

Abstract
Obstructive sleep apnea (OSA) may lead to increased circulating concentrations of inflammatory biomarkers and treatment may change these. We aimed to assess the effect of oral appliance (OA) therapy on inflammatory biomarkers in a randomised controlled pilot trial. A total of 71 patients with OSA and systemic hypertension were randomly allocated to an active, mandible protruded (OAa) or a passive, mandible non‐protruded device (OAp) treatment. Serum concentrations of the inflammatory biomarkers white blood cells, high‐sensitivity C‐reactive protein, interleukin 6, interleukin 10, and tumour necrosis factor‐α were measured at baseline and after 3 months of OA treatment. The differences between treatment groups in biomarker concentration change during the treatment were presented as the Vargha and Delaney effect size and evaluated with the Wilcoxon–Mann–Whitney test. This effect size expresses the probability of a higher value in a random participant from one group compared with a random patient from the other group, and a value of 0.5 means stochastically equal groups. After 3 months of treatment, there was a significant reduction of the apnea–hypopnea index in the OAa group compared with the OAp group (effect size 0.258, 95% confidence interval 0.146–0.386, p < .001). There were no significant differences between the groups in any of the inflammatory markers’ concentration changes during the treatment period (effect sizes between 0.488 and 0.524; all p values ≥.737). Thus, OA treatment for 3 months did not affect circulating concentrations of some common inflammatory markers in patients with OSA and systemic hypertension.
Keywords: C‐reactive protein, inflammation, interleukins, mandibular advancement device, obstructive sleep apnea
1. INTRODUCTION
Obstructive sleep apnea (OSA) is widespread in the general population, with a prevalence reaching approximately 3%–7% in men and 2%–5% in women (Punjabi, 2008). Untreated OSA increases the risk of cardiovascular diseases (CVDs), including arterial hypertension, ischaemic heart disease, and ischaemic stroke (Gottlieb et al., 2010). The relationships between OSA and hypertension, several CVDs, and death are well known (Bradley & Floras, 2009).
Inflammation is an essential player in the development of atherosclerosis and subsequent clinical cardiovascular manifestations (Libby, 2012). Several circulating inflammatory markers, including white blood cell count (WBCC), C‐reactive protein (CRP), interleukins, and tumour necrosis‐α (TNF‐α), have been associated with the development and prognostic impact of CVD (Koenig et al., 2004; Pai et al., 2004; Sabatine et al., 2002; Swerdlow et al., 2012).
Although not fully known, inflammation may be a mediator in the association between OSA and atherosclerotic disease. Increased circulating concentrations of pro‐inflammatory molecules such as CRP, interleukin 6 (IL‐6), and TNF‐α, and decreased concentrations of the anti‐inflammatory marker IL‐10 have been demonstrated in patients with OSA (Li et al., 2009; Nadeem et al., 2013).
Although ambiguous, a majority of studies with a randomised controlled trial (RCT) approach have failed to show an effect on circulating inflammatory marker levels with continuous positive airway pressure (CPAP) in patients with OSA (Jullian‐Desayes et al., 2015). Research on the effect of oral appliance (OA) therapy on circulating inflammatory markers is scarce. Conflicting findings have been reported in observational studies with small sample sizes (Niżankowska‐Jędrzejczyk et al., 2014; Yalamanchali et al., 2015). In an RCT, Recoquillon et al. allocated patients with severe OSA to receive 2 months of treatment with either an effective OA or a sham device (Recoquillon et al., 2019). At follow‐up, there were no detectable net effects on the circulating levels of CRP, IL‐6, or TNF‐α.
In the present study, we aimed to extend previous research by assessing the effects of OA treatment for 3 months on the inflammatory biomarkers WBCC, high‐sensitivity CRP (hsCRP), IL‐6, IL‐10, and TNF‐α in patients with OSA and arterial hypertension.
2. METHODS
2.1. Patients
The present study population was based on an unregistered RCT, where 72 patients with OSA and concomitant systemic hypertension were randomised to treatment with a mandibular protruded (active OA, OAa) or a non‐protruded (passive OA, OAp) OA for 3 months to evaluate the effect on blood pressure (Andrén et al., 2013). The patients were consecutively recruited from the Department of Clinical Physiology, Västmanland County Hospital, Västerås, Sweden, to where they had been referred for an ambulatory somnographic recording.
Patients were eligible for the trial if they had a verified OSA diagnosis defined as an apnea–hypopnea index (AHI) of ≥10 events/hr, systemic hypertension defined as office systolic blood pressure (BP) of >140 mmHg or diastolic BP of >90 mmHg on two separate occasions, and were not currently being treated with an OA or CPAP. Patients also had to possess enough teeth for the retention of an OA. Exclusion criteria included office systolic BP of >180 mmHg or diastolic BP of >110 mmHg, body mass index (BMI) of >35 kg/m2, atrial fibrillation, chronic obstructive lung disease, epilepsy, severe psychiatric disease, maximal protrusion capacity of the mandible of <6 mm, and an inability to speak or understand the national language.
Among 1,623 consecutive patients screened (Figure 1), 137 (8.4%) fulfilled the inclusion criteria, amongst whom 65 were excluded because of an office BP of >180/110 mmHg (five patients), severe psychiatric disease (one), and declining participation (59). The remaining 72 patients were randomly allocated to treatment with OAa or OAp. Two patients in the OAa group did not use their appliance, but attended follow‐up and were analysed as members of the OAa group according to the intention‐to‐treat approach. One patient in the OAp group withdrew before follow‐up, leaving 71 patients for the present analyses.
FIGURE 1.
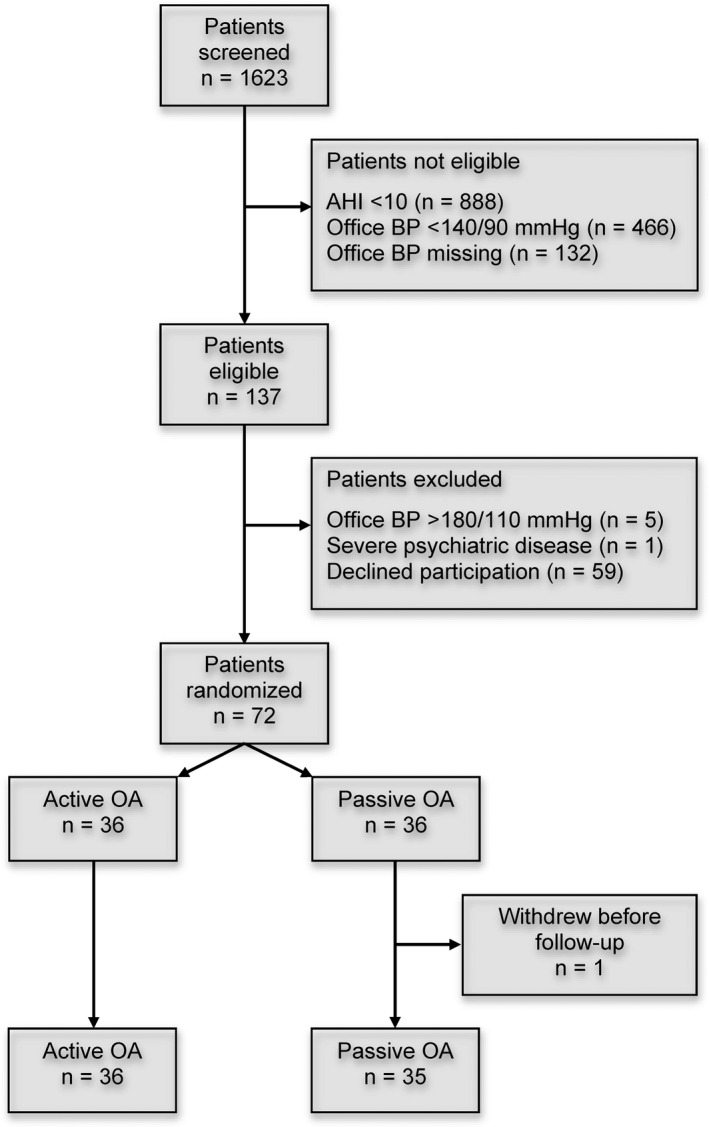
Participant flowchart. AHI, apnea–hypopnea index; BP, blood pressure; OA, oral appliance
All patients gave their written informed consent in accordance with the Helsinki declaration and were also asked not to alter their medication during the 3‐month study period. The Regional Ethical Review Board in Uppsala, Sweden, approved the study (Dnr 2005:227).
2.2. Study protocol
After inclusion, the patients were allocated to intervention by randomisation made in blocks of four. Sequence allocation was determined by a random number generator. At baseline, all patients underwent an ambulatory nocturnal somnographic recording, blood sampling, and completed a questionnaire concerning their general medical condition. The Epworth Sleepiness Scale (ESS) was used to assess daytime sleepiness (Johns, 1991). After 3 months of OA treatment, the patients underwent a repeated ambulatory somnographic recording and blood sampling.
2.3. Intervention
The treatment was performed with two types of monobloc OA. The OAa with mandibular advancement was custom‐made, as previously described by Tegelberg et al. (1999) (Figure 2). The OAa protruded the mandible to 70%–75% of the patient’s maximum mandibular protrusive capacity (>4 mm). The OAp possessed the same feature as the active appliance, except for the lack of mandibular advancement (<0.5 mm). All treatment and oral measurements were performed by the same dentist, not involved in the outcome assessments. Compliance with the treatment was assessed as the self‐reported use of the OA in the number of nights per week during the last month of follow‐up.
FIGURE 2.
Illustration of the oral appliance with monobloc design used in the study
2.4. Sleep analyses
Ambulatory somnographic recordings were made with a portable digital recording unit with sensors for the registration of airflow, saturation, respiratory movements of the chest, body position, and snoring sounds (Embletta PDS device, Medcare Flaga, Iceland). The recordings were undertaken in the patients’ home, transmitted to a computer, and analysed manually by one experienced biomedical analyst blinded to the intervention type. At the 3‐month follow‐up, the patients slept with the OA in situ during registration.
Apnea was defined as a cessation of airflow for ≥10 s. Hypopnea was defined as a reduction in airflow of ≥50% for ≥10 s with an accompanying desaturation of ≥4%. The number of apneas and hypopneas per hour of sleep was calculated to obtain the AHI. Mild, moderate, and severe OSA was defined as an AHI of ≤15, 15–29, and ≥30 events/hr, respectively (Flemons et al., 1999). The oxygen desaturation index (ODI) was defined as the number of desaturations of ≤4% per hour of sleep.
2.5. Inflammatory biomarkers
Peripheral venous blood samples were collected from each patient on two occasions: before treatment start and after 3 months of treatment at a similar time in the morning. All collected samples were stored at −70°C until analysis. The WBCC and serum concentrations of hsCRP, IL‐6, IL‐10, and TNF‐α were included in the analysis. The biomedical analysts were blinded to the choice of treatment modality, and all samples were analysed with the same batches at Clinical Chemistry, Karolinska University Hospital, Stockholm, Sweden.
2.6. Outcomes
The outcomes were the differences between the OAa and OAp groups in the change of inflammatory biomarker concentrations from baseline to the 3‐month follow‐up.
2.7. Statistical analysis
We present continuous variables as median (interquartile range, IQR [25th, 75th percentiles]) and categorical variables as number and percentage. We used the Wilcoxon–Mann–Whitney test to evaluate differences between groups for continuous variables and Pearson’s chi‐square or Fisher’s exact test for categorical variables. We used the Spearman rank‐order correlation (rho) to evaluate correlations between variables.
Due to skewed or multimodal distributions, the outcomes were evaluated with nonparametric methods. We calculated effect size according to Vargha and Delaney for the outcomes (Vargha & Delaney, 2000). This effect size is a standardised quantification of the difference between groups. It presents the probability that a value from a random patient in one group is greater than a value from a random patient in the other group. A value of 0.5 suggests that the two groups are stochastically equal. A value of 1 indicates a complete stochastic dominance of the first group, whereas a value of 0 indicates complete dominance of the second group. Vargha and Delaney suggested an effect size of 0.45–0.55 as a negligible effect, 0.56–0.63 (or 0.35–0.44) as a small effect, 0.64–0.70 (or 0.30–0.34) as a medium effect, and >0.70 (or <0.30) as a large effect (Vargha & Delaney, 2000). We also calculated the smallest total sample size required to detect a statistically significant difference (α = 0.05) at the found effect size and a power of 0.8 (Noether, 1987).
In an additional analysis, we compared the changes in inflammatory markers according to the AHI response during treatment. In this analysis, responders and non‐responders were defined as patients demonstrating a reduction in the AHI of >50% and ≤50%, respectively, during the 3‐month treatment period. The statistical analyses were performed using R version 4.0.2 with the packages compareGroups, effsize, and WMWssp (R Core Team, 2020). Statistical significance was set to p < .05.
3. RESULTS
The baseline characteristics were similar in the two treatment groups (Table 1). There were no significant differences between the treatment groups in the AHI, ODI, ESS, or circulating concentrations of the measured inflammatory markers at baseline. When evaluated in all 71 patients, there were no significant correlations between indices of OSA severity (AHI or ODI) and inflammatory marker concentrations at baseline. The strongest positive correlation was shown between the ODI and hsCRP (rho 0.067, p = .578) and the strongest negative correlation between the ODI and IL‐6 (rho −0.190, p = .119).
TABLE 1.
Baseline characteristics of the included patients according to treatment allocation
| Characteristic | All patients, n = 71 | Active OA, n = 36 | Passive OA, n = 35 | p a |
|---|---|---|---|---|
| Age, years, median (IQR) | 59 (52, 64) | 60 (53, 63) | 59 (52, 66) | .407 |
| Male sex, n (%) | 56 (78.9) | 30 (83.3) | 26 (74.3) | .520 |
| Body mass index, kg/m2, median (IQR) | 28.6 (26.4, 32.4) | 28.8 (26.8, 31.8) | 28.1 (25.7, 32.4) | .597 |
| Current smoker, n (%) | 13 (18.3) | 7 (19.4) | 6 (17.1) | 1.00 |
| Diabetes, n (%) | 1 (1.4) | 1 (2.8) | 0 (0.0) | 1.00 |
| Systolic BP, mmHg, median (IQR) | 150 (138, 160) | 144 (136, 158) | 155 (140, 167) | .137 |
| Diastolic BP, mmHg, median (IQR) | 90 (85, 95) | 90 (85, 95) | 90 (85, 95) | .917 |
| Cholesterol, mmol/L, median (IQR) | 5.7 (5.1, 6.5) | 5.7 (5.1, 6.2) | 5.9 (5.4, 6.7) | .269 |
| Triglycerides, mmol/L, median (IQR) | 1.3 (1.0, 2.0) | 1.4 (0.9, 2.2) | 1.3 (1.0, 1.8) | .833 |
| ESS, median (IQR) | 11 (8, 14) | 10 (8, 17) | 11 (8, 14) | .982 |
| AHI at baseline, events/hr, median (IQR) | 19 (14, 28) | 19 (15, 27) | 19 (12, 30) | .982 |
| OSA grade, n (%) | ||||
| Mild OSA (AHI <15 events/hr) | 20 (28.2) | 9 (25.0) | 11 (31.4) | .274 |
| Moderate OSA (AHI 15–29 events/hr) | 35 (49.3) | 21 (58.3) | 14 (40.0) | |
| Severe OSA (AHI ≥30 events/hr) | 16 (22.5) | 6 (16.7) | 10 (28.6) | |
| ODI at baseline, median (IQR) | 16 (10, 24) | 18 (11, 22) | 15 (10, 26) | .849 |
| Medication, n (%) | ||||
| ACE‐I or ARB | 12 (16.9) | 5 (13.9) | 7 (20.0) | .711 |
| β‐blocker | 10 (14.1) | 4 (11.1) | 6 (17.1) | .514 |
| Ca‐I | 4 (5.6) | 1 (2.8) | 3 (8.6) | .357 |
| Diuretic | 6 (8.5) | 3 (8.3) | 3 (8.6) | 1.00 |
| Biomarkers, median (IQR) | ||||
| WBCC, ×109/L | 6.2 (5.4, 7.3) | 6.1 (5.4, 7.0) | 6.3 (5.4, 7.4) | .968 |
| hsCRP, mg/L | 1.9 (0.9, 3.8) | 2.0 (1.0, 4.0) | 1.6 (0.8, 3.8) | .617 |
| IL‐6, ng/L | 1.01 (0.71, 1.77) | 1.00 (0.70, 1.84) | 1.04 (0.79, 1.48) | 0.966 |
| IL‐10, ng/L | 0.95 (0.68, 1.53) | 1.04 (0.83, 1.54) | 0.91 (0.58, 1.48) | .180 |
| TNF‐α, ng/L | 1.19 (1.00, 1.49) | 1.25 (1.10, 1.50) | 1.10 (0.98, 1.41) | .180 |
ACE‐I, angiotensin‐converting enzyme inhibitor; AHI, apnea–hypopnea index; ARB, angiotensin receptor blocker; BP, blood pressure; Ca‐I, calcium inhibitor; ESS, Epworth Sleepiness Score; hsCRP, high‐sensitivity C‐reactive protein; IL, interleukin; IQR, interquartile range (25th, 75th percentiles); OA, oral appliance; ODI, oxygen desaturation index; OSA, obstructive sleep apnea; TNF, tumour necrosis factor; WBCC, white blood cell count
Baseline values for IL‐6, IL‐10, and TNF‐α were missing in two, two, and one patients, respectively, in the passive group
p value for the difference between active and passive groups
After 3 months of treatment, there was a large reduction in the AHI in the OAa group compared with the OAp group (effect size 0.258, 95% CI 0.153–0.390, p < .001; Table 2). To have an AHI of <10 events/hr at follow‐up was significantly more common in the OAa group than in the OAp group (78% versus 29%, p < .001) and to be a responder (i.e. reduction of the AHI by >50% compared with baseline) was significantly more common in the OAa group (69% versus 34%; p = .006).
TABLE 2.
Changes in the apnea–hypopnea index, white blood cell count, and serum concentration of inflammatory biomarkers after 3 months of oral appliance (OA) treatment in the active and passive groups
| Active OA, median (IQR) | Passive OA, median (IQR) | Effect size (95% CI)a | p b | Required sample sizec | |
|---|---|---|---|---|---|
| AHI | −12 (−20, −8) | −2 (−12, 6) | 0.258 (0.153–0.390) | <.001 | 46 |
| ODI | −9.5 (−16.2, −5) | −1 (−10, 6) | 0.261 (0.155–0.396) | <.001 | 46 |
| WBCC | −0.40 (−0.62, 0.28) | −0.20 (−0.75, 0.30) | 0.502 (0.364–0.647) | .982 | 1,038,408 |
| hsCRP | −0.02 (−0.52, 0.26) | −0.01 (−0.69, 0.24) | 0.497 (0.353–0.634) | .963 | 259,602 |
| IL‐6 | 0.02 (−0.24, 0.41) | −0.06 (−0.37, 0.49) | 0.524 (0.370–0.663) | .737 | 4,710 |
| IL‐10 | 0.09 (−0.43, 0.58) | 0.08 (−0.39, 0.52) | 0.499 (0.356–0.642) | .989 | 2,738,028 |
| TNF‐α | 0.08 (−0.17, 0.23) | 0.04 (−0.13, 0.26) | 0.488 (0.356–0.626) | .865 | 18,644 |
AHI, apnea–hypopnea index; hsCRP, high‐sensitivity C‐reactive protein; IL, interleukin; IQR, interquartile range (25th, 75th percentiles); OA, oral appliance; TNF, tumour necrosis factor; WBCC, white blood cell count
Values for IL‐6, IL‐10, and TNF‐α were missing in two, four, and one patients, respectively, in the passive group; and for IL‐10 in three patients in the active group
Effect size according to Vargha and Delaney (see Methods for details)
p value for the difference between active and passive groups in AHI and biomarker level change during treatment.
Required total sample size to detect a statistically significant difference based on the found effect size, power of 80%, and α = 0.05
There was a significant reduction in the ODI in the OAa group compared with the OAp group (effect size 0.261, 95% CI 0.155–0.396, p < .001).
There was no significant difference between the OAa and OAp groups regarding the self‐reported number of nights use of the OA during the last month of treatment (median [IQR] 7.0 [6.0, 7.0] versus 7.0 [5.0, 7.0], effect size [95% CI] 0.562 [0.451–0.679], p = .311).
There were no significant differences between the treatment groups regarding the changes in WBCC and serum concentrations of the other inflammatory markers during the treatment period (Table 2). Effect sizes of ~0.5 were seen for all the inflammatory markers indicating negligible effects of the 3‐month OA therapy on the measured markers.
When outcomes were re‐analysed with respect to the AHI response status, there were no significant differences between responders and non‐responders in any of the biomarkers (Table 3). However, there was a trend towards a net reduction in TNF‐α concentrations in the responder group (p = .057).
TABLE 3.
Changes in white blood cell count and serum concentration of inflammatory biomarkers after 3 months of oral appliance (OA) treatment in the responders and non‐responders. Responders and non‐responders were defined as patients with an AHI reduction of >50% and ≤50%, respectively, after 3 months of OA treatment
| Responders, median (IQR) | Non‐responders, median (IQR) | Effect size (95% CI)a | p b | |
|---|---|---|---|---|
| WBCC | −0.20 (−0.50, 0.50) | −0.35 (−0.78, 0.08) | 0.587 (0.451–0.727) | .207 |
| hsCRP | −0.07 (−0.80, 0.10) | 0.00 (−0.53, 0.40) | 0.433 (0.298–0.568) | .333 |
| IL‐6 | −0.09 (−0.39, 0.48) | 0.10 (−0.20, 0.46) | 0.410 (0.280–0.557) | .202 |
| IL‐10 | 0.23 (−0.43, 0.66) | −0.02 (−0.39, 0.36) | 0.546 (0.408–0.680) | .528 |
| TNF‐α | −0.03 (−0.18, 0.19) | 0.13 (−0.07, 0.29) | 0.368 (0.243–0.519) | .057 |
AHI, apnea–hypopnea index; hsCRP, high‐sensitivity C‐reactive protein; IL, interleukin; IQR, interquartile range (25th, 75th percentiles); TNF, tumour necrosis factor; WBCC, white blood cell count
Values for IL‐6, IL‐10, and TNF‐α were missing in two, three, and one patients, respectively, in the non‐responder group; and for IL‐10 in four patients in the responder group
Effect size, according to Vargha and Delaney (see Methods for details)
p value for the difference between responders and non‐responders in biomarker level change during treatment
4. DISCUSSION
Our present findings suggest that 3 months of treatment with an OA has no effect on the circulating concentrations of WBCs, hsCRP, IL‐6, IL‐10, and TNF‐α, despite a significant improvement in the AHI and ODI in hypertensive patients with OSA.
The development of atherosclerosis and its manifestation in CVD is closely associated with inflammatory processes (Libby, 2012). One potential mechanism of CVD in OSA may be low‐grade inflammation initiated by the episodes of intermittent hypoxia seen in OSA. Animal studies have shown intermittent hypoxia to be a strong stimulus to systemic and vascular inflammation, leading to aggravation in atherosclerotic changes (Arnaud et al., 2011). Further, normoxic breathing can revert structural vascular remodelling induced by a period of chronic intermittent hypoxia in mice (Castro‐Grattoni et al., 2016), supporting the reduction of intermittent hypoxia as a therapeutic target. Despite a large decrease in the ODI with OA therapy in our patients, there were negligible effects on the levels of the inflammatory markers.
Studies on the effect of OA treatment on inflammatory markers in OSA are scarce. A work comprising 44 patients with mild‐to‐severe OSA, but no control group, reported a significant reduction in hsCRP after a minimum of 30 days on OA treatment (Yalamanchali et al., 2015). In another small non‐RCT study, 22 patients with mild‐to‐moderate OSA showed similar circulating concentrations of CRP, IL‐6, and IL‐10 at baseline compared with a healthy control group (Niżankowska‐Jędrzejczyk et al., 2014). After 6 months of OA treatment, there was a significant reduction in IL‐10 levels, but not in the other aforementioned inflammatory markers. Our present findings confirm the results from a recent RCT showing that OAa treatment compared with a sham device has negligible effects on serum concentrations of CRP, IL‐6, and TNF‐α (Recoquillon et al., 2019). We extend these findings by a slightly longer treatment period of 3 months (2 months in the Recoquillon study) and by presenting data for WBCC and IL‐10.
Assessment of the AHI is the most used criterion of effectiveness in OSA therapy. An AHI reduction of ≥50% is considered to be the criterion for responding to the treatment (Flemons et al., 1999). In previous studies, ~50% of patients have reached this AHI threshold with OA treatment, which corresponds well with the present results (Lim et al., 2004).
On the market, there are different kinds of mandibular advancement devices available for use. In the present study, we used one type of custom‐made OA with a monobloc design (Tegelberg et al., 1999) with high treatment adherence; median 7 nights per week. The custom‐made appliances have been reported to be preferable with higher treatment compliance than a thermoplastic “boil and bite” OA (Vanderveken et al., 2008).
Compared with an OA, CPAP is more efficient in reducing the AHI (Li et al., 2013). However, there seem to be no differences between these treatment modalities regarding effects on quality of life, cognitive or physical function (Schwartz et al., 2018), which possibly could be due to better compliance with an OA than with CPAP (Schwartz et al., 2018).
The effect of CPAP treatment on inflammatory markers is uncertain. A reduction in circulating concentrations of hsCRP (Schiza et al., 2010; Steiropoulos et al., 2007; Yokoe et al., 2003), IL‐6 (Ye et al., 2010; Yokoe et al., 2003), and TNF‐α (Arias et al., 2008; Ryan et al., 2006) has been reported in some studies, whereas others have failed to detect changes in all or some of these biomarkers (Arias et al., 2008; Borges et al., 2020; Ryan et al., 2006; Stradling et al., 2015). Most of these studies were observational in design, and some were flawed by the lack of a control group. In a recent RCT, 220 patients with coronary artery disease and non‐sleepy OSA with a mean AHI of 29 events/hr and an ESS of <10 were allocated to CPAP or no CPAP (Thunström et al., 2017). During a follow‐up of 1 year, no net effects of the CPAP treatment could be detected on the circulating concentrations of hsCRP, IL‐6, IL‐8, or TNF‐α.
The strengths of the present work include the RCT design, the well‐characterised study population, and that the evaluation of biomarker data was blinded from the treatment allocation and outcome data.
There are several limitations to be considered. At the time of planning and inclusion start of the original study in 2005, registration of RCTs was not yet an established recommendation. Therefore, unfortunately, no registration was made on ClinicalTrials.gov. The results are limited to patients of European origin with OSA and systemic hypertension. The sample size of the present study was primarily dimensioned for evaluation of blood pressure as the outcome and not for inflammatory biomarkers. We acknowledge that the sample size in our present study is small and that the analysis is underpowered. However, considering the tiny effect sizes detected, it is unlikely that a better‐powered study with the same time to follow‐up could detect a clinically meaningful effect on the examined biomarkers.
The follow‐up time in the present study was 3 months. Possibly, an extended treatment period might be required to achieve an impact on the circulating biomarkers. However, considering that a previous RCT on CPAP treatment for 1 year failed to detect a net reduction in the concentrations of circulating inflammatory markers (Thunström et al., 2017), it is uncertain if an extended follow‐up with OA treatment would detect an effect.
Most of our patients had inflammatory marker concentrations within the normal range at baseline which, due to a floor effect, might be a cause of the failure to show any treatment effects. Subgroup analyses, evaluating treatment effects by OSA severity were considered. However, due to the small sample size and the lack of correlation between OSA severity and biomarker concentrations at baseline, such subgroup analyses were deemed futile.
In conclusion, a 3‐month period of OA treatment had no effect on circulating inflammatory markers, despite a significant improvement in the AHI and ODI in patients with OSA and concomitant systemic hypertension.
CONFLICT OF INTERESTS
The authors report no conflict of interest.
AUTHOR CONTRIBUTIONS
PH and ÅT contributed to the study design, planning, data collection, and statistical analysis. All authors contributed to data interpretation, writing, critical revision of the manuscript, and all gave their final approval for publication. All authors participated sufficiently in the work to take public responsibility for its content.
Hedberg P, Nohlert E, Tegelberg Å. Effects of oral appliance treatment on inflammatory biomarkers in obstructive sleep apnea: A randomised controlled trial. J Sleep Res.2021;30:e13253. 10.1111/jsr.13253
DATA AVAILABILITY STATEMENT
Data available on request due to privacy/ethical restrictions
REFERENCES
- Andrén, A., Hedberg, P., Walker‐Engström, M.‐L., Wahlén, P., & Tegelberg, A. (2013). Effects of treatment with oral appliance on 24‐h blood pressure in patients with obstructive sleep apnea and hypertension: A randomized clinical trial. Sleep and Breathing, 17(2), 705–712. 10.1007/s11325-012-0746-7 [DOI] [PubMed] [Google Scholar]
- Arias, M. A., García‐Río, F., Alonso‐Fernández, A., Hernanz, Á., Hidalgo, R., Martínez‐Mateo, V., Bartolomé, S., & Rodríguez‐Padial, L. (2008). CPAP decreases plasma levels of soluble tumour necrosis factor‐α receptor 1 in obstructive sleep apnoea. European Respiratory Journal, 32(4), 1009–1015. 10.1183/09031936.00007008 [DOI] [PubMed] [Google Scholar]
- Arnaud, C., Poulain, L., Lévy, P., & Dematteis, M. (2011). Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein‐E knock out mice. Atherosclerosis, 219(2), 425–431. 10.1016/j.atherosclerosis.2011.07.122 [DOI] [PubMed] [Google Scholar]
- Borges, Y. G., Cipriano, L. H. C., Aires, R., Zovico, P. V. C., Campos, F. V., de Araújo, M. T. M., & Gouvea, S. A. (2020). Oxidative stress and inflammatory profiles in obstructive sleep apnea: Are short‐term CPAP or aerobic exercise therapies effective? Sleep and Breathing, 24(2), 541–549. 10.1007/s11325-019-01898-0 [DOI] [PubMed] [Google Scholar]
- Bradley, T. D., & Floras, J. S. (2009). Obstructive sleep apnoea and its cardiovascular consequences. Lancet, 373(9657), 82–93. 10.1016/S0140-6736(08)61622-0 [DOI] [PubMed] [Google Scholar]
- Castro‐Grattoni, A. L., Alvarez‐Buvé, R., Torres, M., Farré, R., Montserrat, J. M., Dalmases, M., Almendros, I., Barbé, F., & Sánchez‐De‐La‐Torre, M. (2016). Intermittent Hypoxia‐Induced Cardiovascular Remodeling Is Reversed by Normoxia in a Mouse Model of Sleep Apnea. Chest, 149(6), 1400–1408. 10.1016/j.chest.2015.11.010 [DOI] [PubMed] [Google Scholar]
- Flemons, W. W., Buysse, D., Redline, S., Oack, A., Strohl, K., Wheatley, J., Young, T., Douglas, N., Levy, P., McNicolas, W., Fleetham, J., White, D., Schmidt‐Nowarra, W., Carley, D., & Romaniuk, J. (1999). Sleep‐related breathing disorders in adults: Recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep, 22(5), 667–689. 10.1093/sleep/22.5.667 [DOI] [PubMed] [Google Scholar]
- Gottlieb, D. J., Yenokyan, G., Newman, A. B., O’Connor, G. T., Punjabi, N. M., Quan, S. F., Redline, S., Resnick, H. E., Tong, E. K., Diener‐West, M., & Shahar, E. (2010). Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: The sleep heart health study. Circulation, 122(4), 352–360. 10.1161/CIRCULATIONAHA.109.901801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns, M. W. (1991). A new method for measuring daytime sleepiness: The Epworth sleepiness scale. Sleep, 14(6), 540–545. 10.1093/sleep/14.6.540 [DOI] [PubMed] [Google Scholar]
- Jullian‐Desayes, I., Joyeux‐Faure, M., Tamisier, R., Launois, S., Borel, A. L., Levy, P., & Pepin, J. L. (2015). Impact of obstructive sleep apnea treatment by continuous positive airway pressure on cardiometabolic biomarkers: A systematic review from sham CPAP randomized controlled trials. Sleep Medicine Reviews, 21, 23–38. 10.1016/j.smrv.2014.07.004 [DOI] [PubMed] [Google Scholar]
- Koenig, W., Löwel, H., Baumert, J., & Meisinger, C. (2004). C‐Reactive Protein Modulates Risk Prediction Based on the Framingham Score ‐ Implications for Future Risk Assessment: Results from a Large Cohort Study in Southern Germany. Circulation, 109(11), 1349–1353. 10.1161/01.CIR.0000120707.98922.E3 [DOI] [PubMed] [Google Scholar]
- Li, W., Xiao, L., & Hu, J. (2013). The comparison of CPAP and oral appliances in treatment of patients with OSA: A systematic review and meta‐analysis. Respiratory Care, 58(7), 1184–1195. 10.4187/respcare.02245 [DOI] [PubMed] [Google Scholar]
- Li, Y., Chongsuvivatwong, V., Geater, A., & Liu, A. (2009). Exhaled breath condensate cytokine level as a diagnostic tool for obstructive sleep apnea syndrome. Sleep Medicine, 10(1), 95–103. 10.1016/j.sleep.2007.11.013 [DOI] [PubMed] [Google Scholar]
- Libby, P. (2012). Inflammation in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 32(9), 2045–2051. 10.1161/ATVBAHA.108.179705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, J., Lasserson, T., Fleetham, J., & Wright, J. (2004). Oral appliances for obstructive sleep apnoea. In Lim J. (Ed.), Cochrane Database of Systematic Reviews (Issue 4). Chichester, UK: John Wiley & Sons Ltd. 10.1002/14651858.CD004435.pub2 [DOI] [PubMed] [Google Scholar]
- Nadeem, R., Molnar, J., Madbouly, E. M., Nida, M., Aggarwal, S., Sajid, H., Naseem, J., & Loomba, R. (2013). Serum inflammatory markers in obstructive sleep apnea: A meta‐analysis. Journal of Clinical Sleep Medicine, 9(10), 1003–1012. 10.5664/jcsm.3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niżankowska‐Jędrzejczyk, A., Almeida, F. R., Lowe, A. A., Kania, A., Nastałek, P., Mejza, F., Foley, J. H., Niżankowska‐Mogilnicka, E., & Undas, A. (2014). Modulation of inflammatory and hemostatic markers in obstructive sleep apnea patients treated with mandibular advancement splints: A parallel, controlled trial. Journal of Clinical Sleep Medicine, 10(3), 255–262. 10.5664/jcsm.3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noether, G. E. (1987). Sample Size Determination for Some Common Nonparametric Tests. Journal of the American Statistical Association, 82(398), 645–647. 10.1080/01621459.1987.10478478 [DOI] [Google Scholar]
- Pai, J. K., Pischon, T., Ma, J., Manson, J. E., Hankinson, S. E., Joshipura, K., Curhan, G. C., Rifai, N., Cannuscio, C. C., Stampfer, M. J., & Rimm, E. B. (2004). Inflammatory markers and the risk of coronary heart disease in men and women. New England Journal of Medicine, 351(25), 2599–2610. 10.1056/NEJMoa040967 [DOI] [PubMed] [Google Scholar]
- Punjabi, N. M. (2008). The epidemiology of adult obstructive sleep apnea. Proceedings of the American Thoracic Society, 5(2), 136–143. 10.1513/pats.200709-155MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2020). R: A Language and Environment for Statistical Computing. Retrieved from https://www.r‐project.org/ [Google Scholar]
- Recoquillon, S., Pépin, J. L., Vielle, B., Andriantsitohaina, R., Bironneau, V., Chouet‐Girard, F., Fleury, B., Goupil, F., Launois, S., Martinez, M. C., Meslier, N., Nguyen, X. L., Paris, A., Priou, P., Tamisier, R., Trzepizur, W., & Gagnadoux, F. (2019). Effect of mandibular advancement therapy on inflammatory and metabolic biomarkers in patients with severe obstructive sleep apnoea: A randomised controlled trial. Thorax, 74(5), 496–499. 10.1136/thoraxjnl-2018-212609 [DOI] [PubMed] [Google Scholar]
- Ryan, S., Taylor, C. T., & McNicholas, W. T. (2006). Predictors of elevated nuclear factor‐κB‐dependent genes in obstructive sleep apnea syndrome. American Journal of Respiratory and Critical Care Medicine, 174(7), 824–830. 10.1164/rccm.200601-066OC [DOI] [PubMed] [Google Scholar]
- Sabatine, M. S., Morrow, D. A., Cannon, C. P., Murphy, S. A., Demopoulos, L. A., DiBattiste, P. M., McCabe, C. H., Braunwald, E., & Gibson, C. M. (2002). Relationship between baseline white blood cell count and degree of coronary artery disease and mortality in patients with acute coronary syndromes: A TACTICS‐TIMI 18 (Treat Angina with Aggrastat and determine Cost of Therapy with an Invasive or Conservati. Journal of the American College of Cardiology, 40(10), 1761–1768. 10.1016/S0735-1097(02)02484-1 [DOI] [PubMed] [Google Scholar]
- Schiza, S. E., Mermigkis, C., Panagiotis, P., Bouloukaki, I., Kallergis, E., Tzanakis, N., Tzortzaki, E., Vlachaki, E., & Siafakas, N. M. (2010). C‐reactive protein evolution in obstructive sleep apnoea patients under CPAP therapy. European Journal of Clinical Investigation, 40(11), 968–975. 10.1111/j.1365-2362.2010.02348.x [DOI] [PubMed] [Google Scholar]
- Schwartz, M., Acosta, L., Hung, Y. L., Padilla, M., & Enciso, R. (2018). Effects of CPAP and mandibular advancement device treatment in obstructive sleep apnea patients: A systematic review and meta‐analysis. Sleep and Breathing, 22(3), 555–568. 10.1007/s11325-017-1590-6 [DOI] [PubMed] [Google Scholar]
- Steiropoulos, P., Tsara, V., Nena, E., Fitili, C., Kataropoulou, M., Froudarakis, M., Christaki, P., & Bouros, D. (2007). Effect of continuous positive airway pressure treatment on serum cardiovascular risk factors in patients with obstructive sleep apnea‐hypopnea syndrome. Chest, 132(3), 843–851. 10.1378/chest.07-0074 [DOI] [PubMed] [Google Scholar]
- Stradling, J. R., Craig, S. E., Kohler, M., Nicoll, D., Ayers, L., Nunn, A. J., & Bratton, D. J. (2015). Markers of inflammation: Data from the MOSAIC randomised trial of CPAP for minimally symptomatic OSA. Thorax, 70(2), 181–182. 10.1136/thoraxjnl-2014-205958 [DOI] [PubMed] [Google Scholar]
- Swerdlow, D. I., Holmes, M. V., Kuchenbaecker, K. B., Engmann, J. E. L., Shah, T., Sofat, R., Guo, Y., Chung, C., Peasey, A., Pfister, R., Mooijaart, S. P., Ireland, H. A., Leusink, M., Langenberg, C., Li, K. W., Palmen, J., Howard, P., Cooper, J. A., Drenos, F., … Casas, J. P. (2012). The interleukin‐6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet, 379(9822), 1214–1224. 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegelberg, A., Wilhelmsson, B., Walker‐Engström, M. L., Ringqvist, M., Andersson, L., Krekmanov, L., & Ringqvist, I. (1999). Effects and adverse events of a dental appliance for treatment of obstructive sleep apnoea. Swedish Dental Journal, 23(4), 117–126. [PubMed] [Google Scholar]
- Thunström, E., Glantz, H., Yucel‐Lindberg, T., Lindberg, K., Saygin, M., & Peker, Y. (2017). CPAP does not reduce inflammatory biomarkers in patients with coronary artery disease and nonsleepy obstructive sleep apnea: A randomized controlled trial. Sleep, 40(11), zsx157. 10.1093/sleep/zsx157 [DOI] [PubMed] [Google Scholar]
- Vanderveken, O. M., Devolder, A., Marklund, M., Boudewyns, A. N., Braem, M. J., Okkerse, W., Verbraecken, J. A., Franklin, K. A., De Backer, W. A., & Van de Heyning, P. H. (2008). Comparison of a custom‐made and a thermoplastic oral appliance for the treatment of mild sleep apnea. American Journal of Respiratory and Critical Care Medicine, 178(2), 197–202. 10.1164/rccm.200701-114OC [DOI] [PubMed] [Google Scholar]
- Vargha, A., & Delaney, H. D. (2000). A critique and improvement of the CL common language effect size statistics of McGraw and Wong. Journal of Educational and Behavioral Statistics, 25(2), 101–132. 10.3102/10769986025002101 [DOI] [Google Scholar]
- Yalamanchali, S., Salapatas, A. M., Hwang, M. S., Pott, T. R., Lundgren, M. E., Joseph, N. J., & Friedman, M. (2015). Impact of mandibular advancement devices on C‐reactive protein levels in patients with obstructive sleep apnea. Laryngoscope, 125(7), 1733–1736. 10.1002/lary.25061 [DOI] [PubMed] [Google Scholar]
- Ye, L., Ma, G. H., Chen, L., Li, M., Liu, J. L., Yang, K., Li, Q. Y., Li, N., & Wan, H. Y. (2010). Quantification of circulating cell‐free DNA in the serum of patients with obstructive sleep apnea‐hypopnea syndrome. Lung, 188(6), 469–474. 10.1007/s00408-010-9253-4 [DOI] [PubMed] [Google Scholar]
- Yokoe, T., Minoguchi, K., Matsuo, H., Oda, N., Minoguchi, H., Yoshino, G., Hirano, T., & Adachi, M. (2003). Elevated levels of C‐reactive protein and interleukin‐6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation, 107(8), 1129–1134. 10.1161/01.cir.0000052627.99976.18 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request due to privacy/ethical restrictions