are shown in Table 5.
Table 5. Crystallization Conditions and Data Collection Statistics.
| ligand | 13g | UNC10244803 |
|---|---|---|
| data collection | ||
| X-ray source | APS 24-ID-C | DLS I03 |
| wavelength (Å) | 0.9791 | 0.9763 |
| space group | P43212 | P43212 |
| cell dimensions | ||
| a, b, c (Å) | 73.3, 73.3, 122.0 | 73.2, 73.2, 120.3 |
| α, β, γ (deg) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| resolution (Å) | 19.74–1.70 (1.73–1.70) | 19.61–1.60 (1.63–1.60) |
| no. of unique reflections | 37,374 (1,767) | 43,864 (3,161) |
| Rmerge (%) | 14.7 (183.7) | 12.2 (31.5) |
| mean I/σI | 14.2 (2.0) | 21.3 (10.9) |
| mean CC(1/2) | 1.0 (0.75) | 1.0 (0.99) |
| completeness (%) | 99.9 (100) | 99.9 (100) |
| redundancy | 21.7 (22.1) | 25.8 (25.0) |
| refinement | ||
| resolution (Å) | 19.75–1.70 (1.75–1.70) | 19.61–1.60 (1.64–1.60) |
| Rcryst/Rfree (%) | 16.0/18.2 | 15.3/17.1 |
| no. of non-hydrogen atoms/mean B-factor (Å) | ||
| protein atoms | 2230/28.1 | 2229/21.3 |
| solvent atoms | 370/44.3 | 338/33.6 |
| ligand atoms | 29/19.8 | 26/12.5 |
| rmsd bond lengths (Å) | 0.010 | 0.01 |
| rmsd bond angles (deg) | 1.05 | 1.04 |
| Ramachandran statistics (%) | ||
| favored | 98.2 | 98.2 |
| allowed | 1.8 | 1.8 |
| PDB ID | 5UY6 | 5UYJ |
| crystallization conditions | 25% PEG 3350, 0.2 M ammonium sulfate, 0.1 M bis–tris buffer, pH 6.5 | 20% PEG 3350, 0.02 M ammonium sulfate, 0.1 M CHC buffer system, pH 8.5 |
Furopyridine 13g displays a similar binding mode to GSK650394. The 5,6-fused ring systems are oriented in the same way in the CAMKK2 active site, and the pyridine moieties of the two heterocycles form comparable hydrogen bond interactions with the NH group of Val270, and the furan oxygen atom and pyrrole NH group are in the same region of space. Although we have no crystal structure of thienopyridine 14g, we hypothesize that it would likely bind in a similar fashion.
To assess the binding modes of 14g and other compounds, we performed in silico docking studies with the docking software Glide version 2014 (Schrödinger).52 For our docking studies, we utilized three of the X-ray crystal structures of CAMKK2 (PDB-ID: 6BKU, 5UY6, and 5UYJ). We based our choice of the CAMKK2 PDB protein structure to use for our modeling on the structural similarity between the co-crystallized ligand and the compound we planned to use in the in silico docking. In order to ensure reliability of the docking protocol and the ensuing results for hypothesis generation, the co-crystallized ligands GSK650394, 13g, and UNC10244803 [2-cyclopentyl-4-(7-methoxyquinolin-4-yl)benzoic acid, CAMKK2 enzyme IC50 = 33 nM] were removed from the binding site and the ligands were re-docked into the binding pocket. The docking poses were then compared with the original pose from the crystal structures. The root-mean-square deviation (rmsd) value was lower than 1 Å, indicating a reliable docking procedure. The predicted binding modes of active compounds (Figure 3) show conserved H-bonds or close proximity between the compound’s carboxylate group and both the protonated amine of Lys194 and the carboxylate group of Glu236 in a water-mediated manner, analogous to what was observed in the crystal structures. The overall orientation of the tested compounds and their interactions with the hinge region is also of interest.
As expected, the predicted binding mode of thienopyridine 14g overlaid almost perfectly with the pose of furopyridine 13g in the X-ray structure (Figure 4a). The backbone NH group of Val270 and the thienopyridine N-atom are well positioned for a hydrogen bond interaction. Interestingly, there appears to be an additional favorable interaction between the sulfur atom of 14g and the carbonyl group of Glu268. Inter- and intra-molecular interactions between sulfur and oxygen atoms are relatively common and can be used to the medicinal chemists’ advantage.53,54
Figure 4.

X-ray structures and in silico docking. In silico docking was performed with Glide, and images were generated with PyMOL. The protein is colored in gray. Blue-dashed lines indicate H-bond interactions, while green-dashed lines display CH−π interactions and orange-dashed lines refer to sulfur−σ hole bonding. Docked ligands are shown as yellow sticks or orange sticks, co-crystallized ligands as purple sticks, and the water molecule as a red sphere. Oxygen and nitrogen atoms are colored in red and blue, respectively. (a) Predicted binding mode of 14g (yellow) using the protein structure from PDB-ID 5UY6 compared to the co-crystallized ligand 13g (purple). (b) Predicted binding modes of 46 (yellow) using the protein structure from PDB-ID 5UYJ and the co-crystallized ligand UNC10244803 (purple). (c) Predicted binding mode of 56 using the protein structure from PDB-ID 5UYJ. (d) Predicted binding mode of 22 using the protein structure from PDB-ID 6BKU. (e) Predicted binding modes of 10 (orange) and 29 (yellow) using the protein structure from PDB-ID 6BKU (light gray) and 5UYJ (dark gray), respectively. (f) Predicted binding mode of 38 using the protein structure from PDB-ID 5UYJ.
We were also able to obtain a crystal structure of quinoline UNC10244803 (PDB ID: 5UYJ). The quinoline nitrogen atom is positioned to form a hydrogen bond with the NH group of Val270. The predicted binding pose of 46 (Figure 4b) is highly comparable to the binding mode of the co-crystallized ligand UNC10244803. Similar to quinoline 46, 2-phenyl-pyrimidine 56 has only one heteroatom that is capable of forming hydrogen bonds with the hinge (Figure 4c). The N-atom of pyridine in the 1-position of 56 shows a H-bond with the NH group of Val270. Together with the presumed H-bond formed between the protonated N-atom of Lys194 and the carboxylate group of 56, these two interactions anchor 56 in the active site. In this orientation, the phenyl ring in the 2-position is tolerated by the binding pocket. 2-Aryl-pyrimidines are much less common as kinase inhibitors than 2-anilinopyrimidines and so warrant further exploration. The 2-position phenyl ring of 56 is adjacent to Leu269 of CAMKK2. We speculate that kinases incorporating amino acids with larger side chains such as Phe or Tyr in this location at the hinge region will be less tolerant of this scaffold, perhaps offering a path to enhance selectivity.
Pyrazolopyrimidine 22 (Figure 4d) forms one hinge-binding interaction between the N-atom in position 1 and the backbone NH group of Val270. The distance between the NH3+ group of Lys194 and the carboxylate group of 22 is greater compared to the predicted binding modes of the other compounds (Figure 4b,e). The N-methyl group of compound 10 (Figure 4e) appears to preclude binding of this compound in the same orientation as GSK650394. This compound is inactive in the enzyme assay, and the docking predicts a flipped orientation of the compound. N-methyl compound 29, however, retains activity, and the in silico docking result suggests a conformation that allows the pyridyl nitrogen atom to interact with the NH group of Val270. The methyl group in this orientation is tolerated, perhaps analogously to the 2-phenyl of compound 56. This flipped binding mode is possibly due to the shift of the attachment position of the cyclopentyl benzoic acid moiety from the 5-position of 10 to the 4-position in 29. The hinge-binding interactions shown in the predicted binding mode of thienopyrimidine 38 are comparable (Figure 4f) to those of 56. Additionally, the orientation of 38 is further stabilized by an interaction that is formed between the O-atom of the carbonyl moiety of Ile171 and the S-atom of the thienopyrimidine scaffold.
Compound Selectivity
In order to begin to understand kinome-wide selectivity for these new compounds, we profiled 9 exemplars in a panel of over 400 wild-type human kinases using the Eurofins DiscoverX’s KINOMEscan technology50 (Freemont, CA USA). A summary of the kinome selectivity results is depicted in Table 6, and the complete KINOMEscan data sets are available in the Supporting Information. Table 6 contains the structures of the compounds profiled, the PoC values for CAMKK1 and CAMKK2 at the 1 μM screening concentration, and a list of all the kinases in the panel that bound with a PoC < 10. The S-score is a quantitative measure of a compound’s selectivity at a particular screening concentration, calculated by dividing the number of kinases that a particular compound binds to at a chosen threshold by the total number of distinct kinases tested. All the compounds we tested from this scaffold-hopping effort have an S10 (1 μM) < 0.05, which implies that they bind to fewer than 5% of the wild-type kinases tested in this panel with a PoC of 10 or less, thus offering promising selectivity for a starting point for further optimization.
Table 6. Selectivity Screening Summary Results for Nine Different Exemplars.
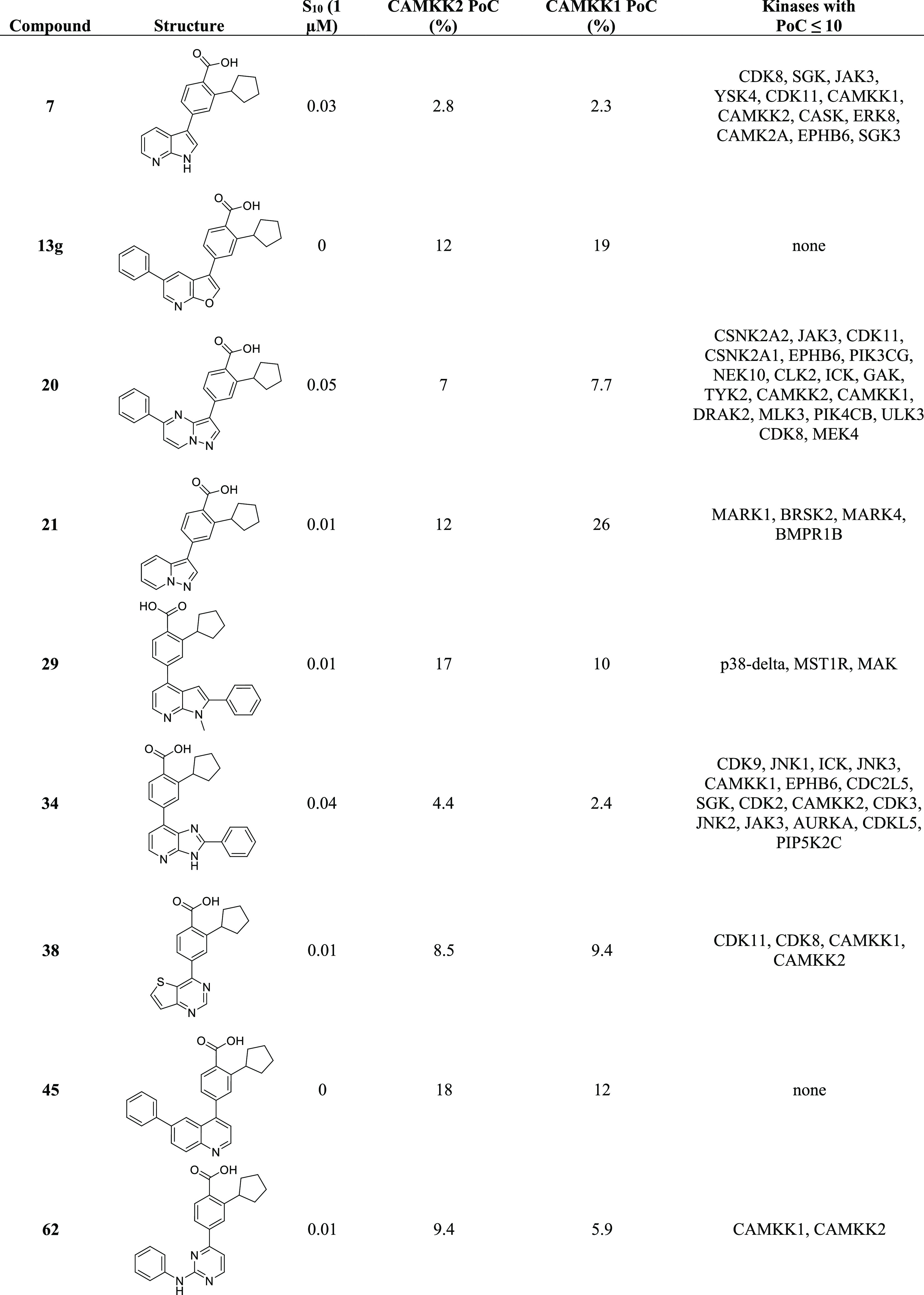
Figure 5 is a visual representation of the KINOMEscan data for two of the compounds, azaindole compound 7 and 2-anilino-pyrimidine compound 62. All kinases bound with a PoC < 10 are depicted as red dots. These kinases are listed in Table 5, and the full data sets are available in the Supporting Information. For compound 7, 12 kinases have a PoC less than or equal to 10, including CAMKK2 and CAMKK1. For compound 7, S10 (1 μM) = 0.03 implies that 3% of the kinases tested had a PoC < 10 at an inhibitor screening concentration of 1 μM. For compound 62, only two kinases (CAMKK1 and CAMKK2) have a PoC < 10. S10 (1 μM) = 0.005 implies that 0.5% of the kinases tested had a PoC < 10 at an inhibitor screening concentration of 1 μM. Next steps will need to include testing of the compounds in enzyme inhibition assays and cell-based assays for the identified kinases for each scaffold.
Figure 5.

In vitro kinase selectivity profile of compound 7 (5a) and compound 62 (5b) at 1 μM (Eurofins DiscoverX KINOMEscan). TREEspot interaction maps for 7 and 62 profiled against >400 human kinase targets. In this assay, primary screen binding interactions are reported as PoC, where lower values are an indication of stronger hits. Red dots depict the kinases for which PoC at 1 μM < 10. The size of the dot is based on the PoC values, with larger dots having smaller PoC values (more potent binders). PoC values for all kinases can be found in the Supporting Information.
CAMKK1 activity
CAMKK1 is a closely related and also understudied kinase. In order to learn if selectivity between CAMKK1 and CAMKK2 may be attainable in these series, we determined IC50 values for CAMKK1 inhibition for the 18 compounds with CAMKK2 IC50 < 150 nM, with the results depicted in Table 7. STO-609 is reported to have some selectivity for CAMKK2 over CAMKK1, and this is recapitulated in our assays.55 In general, the compounds are more potent inhibitors of CAMKK2 than CAMKK1. Compound 34 (CAMKK1 IC50 = 27 nM, CAMKK2 IC50 = 23 nM) and compound 62 (CAMKK1 IC50 = 31 nM, CAMKK2 IC50 = 51 nM) are exceptions, being equipotent or slightly more potent on CAMKK1. For STO-609, a switch in a residue near the inhibitor hinge-binding group from a valine in CAMKK2 (Val269) to a leucine in CAMKK1 (Leu233) has been hypothesized as the source of selectivity.56 Further experiments will need to be done to understand and exploit the differences in CAMKK1 and CAMKK2 activity observed for the inhibitors described here.
Table 7. CAMKK1 and CAMKK2 Enzyme Inhibition Data, CAMKK2 NanoBRET in Cell Target Engagement, LE Metrics, and cLog P and Solubility Data for Potent CAMKK2 Inhibitorsa.
| ID | CAMKK1 enzyme IC50 [nM] | CAMKK2 enzyme IC50 [nM] | CAMKK2 NB IC50 [nM] | cLog P | LE | LLE | solubility [μg/mL] |
|---|---|---|---|---|---|---|---|
| STO-609 | 408 ± 22 | 58 ± 5.7 | NG | 3.59 | 0.41 | 3.61 | 60 |
| GSK650394 | 33 ± 6.9 | 3 ± 0.4 | <3 | 5.67 | 0.40 | 2.83 | 2 |
| 7 | 195 ± 25 | 26 ± 6.1 | NG | 3.78 | 0.45 | 3.82 | |
| 13g | 961 ± 132 | 65 ± 15 | 700 ± 9.3 | 5.68 | 0.34 | 1.52 | 77 |
| 14g | 384 ± 53 | 5 ± 1.1 | 210 ± 24 | 6.32 | 0.39 | 1.98 | 1.5 |
| 19 | 1480 ± 120 | 145 ± 30 | 530 ± 95 | 6.01 | 0.32 | 0.79 | 42 |
| 20 | 238 ± 32 | 21 ± 1.3 | 190 ± 19 | 5.17 | 0.36 | 2.53 | 50.5 |
| 21 | 2013 ± 174 | 44 ± 9.1 | 200 ± 11 | 4.13 | 0.44 | 3.27 | 48.3 |
| 29 | >10,000 | 120 ± 21 | 240 ± 7 | 6.06 | 0.32 | 0.84 | |
| 30 | 1345 ± 222 | 56 ± 14 | 470 ± 44 | 3.97 | 0.42 | 3.33 | 40 |
| 34 | 27 ± 4.5 | 23 ± 4.2 | 8.1 ± 0.8 | 5.42 | 0.36 | 2.18 | 55.1 |
| 38 | 407 ± 25 | 24 ± 1.1 | 170 ± 18 | 3.99 | 0.45 | 3.61 | 55.4 |
| 42 | 7280 ± 216 | 31 ± 4.3 | 1400 ± 250 | 3.78 | 0.45 | 3.72 | 62.6 |
| 45 | 1838 ± 348 | 137 ± 12 | 540 ± 62 | 6.59 | 0.32 | 0.31 | 38 |
| 46 | 239 ± 20 | 13 ± 1.0 | 140 ± 12 | 4.7 | 0.45 | 3.2 | 64.4 |
| 49 | 2614 ± 349 | 12 ± 3.9 | 890 ± 71 | 5.86 | 0.36 | 2.04 | 70.4 |
| 50 | 5481 ± 130 | 96 ± 14 | 1900 ± 200 | 3.97 | 0.40 | 3.03 | |
| 56 | >10,000 | 21 ± 2.6 | 290 ± 23 | 4.68 | 0.41 | 3.02 | 19.4 |
| 61 | >10,000 | 108 ± 6.9 | 950 ± 110 | 2.57 | 0.46 | 4.43 | |
| 62 | 31 ± 6.5 | 51 ± 13 | <3 | 5.38 | 0.37 | 1.92 | 10.6 |
NB = data from the NanoBRET in the cell target engagement assay; NG = data not generated.
Compound Properties
A number of key physicochemical screens and calculations are considered in early-stage drug discovery, including lipophilicity, pKa, solubility, permeability, and stability. Solubility is one of the most critical physicochemical properties as poor solubility can lead to an underestimation of activity, inaccurate SAR, inaccurate in vitro ADMET test results, and downstream compound development issues.57,58 Highlighting the critical importance of solubility, it has been shown that 87% of commercial drugs have solubility ≥65 μg/mL, but only 7% had solubility ≤20 μg/mL.59 Therefore, solubility data can identify a series liability that needs to be fixed and highlight promising series with enhanced solubility. This information can help guide optimization strategies. LE metrics are also a commonly used tool that can guide hit-to-lead optimization for drug candidates.60,61 To this end, we calculated LE and lipophilic ligand efficiency (LLE) and collected kinetic solubility data for compounds with IC50 values <150 nM in the CAMKK2 enzyme assay (Table 7).62
LE values are influenced by molecular size; hence, it was unsurprising to see two clear ranges depending on the presence or absence of the phenyl ring. The hinge binders without the phenyl ring had LE values ranging from 0.41 to 0.46. These are smaller compounds with molecular weights between 300 and 325, and generally, LE values above 0.3 are acceptable for this MW range.60 The LLE values for these compounds were between 3.02 and 4.43. Aminopyridine 61 had the highest LE and LLE values of 0.46 and 4.43, respectively. Both 7 and 38 had comparable LE values of 0.45 but significantly lower LLE values of 3.82 and 3.61, respectively.
Both the LE and LLE metrics of the phenyl-substituted analogues have lower values. This is reflective of the increased atom count and higher cLog P values due to the additional lipophilicity of the phenyl ring. The LE values are between 0.32 and 0.40 for these compounds. GSK650394 has the highest LE and LLE values of 0.40 and 2.83, respectively. The analogous thienopyridine 14g has the next highest LE value of 0.39 but a lower LLE value of 1.98. Quinoline 45 had the lowest LE and LLE values of 0.32 and 0.31, respectively. Solely relying on LE metrics to select compounds is unadvisable as many other factors, such as solubility, underpin successful drug development. A future direction could be to focus on polar substituents on the pendant phenyl to lower log P, enhance solubility, and mitigate the metabolic liability that may be seen in high-logP compounds. Similarly, non-aromatic substituents could be explored to access this same region of the active site.
Of the phenyl-substituted analogues, GSK650394 and 14g were the most potent, with IC50 values of 3 and 5 nM, respectively, and had the best LE and LLE values. Although promising in this regard, these compounds have extremely poor solubility values of 2 and 1.5 μg/mL, respectively. Optimization of these compounds will thus require extensive structure–property-relationship studies in parallel to SAR studies to develop them into useful tools with adequate solubility for further study. Interestingly, furopyridine 13g had the highest measured solubility value of 77 μg/mL. Substitution of the azaindole’s NH with an oxygen greatly improved solubility. The effect was reversed when a more lipophilic sulfur atom is incorporated into the same position, 14g. Pyrimidine analogues 56 and 62 are also only modestly soluble (10.6 and 19.4 μg/mL). Optimization to tool compounds will likely require the introduction of solubilizing functional groups. A number of the compounds tested had reasonable solubility ranging from 38 to 55 μg/mL. Four compounds had a promising solubility above that recorded for STO-609, 60 μg/mL. These were the thienopyrimidine 42 (62.6 μg/mL) and quinoline 46 (64.4 μg/mL), both without the pendant phenyl ring. The phenyl-substituted quinazoline 49 has a solubility of 70.4 μg/mL, second only to furopyridine 13g. Overall, the solubility of several potent compounds was encouraging but will need to be monitored as optimization proceeds.
CAMKK2 NanoBRET Cellular Target Engagement
Having demonstrated potent CAMKK2 inhibition and promising selectivity profiles for these compounds we tested in kinome-wide profiling, we turned our attention to the cellular activity of the compounds (Table 7). To this end, we developed a CAMKK2 NanoBRET target engagement assay.63 This assay uses bioluminescence resonance energy transfer (BRET) between nanoluciferase (NL) fused to the kinase domain of CAMKK2 (BRET donor) and a tracer molecule that binds to the ATP-binding site of the kinase (BRET acceptor). The addition of cell penetrant CAMKK2 inhibitors leads to displacement of the tracer molecule and a quantifiable reduction in BRET signal. This assay is conducted in living cells, and the observation of binding not only depends on affinity between the compound and CAMKK2 but also on compound cell penetrance. The parent molecule, GSK650394, was very potent in this assay (NB IC50 < 3 nM). Several additional key compounds were evaluated in this assay (Table 7 and the Supporting Information). A key takeaway here is that all the compounds that advanced to this assay (those with CAMKK2 enzyme IC50 < 150 nM) demonstrated measurable CAMKK2 target engagement in cells. The potency was below 500 nM for thienopyridine 14g (210 nM), pyrazolopyrimidines 20 and 21 (190 and 200 nM), N-methyl azaindoles 29 and 30 (240 and 470 nM), imidazopyridine 34 (8 nM), thienopyrimidine 38 (170 nM), quinoline 46 (140 nM), 2-phenylpyrimidine 56 (290 nM), and 2-amino-pyrimidine 62 (<3 nM). The results for exemplar compounds from five different scaffolds (13g, 45, 46, 56, and 62) are depicted in Figure 6.
Figure 6.

CAMKK2 NanoBRET dose–response curves for compounds 13g, 45, 46, 56, and 62.
On-Target Cellular Effect (Phosphorylation Inhibition Assay)
We also assessed the impact of our inhibitors on phosphorylation downstream from CAMKK2 using Western blot analysis with C4-2 prostate cancer cells to provide evidence of a phenotypic and on-target effect of our CAMKK2 inhibitors in intact cells. AMPK (Thr172) was chosen because in intact cells, CAMKK2, but not the related CAMKK1, can readily phosphorylate this substrate.64 We performed preliminary Western blot analysis at a single inhibitor concentration of 1 μM for the 18 compounds with CAMKK2 enzyme IC50 < 150 nM along with STO-609. These results are shown in Figure 7.
Figure 7.

Western blots of the 18 compounds with IC50 < 150 nM in the CAMKK2 enzyme assay, together with STO-609, screened at a single inhibitor concentration = 1 μM.
Based on these results from screening at a single concentration, compounds 7, 20, 29, 34, 38, 46, 56, and 62 showed the most robust reduction of the p-AMPK band and were advanced into full dose response experiments in this same assay format to provide IC50 estimates for inhibition of phosphorylation of AMPK at Thr172. The IC50 values were determined from dose response experiments using 0–10 μM of the compound. The Western blot dose response experiments for the most potent compound, 62, are depicted in Figure 8a. The Western blot dose response data for compounds 7, 20, 29, 34, 38, 46, and 56 can be found in the Supporting Information. In addition, dose response experiments for compound 62 utilizing lower inhibitor concentrations can be found in the Supporting Information. The calculated IC50 values for the dose response experiments for all eight compounds are shown in Figure 8b. Compounds 62 (IC50 = 10 nM), 46 (IC50 = 390 nM), 56 (IC50 = 790 nM), 20 (IC50 = 900 nM), and 29 (IC50 980 nM) all have IC50 values for inhibition of phosphorylation of AMPK at Thr172 below 1 μM, suggesting that optimization into compounds with potent cellular activity will be achievable for these series.
Figure 8.

(a) Dose response for inhibition of phosphorylation of AMPK at Thr172 in C4-2 prostate cancer cells by compound 62. (b). Calculated IC50 values for inhibition of phosphorylation of AMPK at Thr172 in C4-2 prostate cancer cells for the top compounds from the single concentration experiment depicted in Figure 7.
Discussion and Conclusions
Through a hinge-binder scaffold hopping strategy, we have designed, synthesized, and biologically evaluated as CAMKK2 inhibitors a series of 32 compounds that utilize 5,6-bicyclic, 6,6-bicyclic, and single-ring hinge-binding moieties that are based on the 7-azaindole GSK650394. These inhibitors were designed with the aim of retaining CAMKK2 potency and improving the kinase selectivity of the promiscuous azaindole by changing the strong H-bond interactions that the parent azaindole makes with the kinase hinge-binding residues. Additionally, we sought to identify inhibitors with improved physicochemical properties, drug likeness, and selectivity all while retaining CAMKK2 inhibitory potency. Several compounds with similar or better potency than GSK650394 and STO-609 have been identified, some of which also showed improved physicochemical properties. We found that a number of these compounds, but not all, have some selectivity over the closely related enzyme CAMKK1 in enzyme assays. Further work will be needed to understand selectivity determinants, to optimize this selectivity, and to determine if the observed selectivity holds in a cellular context. Kinome-wide profiling revealed that the nine exemplars we screened in the Eurofins DiscoverX KINOMEscan platform are not broadly promiscuous, with S10 (1 μM) < 0.05. In addition, we have demonstrated that exemplars have cellular activity in two orthogonal assays, a CAMKK2 NanoBRET in cell target engagement assay, and Western blot experiments looking at inhibition of the production of p-AMPK. Our work has shown that kinase inhibitors with weakened (non-ideal) hinge-binding interactions can show highly selective kinase inhibition and still have useful on-target enzyme potency and cellular potency. This work has led to the identification of several CAMKK2 scaffolds with alternate hinge-binding moieties that hold promise as starting points for the discovery of potent, selective, and cell-active CAMKK2 inhibitors.
Experimental Section
Biology
Protein Expression and Purification/DSF Assay
Small-molecule screening by DSF was performed as described previously.48,65 Briefly, the DSF assay was performed in the 96-well format. Purified CAMKK1 or CAMKK2 was diluted to 2 μM kinase in 100 mM potassium phosphate pH 7.5, 150 mM NaCl, and 10% glycerol supplemented with 5 × SYPRO Orange (Invitrogen, Carlsbad, CA, USA). All assay experiments used 19.5 μL of 2 μM kinase and SYPRO Orange mixture. Compounds solubilized in dimethyl sulfoxide (DMSO) were used at a 12.5 μM final concentration, with a 2.5% concentration of DMSO per well. PCR plates were sealed using optically clear films and transferred to a C1000 thermal cycler with CFX-96 RT-PCR head (BioRad, Hercules, CA, USA). The fluorescence intensity was measured over a temperature gradient from 25 to 95 °C at a constant rate of 0.05 °C/s. Curve fitting and protein melting temperatures were calculated based on a Boltzmann function fitting to experimental data (GraphPad Prism 8). Protein with the addition of 2.5% DMSO was used as a reference. All experiments were carried out in triplicate, and the mean of the ΔTm is reported. Compounds that provided negative values are presented as having a ΔTm of 0 °C.
CAMKK1 and CAMKK2 Enzyme Assays
CAMKK1 and CAMKK2 activity was determined by measuring the transfer of radiolabeled phosphate from [γ-32P]-ATP to a synthetic peptide substrate (CaMKKtide) as previously described.66 Briefly, purified recombinant CAMKK1 or CAMKK2 (100 pM) was incubated in the assay buffer [50 mM HEPES (pH 7.4), 1 mM dithiothreitol, 0.02% (v/v) Brij-35] containing 200 μM CaMKKtide (Genscript), 100 μM CaCl2, 1 μM CaM (Sigma-Aldrich, Castle Hill, NSW, Australia), 200 μM [γ-32P]-ATP (Perkin Elmer, Boston, MA, USA), 5 mM MgCl2 (Sigma-Aldrich, Castle Hill, NSW, Australia), and various concentrations of inhibitors (0–1 μM) in a standard 30 μL assay for 10 min at 30 °C. Reactions were terminated by spotting 15 μl onto P81 phosphocellulose paper (GE Lifesciences, Paramatta, NSW, Australia) and washing extensively in 1% phosphoric acid (Sigma-Aldrich, Castle Hill, NSW, Australia). Radioactivity was quantified by liquid scintillation counting.
CAMKK2 NanoBRET Assay
To quantify the cellular activity of these inhibitors, we developed a CAMKK2 NanoBRET target engagement assay.63 Briefly, this assay utilizes an NL fused to the kinase domain of CAMKK2. This NL kinase fusion is then transiently transfected into HEK293 cells, and after 24 h, the tracer is added to the cells. When the tracer and the NL-CAMKK2 fusion come into proximity, they create a BRET signal that can be competed in a dose-dependent manner by the addition of cell-penetrant CAMKK2 inhibitors.
Western Blot Analysis
C4-2 cells were plated in 6-well plates in the IMEM medium containing 0.5% fetal bovine serum. After 72 h, the cells were then treated with the compounds for 24 h before the media were aspirated and wells were washed twice in ice-cold phosphate-buffered saline. Cells were lysed using RIPA buffer containing phosphatase and the protease inhibitor cocktail while rotating for 30 min at 4 °C. In each lane, 30 μg/well of protein lysate was loaded into a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and run for 1 h and 30 min. Gels were then transferred overnight in a TRIS-glycine/methanol transfer buffer onto a poly(vinylidene difluoride) membrane at 4 °C. Membranes were blocked, incubated with primary overnight at 4 °C, washed, incubated with secondary at room temperature (rt) for 1 h, washed, and then developed on an Azure Biosystems C-600 imager.
[Cell signaling: phospho-AMPKα (Thr172) (40H9) rabbit mAb: Cat#: 2535; AMPKα (D5A2) Rabbit mAb Cat#: 5831; BD Bioscience: CAMKK mouse mAb Cat# 610544; Sigma: GAPDH rabbit pAb: Cat# G9545; secondary antibody: goat anti-rabbit IgG (H + L)-HRP conjugate was from Bio-Rad (Cat#:1706515)].
Crystallization, Data Collection, and Structure Determination
Crystallization of the CAMKK2-kinase domain bound to 13g or UNC10244803 followed a previously established protocol.38 Briefly, the inhibitor (dissolved in 100% DMSO) was added to the protein in 3-fold molar excess and incubated on ice for approximately 30 min. The mixture was centrifuged at 21,000g for 10 minutes at 4 °C before setting up 150 nL volume sitting drops at three ratios (2:1, 1:1, or 1:2 protein–inhibitor complex to reservoir solution). Crystallization experiments were performed at 20 °C. Crystal optimization used Newman’s buffer system.67 Crystals were cryoprotected in the mother liquor supplemented with 25–30% glycerol before flash cooling in liquid nitrogen for data collection. Diffraction data were collected at 100 K at the Advanced Photon Source 24ID-C or at the Diamond Light Source (DLS) I03 beamline.
Diffraction data were integrated with XDS68 and scaled using AIMLESS from the CCP4 software suite.69 The structure was solved by molecular replacement using Phaser70 and the kinase domain of CAMKK2 as the search model (PDB ID 2ZV2).71 Refinement was performed using REFMAC5,72 and Coot73 was used for model building. Structure validation was performed using MolProbity.74 Structure factors and coordinates for the structure were deposited at the PDB (PDB ID 5UY6).
Chemistry
General Chemistry Information
All reagents and solvents, unless specifically stated, were used as obtained from their commercial sources without further purification. Solvents were degassed with nitrogen for cross-coupling reactions. Air- and moisture-sensitive reactions were performed under an inert atmosphere using nitrogen in a previously oven-dried or flame-dried reaction flask, and addition of reagents was done using a syringe. All microwave (μW) reactions were carried out in a Biotage Initiator EXP US 400W microwave synthesizer. Thin-layer chromatography (TLC) analyses were performed using 200 μm pre-coated sorbtech fluorescent TLC plates, and spots were visualized using UV light. High-resolution mass spectrometry samples were analyzed with a ThermoFisher Q Exactive HF-X (ThermoFisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. All high-resolution mass spectrometry (HRMS) spectra were recorded via electrospray ionization (ESI). Column chromatography was undertaken with a Biotage Isolera One or Prime instrument. Nuclear magnetic resonance (NMR) spectrometry was run on a Varian Inova 400 MHz or Bruker AVANCE III 700 MHz spectrometer equipped with a TCI H-C/N-D 5 mm cryoprobe, and data were processed using the MestReNova processor. Chemical shifts are reported in ppm with residual solvent peaks referenced as the internal standard. All compounds were >95% pure by analytical LC.
4-Bromo-2-cyclopentylbenzoic Acid (2)
4-Bromo-2-fluorobenzoic acid (2.00 g, 9.00 mmol) was dissolved in tetrahydrofuran (THF) (20.0 mL) and cooled to 0 °C. Cyclopentylmagnesium bromide solution (16.0 mL of a 2 M solution, 32.0 mmol) was added dropwise. The reaction was stirred at 0 °C for 4 h. 2 M HCl (25.0 mL) was then slowly added to the solution, followed by EtOAc (40.0 mL). The organic phase was separated, concentrated, and purified using column chromatography (10% EtOAc/hexane) to afford 4-bromo-2-cyclopentylbenzoic acid 2 (1.70 g, 71%) as a pure white solid. The NMR data for this compound match those previously reported.38
1H NMR (400 MHz, DMSO-d6): δ 13.07 (s, 1H), 7.60–7.56 (m, 2H), 7.45 (dd, J = 8.3, 2.1 Hz, 1H), 3.68 (tt, J = 9.5, 7.5 Hz, 1H), 2.03–1.94 (m, 2H), 1.82–1.72 (m, 2H), 1.67–1.46 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 168.9, 148.6, 131.2, 131.1, 129.5, 128.6, 125.1, 41.2, 34.2, 25.2; LCMS: [M + H]+m/z, 269.0.
2-Cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic Acid (3)
To a stirred solution of 4-bromo-2-cyclopentylbenzoic acid 2 (2.50 g, 9.30 mmol) and bis(pinacolato)diboron (2.80 g, 11.0 mmol) in dioxane (50.0 mL) were added PdCl2(dppf)·CH2Cl2 (0.76 g, 0.96 mmol) and KOAc (3.60 g, 37.0 mmol). The solution was heated to 95 °C for 2 h. Once cooled to rt, the solution was diluted with EtOAc and filtered through a celite pad. The solution was concentrated, and the crude product was purified by column chromatography (10% EtOAc/hexane) to afford 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 3 (2.30 g, 78%) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 13.00 (s, 1H), 7.69 (d, J = 1.1 Hz, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.53 (dd, J = 7.6, 1.1 Hz, 1H), 3.62 (tt, J = 9.8, 7.4 Hz, 1H), 2.05–1.95 (m, 2H), 1.83–1.73 (m, 2H), 1.69–1.45 (m, 4H), 1.29 (s, 12H); 13C NMR (100 MHz, DMSO-d6): δ 169.7, 144.4, 134.9, 132.1, 131.5, 128.2, 83.9, 41.2, 34.4, 25.2, 24.7; LCMS: [M + H]+m/z, 317.2.
Methyl 2-Cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (4)
To a stirred solution of methyl 4-bromo-2-cyclopentylbenzoate SI1 (1.50 g, 5.29 mmol) and bis(pinacolato)diboron (1.88 g, 7.42 mmol) in dioxane (35.0 mL) were added Pd(dppf)Cl2 (194 mg, 0.26 mmol) and KOAc (1.56 g, 15.9 mmol). The solution was heated to 100 °C for 2 h. Once cooled to rt, the solution was filtered through a pad of Celite and washed with 100 mL of EtOAc. The volatiles were removed in vacuo, and the crude residue was purified via column chromatography (0–5% EtOAc/hexane) to afford methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (1.37 g, 78%) as oil which slowly solidified into a white solid under vacuum.
1H NMR (400 MHz, DMSO-d6): δ 7.70 (d, J = 1.1 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.56 (d, J = 1.1 Hz, 1H), 3.83 (s, 3H), 3.50 (tt, J = 9.9, 7.5 Hz, 1H), 2.04–1.93 (m, 2H), 1.85–1.70 (m, 2H), 1.70–1.44 (m, 4H), 1.30 (s, 12H); 13C NMR (100 MHz, DMSO-d6): δ 168.3, 144.6, 133.6, 132.2, 131.6, 128.3, 84.0, 52.2, 41.3, 34.3, 25.2, 24.7; LCMS: [M + H]+m/z, 330.2.
Methyl 2-Cyclopentyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoate (6)
To a stirred solution of 3-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (450 mg, 1.37 mmol) and methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (681 mg, 2.06 mmol) in PhMe (8.0 mL) and H2O (1.00 mL) were added Pd(OAc)2 (15.4 mg, 68.7 μmol), K3PO4 (934 mg, 4.40 mmol), and P(Cy)3 (38.6 mg, 137 μmol). The reaction mixture was heated to 100 °C for 18 h and allowed to cool to rt, filtered through a pad of Celite, and washed with EtOAc (10 mL). The filtrate was concentrated in vacuo, and the crude was purified by column chromatography eluting with 0–30% EtOAc/hexane to afford methyl 2-cyclopentyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoate (545 mg, 88%) as a light-yellow solid (LCMS: [M + H]+m/z, 327.3). A solution of the above material (“SEM-protected 6”), 2-cyclopentyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoate (500 mg, 1.11 mmol), in CH2Cl2 (10 mL) was treated with trifluoroacetyl (3.50 mL, 45.5 mmol). The solution was stirred at 25 °C for 2 h and then neutralized with sat. NaHCO3 (50 mL). The solution was extracted with EtOAc (3 × 100 mL), and the combined organic phases were washed with sat. NaCl solution, dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in ethanol (40.0 mL), and NaOAc (1.82 mg, 22.2 mmol) was added. The mixture was stirred at 50 °C for 20 h and allowed to cool to rt. The solution was diluted with H2O (50 mL) and EtOAc (75.0 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 75 mL). The combined organics were washed with a saturated NaCl solution (50 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The crude material was purified by column chromatography (0–30% EtOAc/hexane) to afford methyl 2-cyclopentyl-4-(1H-pyrrolo[2,3-b]pyridin-3-yl)benzoate 6 (245 mg, 69%) as a deep-yellow solid.
1H NMR (400 MHz, DMSO-d6): δ 12.10–12.05 (m, 1H), 8.32–8.24 (m, 2H), 8.05 (d, J = 2.7 Hz, 1H), 7.78–7.72 (m, 2H), 7.63 (dd, J = 8.1, 1.7 Hz, 1H), 7.20 (dd, J = 8.0, 4.6 Hz, 1H), 3.84 (s, 3H), 3.79–3.67 (m, 1H), 2.11–1.98 (m, 2H), 1.88–1.77 (m, 2H), 1.74–1.58 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 167.9, 149.2, 147.2, 143.1, 138.7, 130.3, 127.4, 127.2, 125.1, 124.1, 123.2, 117.2, 116.4, 113.4, 51.9, 41.3, 34.3, 25.2; LCMS: [M + H]+m/z, 320.4.
2-Cyclopentyl-4-(1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic Acid (7)
Methyl 2-cyclopentyl-4-(1H-pyrrolo[2,3-b]pyridin-3-yl)benzoate (100 mg, 0.312 mmol) was dissolved in methanol (8.00 mL), followed by the addition of 50% NaOH solution (170 μL, 3.12 mmol). The mixture was heated to 75 °C for 1 h, allowed to cool, and extracted with diethyl ether (20.0 mL). The aqueous layer was then acidified to pH 5 with 1 M HCl and extracted with CH2Cl2 (15 mL). The organic layer was then dried (Na2SO4), filtered, and concentrated in vacuo to afford 2-cyclopentyl-4-(1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid 7 (85.0 mg, 89%) as a pale-yellow solid.
1H NMR (400 MHz, DMSO-d6): δ 12.64 (s, 1H), 12.12–12.08 (m, 1H), 8.32–8.27 (m, 2H), 8.03 (d, J = 2.6 Hz, 1H), 7.78–7.71 (m, 2H), 7.60 (dd, J = 8.1, 1.8 Hz, 1H), 7.22 (dd, J = 8.0, 4.7 Hz, 1H), 3.92–3.81 (m, 1H), 2.07 (d, J = 14.0 Hz, 2H), 1.82 (t, J = 5.8 Hz, 2H), 1.73–1.61 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 169.3, 148.3, 147.1, 142.3, 138.1, 130.4, 128.6, 128.2, 125.2, 124.2, 123.2, 117.7, 116.4, 113.8, 41.2, 34.4, 25.3; HRMS: calcd for C19H18N2O2 [M + H]+m/z, 307.1447; found, 307.1426; mp range 238–242 °C.
Methyl 4-(5-Chloro-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate (9)
5-Chloro-3-iodo-1-methyl-1H-pyrrolo[2,3-b]pyridine (100 mg, 0.34 mmol), methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (113 mg, 0.34 mmol), Pd(dppf)Cl2·CH2Cl2 (28.0 mg, 34.0 μmol), and Cs2CO3 (334 mg, 1.00 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (2 mL) was added before the vial was flushed with nitrogen and capped. The solution was stirred at rt for 16 h. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (5–20% EtOAc/hexane) to afford methyl 4-(5-chloro-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate 9 (217 mg, 69%) as a white solid.
1H NMR (400 MHz, CDCl3): δ 8.34 (d, J = 2.2 Hz, 1H), 8.15 (d, J = 2.0 Hz, 1H), 7.86 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 1.6 Hz, 1H), 7.49 (s, 1H), 7.42 (dd, J = 8.1, 1.7 Hz, 1H), 3.97 (s, 3H), 3.93 (s, 3H), 3.92–3.84 (m, 1H), 2.31–2.06 (m, 2H), 1.92–1.80 (m, 2H), 1.80–1.71 (m, 2H), 1.71–1.59 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 168.5, 148.6, 146.3, 141.7, 137.6, 130.8, 128.5, 128.3, 127.6, 125.0, 124.4, 123.6, 119.3, 114.2, 52.0, 41.7, 34.9, 31.8, 25.7; HRMS: calcd for C21H22N2O2Cl [M + H]+m/z, 369.1369; found: m/z, 369.1348; mp range 144–148 °C.
2-Cyclopentyl-4-(1-methyl-5-phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic Acid (10)
Methyl 4-(5-chloro-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate 9 (100 mg, 0.27 mmol), phenylboronic acid (40.0 mg, 0.32 mmol), Pd2(dba)3 (12.0 mg, 13.0 μmol), XPhos (13.0 mg, 27.0 μmol), and Cs2CO3 (265 mg, 0.81 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (2 ml) was added, and the vial was flushed with nitrogen and capped. The solution was heated to 120 °C for 16 h. Once cooled, the solution was diluted with EtOAc (2 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (5–10% EtOAc/hexane) to afford the methyl ester intermediate. The methyl ester was saponified in a solution of aq LiOH (1.00 mL, 1 M) and dioxane (1.00 mL) at 100 °C for 16 h. The solution was cooled to rt, diluted with water (2 mL), and then acidified with aq HCl (1 M) until pH 4. The solution was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The solid was washed with cold water (5.00 mL), followed by hexane (10.0 mL), and dried to afford 2-cyclopentyl-4-(1-methyl-5-phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid 10 (77.0 mg, 72%) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 12.75 (br s, 1H), 8.63 (d, J = 2.1 Hz, 1H), 8.43 (d, J = 2.1 Hz, 1H), 8.15 (s, 1H), 7.83–7.67 (m, 5H), 7.50 (dd, J = 8.4, 7.0 Hz, 2H), 7.43–7.34 (m, 1H), 3.94–3.85 (m, 1H), 3.92 (s, 3H), 2.12–1.99 (m, 2H), 1.88–1.76 (m, 2H), 1.75–1.58 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.7, 148.0, 147.7, 142.4, 139.2, 138.2, 131.0, 130.0, 129.6, 129.5, 128.9, 127.6, 126.1, 124.5, 123.6, 117.9, 113.3, 41.4, 34.8, 31.6, 25.8; HRMS: calcd for C26H25N2O2 [M + H]+m/z, 397.1916; found m/z, 397.1911; mp range 199–201 °C.
2-Cyclopentyl-4-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic Acid (11)
3-Bromo-1-methyl-1H-pyrrolo[2,3-b]pyridine (100 mg, 0.47 mmol), methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (180 mg, 0.54 mmol), Pd2(dba)3 (22.0 mg, 24 μmol), XPhos (23.0 mg, 48.0 μmol), and Cs2CO3 (540 mg, 1.66 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (2.00 mL) was added, and the vial was flushed with nitrogen and capped. The solution was heated to 120 °C for 16 h. Once cooled, the solution was diluted with EtOAc (2.0 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (0–20% EtOAc/hexane) to afford the methyl ester intermediate. The methyl ester was saponified in a solution of aq LiOH (2.00 mL, 1.00 M) and dioxane (2.00 mL) at 100 °C for 16 h. The solution was cooled to rt, diluted with water (2.00 mL), and then acidified with aq HCl (1.00 M) until pH 4. The solution was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The solid was washed with cold water (5.00 mL), followed by hexane (10.0 mL), and dried to afford 11 (113 mg, 75%) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.33 (d, J = 2.2 Hz, 1H), 8.29 (d, J = 2.2 Hz, 1H), 8.20 (s, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.65 (d, J = 1.8 Hz, 1H), 7.59 (dd, J = 8.1, 1.8 Hz, 1H), 3.91–3.80 (m, 1H) 3.87 (s, 3H), 2.12–1.96 (m, 2H), 1.82 (m, 2H), 1.71–159 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.3, 147.0, 146.3, 141.0, 136.9, 130.7, 130.5, 129.1, 126.8, 124.0, 123.4, 123.1, 118.2, 112.3, 41.1, 34.3, 31.3, 25.3; HRMS: calcd for C20H21N2O2 [M + H]+m/z, 321.1603; found m/z, 321.1596; mp range 181–184 °C.
Ethyl 2-Chloronicotinate (12b)
To a solution of 2-chloronicotinic acid (5.00 g, 31.7 mmol) in toluene (35.0 mL) was added dropwise with stirring triethyl orthoacetate (17.5 mL, 95.2 mmol). The mixture was heated to reflux for 16 h and allowed to cool to rt, and the resultant solution was washed with sat. NaHCO3 (50.0 mL). The organic phase was dried with MgSO4, and the solvent was removed in vacuo to afford the title compound, ethyl 2-chloronicotinate 12b (5.40 g, 92%), as a clear oil.
1H NMR (400 MHz, DMSO-d6): δ 8.58 (dd, J = 4.8, 2.0 Hz, 1H), 8.24 (dd, J = 7.7, 2.0 Hz, 1H), 7.57 (dd, J = 7.7, 4.8 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6): δ 164.1, 152.2, 147.8, 140.2, 127.1, 123.2, 61.8, 13.9; LCMS: [M + H]+m/z, 186.0.
Ethyl 5-Bromo-2-chloronicotinate (13b)
To a solution of 5-bromo-2-chloronicotinic acid (10.0 g, 42.0 mmol) in ethanol (60.0 mL) was added slowly H2SO4 (9.10 g, 5.0 mL, 93.0 mmol). The reaction mixture was heated to reflux for 16 h. The mixture was concentrated in vacuo, re-dissolved in aq NaHCO3 (150 mL), and extracted with EtOAc (2 × 300 mL). The combined organic layers were dried over Na2SO4 and concentrated to give the desired product, 13b (10.5 g, 92%), as a light-yellowish oil.
1H NMR (400 MHz, DMSO-d6): δ 8.76 (d, J = 2.5 Hz, 1H), 8.47 (d, J = 2.5 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 1.33 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 162.88, 152.79, 146.65, 142.12, 128.34, 118.85, 62.24, 13.83; LCMS: [M + H]+m/z, 186.0.
Ethyl 2,5-Dichloronicotinate (14b)
Prepared following the procedure for 12b using 2,5-dichloronicotinic acid (10.0 g, 42.0 mmol). After purification, ethyl 2,5-dichloronicotinate 14b (11.0 g, 99%) was afforded as a clear oil.
1H NMR (400 MHz, DMSO-d6): δ 8.69 (d, J = 2.6 Hz, 1H), 8.38 (d, J = 2.6 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 1.33 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 162.9, 150.6, 146.1, 139.5, 130.3, 128.0, 62.3, 13.8; LCMS: [M + H]+m/z, 220.1.
Ethyl 3-Hydroxyfuro[2,3-b]pyridine-2-carboxylate (12c)
To a suspension of sodium hydride, 60% dispersed in mineral oil, (5.60 g, 0.14 mol) in 1,2-dimethoxyethane (60.0 mL) was added ethyl 2-hydroxyacetate (13.0 mL, 0.13 mol) under ice cooling and stirring. The ice bath was removed, and the solution was allowed warm to rt. After 30 min, a solution of ethyl 2-chloronicotinate (10.0 g, 54.0 mmol) in 100 mL of 1,2-dimethoxyethane was slowly added and the mixture was heated to 75 °C for 2 h. The solvent was removed in vacuo, and the residual solid was re-dissolved in aq NaHCO3 solution (150 mL) and EtOAc (250 mL). The aq layer was acidified with AcOH (pH 4) and extracted with CH2Cl2 (3 × 200 mL). The combined organic phases were dried over Na2SO4 and concentrated in vacuo. Purification by flash column chromatography (10% EtOAc/hexane) afforded ethyl 3-hydroxyfuro[2,3-b]pyridine-2-carboxylate 12c (8.5 g, 76%) as a pale-white solid.
1H NMR (400 MHz, CDCl3): δ 8.53 (dd, J = 4.8, 1.8 Hz, 1H), 8.11 (dd, J = 7.8, 1.8 Hz, 1H), 7.30 (dd, J = 7.8, 4.8 Hz, 1H), 4.48 (q, J = 7.1 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H).
LCMS: [M + H]+m/z, 208.1.
Ethyl 5-Bromo-3-hydroxyfuro[2,3-b]pyridine-2-carboxylate (13c)
Prepared following the procedure for 12c using ethyl 5-bromo-2-chloronicotinate 13b (18.0 g, 68.0 mmol). After purification, ethyl 5-bromo-3-hydroxyfuro[2,3-b]pyridine-2-carboxylate 13c (19.0 g, 76%) was afforded as a light-brown solid.
1H NMR (400 MHz, DMSO-d6): δ 11.34–11.30 (m, 1H), 8.58 (d, J = 2.3 Hz, 1H), 8.53 (d, J = 2.3 Hz, 1H), 4.32 (q, J = 7.1 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 158.7, 156.7, 148.8, 144.7, 133.5, 127.2, 115.9, 114.3, 60.4, 14.3; LCMS: [M + H]+m/z, 286.0.
Ethyl 5-Chloro-3-hydroxythieno[2,3-b]pyridine-2-carboxylate (14c)
Prepared following the procedure for 12c using ethyl 2,5-dichloronicotinate 14b (15.0 g, 68.0 mmol) and ethyl 2-mercaptoacetate (15.0 mL, 0.14 mmol). After purification, ethyl 5-chloro-3-hydroxythieno[2,3-b]pyridine-2-carboxylate 14c (12.4 g, 71%) was afforded as a light-orange solid.
1H NMR (400 MHz, DMSO-d6): δ 11.04 (s, 1H), 8.73 (d, J = 2.4 Hz, 1H), 8.41 (d, J = 2.4 Hz, 1H), 4.33 (q, J = 7.1 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 162.6, 155.7, 152.2, 149.4, 130.3, 127.7, 126.7, 105.5, 61.1, 14.2; LCMS: [M + H]+m/z, 258.0.
Furo[2,3-b]pyridin-3(2H)-one (12d)
A mixture of ethyl 3-hydroxyfuro[2,3-b]pyridine-2-carboxylate 12c (6.00 g, 30.0 mmol) in ethanol (100 mL) and sodium hydroxide (5.00 g, 100 mmol) dissolved in 15.0 mL of water was refluxed for 1 h. After evaporation of the solvent, the yellow-orange crystalline mass was dissolved in water (100 mL), acidified to pH 2–3 with conc. HCl, and heated to reflux for 45 min. Once cooled, the solution was neutralized with sodium bicarbonate and extracted with chloroform. The organic phase was dried (Na2SO4) and filtered, and the solvent was removed in vacuo to afford furo[2,3-b]pyridin-3(2H)-one 12d (2.70 g, 67%) as a light-brown solid. The title compound was isolated as a mixture of keto–enol tautomers (9:1).
1H NMR (400 MHz, DMSO-d6): δ 9.71 (s, 0H), 8.60 (dd, J = 4.9, 2.0 Hz, 1H), 8.15 (dd, J = 7.5, 1.9 Hz, 1H), 7.27–7.22 (m, 1H), 4.89 (s, 2H); 13C NMR (101 MHz, DMSO-d6): δ 197.7, 177.1, 156.9, 134.3, 118.8, 113.6, 74.6; LCMS: [M + H]+m/z, 136.1.
5-Bromofuro[2,3-b]pyridin-3(2H)-one (13d)
Prepared following the procedure for 12d using ethyl 5-bromo-3-hydroxyfuro[2,3-b]pyridine-2-carboxylate 13c (10.0 g, 35.0 mmol). After purification, 5-bromofuro[2,3-b]pyridin-3(2H)-one 13d (6.30 g, 84%) was afforded as a brown solid.
1H NMR (400 MHz, DMSO-d6): δ 8.71 (d, J = 2.5 Hz, 1H), 8.42 (d, J = 2.5 Hz, 1H), 4.96 (s, 2H); 13C NMR (100 MHz, DMSO): δ 196.4, 175.6, 156.9, 136.3, 115.5, 113.2, 75.8; LCMS: [M + H]+m/z, 213.4.
5-Chlorothieno[2,3-b]pyridin-3(2H)-one (14d)
Prepared following the procedure for 12d using ethyl 5-chloro-3-hydroxythieno[2,3-b]pyridine-2-carboxylate 14c (3.0g, 12 mmol). After purification, 5-chlorothieno[2,3-b]pyridin-3(2H)-one 14d (1.0 g, 50%) was isolated as an orange solid composed of keto- and enol-isomers.
1H NMR (400 MHz, DMSO-d6): δ 10.46 (s, 1H), 8.77 (d, J = 2.5 Hz, 1H), 8.56 (dd, J = 2.3, 0.5 Hz, 1H), 8.16 (d, J = 2.4 Hz, 1H), 8.11 (d, J = 2.5 Hz, 1H), 6.70 (d, J = 0.5 Hz, 1H), 4.18 (s, 2H); 13C NMR (101 MHz, DMSO-d6): δ 196.8, 172.1, 156.5, 154.7, 145.4, 145.3, 133.2, 127.9, 127.8, 126.9, 126.0, 100.9, 40.8; LCMS: [M + H]+m/z, 186.4.
Furo[2,3-b]pyridin-3-yl Trifluoromethanesulfonate (12e)
A solution of 12d (450 mg, 3.33 mmol) in CH2Cl2 (8.00 mL) was cooled to 0 °C. DIPEA (0.70 mL, 4.00 mmol) was added, followed by dropwise addition of trifluoromethanesulfonic anhydride (788 μL, 4.66 mmol). The reaction mixture was slowly warmed to rt and stirred for 2 h and then quenched with water. The aqueous phase was extracted with CH2Cl2, and the combined organic extracts were dried, filtered, and concentrated in vacuo. The residue was purified by column chromatography (10% EtOAc/hexane) to afford furo[2,3-b]pyridin-3-yl trifluoromethanesulfonate 12e (640 mg, 72%) as a brown oil.
1H NMR (400 MHz, CDCl3): δ 8.55–8.41 (m, 1H), 8.03 (dd, J = 7.8, 1.7 Hz, 1H), 7.91 (s, 1H), 7.43–7.35 (m, 1H); 13C NMR (101 MHz, CDCl3): δ 158.81, 146.35, 135.28, 132.54, 129.95, 128.17, 120.11, 118.71 (q, J = 321.6 Hz); LCMS: [M + H]+m/z, 268.2.
5-Bromofuro[2,3-b]pyridin-3-yl Trifluoromethanesulfonate (13e)
Prepared following the procedure for 12e using 5-bromofuro[2,3-b]pyridin-3(2H)-one 13d (10.0 g, 47.0 mmol). After purification, 5-bromofuro[2,3-b]pyridin-3-yl trifluoromethanesulfonate 13e (12.8 g, 79%) was isolated as a light-brown oil which solidified upon standing.
1H NMR (400 MHz, CDCl3): δ 8.50 (dd, J = 2.2, 0.4 Hz, 1H), 8.14 (d, J = 2.2 Hz, 1H), 7.92 (s, 1H); LCMS: [M + H]+m/z, 347.1.
5-Chlorothieno[2,3-b]pyridin-3-yl Trifluoromethanesulfonate (14e)
Prepared following the procedure for 12e using 5-chlorothieno[2,3-b]pyridin-3(2H)-one 14d (800 mg, 4.31 mmol). After purification, 5-Chlorothieno[2,3-b]pyridin-3-yl trifluoromethanesulfonate 14e (1.00 g, 74%) was isolated as a brown oil.
1H NMR (400 MHz, CDCl3): δ 8.60 (d, J = 2.3 Hz, 1H), 8.02 (d, J = 2.3 Hz, 1H), 7.55 (d, J = 0.5 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 155.34, 147.62, 134.57, 129.60, 127.41, 125.23, 118.67 (q, J = 321.2 Hz), 118.13; LCMS: [M + H]+m/z, 318.4.
2-Cyclopentyl-4-(furo[2,3-b]pyridin-3-yl)benzoic Acid (12f)
A microwave vial was loaded with furo[2,3-b]pyridin-3-yl trifluoromethanesulfonate 12e (250 mg, 936 μmol), 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 3 (444 mg, 1.40 mmol), Pd(PPh3)4 (11.0 mg, 9.50 μmol), sodium carbonate (397 mg, 3.74 mmol), MeOH (2.00 mL), and CH2Cl2 (0.50 mL). The vial was sealed and heated to 90 °C for 2 h. The solution was neutralized with aq HCl (10 mL) and then extracted with ethyl acetate (2 × 20.0 mL). The organic phases were combined and dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (30% EtOAc/hexane) to afford 12f (217 mg, 76%) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 12.94 (s, 1H), 8.67 (s, 1H), 8.41 (d, J = 6.3 Hz, 2H), 7.81–7.75 (m, 2H), 7.67 (dd, J = 8.0, 1.8 Hz, 1H), 7.52–7.46 (m, 1H), 3.84–3.73 (m, 1H), 2.07–2.03 (m, 2H), 1.86–1.81 (m, 2H), 1.73–1.62 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 169.2, 161.9, 147.0, 144.5, 143.0, 133.7, 131.0, 130.2, 124.8, 123.9, 120.2, 119.9, 117.4, 41.3, 34.3, 25.2; HRMS calcd for C19H17NO3: [M + H]+m/z, 308.1287; found, 308.1266; mp range 164–168 °C.
Methyl 4-(5-Bromofuro[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate (13f)
Prepared following the procedure for 12f using 5-bromofuro[2,3-b]pyridin-3-yl trifluoromethanesulfonate 13e (100 mg, 0.29 mmol) and 4 (95.4 mg, 0.29 mmol). After purification, methyl 4-(5-bromofuro[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate 13f (68 mg, 59%) was isolated as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.74 (s, 1H), 8.56 (d, J = 1.8 Hz, 1H), 8.48 (d, J = 1.6 Hz, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.69 (dd, J = 7.9, 1.7 Hz, 1H), 3.85 (s, 3H), 3.71–3.58 (m, 1H), 2.08–1.96 (m, 2H), 1.87–1.76 (m, 2H), 1.73–1.61 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 167.9, 160.5, 147.0, 144.9, 144.8, 133.4, 132.3, 130.2, 130.0, 125.0, 124.1, 119.6, 119.5, 115.3, 52.2, 41.4, 34.2, 25.2.
Methyl 4-(5-Chlorothieno[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate (14f)
Prepared following the procedure for 12f using 5-chlorothieno[2,3-b]pyridin-3-yl trifluoromethanesulfonate 14e (950 mg, 2.99 mmol) and 4 (988 mg, 2.99 mmol). After purification, methyl 4-(5-chlorothieno[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate 14f (870 mg, 78%) was isolated as a near-white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.69 (d, J = 2.3 Hz, 1H), 8.27–8.24 (m, 2H), 7.79 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 1.8 Hz, 1H), 7.57 (dd, J = 8.0, 1.7 Hz, 1H), 3.87 (s, 3H), 3.71–3.61 (m, 1H), 2.08–2.01 (m, 2H), 1.85–1.75 (m, 2H), 1.69–1.62 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 168.0, 159.5, 147.1, 145.4, 137.1, 133.6, 131.3, 130.2, 130.0, 129.7, 128.1, 128.0, 126.6, 125.3, 52.2, 41.4, 34.3, 25.2.
2-Cyclopentyl-4-(5-phenylfuro[2,3-b]pyridin-3-yl)benzoic Acid (13g)
4-(5-Bromofuro[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoic acid, 13f (48.0 mg, 0.12 mmol), phenylboronic acid (30.0 mg, 0.25 mmol), Na2CO3 (40.0 mg, 0.37 mmol), and Pd(PPh3)4 (14.0 mg, 12 μmol) were dissolved in a 4:1 mixture of dioxane (2.5 mL) and heated to 90 °C under nitrogen for 16 h. The mixture was neutralized to pH 6–7 with aq HCl and extracted with ethyl acetate (5.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude residue was purified using flash column chromatography (30% EtOAc/hexane) to obtain the methyl ester intermediate which was immediately dissolved in MeOH (3.0 mL), added aq NaOH (small excess), and heated to 70 °C for 1 h. After cooling, H2O (4.0 mL) was added and the mixture was extracted with diethyl ether (2 × 4.0 mL). The aqueous phase was acidified to pH 6 with aq HCl, extracted with CH2Cl2 (6.00 mL), and concentrated to afford 2-cyclopentyl-4-(5-phenylfuro[2,3-b]pyridin-3-yl)benzoic acid 13g (39 mg, 82%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 12.95 (s, 1H), 8.72 (s, 1H), 8.67 (d, J = 2.2 Hz, 1H), 8.53 (d, J = 2.2 Hz, 1H), 7.82 (d, J = 1.5 Hz, 1H), 7.81–7.79 (m, 4H), 7.56–7.50 (m, 2H), 7.47–7.41 (m, 1H), 3.86–3.77 (m, 1H), 2.11–2.01 (m, 2H), 1.87–1.79 (m, 2H), 1.73–1.62 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 169.3, 161.6, 147.0, 143.8, 143.2, 137.5, 133.6, 132.9, 131.0, 130.3, 129.1, 128.2, 127.8, 127.5, 125.0, 124.1, 120.2, 117.6, 41.2, 34.4, 25.3; HRMS: calcd for C25H21NO3 [M + H]+m/z, 384.1600; found, 384.1593; mp range 259–262 °C.
2-Cyclopentyl-4-(5-phenylthieno[2,3-b]pyridin-3-yl)benzoic Acid (14g)
Methyl 4-(5-chlorothieno[2,3-b]pyridin-3-yl)-2-cyclopentylbenzoate 14f (150 mg, 403 μmol), phenylboronic acid (73.8 mg, 605 μmol), Pd2(dba)3 (18.5 mg, 20.2 μmol), dicyclohexyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphane (28.8 mg, 60.5 μmol), and Cs2CO3 (394 mg, 1.21 mmol) were dissolved in dioxane (3.00 mL) and H2O (0.8 mL). The solution was heated to 90 °C for 16 h. The reaction mixture was allowed to cool to rt, neutralized to pH 7 with aq HCl, and extracted with ethyl acetate (15 mL). The combined organic phases were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified by column chromatography (25% EtOAc/hexane) to afford the methyl ester intermediate. This material was dissolved in MeOH (6 mL), added aq NaOH (small excess), and heated to 70 °C for 1 h. After cooling, H2O (8 mL) was added and the mixture was extracted with diethyl ether (2 × 10.0 mL). The aqueous phase was acidified to pH 6, extracted with CH2Cl2 (10 mL), and concentrated to afford 2-cyclopentyl-4-(5-phenylthieno[2,3-b]pyridin-3-yl)benzoic acid 14g (116 mg, 72%) as a yellow solid.
1H NMR (400 MHz, DMSO-d6): δ 12.94 (s, 1H), 8.96 (d, J = 2.1 Hz, 1H), 8.38 (d, J = 2.2 Hz, 1H), 8.17 (s, 1H), 7.83–7.77 (m, 3H), 7.72 (d, J = 1.7 Hz, 1H), 7.62 (dd, J = 8.0, 1.7 Hz, 1H), 7.55–7.50 (m, 2H), 7.47–7.41 (m, 1H), 3.87–3.78 (m, 1H), 2.09–2.04 (m, 2H), 1.83–1.78 (m, 2H), 1.71–1.63 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.4, 160.6, 147.0, 145.8, 137.3, 137.3, 134.4, 132.5, 131.2, 130.4, 130.1, 129.2, 128.1, 128.1, 127.3, 126.5, 126.2, 125.3, 41.2, 34.5, 25.3; HRMS: calcd for C25H21NO2S [M + H]+m/z, 400.1371; found, 400.1365; mp range 269–273 °C.
2-Cyclopentyl-4-(thieno[2,3-b]pyridin-3-yl)benzoic Acid (12g)
Prepared following the procedure for 12f using 3-bromothieno[2,3-b]pyridine 15e (50.0 mg, 0.23 mmol). After purification, 2-cyclopentyl-4-(thieno[2,3-b]pyridin-3-yl)benzoic acid 12g (66.0 mg, 88%) was afforded as a light-brown solid.
1H NMR (400 MHz, DMSO-d6): δ 12.99 (s, 1H), 8.65 (dd, J = 4.6, 1.6 Hz, 1H), 8.27 (dd, J = 8.2, 1.6 Hz, 1H), 8.10 (s, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 1.8 Hz, 1H), 7.56–7.50 (m, 2H), 3.85–3.76 (m, 1H), 2.08–2.03 (m, 2H), 1.82–1.78 (m, 2H), 1.72–1.58 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 172.6, 164.7, 150.1, 149.9, 140.4, 137.4, 134.5, 133.7, 133.4, 133.1, 129.5, 128.4, 128.4, 123.4, 44.4, 37.5, 28.4; HRMS: calcd for C19H17NO2S [M + H]+m/z, 324.1058; found, 324.1054; mp range 194–197 °C.
Methyl 4-(5-Bromopyrazolo[1,5-a]pyridin-3-yl)-2-cyclopentylbenzoate (17)
5-Bromo-3-iodopyrazolo[1,5-a]pyridine (200 mg, 0.62 mmol) and methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (194 mg, 0.60 mmol) were dissolved in a 3:1 solution of dioxane/water (4.00 mL). Pd2(dba)3 (28 mg, 31 μmol), XPhos (30.0 mg, 62.0 μmol), and Cs2CO3 (605 mg, 1.90 mmol) were added. The mixture was stirred at rt for 16 h. The aqueous phase was removed, and the solution was concentrated in vacuo. The crude was purified via column chromatography (0–20% EtOAc/hexane) to afford ethyl 4-(5-bromopyrazolo[1,5-a]pyridin-3-yl)-2-cyclopentylbenzoate 17 (189 mg, 76%) as a mustard yellow solid.
1H NMR (400 MHz, CDCl3): δ 8.36 (dd, J = 7.3, 0.8 Hz, 1H), 8.16 (s, 1H), 7.94 (dd, J = 2.1, 0.8 Hz, 1H), 7.86 (dd, J = 8.1, 0.4 Hz, 1H), 7.57 (d, J = 1.8 Hz, 1H), 7.40 (dd, J = 8.1, 1.8 Hz, 1H), 6.91 (ddd, J = 7.3, 2.1, 0.4 Hz, 1H), 3.93 (s, 3H), 3.91–3.83 (m, 1H), 2.24–2.11 (m, 2H), 1.93–1.81 (m, 2H), 1.81–1.72 (m, 2H), 1.71–1.61 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 168.5, 148.7, 141.6, 137.6, 135.8, 130.9, 129.7, 128.4, 125.3, 123.7, 119.7, 118.7, 116.0, 112.4, 52.0, 41.7, 34.8, 25.7; HRMS: calcd for C20H20N2O2Br [M + H]+m/z, 399.0708; found m/z, 399.0705; mp range 182–184 °C.
Methyl 4-(5-Chloropyrazolo[1,5-a]pyrimidin-3-yl)-2-cyclopentylbenzoate (18)
5-Chloro-3-iodopyrazolo[1,5-a]pyrimidine (500 mg, 1.79 mmol) and methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (591 mg) were dissolved in a 10:1 mixture of dioxane/water (10.0 mL). Pd(dppf)Cl2·CH2Cl2 (73.0 mg, 0.9 mmol) and Cs2CO3 (1.75 g, 5.37 mmol) were added, and the solution was heated to 80 °C for 16 h. The solution was allowed to cool to rt, the aqueous phase was removed, and the organic phase was concentrated in vacuo. The crude was purified via column chromatography (10–20% EtOAc/hexane) to afford methyl 4-(5-chloropyrazolo[1,5-a]pyrimidin-3-yl)-2-cyclopentylbenzoate 18 (270 mg, 44%) as a bright-yellow solid.
1H NMR (400 MHz, CDCl3): δ 8.59 (d, J = 7.2 Hz, 1H), 8.48 (s, 1H), 8.12 (d, J = 1.5 Hz, 1H), 7.92–7.78 (m, 2H), 6.86 (d, J = 7.2 Hz, 1H), 3.99–3.86 (m, 4H), 2.28–2.04 (m, 2H), 1.97–1.84 (m, 2H), 1.84–1.61 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 168.6, 150.8, 148.6, 143.9, 136.5, 134.4, 130.6, 128.3, 124.5, 122.8, 110.1, 109.4, 51.9, 41.7, 34.8, 25.8; HRMS: calcd for C19H19N3O2Cl [M + H]+m/z, 356.1165; found m/z, 356.1161; mp range 163–166 °C.
2-Cyclopentyl-4-(5-phenylpyrazolo[1,5-a]pyridin-3-yl)benzoic Acid (19)
Methyl 4-(5-bromopyrazolo[1,5-a]pyridin-3-yl)-2-cyclopentylbenzoate 17 (100 mg, 0.25 mmol), phenylboronic acid (37.0 mg, 0.30 mmol), Pd(PPh3)4 (29.0 mg, 25.0 μmol), and Cs2CO3 (245 mg, 0.75 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (1.50 mL) was added, and the vial was flushed with nitrogen and sealed. The mixture was irradiated at 120 °C for 30 min. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (5–10% EtOAc/hexane) to afford the methyl ester intermediate. The methyl ester was saponified in a solution of aq LiOH (1.00 mL, 1.00 M) and dioxane (1.00 mL) at 100 °C for 16 h. The solution was cooled to rt, diluted with water (2.00 mL), and then acidified with aq HCl (1 M) until pH 4. The solution was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The solid was washed with cold water (5.00 mL), followed by hexane (10.0 mL), and dried to afford the acid (68.0 mg, 71%) as a yellow solid.
1H NMR (700 MHz, DMSO-d6): δ 8.83 (d, J = 6.9 Hz, 1H), 8.50 (s, 1H), 8.10 (s, 1H), 8.02 (s, 1H), 7.85 (d, J = 7.1 Hz, 2H), 7.79 (d, J = 7.8 Hz, 1H), 7.72 (s, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.53 (t, J = 6.9 Hz, 2H), 7.45 (t, J = 6.8 Hz, 1H), 7.33 (d, J = 6.8 Hz, 1H), 3.91–3.85 (m, 1H), 2.10–2.01 (m, 2H), 1.88–1.78 (m, 2H), 1.73–1.60 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.2, 147.2, 141.4, 137.6, 136.9, 136.5, 135.7, 131.5, 131.4, 130.5, 129.7, 129.3, 129.1, 128.9, 128.7, 128.5, 126.7, 124.5, 123.4, 113.5, 111.9, 111.7, 41.1, 34.3, 25.3; HRMS: calcd for C25H23N2O2 [M + H]+m/z, 383.1759; found m/z, 383.1754; mp range 153–155 °C.
2-Cyclopentyl-4-(5-phenylpyrazolo[1,5-a]pyrimidin-3-yl)benzoic Acid (20)
Methyl 4-(5-chloropyrazolo[1,5-a]pyrimidin-3-yl)-2-cyclopentylbenzoate 18 (100 mg, 0.28 mmol), phenylboronic acid (41.0 mg, 0.33 mmol), Pd(PPh3)4 (32.0 mg, 28.0 μmol), and Cs2CO3 (275 mg, 0.84 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (2.00 mL) was added, and the vial was flushed with nitrogen and capped. The solution was irradiated at 120 °C for 30 min. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (5–10% EtOAc/hexane) to afford the methyl ester intermediate. The methyl ester was saponified in a solution of aq LiOH (1.00 mL, 1.00 M) and dioxane (1.00 mL) at 100 °C for 16 h. The solution was cooled to rt, diluted with water (2.00 mL), and then acidified with aq HCl (1.00 M) until pH 4. The solution was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The solid was washed with cold water (5.00 mL), followed by hexane (10.0 mL), and dried to afford 2-cyclopentyl-4-(5-phenylpyrazolo[1,5-a]pyrimidin-3-yl)benzoic acid 20 (87.0 mg, 81%) as a bright-yellow solid.
1H NMR (700 MHz, DMSO-d6): δ 12.94 (s, 1H), 9.24 (dd, J = 7.6, 2.8 Hz, 1H), 8.87 (s, 1H), 8.65 (s, 1H), 8.39–8.28 (m, 2H), 7.95 (d, J = 8.1 Hz, 1H), 7.84–7.72 (m, 2H), 7.61–7.55 (m, 3H), 3.98–3.91 (m, 1H), 2.15–2.09 (m, 2H), 1.93–1.86 (m, 2H), 1.77–1.68 (m, 4H); 13C NMR (176 MHz, DMSO-d6): 169.2, 155.9, 147.3, 144.3, 143.6, 137.0, 136.6, 135.3, 131.0, 130.1, 129.0 (2 × ArCH), 128.4, 127.2 (2 × ArCH), 123.8, 122.1, 108.2, 105.9, 41.0, 34.7, 25.6; HRMS: calcd for C24H22N3O2 [M + H]+m/z, 384.1712; found, m/z 384.1707; mp range 210–214 °C.
2-Cyclopentyl-4-(pyrazolo[1,5-a]pyridin-3-yl)benzoic Acid (21)
3-Bromopyrazolo[1,5-a]pyridine (100 mg, 0.50 mmol), 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 3 (193 mg, 0.61 mmol), Pd2(dba)3 (23.0 mg, 25.0 μmol), XPhos (24.0 mg, 51.0 μmol), and Cs2CO3 (579 mg, 1.80 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (3.00 mL) was added, and the vial was flushed with nitrogen and sealed. The mixture was heated to 120 °C for 16 h. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The mixture was acidified to pH 4 with 1.00 M aq HCl solution. The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (50–100% EtOAc/hexane) to afford 2-cyclopentyl-4-(pyrazolo[1,5-a]pyridin-3-yl)benzoic acid 21 (64.0 mg, 41%) as a yellow solid.
1H NMR (400 MHz, DMSO-d6): δ 12.78 (br s, 1H), 8.76 (d, J = 7.0 Hz, 1H), 8.47 (s, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.67 (d, J = 1.6 Hz, 1H), 7.57 (dd, J = 8.1, 1.7 Hz, 1H), 7.39 (ddd, J = 8.9, 6.7, 1.0 Hz, 1H), 6.99 (td, J = 6.9, 1.1 Hz, 1H), 3.90–3.79 (m, 1H), 2.09–1.97 (m, 2H), 1.88–1.76 (m, 2H), 1.73–1.58 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 169.2, 147.1, 140.8, 136.3, 135.8, 130.5, 129.6, 128.7, 125.4, 124.2, 123.2, 117.1, 112.7, 110.9, 41.2, 34.3, 25.2; HRMS (ESI): calcd for C19H19N2O2 [M + H]+m/z, 307.1446; found m/z, 307.1439; mp range 167–169 °C.
2-Cyclopentyl-4-(pyrazolo[1,5-a]pyrimidin-3-yl)benzoic Acid (22)
3-Bromopyrazolo[1,5-a]pyrimidine (150 mg, 0.75 mmol), 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 3 (287 mg, 0.90 mmol), Pd(PPh3)4 (87.0 mg, 75.0 μmol), and Cs2CO3 (864 mg, 2.65 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (2.00 mL) was added, and the vial was flushed with nitrogen and capped. The solution was irradiated at 120 °C for 30 min. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The mixture was acidified to pH 4 with 1 M aq HCl solution. The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (50–100% EtOAc/hexane) to afford 2-cyclopentyl-4-(pyrazolo[1,5-a]pyrimidin-3-yl)benzoic acid 22 (137 mg, 59%) as a bright-yellow solid.
1H NMR (700 MHz, DMSO-d6): δ 12.74 (br s, 1H), 9.24–9.12 (d, J = 6.0 Hz, 1H), 8.87 (d, J = 2.9 Hz, 1H), 8.72 (s, 1H), 8.25 (s, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.75 (dd, J = 8.1, 2.8 Hz, 1H), 7.14 (dt, J = 7.4, 3.5 Hz, 1H), 3.89–3.83 (m, 1H), 2.08–2.01 (m, 2H), 1.88–1.82 (m, 2H), 1.71–1.63 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.3, 150.9, 146.9, 144.5, 143.2, 136.6, 135.0, 130.1, 128.6, 123.6, 122.5, 109.0, 108.2, 41.2, 34.3, 25.2; HRMS: calcd for C18H18N3O2 [M + H]+m/z, 308.1399; found m/z, 308.1393; mp range 196–199 °C.
Methyl 4-(6-Chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-2-cyclopentylbenzoate (24)
3-Bromo-6-chloro-[1,2,4]triazolo[4,3-b]pyridazine (924 mg, 3.96 mmol), methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (1.31 g, 3.96 mmol), K2CO3 (1.64 g, 11.9 mmol), and Pd(PPh3)4 (320 mg, 0.28 mmol) in a mixture of dioxane (12.0 mL) and water (3.00 mL) were degassed and put under a N2 atmosphere. The reaction mixture was heated to 90 °C and stirred for 16 h. The reaction mixture was diluted with aq HCl (50.0 mL) to pH 6 and extracted with EtOAc (2 × 75.0 mL), and the combined organic phases were concentrated in vacuo. The crude residue was purified via column chromatography (35% EtOAc/hexane) to afford methyl 4-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-2-cyclopentylbenzoate 24 (1.20 g, 85%) as a grayish solid. 1H NMR (400 MHz, DMSO-d6): δ 8.58 (d, J = 9.7 Hz, 1H), 8.47 (d, J = 1.7 Hz, 1H), 8.21 (dd, J = 8.2, 1.7 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 2.8 Hz, 1H), 3.88 (s, 3H), 3.77–3.66 (m, 1H), 2.14–2.03 (m, 2H), 1.91–1.79 (m, 2H), 1.75–1.58 (m, 4H). LCMS: [M + H]+m/z, 356.2.
2-Cyclopentyl-4-(6-phenyl-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)benzoic Acid (25)
Following the procedure for 14g using 4-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-2-cyclopentylbenzoate 24 (220 mg, 0.62 mmol). After purification, cyclopentyl-4-(6-phenyl-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)benzoic acid 25 (200 mg, 84%) was afforded as a near-yellow solid.
1H NMR (400 MHz, DMSO-d6): δ 13.14 (s, 1H), 8.74 (d, J = 1.7 Hz, 1H), 8.58 (d, J = 9.8 Hz, 1H), 8.32 (dd, J = 8.2, 1.7 Hz, 1H), 8.21–8.18 (m, 2H), 8.07 (d, J = 9.8 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.65–7.59 (m, 3H), 3.91–3.82 (m, 1H), 2.15–2.11 (m, 2H), 1.88–1.83 (m, 2H), 1.72–1.66 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.2, 153.5, 146.6, 146.4, 144.5, 134.1, 133.0, 131.1, 129.7, 129.1, 128.7, 127.4, 125.6, 125.2, 124.2, 119.9, 41.1, 34.7, 25.6; HRMS: calcd for C23H20N4O2 [M + H]+m/z, 385.1665; found, 385.1659; mp range 228–231 °C.
4-([1,2,4]Triazolo[4,3-b]pyridazin-3-yl)-2-cyclopentylbenzoic Acid (26)
3-Bromo-[1,2,4]triazolo[4,3-b]pyridazine (150 mg, 754 μmol), methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (373 mg, 1.13 mmol), XPhos (53.9 mg, 113 μmol), Cs2CO3 (516 mg, 1.58 mmol), and Pd2(dba)3 (34.5 mg, 0.04 mmol) in a 3:1 mixture of dioxane/water (4.00 mL) were heated to 90 °C under a N2 atmosphere for 16 h. The reaction mixture was diluted with aq HCl (25.0 mL, pH 6) and extracted with EtOAc (2 × 50.0 mL); the organic phases were combined, concentrated, and purified using flash column chromatography (40% EtOAc/hexane) to afford 4-([1,2,4]triazolo[4,3-b]pyridazin-3-yl)-2-cyclopentylbenzoic acid 26 (178.0 mg, 77%) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 13.14 (s, 1H), 8.79 (dd, J = 4.3, 1.5 Hz, 1H), 8.50–8.46 (m, 2H), 8.29 (dd, J = 8.2, 1.7 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.44 (dd, J = 9.4, 4.3 Hz, 1H), 3.86–3.75 (m, 1H), 2.14–2.04 (m, 2H), 1.89–1.79 (m, 2H), 1.74–1.58 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.2, 146.8, 146.4, 146.3, 145.1, 133.1, 129.6, 128.6, 125.4, 125.3, 124.1, 120.9, 41.3, 34.4, 25.3; HRMS: calcd for C17H16N4O2 [M + H]+m/z, 309.1352; found, 309.1346; mp range 208–211 °C.
4-Chloro-1-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridine (28)
4-Chloro-2-iodo-1-methyl-1H-pyrrolo[2,3-b]pyridine (75.0 mg, 0.25 mmol), phenylboronic acid (31.0 mg, 0.25 mmol), Cs2CO3 (251 mg, 0.77 mmol), and Pd(PPh3)4 (30.0 mg, 0.02 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (2.00 mL) was added, and the vial was flushed with nitrogen and capped. The solution was irradiated at 120 °C for 30 min. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (10% EtOAc/hexane) to afford 4-chloro-1-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridine 28 (58.0 mg, 93%) as a white-gray solid.
1H NMR (400 MHz, CDCl3): δ 8.23 (d, J = 5.2 Hz, 1H), 7.63–7.40 (m, 5H), 7.12 (d, J = 5.2 Hz, 1H), 6.63 (s, 1H), 3.89 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 149.6, 142.6, 142.5, 135.3, 131.7, 129.1 (2 × ArCH), 128.7 (2 × ArCH), 128.6, 119.9, 116.2, 97.9, 30.3; mp range 113–116 °C.
2-Cyclopentyl-4-(1-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridin-4-yl)benzoic Acid (29)
4-Chloro-1-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridine 28 (60.0 mg, 0.25 mmol), methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (98.0 mg, 0.30 mmol), Pd(PPh3)4 (29.0 mg, 25.0 μmol), and Cs2CO3 (242 mg, 0.74 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (2.00 mL) was added, and the vial was flushed with nitrogen and capped. The solution was irradiated at 120 °C for 30 min. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (5–10% EtOAc/hexane) to afford the methyl-substituted ester intermediate. The methyl ester was saponified in a solution of aq LiOH (1.00 mL, 1.00 M) and dioxane (1.00 mL) at 100 °C for 16 h. The solution was cooled to rt, diluted with water (2.00 mL), and then acidified with aq HCl (1.00 M) until pH 4. The solution was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The solid was washed with cold water (5.00 mL), followed by hexane (10.0 mL), and dried to afford 2-cyclopentyl-4-(1-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridin-4-yl)benzoic acid 29 (79.0 mg, 80%) as a brown-yellow solid.
1H NMR (700 MHz, DMSO-d6): δ 8.40 (d, J = 4.9 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 1.7 Hz, 1H), 7.71 (t, J = 7.7 Hz, 3H), 7.56 (t, J = 7.5 Hz, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.33 (d, J = 4.9 Hz, 1H), 6.73 (s, 1H), 3.89 (s, 3H), 3.86–3.76 (m, 1H), 2.12–2.04 (m, 2H), 1.84–1.77 (m, 2H), 1.70–1.59 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.3, 149.5, 146.7, 143.0, 142.2, 140.8, 139.4, 131.8, 131.6, 130.0, 128.9 (2 × ArCH), 128.8 (2 × ArCH), 128.6, 126.5, 125.5, 117.5, 115.1, 98.1, 41.2, 34.4, 29.9, 25.3; HRMS: calcd for C26H25N2O2 [M + H]+m/z, 397.1916; found m/z, 397.1908; mp range 187–189 °C.
2-Cyclopentyl-4-(1-methyl-1H-pyrrolo[2,3-b]pyridin-4-yl)benzoic Acid (30)
4-Chloro-1-methyl-1H-pyrrolo[2,3-b]pyridine (75.0 mg, 0.45 mmol), methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (180 mg, 0.54 mmol), Pd(PPh3)4 (52.0 mg, 45.0 μmol), and Cs2CO3 (440 mg, 1.40 mmol) were loaded into a microwave vial. A 3:1 mixture of dioxane/water (3.00 mL) was added, and the vial was flushed with nitrogen and capped. The solution was irradiated at 120 °C for 30 min. Once cooled, the solution was diluted with EtOAc (2.00 mL) and water (2.00 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (5–10% EtOAc/hexane) to afford the methyl ester intermediate. The methyl ester was saponified in a solution of aq LiOH (1.00 mL, 1.00 M) and dioxane (1.00 mL) at 100 °C for 16 h. The solution was cooled to rt, diluted with water (2.00 mL), and then acidified with aq HCl (1.00 M) until pH 4. The solution was extracted with EtOAc (2 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The solid was washed with cold water (5.00 mL), followed by hexane (10.0 mL), and dried to afford 2-cyclopentyl-4-(1-methyl-1H-pyrrolo[2,3-b]pyridin-4-yl)benzoic acid 30 (97.0 mg, 67%) as a pale-yellow solid.
1H NMR (700 MHz, DMSO-d6): δ 13.01 (s, 1H), 8.36 (s, 1H), 7.81 (d, J = 7.6 Hz, 1H), 7.76 (s, 1H), 7.67–7.61 (m, 2H), 7.27 (s, 1H), 6.57 (s, 1H), 3.87 (s, 3H), 3.80 (dd, J = 19.7, 12.5 Hz, 1H), 2.13–2.00 (m, 2H), 1.87–1.74 (m, 2H), 1.71–1.56 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.3, 147.9, 146.6, 142.6, 140.8, 139.9, 131.7, 131.0, 129.9, 126.5, 125.4, 117.5, 114.3, 97.8, 41.1, 34.4, 31.2, 25.3; HRMS: calcd for C20H21N2O2 [M + H]+m/z, 321.1603; found m/z, 321.1595; mp range 201–203 °C.
Methyl 2-Cyclopentyl-4-(2,3-diaminopyridin-4-yl)benzoate (32)
4-Chloropyridine-2,3-diamine (103 mg, 0.71 mmol) and methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (250 mg, 0.75 mmol) were dissolved in a 10:1 mixture of dioxane/H2O (10.0 mL). Pd2(dba)3 (69.0 mg, 75 μmol), XPhos (72.0 mg, 150 μmol), and Cs2CO3 (740 mg, 2.30 mmol) were added. The solution was heated to 100 °C for 16 h. Once cooled to rt, the aqueous phase was removed and the organic phase was concentrated in vacuo. The crude was purified via column chromatography (25–75% EtOAc/hexane) to afford methyl 2-cyclopentyl-4-(2,3-diaminopyridin-4-yl)benzoate 32 (169 mg, 71%) as a dark-brown solid.
1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 5.3 Hz, 1H), 7.47 (d, J = 1.8 Hz, 1H), 7.31–7.23 (dd, 1H), 6.61 (d, J = 5.3 Hz, 1H), 4.63 (br s, 2H), 3.93 (s, 3H), 3.86–3.71 (m, 1H), 3.61 (br s, 2H), 2.19–2.07 (m, 2H), 1.90–1.73 (m, 2H), 1.75–1.65 (m, 2H), 1.65–1.50 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 168.5, 149.1, 148.4, 140.6, 136.8, 133.8, 130.4, 130.4, 127.2, 126.6, 125.5, 116.3, 52.2, 41.7, 35.0, 25.7; HRMS: calcd for C18H22N3O2 [M + H]+m/z, 312.1712; found m/z, 312.1704; mp range 125–129 °C.
2-Cyclopentyl-4-(3H-imidazo[4,5-b]pyridin-7-yl)benzoic Acid (33)
Methyl 2-cyclopentyl-4-(2,3-diaminopyridin-4-yl)benzoate 32 (100 mg, 0.32 mmol) was dissolved in MeOH (1 mL). Sulfamic acid (3.00 mg, 32.0 μmol) and trimethyl orthoformate (85.0 mg, 0.80 mmol) were added, and the mixture was stirred at rt for 1 h. The solvent was removed in vacuo, and the residue was dissolved in EtOAc (10.0 mL) and water (10.0 mL). The phases were separated, and the aqueous phase was extracted with EtOAc (2 × 10.0 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude residue was purified by column chromatography (50–100% EtOAc/hexane). The isolated methyl ester was dissolved in dioxane (1.00 mL) and aq LiOH (1.00 mL of a 1.00 M solution). The solution was heated to 100 °C for 16 h, cooled, and diluted with EtOAc (3.00 mL) and water (3.00 mL). The mixture was acidified to pH 4 with 1.00 M aq HCl solution. The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo to afford 2-cyclopentyl-4-(3H-imidazo[4,5-b]pyridin-7-yl)benzoic acid 33 (45 mg, 46%) as a white solid.
1H NMR (700 MHz, DMSO-d6): δ 13.23 (s, 1H), 8.51 (s, 1H), 8.45 (s, 1H), 8.41 (d, J = 4.7 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.59 (d, J = 4.7 Hz, 1H), 3.80 (t, J = 8.7 Hz, 1H), 2.10–2.03 (br m, 2H), 1.87–1.73 (br m, 2H), 1.67 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 171.9, 169.4, 148.8, 146.1, 144.2, 143.8, 138.2, 136.5, 132.1, 131.8, 129.3, 127.6, 125.8, 115.3, 41.3, 34.4, 25.2; HRMS: calcd for C18H18N3O2 [M + H]+m/z, 308.1399; found, m/z 308.1392; mp range 183–186 °C.
2-Cyclopentyl-4-(2-phenyl-3H-imidazo[4,5-b]pyridin-7-yl)benzoic Acid (34)
Benzoic acid (47.0 mg, 0.38 mmol) and CDI (63.0 mg, 0.38 mmol) were loaded into a microwave vial and dissolved in dimethylformamide (DMF) (1.0 mL). The solution was cooled to 0 °C and stirred for 30 min. Methyl 2-cyclopentyl-4-(2,3-diaminopyridin-4-yl)benzoate 4 (100 mg, 0.32 mmol) was added, and the vial was sealed. The mixture was heated to 200 °C for 10 min and allowed to cool to rt, and water (3.00 mL) was added, causing a precipitate to form. The precipitate was filtered and washed with cold water (10.0 mL) and hexane (10.0 mL). The precipitate was dissolved in dioxane (1.00 mL) and aq LiOH (1.00 mL of a 1.00 M solution). The solution was heated to 100 °C for 16 h and then diluted with EtOAc (3.00 mL) and water (3.00 mL). The mixture was acidified to pH 4 with 1 M aq HCl solution. The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 3.00 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (50–100% EtOAc/hexane) to afford 2-cyclopentyl-4-(2-phenyl-3H-imidazo[4,5-b]pyridin-7-yl)benzoic acid 34 (93.0 mg, 76%) as a white solid.
1H NMR (700 MHz, DMSO-d6): δ 8.55–8.44 (m, 2H), 8.33 (d, J = 6.3 Hz, 2H), 8.05 (d, J = 6.7 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.67 (s, 1H), 7.61 (d, J = 6.6 Hz, 3H), 3.93–3.79 (m, 1H), 2.18–2.05 (m, 2H), 1.93–1.83 (m, 2H), 1.82–1.66 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.4, 150.2, 146.6, 137.7, 132.4, 131.1, 129.5, 129.1, 128.0, 127.1, 125.6, 116.2, 41.1, 39.5, 34.5, 25.4; HRMS: calcd for C24H22N3O2 [M + H]+m/z, 384.1712; found m/z, 384.1704; mp range 196–199 °C.
4-Chloro-6-iodothieno[3,2-d]pyrimidine (35b)
An oven-dried 250 mL three-neck round-bottom flask provided with a pressure-equalizing addition funnel was cooled under N2. 4-Chlorothieno[3,2-d]pyrimidine 35a (3.40 g, 20.0 mmol) was added under positive N2 pressure, followed by the addition of anhydrous THF (65.0 mL). The solution was cooled down to −78 °C; LDA (12.0 mL, 24.0 mmol) was added dropwise over 5 min, and the mixture was stirred at −78 °C for 20 min. A solution of I2 (6.08 g, 24.0 mmol) in anhydrous THF (15.0 mL) was added dropwise over 10 min using the equalizing addition funnel. The resulting mixture was stirred at −78 °C for 30 min, allowed to warm up to rt over 1 h, and then stirred for 16 h. The reaction was quenched with saturated NH4Cl/ice and diluted with CHCl3 (200 mL). The layers were separated, and the aqueous layer was extracted with CHCl3 (2 × 50.0 mL). The combined organics were rinsed with aq Na2S2O3 (2 × 25.0 mL) and sat. NaCl solution (2 × 25.0 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (0–100% hexane/EtOAc), affording 4-chloro-6-iodothieno[3,2-d]pyrimidine 35b (4.60 g, 68%) as a clear brown solid.
1H NMR (400 MHz, CDCl3): δ 8.90 (s, 1H), 7.84 (s, 1H); 13C NMR (176 MHz, DMSO-d6): δ 162.6, 154.9, 154.8, 151.7, 134.8, 134.2, 97.4.
4-Chloro-6-phenylthieno[3,2-d]pyrimidine (36)
4-Chloro-6-iodothieno[3,2-d]pyrimidine 35b (1.01 mmol, 300 mg), phenylboronic acid (1.01 mmol, 125 mg), NaHCO3 (1.01 mmol, 85.0 mg), CsF (1.01 mmol, 155 mg), and Pd(Ph3)4 (35.0 mg, 0.03 mmol) were dissolved in a 3:1 mixture of dioxane/water (7.00 mL). The reaction mixture was stirred at reflux for 3 h. The volatiles were removed under reduced pressure, and the crude was purified via column chromatography (hexane/EtOAc 0–30%) to afford 4-chloro-6-phenylthieno[3,2-d]pyrimidine 36 (175 mg, 70%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 9.04 (s, 1H), 8.26 (s, 1H), 8.05–7.97 (m, 2H), 7.62–7.52 (m, 3H).
2-Cyclopentyl-4-(6-phenylthieno[3,2-d]pyrimidin-4-yl)benzoic Acid (37)
A 5 mL microwave vial was loaded with pyrimidine 36 (55.0 mg, 0.22 mmol) and 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 3 (70.0 mg, 0.22 mmol), Cs2CO3 (0.45 mmol, 150 mg), and Pd(Ph3)4 (5 mol %, 13.0 mg). The solids were dissolved in a solution of 1,4-dioxane/water (3:1, 1.50 mL). The vial was sealed, and the reaction mixture was irradiated at 125 °C for 15 min. The volatiles were removed in vacuo, and remaining solids were suspended in ice/water before the pH was adjusted to 2–3 with 2 M HCl. After an initial purification over silica gel, the compound was further purified by recrystallization from diethyl ether, affording 2-cyclopentyl-4-(6-phenylthieno[3,2-d]pyrimidin-4-yl)benzoic acid 37 (12.0 mg, 15%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 13.25 (s, 1H), 9.30 (s, 1H), 8.23 (s, 2H), 8.07 (dd, J = 8.1, 1.8 Hz, 1H), 8.00 (dd, J = 7.1, 2.5 Hz, 2H), 7.89 (d, J = 8.1 Hz, 1H), 7.62–7.52 (m, 3H), 3.80 (p, J = 8.3 Hz, 1H), 2.18–2.06 (m, 2H), 1.92–1.78 (m, 2H), 1.75–1.60 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.2, 162.98, 157.7, 155.2, 153.6, 146.6, 138.9, 134.4, 131.9, 130.6, 129.8, 129.5, 127.4, 126.8, 126.6, 125.4, 120.3, 41.3, 34.5, 25.3; HRMS calcd for C24H20N2O2S [M + H]+, 401.1324; found, 401.1329; mp range >250 °C.
2-Cyclopentyl-4-(thieno[3,2-d]pyrimidin-4-yl)benzoic Acid (38)
A 5 mL microwave vial was loaded with 4-chlorothieno[3,2-d]pyrimidine 35a (0.41 mmol, 70.0 mg), 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 3 (0.41 mmol, 130 mg), Cs2CO3 (0.82 mmol, 270 mg), and Pd(Ph3)4 (5 mol %, 25.0 mg). The solids were dissolved in a solution of 1,4-dioxane/water (3:1, 1.50 mL). The vial was sealed, and the reaction mixture was irradiated at 125 °C for 15 min. The volatiles were removed in vacuo, and the remaining solids were suspended with ice/water before the pH was adjusted to 2–3 with 2 M HCl(aq). The crude was purified via column chromatography (CH2Cl2/MeOH/HOAc, 95:4:1–90:9:1), affording 2-cyclopentyl-4-(thieno[3,2-d]pyrimidin-4-yl)benzoic acid 38 (70.0 mg, 48%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 13.24 (s, 1H), 9.31 (s, 1H), 8.60 (d, J = 5.5 Hz, 1H), 8.22 (d, J = 1.7 Hz, 1H), 8.04 (dd, J = 8.1, 1.7 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.76 (d, J = 5.5 Hz, 1H), 3.80 (p, J = 8.4, 7.9 Hz, 1H), 2.16–2.06 (m, 2H), 1.89–1.79 (m, 2H), 1.74–1.60 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.2, 162.1, 158.3, 154.7, 146.6, 139.0, 138.9, 134.4, 129.8, 127.6, 126.6, 125.5, 124.5, 41.2, 34.5, 25.4; HRMS calcd for C18H16N2O2S [M + H]+, 325.1011; found, 325.1011; mp range 205–209 °C.
Methyl 4-(6-Bromothieno[2,3-d]pyrimidin-4-yl)-2-cyclopentylbenzoate (40)
A 5 mL microwave vial was loaded with 6-bromo-4-chlorothieno[2,3-d]pyrimidine 39b (0.40 mmol, 100 mg), methyl 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (0.36 mmol, 120 mg), Cs2CO3 (0.60 mmol, 195 mg), and Pd(Ph3)4 (5 mol %, 23.0 mg). The solids were dissolved in a solution of 1,4-dioxane/water (3:1, 1.5 mL). The vial was sealed, and the reaction mixture was irradiated at 145 °C for 15 min. The volatiles were removed in vacuo, and the crude was purified via column chromatography (0–5% hexane/EtOAc) to afford methyl 4-(6-bromothieno[2,3-d]pyrimidin-4-yl)-2-cyclopentylbenzoate 40 (50.0 mg, 30%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.95 (s, 1H), 8.22 (s, 1H), 7.98 (d, J = 1.9 Hz, 1H), 7.83–7.76 (m, 2H), 3.86 (s, 3H), 3.63–3.57 (m, 1H), 2.08–2.00 (m, 2H), 1.90–1.81 (m, 2H), 1.75–1.63 (m, 4H).
2-Cyclopentyl-4-(6-phenylthieno[2,3-d]pyrimidin-4-yl)benzoic Acid (41)
Methyl 4-(6-bromothieno[2,3-d]pyrimidin-4-yl)-2-cyclopentylbenzoate 40 (0.11 mmol, 45.0 mg), phenylboronic acid (0.12 mmol, 15.0 mg), Cs2CO3 (0.38 mmol, 52.0 mg), and Pd(Ph3)4 (5 mol %, 6.00 mg) were dissolved in a solution of 1,4-dioxane/water (3:1, 2.00 mL). The reaction mixture was stirred at reflux for 3 h. The volatiles were removed in vacuo, and the crude was purified via column chromatography (0–10% EtOAc/hexane). The material was further purified using preparative TLC silica plates, affording methyl 2-cyclopentyl-4-(6-phenylthieno[2,3-d]pyrimidin-4-yl)benzoate (25 mg, 55%) as a white solid. The methyl ester was saponified as follows: methyl 2-cyclopentyl-4-(6-phenylthieno[2,3-d]pyrimidin-4-yl)benzoate (0.06 mmol, 25.0 mg) was dissolved in 1,4-dioxane (2.50 mL). LiOH (0.30 mmol, 7.00 mg) was dissolved in H2O (0.50 mL) and added to the solution. This mixture was stirred at 80 °C for 4 h. Solvents were removed in vacuo; the material was suspended in ice/water, and the pH was adjusted to 2–3 with 2 M HCl. The resulting solid was filtrated, thoroughly rinsed with water, and air- and vacuum-dried, affording 2-cyclopentyl-4-(6-phenylthieno[2,3-d]pyrimidin-4-yl)benzoic acid 41 (17.0 mg, 70%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 13.11 (s, 1H), 9.16 (s, 1H), 8.16 (s, 1H), 8.13–8.06 (m, 2H), 7.83 (dd, J = 4.3, 2.4 Hz, 2H), 7.78–7.74 (m, 1H), 7.68–7.63 (m, 3H), 3.68–3.62 (m, 1H), 2.10–1.98 (m, 2H), 1.87–1.79 (m, 2H), 1.70–1.62 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.1, 168.8, 159.9, 153.4, 146.8, 142.8, 136.9, 134.9, 130.7, 130.2, 129.4, 129.0, 128.9, 126.7, 124.9, 124.4, 117.7, 41.4, 34.2, 25.1; HRMS (HESI) calcd for C24H20N2O2S [M + H]+, 401.1324; found, 401.1330; mp range >250 °C.
2-Cyclopentyl-4-(thieno[2,3-d]pyrimidin-4-yl)benzoic Acid (42)
Prepared following the procedure for 38 using 4-chlorothieno[2,3-d]pyrimidine 35a (70.0 mg, 0.41 mmol) and 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 3 (130 mg, 0.41 mmol). After purification, 2-cyclopentyl-4-(thieno[2,3-d]pyrimidin-4-yl)benzoic acid 42 (72.0 mg, 55%) was afforded as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 9.19 (s, 1H), 8.09 (d, J = 6.1 Hz, 1H), 7.98 (d, J = 1.6 Hz, 1H), 7.87–7.79 (m, 2H), 7.68 (d, J = 6.1 Hz, 1H), 3.83–3.70 (m, 1H), 2.12–2.02 (m, 2H), 1.86–1.74 (m, 2H), 1.65–1.56 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.3, 169.3, 159.2, 153.1, 146.2, 139.4, 133.9, 129.5, 129.3, 127.4, 127.4, 126.4, 120.7, 41.3, 34.4, 25.3; HRMS (HESI) calcd for C18H16N2O2S [M + H]+, 325.1011; found, 325.1013; mp range 146–149.5 °C.
Methyl 4-(6-Chloroquinolin-4-yl)-2-cyclopentylbenzoate (44)
4,6-Dichloroquinoline (1.00 g, 5.05 mmol) and 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (1.58 g, 4.80 mmol) were dissolved in a 10:1 solution of dioxane/water (27.5 mL). Pd2(dba)3 (231 mg, 0.25 mmol), XPhos (240 mg, 0.51 mmol), and Cs2CO3 (4.94 g, 15.2 mmol) were added, and the mixture was stirred for 16 h at 25 °C. The solution was diluted with EtOAc (20.0 mL) and water (25.0 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 20.0 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (10–30% EtOAc/hexane) to afford methyl 4-(6-chloroquinolin-4-yl)-2-cyclopentylbenzoate 44 (1.54 g, 83%) as a white solid.
1H NMR (400 MHz, CDCl3): δ 8.96 (d, J = 4.4 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 2.3 Hz, 1H), 7.69 (dd, J = 9.0, 2.3 Hz, 1H), 7.51 (d, J = 1.6 Hz, 1H), 7.42–7.33 (m, 2H), 3.97 (s, 3H), 3.89–3.78 (m, 1H), 2.23–2.12 (m, 2H), 1.86–1.69 (m, 4H), 1.63 (ddd, J = 16.2, 12.9, 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 168.5, 145.0, 148.1, 147.2, 146.9, 140.4, 133.0, 131.5, 131.1, 130.5, 130.0, 128.1, 127.2, 126.4, 124.5, 121.8, 77.0, 52.3, 41.8, 35.9, 25.7; HRMS: calcd for C22H21ClNO2 [M + H]+m/z, 366.1260; found m/z, 366.1246; mp range 137–139 °C.
2-Cyclopentyl-4-(6-phenylquinolin-4-yl)benzoic Acid (45)
Methyl 4-(6-chloroquinolin-4-yl)-2-cyclopentylbenzoate 44 (100 mg, 0.27 mmol) and phenylboronic acid (66.0 mg, 0.32 mmol) were dissolved in a 10:1 solution of dioxane/water (12 mL). Pd2(dba)3 (25 mg, 0.03 mmol), XPhos (29.0 mg, 0.06 mmol), and Cs2CO3 (264 mg, 0.81 mmol) were added, and the solution was heated to 80 °C for 16 h. Once cooled, the solution was diluted with EtOAc (10 mL) and water (10 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 10 mL). The combined organics were dried (Na2SO4), filtered, and concentrated in vacuo. The crude was purified via column chromatography (10–30% EtOAc/hexane) to afford methyl 2-cyclopentyl-4-(6-phenylquinolin-4-yl)benzoate as an intermediate. The intermediate methyl ester was dissolved in dioxane (1 mL) and aq LiOH (1 mL of a 1 M solution). The solution was heated to 100 °C for 16 h and then acidified to pH 4 with 1 M aq HCl, causing a precipitate to form. The solid was collected by filtration, washed with cold water (4 mL) and Et2O (10 mL), and dried under vacuum to afford 2-cyclopentyl-4-(6-phenylquinolin-4-yl)benzoic acid 45 (81 mg, 76%) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 9.15 (d, J = 4.8 Hz, 1H), 8.39 (d, J = 8.6 Hz, 1H), 8.32 (d, J = 8.7 Hz, 1H), 8.13 (d, J = 1.7 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.84–7.79 (m, 1H), 7.74–7.69 (m, 3H), 7.59 (dd, J = 8.0, 1.6 Hz, 1H), 7.51 (t, J = 7.4 Hz, 2H), 7.47–7.41 (m, 1H), 3.88–3.72 (m, 1H), 2.14–2.01 (m, 2H), 1.84–1.71 (m, 2H), 1.71–1.50 (m, 4H); 13C NMR (176 MHz, DMSO): δ 169.3, 146.4, 145.2, 139.8, 138.9, 138.8, 132.8, 130.7, 129.6, 129.3, 129.2, 128.3, 128.1, 127.1, 126.8, 126.2, 123.0, 122.3, 119.4, 99.5, 41.2, 34.5, 25.3; HRMS: calcd for C27H24NO2 [M + H]+m/z, 394.1807; found m/z, 394.1786; mp range 164–167 °C.
2-Cyclopentyl-4-(quinolin-4-yl)benzoic Acid (46)
Prepared following the procedure for 21 using 4-chloroquinoline (100 mg, 611 μmol). After purification, 2-cyclopentyl-4-(quinolin-4-yl)benzoic acid 46 (l60 mg, 83%) was afforded as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 13.09 (s, 1H), 8.97 (d, J = 4.4 Hz, 1H), 8.14–8.11 (m, 1H), 7.8–7.78 (m, 3H), 7.62 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 7.56 (d, J = 1.7 Hz, 1H), 7.50 (d, J = 4.4 Hz, 1H), 7.44 (dd, J = 7.9, 1.7 Hz, 1H), 3.85–3.73 (m, 1H), 2.11–2.00 (m, 2H), 1.81–1.71 (m, 2H), 1.70–1.54 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.4, 150.2, 148.1, 146.8, 146.6, 140.0, 132.1, 129.6, 129.6, 129.5, 127.8, 127.2, 126.7, 125.7, 125.2, 121.5, 41.2, 34.5, 25.3; HRMS: calcd for C21H19NO2 [M – H]−m/z, 316.1338; found, 316.1339; mp range 259–263 °C.
Methyl 2-Cyclopentyl-4-(6-hydroxyquinazolin-4-yl)benzoate (48)
Prepared following the procedure for 32 using 4-chloroquinazolin-6-ol 47b (50.0 mg, 0.28 mmol). After purification, methyl 2-cyclopentyl-4-(6-hydroxyquinazolin-4-yl)benzoate 48 (63.0 mg, 65%) was afforded as a thick oil.
1H NMR (400 MHz, DMSO-d6): δ 9.42 (s, 1H), 8.16 (d, J = 9.0 Hz, 1H), 8.10 (dd, J = 9.0, 2.3 Hz, 1H), 7.99–7.98 (m, 1H), 7.88–7.84 (m, 3H), 7.71 (dd, J = 7.9, 1.7 Hz, 1H), 3.70–3.62 (m, 1H), 2.13–2.01 (m, 2H), 1.81–1.75 (m, 2H), 1.72–1.57 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 167.9, 166.2, 154.7, 149.0, 146.4, 138.9, 134.8, 132.6, 132.3, 130.8, 129.4, 128.4, 127.1, 125.2, 122.9, 52.4, 41.3, 34.4, 25.2.
2-Cyclopentyl-4-(6-phenylquinazolin-4-yl)benzoic Acid (49)
Methyl 2-cyclopentyl-4-(6-phenylquinazolin-4-yl)benzoate SI4 (170 mg, 0.41 mmol) was dissolved in MeOH (3.5 mL) and aq NaOH (1 mL). The resulting solution was heated to 70 °C for 1 h and allowed to cool to rt, added water (4 mL), and extracted with ether (2 × 10 mL). The aqueous phase was acidified with 1 N HCl (pH 6) and then extracted with EtOAc. The organic phase was dried (Na2CO3) and concentrated to afford 2-cyclopentyl-4-(6-phenylquinazolin-4-yl)benzoic acid 49 (150 mg, 91%) as a light-yellow solid.
1H NMR (400 MHz, DMSO-d6): δ 13.19 (s, 1H), 9.39 (s, 1H), 8.39 (dd, J = 8.8, 2.1 Hz, 1H), 8.24–8.19 (m, 2H), 7.92 (d, J = 1.7 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.78–7.73 (m, 3H), 7.51 (t, J = 7.4 Hz, 2H), 7.45 (d, J = 7.3 Hz, 1H), 3.86–3.75 (m, 1H), 2.12–2.04 (m, 2H), 1.80–1.74 (m, 2H), 1.69–1.60 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 169.4, 167.0, 154.4, 149.9, 146.1, 139.8, 139.0, 138.8, 133.5, 133.4, 129.3, 129.2, 129.2, 128.5, 128.3, 127.2, 127.1, 123.7, 122.5, 41.2, 34.5, 25.3; HRMS: calcd for C26H22N2O2 [M – H]−m/z, 393.1607; found, 393.1603; mp range 265–269 °C.
2-Cyclopentyl-4-(quinazolin-4-yl)benzoic Acid (50)
4-Chloroquinazoline (100 mg, 0.60 mmol), 2-cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 4 (240 mg, 0.72 mmol), Pd(PPh3)4 (70.0 mg, 60.0 μmol), and Cs2CO3 (0.60 g, 1.80 mmol) were loaded into a microwave vial. A solution of dioxane/water (6.0 mL of a 10:1 mix) was added, and the vial was flushed with nitrogen. The mixture was irradiated at 100 °C for 30 min. Once cooled, the aqueous layer was removed and the remaining volatiles were removed in vacuo. The crude was purified via column chromatography, and the intermediate ester was isolated. The ester was dissolved in dioxane (1 mL) and aq LiOH (1 mL of a 1 M solution). The solution was heated to 100 °C for 16 h and then acidified to pH 4 with 1 M aq HCl, causing a precipitate to form. The solid was collected by filtration, washed with cold H2O, and then dried under vacuum to afford the quinazoline (128 mg, 67%) as a pale-white solid.
1H NMR (400 MHz, DMSO-d6): δ 9.38 (s, 1H), 8.15–8.03 (m, 3H), 7.85 (d, J = 7.9 Hz, 1H), 7.78 (ddd, J = 9.7, 6.8, 1.4 Hz, 3H), 7.66 (dd, J = 7.9, 1.7 Hz, 1H), 3.83–3.73 (m, 1H), 2.11–2.02 (m, 2H), 1.81–1.71 (m, 2H), 1.70–1.57 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 169.3, 167.0, 154.3, 150.3, 146.0, 139.0, 134.4, 133.5, 129.1, 128.6, 128.4, 128.2, 127.08, 126.6, 122.3, 41.3, 34.4, 25.2; HRMS: calcd for C20H19N2O2 [M + H]+m/z, 319.1446; found, m/z 319.1427; mp range 120–122 °C.
2-Cyclopentyl-4-(2-methylquinolin-4-yl)benzoic Acid (52)
Prepared following the procedure for 21 using 4-chloro-2-methylquinoline 51 (200 mg, 1.13 mmol). After purification, 2-cyclopentyl-4-(2-methylquinolin-4-yl)benzoic acid 52 (290 mg, 77%) was afforded as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 13.08 (s, 1H), 8.01 (dd, J = 8.5, 1.5 Hz, 1H), 7.81 (d, J = 7.9 Hz, 1H), 7.78–7.72 (m, 2H), 7.56–7.52 (m, 2H), 7.43–7.39 (m, 2H), 3.86–3.74 (m, 1H), 2.70 (s, 3H), 2.12–2.00 (m, 2H), 1.78–1.74 (m, 2H), 1.67–1.56 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 172.6, 161.6, 151.0, 150.0, 149.3, 143.2, 135.4, 132.6, 132.5, 132.0, 130.8, 129.7, 129.3, 128.1, 127.2, 125.3, 44.4, 37.6, 28.4, 27.9; HRMS: calcd for C22H21NO2 [M – H]−m/z, 332.1651; found, 332.1642; mp range 271–275 °C.
2-Cyclopentyl-4-(1,6-naphthyridin-4-yl)benzoic Acid (54)
Prepared following the procedure for 21 using 4-chloro-1,6-naphthyridine 53 (80.0 mg, 0.49 mmol). After purification, 2-cyclopentyl-4-(1,6-naphthyridin-4-yl)benzoic acid 54 (110 mg, 54%) was afforded as a yellow solid.
1H NMR (400 MHz, DMSO-d6): δ 13.14 (s, 1H), 9.24–9.15 (m, 2H), 8.79 (d, J = 5.8 Hz, 1H), 8.01 (dd, J = 5.9, 0.9 Hz, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.71–7.65 (m, 2H), 7.54 (dd, J = 7.9, 1.8 Hz, 1H), 3.84–3.73 (m, 1H), 2.09–2.04 (m, 2H), 1.80–1.71 (m, 2H), 1.71–1.56 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.88, 155.32, 150.85, 150.45, 147.84, 146.88, 146.7, 138.4, 133.1, 129.9, 128.4, 127.3, 123.5, 122.3, 121.5, 41.6, 34.8, 25.6; HRMS calcd for C20H18N2O2 [M – H]−m/z, 319.1447; found, 319.1427; mp range 195–200 °C.
2-Cyclopentyl-4-(pyrimidin-4-yl)benzoic Acid (56)
4-Chloropyrimidine (162 mg, 1.40 mmol) and Cs2CO3 (1.32 g, 4.00 mmol) were dissolved in 1,2-DME (4.00 mL) and H2O (0.70 mL). Pd(PPh3)4 (78 mg, 68 μmol) and 3 (503 mg, 1.60 mmol) were added, and the solution was stirred at 120 °C for 30 min. The resulting suspension was filtered through Celite under vacuum. The filter cake was washed with ethyl acetate, and the filtrate was dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified by column chromatography [0–50% EtOAc(0.01% acetic acid)/hexane] to afford 2-cyclopentyl-4-(pyrimidin-4-yl)benzoic acid 56 (29 mg, 8%) as a white solid.
1H NMR (700 MHz, DMSO-d6): δ 13.14 (br s, 1H), 9.28 (s, 1H), 8.92–8.87 (m, 1H), 8.25 (s, 1H), 8.20–8.16 (m, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 3.72 (p, J = 8.0 Hz, 1H), 2.09–2.01 (m, 2H), 1.87–1.79 (m, 2H), 1.73–1.59 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.3, 158.8, 158.8, 158.3, 146.4, 138.3, 134.5, 129.7, 125.1, 124.2, 117.7, 41.4, 34.4, 25.3; HRMS: calcd for C16H15N2O2 [M – H]−m/z, 267.1138; found, 267.1139; mp range 245 °C.
2-Cyclopentyl-4-(2-phenylpyrimidin-4-yl)benzoic Acid (58)
4-Chloro-2-phenylpyrimidine (155 mg, 0.81 mmol) and Cs2CO3 (775 mg, 2.40 mmol) were dissolved in 1,2-DME (2.40 mL) and H2O (0.40 mL). Pd(PPh3)4 (48.0 mg, 42.0 μmol) and 3 (307 mg, 0.97 mmol) were added, and the solution was stirred at 120 °C for 30 min. The resulting suspension was filtered through Celite under vacuum. The filter cake was washed with ethyl acetate, and the filtrate was dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified by column chromatography [0–10% EtOAc(0.01% acetic acid)/hexane] to afford 2-cyclopentyl-4-(2-phenylpyrimidin-4-yl)benzoic acid 58 (21 mg, 8%) as a white solid.
1H NMR (700 MHz, DMSO-d6): δ 13.16 (br s, 1H), 9.04–8.95 (m, 1H), 8.52 (d, J = 6.0 Hz, 2H), 8.33 (s, 1H), 8.21 (d, J = 7.6 Hz, 1H), 8.15–8.07 (m, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.63–7.53 (m, 3H), 3.76 (p, J = 8.8 Hz, 1H), 2.12–2.03 (m, 2H), 1.90–1.82 (m, 2H), 1.75–1.65 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.4, 163.4, 162.3, 158.9, 146.4, 138.5, 137.2, 134.5, 131.0, 129.7, 128.8, 127.8, 125.3, 124.3, 115.7, 41.4, 34.4, 25.3; HRMS: calcd for C22H21N2O2 [M + H]+m/z, 345.1598; found, 345.1600; mp range >250 °C.
Methyl 4-(2-Aminopyrimidin-4-yl)-2-cyclopentylbenzoate (60)
4-Chloropyrimidin-2-amine (501 mg, 3.90 mmol) and Cs2CO3 (3.81 mg, 12.0 mmol) were dissolved in 1,2-DME (8.00 mL) and H2O (2.00 mL). Pd(PPh3)4 (238 mg, 0.21 mmol) and 4 (1.50 g, 4.50 mmol) were added, and the solution was irradiated at 120 °C for 30 min. The resulting suspension was filtered through Celite under vacuum. The filter cake was washed with ethyl acetate, and the filtrate was dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified by column chromatography (20% EtOAc/hexane) to afford methyl 4-(2-aminopyrimidin-4-yl)-2-cyclopentylbenzoate 60 (523 mg, 45%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.34 (d, J = 5.2 Hz, 1H), 8.13 (d, J = 1.7 Hz, 1H), 7.93 (dd, J = 8.1, 1.7 Hz, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.19 (d, J = 5.2 Hz, 1H), 6.74 (s, 2H), 3.85 (s, 3H), 3.69–3.54 (m, 1H), 2.11–1.96 (m, 2H), 1.90–1.75 (m, 2H), 1.73–1.55 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 168.00, 163.81, 162.68, 159.31, 146.28, 139.90, 132.37, 129.51, 124.83, 124.03, 106.29, 52.23, 41.48, 34.34, 25.20; mp range 178 °C.
4-(2-Aminopyrimidin-4-yl)-2-cyclopentylbenzoic Acid Hydrochloride (61·HCl)
Methyl 4-(2-aminopyrimidin-4-yl)-2-cyclopentylbenzoate 60 (40.0 mg, 0.13 mmol) was dissolved in 1,4-dioxane (1.00 mL). 1 M LiOH (1.00 mL) was added, and the solution was stirred at 100 °C for 17 h. HCl (6.00 M, 6.00 mL) was added, resulting in precipitate formation. The precipitate was filtered and washed with HCl (6.00 M). The precipitate was dried under reduced pressure to afford 4-(2-aminopyrimidin-4-yl)-2-cyclopentylbenzoic acid hydrochloride 61·HCl (35.0 mg, 85%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.54–8.14 (br s, 1H) 8.53 (d, J = 6.3 Hz, 1H), 8.22 (d, J = 1.8 Hz, 1H), 8.05 (dd, J = 8.2, 1.8 Hz, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.61 (d, J = 6.3 Hz, 1H), 3.73–3.60 (m, 1H), 2.10–1.98 (m, 2H), 1.90–1.76 (m, 2H), 1.73–1.57 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.2, 168.3, 157.1, 150.3, 146.1, 136.7, 136.0, 129.5, 126.1, 125.3, 106.5, 41.5, 34.4, 25.2; HRMS: calcd for C16H18N3O2 [M + H]+m/z, 284.1394; found, 284.1394; mp range >250 °C.
2-Cyclopentyl-4-(2-(phenylamino)pyrimidin-4-yl)benzoic Acid Hydrochloride (62·HCl)
Methyl 2-cyclopentyl-4-(2-(phenylamino)pyrimidin-4-yl)benzoate SI5 (20 mg, 54 μmol) was dissolved in 1,4-dioxane (1.0 mL). 1 M LiOH (1.0 mL) was added, and the solution was stirred for at 100 °C for 16 h. HCl (6.0 M, 6.0 mL) was added, resulting in precipitate formation. The precipitate was filtered and washed with HCl (6.0 M). The precipitate was dried under reduced pressure to afford 2-cyclopentyl-4-(2-(phenylamino)pyrimidin-4-yl)benzoic acid hydrochloride (62·HCl) (12 mg, 63%) as a yellow solid.
1H NMR (700 MHz, DMSO-d6): δ 13.10 (br s, 1H), 9.74 (s, 1H), 8.61–8.55 (m, 1H), 8.29 (s, 1H), 8.00 (d, J = 7.4 Hz, 1H), 7.86 (d, J = 7.3 Hz, 2H), 7.77 (d, J = 7.4 Hz, 1H), 7.50–7.46 (m, 1H), 7.33–7.27 (m, 2H), 7.01–6.96 (m, 1H), 3.77 (p, J = 9.6 Hz, 1H), 2.13–2.03 (m, 2H), 1.89–1.80 (m, 2H), 1.73–1.61 (m, 4H); 13C NMR (176 MHz, DMSO-d6): δ 169.3, 162.6, 160.1, 159.3, 146.4, 140.5, 139.0, 134.0, 129.6, 128.4, 125.1, 124.0, 121.5, 119.0, 108.3, 41.2, 34.5, 25.4; HRMS: calcd for C22H22N3O2 [M + H]+m/z, 360.4365; found, 360.1712; mp range >250 °C.
Acknowledgments
We are grateful for LC–MS/HRMS support provided by Dr. Brandie Ehrmann and Diane E. Wallace in the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill. The core is supported by the National Science Foundation under grant no. CHE-1726291.
Glossary
Abbreviations
- CAMKK2
calcium-calmodulin protein kinase kinase 2
- AMPK
AMP-activated protein kinase
- AKT
protein kinase B (PKB)
- siRNA
small interfering RNA
- HCC
hepatocellular carcinoma
- NAFLD
non-alcoholic fatty liver disease
- GRK3
G protein-coupled receptor kinase 3
- CK2
casein kinase 2
- CDKL2
cyclin-dependent kinase-like 3
- AhR
aryl hydrocarbon receptor
- ATP
adenosine triphosphate
- SEM
trimethylsilylethoxymethyl
- CDI
1,1-carbodiimidazole
- DME
dimethoxyethane
- DSF
differential scanning fluorimetry
- PoC
percentage of control
- ADMET
absorption, distribution, metabolism, excretion, and toxicology
- LE
ligand efficiency
- LLE
lipophilic ligand efficiency
- DMSO
dimethylsulfoxide
- BRET
bioluminescence resonance energy transfer
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c02274.
Author Contributions
⊥⊥ B.J.E., S.N.O., and L.T. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was funded in part by the National Cancer Institute of the National Institutes of Health (grant numbers R01CA218442 and R01CA184208) and was supported by the NIH Illuminating the Druggable Genome program (grant number 1U24DK116204-01), FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) (2013/50724-5 and 2014/50897-0), and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) (465651/2014-3). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, the Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome [106169/ZZ14/Z]. J.W.S. and J.S.O. are funded by National Health and Medical Research Council (NHMRC) grants GNT1138102 and GNT1145265, respectively. P.Z.R. received CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) postdoctoral fellowship (88887.136432/2017-00). A.d.S.S. is an FAPESP postdoctoral fellow (2019/14275-8). C.G.L. is an NHMRC early career fellow (GNT1143080). C.L. is supported by an Antje Wuelfrath Gee and Harry Gee, Jr. Family Legacy Scholarship. D.A. is supported by an American Legion Auxiliary Fellowship. Acknowledgements for beamlines/crystallography: this work is based upon research conducted at the Northeastern Collaborative Access Team beamlines (GU51510, GU56413), which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on the 24-ID-C beamline is funded by an NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. We thank Diamond Light Source for access to beam line I03 (proposal number MX10619-74).
The authors declare no competing financial interest.
Notes
PDB ID codes: 5UY6; 5UYJ. Authors will release the atomic coordinates upon article publication.
Supplementary Material
References
- Chin D.; Means A. R. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- Hook S. S.; Means A. R. Ca(2+)/CaM-dependent kinases: from activation to function. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 471–505. 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- Sharma R. K.; Parameswaran S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. 10.1016/j.ceca.2018.09.002. [DOI] [PubMed] [Google Scholar]
- Clapham D. E. Calcium signaling. Cell 2007, 131, 1047–1058. 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Racioppi L.; Means A. R. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. 10.1074/jbc.r112.356485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. A.; Means R. L.; Huang Q.-H.; Kemp B. E.; Goldstein E. G.; Selbert M. A.; Edelman A. M.; Fremeau R. T.; Means A. R. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J. Biol. Chem. 1998, 273, 31880–31889. 10.1074/jbc.273.48.31880. [DOI] [PubMed] [Google Scholar]
- Hawley S. A.; Pan D. A.; Mustard K. J.; Ross L.; Bain J.; Edelman A. M.; Frenguelli B. G.; Hardie D. G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Karacosta L. G.; Foster B. A.; Azabdaftari G.; Feliciano D. M.; Edelman A. M. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 2012, 287, 24832–24843. 10.1074/jbc.m112.370783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F.; Marcelo K. L.; Rajapakshe K.; Coarfa C.; Dean A.; Wilganowski N.; Robinson H.; Sevick E.; Bissig K.-D.; Goldie L. C.; Means A. R.; York B. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology 2015, 62, 505–520. 10.1002/hep.27832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.-M.; Wang H.-J.; Han B.; Meng X.-Q.; Chen M.-H.; Yang D.-B.; Sun Y.; Li Y.-L.; Jiang C.-L. CAMKK2, regulated by promoter methylation, is a prognostic marker in diffuse gliomas. CNS Neurosci. Ther. 2016, 22, 518–524. 10.1111/cns.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massie C. E.; Lynch A.; Ramos-Montoya A.; Boren J.; Stark R.; Fazli L.; Warren A.; Scott H.; Madhu B.; Sharma N.; Bon H.; Zecchini V.; Smith D.-M.; Denicola G. M.; Mathews N.; Osborne M.; Hadfield J.; Macarthur S.; Adryan B.; Lyons S. K.; Brindle K. M.; Griffiths J.; Gleave M. E.; Rennie P. S.; Neal D. E.; Mills I. G. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Mora O. G.; LaHair M. M.; McCubrey J. A.; Franklin R. A. Calcium/calmodulin-dependent kinase I and calcium/calmodulin-dependent kinase kinase participate in the control of cell cycle progression in MCF-7 human breast cancer cells. Cancer Res. 2005, 65, 5408–5416. 10.1158/0008-5472.can-05-0271. [DOI] [PubMed] [Google Scholar]
- Shima T.; Mizokami A.; Miyagi T.; Kawai K.; Izumi K.; Kumaki M.; Ofude M.; Zhang J.; Keller E. T.; Namiki M. Down-regulation of calcium/calmodulin-dependent protein kinase kinase 2 by androgen deprivation induces castration-resistant prostate cancer. Prostate 2012, 72, 1789–1801. 10.1002/pros.22533. [DOI] [PubMed] [Google Scholar]
- Subbannayya Y.; Syed N.; Barbhuiya M. A.; Raja R.; Marimuthu A.; Sahasrabuddhe N.; Pinto S. M.; Manda S. S.; Renuse S.; Manju H.; Zameer M. A. L.; Sharma J.; Brait M.; Srikumar K.; Roa J. C.; Vijaya Kumar M.; Kumar K. V.; Prasad T. K.; Ramaswamy G.; Kumar R. V.; Pandey A.; Gowda H.; Chatterjee A. Calcium calmodulin dependent kinase kinase 2 - a novel therapeutic target for gastric adenocarcinoma. Canc. Biol. Ther. 2015, 16, 336–345. 10.4161/15384047.2014.972264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S.; Tokumitsu H.; Soderling T. R. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 1998, 396, 584–587. 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- Spengler K.; Zibrova D.; Woods A.; Langendorf C. G.; Scott J. W.; Carling D.; Heller R. Protein kinase A negatively regulates VEGF-induced AMPK activation by phosphorylating CaMKK2 at serine 495. Biochem. J. 2020, 477, 3453–3469. 10.1042/bcj20200555. [DOI] [PubMed] [Google Scholar]
- Gocher A. M.; Azabdaftari G.; Euscher L. M.; Dai S.; Karacosta L. G.; Franke T. F.; Edelman A. M. Akt activation by Ca(2+)/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J. Biol. Chem. 2017, 292, 14188–14204. 10.1074/jbc.m117.778464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt J. M.; Abell E.; Wagner A.; Davare M. A. ERK activation and cell growth require CaM kinases in MCF-7 breast cancer cells. Mol. Cell. Biochem. 2010, 335, 155–171. 10.1007/s11010-009-0252-9. [DOI] [PubMed] [Google Scholar]
- Frigo D. E.; Howe M. K.; Wittmann B. M.; Brunner A. M.; Cushman I.; Wang Q.; Brown M.; Means A. R.; McDonnell D. P. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011, 71, 528–537. 10.1158/0008-5472.can-10-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H.; He H.-c.; Han Z.-d.; Wan Y.-p.; Luo H.-w.; Huang Y.-q.; Cai C.; Liang Y.-x.; Dai Q.-s.; Jiang F.-n.; Zhong W.-d. MicroRNA-224 and its target CAMKK2 synergistically influence tumor progression and patient prognosis in prostate cancer. Tumour Biol. 2015, 36, 1983–1991. 10.1007/s13277-014-2805-0. [DOI] [PubMed] [Google Scholar]
- White M. A.; Tsouko E.; Lin C.; Rajapakshe K.; Spencer J. M.; Wilkenfeld S. R.; Vakili S. S.; Pulliam T. L.; Awad D.; Nikolos F.; Katreddy R. R.; Kaipparettu B. A.; Sreekumar A.; Zhang X.; Cheung E.; Coarfa C.; Frigo D. E. GLUT12 promotes prostate cancer cell growth and is regulated by androgens and CaMKK2 signaling. Endocr. Relat. Canc. 2018, 25, 453–469. 10.1530/erc-17-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui K.; Hashimoto E.; Komorizono Y.; Koike K.; Arii S.; Imai Y.; Shima T.; Kanbara Y.; Saibara T.; Mori T.; Kawata S.; Uto H.; Takami S.; Sumida Y.; Takamura T.; Kawanaka M.; Okanoue T.; Japan Nash Study Group M. o. H. L.; Welfare of J. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433. 10.1016/j.cgh.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Takuma Y.; Nouso K. Nonalcoholic steatohepatitis-associated hepatocellular carcinoma: our case series and literature review. World J. Gastroenterol. 2010, 16, 1436–1441. 10.3748/wjg.v16.i12.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York B.; Li F.; Lin F.; Marcelo K. L.; Mao J.; Dean A.; Gonzales N.; Gooden D.; Maity S.; Coarfa C.; Putluri N.; Means A. R. Pharmacological inhibition of CaMKK2 with the selective antagonist STO-609 regresses NAFLD. Sci. Rep. 2017, 7, 11793. 10.1038/s41598-017-12139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price D. J.; Drewry D. H.; Schaller L. T.; Thompson B. D.; Reid P. R.; Maloney P. R.; Liang X.; Banker P.; Buckholz R. G.; Selley P. K.; McDonald O. B.; Smith J. L.; Shearer T. W.; Cox R. F.; Williams S. P.; Reid R. A.; Tacconi S.; Faggioni F.; Piubelli C.; Sartori I.; Tessari M.; Wang T. Y. An orally available, brain-penetrant CAMKK2 inhibitor reduces food intake in rodent model. Bioorg. Med. Chem. Lett. 2018, 28, 1958–1963. 10.1016/j.bmcl.2018.03.034. [DOI] [PubMed] [Google Scholar]
- Anderson K. A.; Ribar T. J.; Lin F.; Noeldner P. K.; Green M. F.; Muehlbauer M. J.; Witters L. A.; Kemp B. E.; Means A. R. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008, 7, 377–388. 10.1016/j.cmet.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Langendorf C. G.; O’Brien M. T.; Ngoei K. R. W.; McAloon L. M.; Dhagat U.; Hoque A.; Ling N. X. Y.; Dite T. A.; Galic S.; Loh K.; Parker M. W.; Oakhill J. S.; Kemp B. E.; Scott J. W. CaMKK2 is inactivated by cAMP-PKA signaling and 14-3-3 adaptor proteins. J. Biol. Chem. 2020, 295, 16239–16250. 10.1074/jbc.ra120.013756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary R. L.; Waddell S.; Racioppi L.; Long F.; Novack D. V.; Voor M. J.; Sankar U. Inhibition of Ca(2)(+)/calmodulin-dependent protein kinase kinase 2 stimulates osteoblast formation and inhibits osteoclast differentiation. J. Bone Miner. Res. 2013, 28, 1599–1610. 10.1002/jbmr.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard Z. J.; Cary R. L.; Yang C.; Novack D. V.; Voor M. J.; Sankar U. Inhibition of CaMKK2 reverses age-associated decline in bone mass. Bone 2015, 75, 120–127. 10.1016/j.bone.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F.; Ribar T. J.; Means A. R. The Ca2+/calmodulin-dependent protein kinase kinase, CaMKK2, inhibits preadipocyte differentiation. Endocrinology 2011, 152, 3668–3679. 10.1210/en.2011-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X.; Cui C.; Wang C.; Wu G.; Chen H.; Lu Z.; Chen X.; Wang L.; Huang J.; Geng H.; Zhao M.; Chen Z.; Müschen M.; Wang H.-Y.; Zhang C. C. CAMKs support development of acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 30. 10.1186/s13045-018-0574-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.; Wen D.; Wang X.; Yang L.; Liu T.; Liu J.; Zhu J.; Fang X. Growth inhibition of human gastric adenocarcinoma cells in vitro by STO-609 is independent of calcium/calmodulin-dependent protein kinase kinase-beta and adenosine monophosphate-activated protein kinase. Am. J. Transl. Res. 2016, 8, 1164–1171. [PMC free article] [PubMed] [Google Scholar]
- O’Byrne S. N.; Scott J. W.; Pilotte J. R.; Santiago A. D. S.; Langendorf C. G.; Oakhill J. S.; Eduful B. J.; Couñago R. M.; Wells C. I.; Zuercher W. J.; Willson T. M.; Drewry D. H. In depth analysis of kinase cross screening data to identify CAMKK2 inhibitory scaffolds. Molecules 2020, 25, 325. 10.3390/molecules25020325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Liu C.; Amato R. J.; Chang J. T.; Du G.; Li W. CDKL2 promotes epithelial-mesenchymal transition and breast cancer progression. Oncotarget 2014, 5, 10840–10853. 10.18632/oncotarget.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billard M. J.; Fitzhugh D. J.; Parker J. S.; Brozowski J. M.; McGinnis M. W.; Timoshchenko R. G.; Serafin D. S.; Lininger R.; Klauber-Demore N.; Sahagian G.; Truong Y. K.; Sassano M. F.; Serody J. S.; Tarrant T. K. G protein coupled receptor kinase 3 regulates breast cancer migration, invasion, and metastasis. PLoS One 2016, 11, e0152856 10.1371/journal.pone.0152856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landesman-Bollag E.; Romieu-Mourez R.; Song D. H.; Sonenshein G. E.; Cardiff R. D.; Seldin D. C. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001, 20, 3247–3257. 10.1038/sj.onc.1204411. [DOI] [PubMed] [Google Scholar]
- Monteiro P.; Gilot D.; Langouet S.; Fardel O. Activation of the aryl hydrocarbon receptor by the calcium/calmodulin-dependent protein kinase kinase inhibitor 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid (STO-609). Drug Metab. Dispos. 2008, 36, 2556–2563. 10.1124/dmd.108.023333. [DOI] [PubMed] [Google Scholar]
- Asquith C. R. M.; Godoi H. P.; Couñago M. R.; Laitinen T.; Scott W. J.; Langendorf G. C.; Oakhill S. J.; Drewry H. D.; Zuercher J. W.; Koutentis A. P.; Willson M. T.; Kalogirou S. A. 1,2,6-Thiadiazinones as novel narrow spectrum calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) inhibitors. Molecules 2018, 23, 1221. 10.3390/molecules23051221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose A. K.; Herbertz T.; Pippin D. A.; Salvino J. M.; Mallamo J. P. Knowledge based prediction of ligand binding modes and rational inhibitor design for kinase drug discovery. J. Med. Chem. 2008, 51, 5149–5171. 10.1021/jm800475y. [DOI] [PubMed] [Google Scholar]
- Aronov A. M.; Murcko M. A. Toward a pharmacophore for kinase frequent hitters. J. Med. Chem. 2004, 47, 5616–5619. 10.1021/jm049793g. [DOI] [PubMed] [Google Scholar]
- Xing L.; Klug-Mcleod J.; Rai B.; Lunney E. A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. 10.1016/j.bmc.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Irie T.; Sawa M. 7-Azaindole: A versatile scaffold for developing kinase inhibitors. Chem. Pharm. Bull. 2018, 66, 29–36. 10.1248/cpb.c17-00380. [DOI] [PubMed] [Google Scholar]
- Sherk A. B.; Frigo D. E.; Schnackenberg C. G.; Bray J. D.; Laping N. J.; Trizna W.; Hammond M.; Patterson J. R.; Thompson S. K.; Kazmin D.; Norris J. D.; McDonnell D. P. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008, 68, 7475–7483. 10.1158/0008-5472.can-08-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama T.; Murata M.; Miyaura N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: a direct procedure for arylboronic esters. J. Org. Chem. 1995, 60, 7508–7510. 10.1021/jo00128a024. [DOI] [Google Scholar]
- Merour J.-Y.; Joseph B. Synthesis and reactivity of 7-azaindoles (1H-pyrrolo(2,3-b)pyridine). Curr. Org. Chem. 2001, 5, 471–506. 10.2174/1385272013375427. [DOI] [Google Scholar]
- Morita H.; Shiotani S. Furopyridines. VI. Preparation and reactions of 2- and 3-substituted furo[2,3-b]pyridines. J. Heterocycl. Chem. 1986, 23, 1465–1469. 10.1002/jhet.5570230542. [DOI] [Google Scholar]
- Taylor S. L.; Martin J. C. Trifluoromethyl triflate: synthesis and reactions. J. Org. Chem. 1987, 52, 4147–4156. 10.1021/jo00228a001. [DOI] [Google Scholar]
- Fedorov O.; Niesen F. H.; Knapp S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. 10.1007/978-1-61779-337-0_7. [DOI] [PubMed] [Google Scholar]
- Santiago A. D. S.; Couñago R. M.; Ramos P. Z.; Godoi P. H. C.; Massirer K. B.; Gileadi O.; Elkins J. M. Structural analysis of inhibitor binding to CAMKK1 Identifies Features Necessary for Design of Specific Inhibitors. Sci. Rep. 2018, 8, 14800. 10.1038/s41598-018-33043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Profeta G. S.; Dos Reis C. V.; Santiago A. D. S.; Godoi P. H. C.; Fala A. M.; Wells C. I.; Sartori R.; Salmazo A. P. T.; Ramos P. Z.; Massirer K. B.; Elkins J. M.; Drewry D. H.; Gileadi O.; Couñago R. M. Binding and structural analyses of potent inhibitors of the human Ca(2+)/calmodulin dependent protein kinase kinase 2 (CAMKK2) identified from a collection of commercially-available kinase inhibitors. Sci. Rep. 2019, 9, 16452. 10.1038/s41598-019-52795-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. A Survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Gong Z.; Li J.; Lu T. Intermolecular sulfur...oxygen interactions: theoretical and statistical investigations. J. Chem. Inf. Model. 2015, 55, 2138–2153. 10.1021/acs.jcim.5b00177. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H.; Inuzuka H.; Ishikawa Y.; Ikeda M.; Saji I.; Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 2002, 277, 15813–15818. 10.1074/jbc.m201075200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H.; Inuzuka H.; Ishikawa Y.; Kobayashi R. A single amino acid difference between alpha and beta Ca2+/calmodulin-dependent protein kinase kinase dictates sensitivity to the specific inhibitor, STO-609. J. Biol. Chem. 2003, 278, 10908–10913. 10.1074/jbc.m213183200. [DOI] [PubMed] [Google Scholar]
- Alsenz J.; Kansy M. High throughput solubility measurement in drug discovery and development. Adv. Drug Deliv. Rev. 2007, 59, 546–567. 10.1016/j.addr.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Hill A. P.; Young R. J. Getting physical in drug discovery: a contemporary perspective on solubility and hydrophobicity. Drug Discov. Today 2010, 15, 648–655. 10.1016/j.drudis.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/s0169-409x(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Mignani S.; Rodrigues J.; Tomas H.; Jalal R.; Singh P. P.; Majoral J.-P.; Vishwakarma R. A. Present drug-likeness filters in medicinal chemistry during the hit and lead optimization process: how far can they be simplified?. Drug Discov. Today 2018, 23, 605–615. 10.1016/j.drudis.2018.01.010. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Saal C.; Petereit A. C. Optimizing solubility: kinetic versus thermodynamic solubility temptations and risks. Eur. J. Pharm. Sci. 2012, 47, 589–595. 10.1016/j.ejps.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Vasta J. D.; Corona C. R.; Wilkinson J.; Zimprich C. A.; Hartnett J. R.; Ingold M. R.; Zimmerman K.; Machleidt T.; Kirkland T. A.; Huwiler K. G.; Ohana R. F.; Slater M.; Otto P.; Cong M.; Wells C. I.; Berger B.-T.; Hanke T.; Glas C.; Ding K.; Drewry D. H.; Huber K. V. M.; Willson T. M.; Knapp S.; Müller S.; Meisenheimer P. L.; Fan F.; Wood K. V.; Robers M. B. Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem. Biol. 2018, 25, 206–214. 10.1016/j.chembiol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. F.; Anderson K. A.; Means A. R. Characterization of the CaMKKbeta-AMPK signaling complex. Cell. Signal. 2011, 23, 2005–2012. 10.1016/j.cellsig.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Xy Ling N.; Langendorf C. G.; Hoque A.; Galic S.; Loh K.; Kemp B. E.; Gundlach A. L.; Oakhill J. S.; Scott J. W. Functional analysis of an R311C variant of Ca(2+) -calmodulin-dependent protein kinase kinase-2 (CaMKK2) found as a de novo mutation in a patient with bipolar disorder. Bipolar Disord. 2020, 22, 841–848. 10.1111/bdi.12901. [DOI] [PubMed] [Google Scholar]
- Newman J. Novel buffer systems for macromolecular crystallization. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 610–612. 10.1107/s0907444903029640. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukimoto-Niino M.; Yoshikawa S.; Takagi T.; Ohsawa N.; Tomabechi Y.; Terada T.; Shirouzu M.; Suzuki A.; Lee S.; Yamauchi T.; Okada-Iwabu M.; Iwabu M.; Kadowaki T.; Minokoshi Y.; Yokoyama S. Crystal structure of the Ca(2)(+)/calmodulin-dependent protein kinase kinase in complex with the inhibitor STO-609. J. Biol. Chem. 2011, 286, 22570–22579. 10.1074/jbc.m111.251710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/s0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/s0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.