

Abstract
Inputting molecules into chemistry software, such as quantum chemistry packages, currently requires domain expertise, expensive software and/or cumbersome procedures. Leveraging recent breakthroughs in machine learning, we develop ChemPix: an offline, hand-drawn hydrocarbon structure recognition tool designed to remove these barriers. A neural image captioning approach consisting of a convolutional neural network (CNN) encoder and a long short-term memory (LSTM) decoder learned a mapping from photographs of hand-drawn hydrocarbon structures to machine-readable SMILES representations. We generated a large auxiliary training dataset, based on RDKit molecular images, by combining image augmentation, image degradation and background addition. Additionally, a small dataset of ∼600 hand-drawn hydrocarbon chemical structures was crowd-sourced using a phone web application. These datasets were used to train the image-to-SMILES neural network with the goal of maximizing the hand-drawn hydrocarbon recognition accuracy. By forming a committee of the trained neural networks where each network casts one vote for the predicted molecule, we achieved a nearly 10 percentage point improvement of the molecule recognition accuracy and were able to assign a confidence value for the prediction based on the number of agreeing votes. The ensemble model achieved an accuracy of 76% on hand-drawn hydrocarbons, increasing to 86% if the top 3 predictions were considered.
Offline recognition of hand-drawn hydrocarbon structures is learned using an image-to-SMILES neural network through the application of synthetic data generation and ensemble learning.
Introduction
Artificial intelligence (AI) refers to the introduction of “human intelligence” into artificial machines. Machine learning is a subfield of AI that focuses specifically on the “learning” aspect of the machine's intelligence, removing the need for manually coding rules. Although Rosenblatt proposed the perceptron in the 1950s,1 it wasn't until the 1990s that machine learning shifted from a knowledge-based to a data-driven approach. A decade later, “deep learning” emerged as subclass of machine learning that employed multilayer neural networks (NNs). The boom of big-data and increasingly powerful computational hardware allowed deep learning algorithms to achieve unprecedented accuracy on a variety of problems. This resulted in much of the AI software used today, such as music/movie recommenders, speech recognition, language translation and email spam filters.
Deep learning algorithms have been adopted by almost every academic field in the hope of solving both novel and age-old problems.2 The natural sciences have historically relied on the development of theoretical models derived from physically-grounded fundamental equations to explain and/or predict experimental observations. This makes data-driven models an interesting, and often novel, approach. In quantum chemistry, for example, to calculate the energy of a molecule one would traditionally solve an approximation to the electronic Schrodinger equation. A machine learning approach to this problem, however, might involve inputting a dataset of molecules and their respective energies into a NN, which would learn a mapping between the two.3–5 The ability to generate accurate models by extracting features directly from data without human input makes machine learning techniques an exciting avenue to explore in all areas of chemistry – from drug discovery and material design to analytical tools and synthesis planning.
Easy-to-use machine learning based tools have the potential to accelerate research and enrich education. Here, we develop a hand-drawn molecule recognition tool to extract a digital representation of the molecule from an image of a hand-drawn hydrocarbon structure. Drawing skeletal chemical structures by hand is a routine task for students and researchers in the chemistry community. Therefore, photographing a hand-drawn chemical structure offers a low-barrier method of entering molecules into software that would normally require time-consuming workflows and domain expertise. Moreover, for the vast majority of the chemistry community, drawing a chemical structure by hand is far less cumbersome than building it with a mouse. The recognition tool could be integrated into a phone application that performs tasks such as quantum chemistry calculations, database lookups and AI synthesis planning directly from the hand-drawn molecule, extending the ChemVox voice-recognition system we recently developed.6
In addition to its potential as a chemical research and education widget, hand-drawn hydrocarbon recognition is an interesting problem from a fundamental science perspective: it serves as a prototypical example of how deep learning can be applied to a well-suited chemical problem. Sourcing a large training dataset for this task is time and resource intensive – a common obstacle encountered in machine learning applications. To address this, we discuss strategies for synthetic data generation and their generalizability to scenarios where there is access to limited real-world data, but abundant similar data.
Hand-drawn chemical structure recognition is, in many ways, similar to the task of handwriting recognition. Large variation in writing styles, poor image quality, lack of labelled data and cursive letters make hand-written text recognition a challenging task.7–10 Hand-writing recognition falls into two camps: online recognition, in which a user writes text on a tablet or phone and it is recognized in real-time, and offline recognition, which refers to static images of hand-written text. Offline recognition poses considerably more challenges than online recognition due largely to the latter's ability to use time dependent strokes in combination with the final image to distinguish between characters.11 In this work, we focus on offline hand-drawn hydrocarbon structure recognition, extending the potential use cases to digitization of lab notebooks.
Automatic extraction of a molecule from an image of its 2D chemical structure to a machine-readable format, termed optical chemical structure recognition, first emerged in the 1990s.12–17 These systems were developed with the intent of mining ChemDraw type diagrams in the chemical literature to utilize the wealth of largely untapped chemical information that lies within publications.17–28 The majority of optical chemical structure recognition packages, including Kekulé,14 IBM's OROCS,15 CLiDE16 and CLiDEPro,21 ChemOCR,20 OSRA,22 ChemReader,23 MolRec,25 ChemEx,26 MLOCSR,27 and ChemSchematicResolver28 rely on a rule-based workflow rather than a data-driven approach. These systems achieve various degrees of accuracy, with the recently developed ChemSchematicResolver reaching 83–100% precision on a range of datasets.
Rule-based systems often involve complex, interdependent workflows, which can make them brittle, and challenging to revise and extend. Therefore, several optical chemical structure recognition packages have been recently proposed based on data-driven, deep learning techniques.29–31 Notably, Staker et al.29 employed end-to-end segmentation and image to molecule neural networks, and ChemGrapher30 used a series of deep neural networks to extract molecules from the chemical literature. These data-driven systems offer a promising alternative to rule-based systems for this task, provided one can obtain an appropriate training dataset.
The optical chemical structure recognition systems mentioned thus far focus on recognition of computer generated, ChemDraw-type structures. A handful of promising online hand-drawn chemical structure recognition programs have recently been developed.32–34 Our goal of offline extraction of molecules from photographs of hand-drawn chemical structures adds a further level of complexity, and is well-suited for data-driven, machine learning models.
In this article, we begin by discussing our chosen deep learning approach for hand-drawn chemical structure recognition and demonstrate proof-of-concept on ChemDraw type images of molecules produced with the RDKit. Next, we describe the generation of two datasets: a small set of real-world photographs of hand-drawn hydrocarbon structures and a large synthetic dataset. We perform a series of experiments with these datasets, aiming to optimize the recognition accuracy on out-of-sample real-world hand-drawn hydrocarbons. We end by forming an ensemble model consisting of a committee of NNs, which leads to a significant boost in recognition accuracy and introduces a confidence value for the prediction. The work serves as a prototypical case study for approaching a chemical problem with machine learning methods, focusing on the explanation of deep learning, synthetic data generation, and ensemble learning techniques.
Methods
Neural network architecture
In this work, we represent molecules as simplified molecular-input line-entry system (SMILES)35 strings in order to leverage recent advances in natural language processing (NLP).36 We employ neural image captioning, in which an image is input into a NN and a caption for the image is produced.37,38 Here, an image of a hydrocarbon molecule is input and the predicted SMILES string is output, as shown in Fig. 1. The NN architecture consists of a convolutional neural network (CNN)39,40 encoder and a long short term memory (LSTM)41 decoder with beam search and attention. CNNs contain ‘convolutional layers’ that apply a convolutional filter over the image and pass the result to the next hidden layer; they are used primarily for encoding images since they conserve the spatial relationship of the pixels. LSTMs are a type of stable recurrent neural network (RNN) popularly used in language models. This is a useful feature in the case of decoding SMILES strings since there are often relations between characters at the start and end of the string, such as closing of a parentheses pair to indicate the end of a branching group. Our image-to-SMILES approach is inspired by the work of Deng et al., who trained a NN to convert images of mathematical formulas to LaTeX code.42 A similar image-to-SMILES approach was also used by Staker et al., which achieved test set recognition accuracies ranging from 41 to 83% on ChemDraw type structures extracted from the chemical literature.29 Details of the NN architecture used in this work are discussed in the ESI.†
Fig. 1. Image-to-SMILES neural network used for hand-drawn hydrocarbon recognition based on neural image captioning. The network consists of a convolutional neural network (CNN) encoder and long short-term memory (LSTM) decoder network.

We define the NN accuracy as the proportion of molecules predicted exactly correctly, i.e., the predicted SMILES matches the target SMILES character-by-character. Error bars were calculated by bootstrapping the accuracy of 1000 sets of 200 data points sampled from the test set with replacement and computing the range that contains the statistical mean with 95% likelihood based on the resampled sets.
Datasets
We extracted a dataset of 500 000 SMILES strings with a ring size of less than eight carbon atoms from the GDB-13 and GDB-11 databases.43–45 The vocabulary was restricted to “Cc=#()1”, where = and # indicate double and triple bonds, parentheses indicate the start and end of a branching group, lower case letters represent aromaticity, and numbers are found at the start and end of rings. To remove ambiguous skeletal structures from our dataset that confuse the NN during training, we only include the number ‘1’, meaning that molecules with multiple conjoined rings are not considered. The SMILES labels were canonicalized using RDKit to give a consistent target output. After canonicalization, molecules outside of the vocabulary were removed, resulting in a ∼10% reduction in size for all datasets used in the experiments presented. RDKit was used to generate images of molecules from the SMILES dataset, by first generating SVG files and then converting to PNG format. The result is a labelled dataset of image and SMILES pairs; representative examples are shown in the Fig. 2 inset. We used this clean RDKit dataset to perform proof-of-concept for the image-to-SMILES network. A synthetic dataset based on RDKit images designed to mimic hand-drawn data was curated for the purpose of this study. We discuss the auxiliary data generation workflow, and experiments performed on this dataset in the coming sections.
Fig. 2. Out-of-sample accuracy of the image-to-SMILES network trained with an increasing number of clean RDKit hydrocarbon structures and their corresponding SMILES label. Representative examples of labelled RDKit training images and SMILES are shown in the inset.

The computer-generated datasets were first split into a 90% training/validation set, and a 10% test set. The test set serves as out-of-sample data used to evaluate the accuracy of the network after finishing the training process. The training/validation set, used during training, was then split further into a training set (90%) and a validation set (10%). The real-world photographs of hand-drawn hydrocarbons consisted of a total of 613 images. We set aside a 200-image test set, with the remaining 413 images being either used entirely as a validation set or split into validation (200 images) and training (213 images) datasets, depending on the experiment. All images were resized to 256 × 256 pixels and converted to PNG format using OpenCV.46
Results and discussion
Synthetic data generation
To test the suitability of our image-to-SMILES network for hand-drawn molecule recognition, we begin by training with clean images of hydrocarbon skeletal structures generated with RDKit and their respective SMILES labels (Fig. 2). In order to determine the dataset size required to achieve a given recognition accuracy, the NN was trained with datasets of size 104, 5 × 104, 105, 2 × 105 and 5 × 105 images (split between training, validation and test sets as described in the methods section). The results of the proof-of-concept training are shown in Fig. 2, illustrating the increasing NN recognition accuracy with dataset size. A dataset of 50 000 labelled RDKit images achieves an out-of-sample (test set) accuracy of over 90%, and a maximum accuracy of 98% is achieved with a dataset of 500 000 images. This demonstrates that the chosen NN architecture is capable of learning SMILES strings from machine-generated images of hydrocarbons.
Although the results from training with synthetic RDKit images suggest that a dataset of 50 000 images obtains 90% out-of-sample accuracy, in reality a much greater number of hand-drawn hydrocarbon molecules are likely needed to achieve this same accuracy. As with handwritten text recognition, variation in drawing style, backgrounds and image quality provide significant challenges. There is noise associated with (i) the chemical structure, such as varying line widths, lengths, angles and distortion, (ii) the background, such as different textures, lighting, colors and surrounding text, and (iii) the photograph, such as blurring, pixel count and image format (Fig. 3). A further challenge of chemical structure recognition is the ability for a molecule to be drawn in any orientation, in contrast to text recognition of languages written in one direction, e.g., left-to-right.
Fig. 3. Comparison between a computer-generated (RDKit) image of a hydrocarbon structure (left) and a photographed hand-drawn hydrocarbon structure (right). The differences between the two images are highlighted, demonstrating the increased complexity of hand-drawn structure recognition.

Since end-to-end NNs learn a model solely from the data presented during training, access to high-quality data is imperative to achieve an accurate model. Unfortunately, a large labelled dataset of real-world hand-drawn molecules does not exist and cannot be easily generated. Therefore, unlike in the case of RDKit images, it is not possible to achieve high recognition accuracy by simply training with hundreds of thousands of hand-drawn structures. Lack of training data is a common hurdle when attempting to apply end-to-end deep learning models to real-world problems, particularly in fields where data generation is time and energy intensive such as the chemical domain. In cases such as these, generating synthetic data can prove more efficient than spending excessive time and resources collecting large amounts of real-world data.
We developed a data collection web app to source a small dataset of hand-drawn chemical structures. In order to capture the large noise in drawing style, photograph quality and background types that are prevalent in real-world data, we collected data from many different drawers by promoting the app to a range of groups in the Stanford University Chemistry Department. Over 100 unique users of the app generated over 5800 photographs of hand-drawn chemical structures, 613 of which were hydrocarbons. Details of the data collection app are shown in Fig. S1† and the collected dataset is released with this paper.47 Based on our earlier RDKit image results (Fig. 2), ∼600 images is several orders of magnitude less data than necessary to train to any reasonable recognition accuracy. As a result, in addition to sourcing real-world data, we also developed a workflow to generate a large synthetic dataset to be used in conjunction with the limited real-world dataset for training. We go on to show that our strategy is able to successfully train an accurate NN with this limited amount of real-world data. This is an encouraging result for machine learning approaches in the chemical sciences, where the availability of accurate data is often problematic.
An ideal synthetic dataset is exactly equivalent to the target data but can be readily generated on large scales (unlike the target data). The desired datatype (of which there is insufficient data for training) could therefore be substituted with synthetic data during training and the weights would be directly transferable to the target data. To discuss how to generate such an auxiliary dataset, we consider a subspace that spans from the desired datatype to a similar machine-scalable datatype. In our case, this is the subspace between photographs of hand-drawn molecules and RDKit images. The aim is to find a mapping that moves both datatypes to the same point in the subspace such that they are indistinguishable. Fig. 4 depicts such a subspace, highlighting possible convergence routes. Perhaps the most obvious pathway transforms raw RDKit data (bottom right) into images that resemble raw hand-drawn data (top left) as closely as possible (or visa versa). This might involve adding in backgrounds, distorting the lines and blurring the image. However, it is also possible to modify both datatypes such that they reach a common point in the subspace that lies away from both of the original data points. As long as the two datatypes are uniquely mapped to the same point, they are equivalent. For example, applying edge detection (or background removal) to both the hand-drawn and computer-generated data would result in movement away from their respective raw datatypes, but closer to one another. In this illustrative example, a model would be trained with an edge-detected synthetic dataset, and later applied to hand-drawn hydrocarbon molecule images that have been pre-processed with edge detection.
Fig. 4. Data subspace that spans from target data (photographs of hand-drawn hydrocarbon chemical structures, top left) to readily available machine-generated data (raw RDKit images, bottom right). Paths to reach similar points in the subspace for the target data and synthetic data are demonstrated. Blue outline: hand-drawn images, red outline: computer-generated images.

Mapping two datatypes to a common point in a subspace is commonly used in deep learning applications since there is often a limited amount of the exact data needed, but a similar readily accessible datatype that can form the basis of a synthetic dataset.10,48,49 It is important to note that a one-to-one mapping between the two datatypes and the output label must exist, i.e., one image should only correspond to exactly one molecule.
Although we did explore auxiliary datasets based on background removal and edge detection algorithms, we abandoned these image processing techniques because they were found to be brittle when applied to real-world hand-drawn data. For example, dark shadows, lined paper and thin pencils made it hard to clearly identify the molecule after applying such algorithms (Fig. S2†). To ensure the recognition software is robust to a wide range of potential images, for the remainder of this study we focus on generating a synthetic dataset that resembles hand-drawn molecules as closely as possible.
Fig. 5a outlines the synthetic data generation workflow developed to transform RDKit images into synthetic photographs of hand-drawn hydrocarbon structures. First, we introduce randomness to bond angles, lengths and widths via modification of the RDKit source code (RDKit'). The image is then passed through an augmentation pipeline which applies a series of random image transformations (RDKit'-aug). The augmented molecule image is then combined with a randomly augmented background image using OpenCV (RDKit'-aug-bkg). Next, the image is passed through a degradation pipeline to form the final synthetic data (RDKit'-aug-bkg-deg). The molecule augmentation, background augmentation and image degradation workflows are outlined in Fig. 5b (the transformations applied in these pipelines are detailed in Table S2†). A comparison of examples from the synthetic dataset and the real-world dataset can be found in Fig. S5.†
Fig. 5. (a) The synthetic data generation workflow with the datatype's assigned name for each stage of the pipeline. (b) The augment molecule, augment background and degradation pipelines used for the synthetic data generation. Each box corresponds to a function that is applied with probability p. A complete list of the image transforms associated with each function is given in the ESI.† (c) Schematic depiction of how the steps in the synthetic data workflow move the synthetic data distribution towards the hand-drawn data distribution by representing the datasets as two-dimensional Gaussians (not to scale).

Generating a synthetic datapoint from a SMILES string takes ∼1 s, hence, over 85 000 labelled images of hydrocarbons can be produced in 24 hours of compute time. For comparison, it takes ∼1 minute for a human to draw, photograph, and label a hydrocarbon chemical structure, meaning that ∼2 months of continuous human effort would be needed to collect a dataset of this size.
The molecule and background image augmentation pipelines (Fig. 5b) introduce noise into the data through rotations, translations, distortion and other image transformations. This acts as a form of regularization during training to reduce overfitting (where the NN reaches high accuracies during training but much lower accuracies on out-of-sample data). The importance of broadening the data distribution can be exemplified with background augmentation: without augmenting backgrounds the NN may become overly familiar with the structure of the background images used during training and learn to remove them from the image. The result is bad generalization when presented with images that have different backgrounds to those seen during training. We also randomly degrade the data to further increase the regularization. This accounts for features like variation in image quality and type. The degradation pipeline was adapted from work by Ingle et al.,10 which leveraged a large dataset of online data for offline hand-written text recognition by applying aggressive degradation. The augmentation and degradation are deliberately more aggressive than what would be found in real-world images to span the maximum dataset subspace, i.e., make the distribution as wide as possible.
As described previously, the stages of the synthetic data generation pipeline are designed to map the synthetic distribution onto the distribution of real-world hand-drawn chemical structures. A simplified schematic of how each step effects the data distribution is shown in Fig. 5c. The datasets are represented as two-dimensional Gaussians, with their amplitude proportional to the quantity of data and their width proportional to the data variation within the distribution. As the data proceeds through the augmentation, background addition and degradation steps, the synthetic distribution approaches the hand-drawn data distribution in the subspace.
Neural network experiments
In the following section we describe a series of experiments designed to understand how our real-world and synthetic datasets can be best utilized to achieve the highest out-of-sample hand-drawn hydrocarbon recognition accuracy. We form an ensemble model from the trained NNs, which allows us to assign a confidence value to the prediction, as well as improve the recognition accuracy. We finish by analysing the success of the model on specific examples from the test set and its performance on chemical subsets.
First, we investigate how the NN performs when exposed only to synthetic data during training. To determine the effect of moving through the synthetic data generation pipeline (Fig. 5), we train the model on data from each stage of the workflow. Fig. S7† shows that image augmentation and degradation result in large increases in recognition of hydrocarbons in the hand-drawn test set, and somewhat surprisingly, the addition of backgrounds has an insignificant effect on the accuracy. By training with 500 000 synthetic images (RDKit'-aug-bkg-deg), we are able to correctly recognise an out-of-sample photograph of a hand-drawn hydrocarbon structure with over 50% accuracy. Although this accuracy is insufficient, at this stage the neural network has never seen a real-life hand-drawn image. We improve the accuracy by introducing our limited hand-drawn dataset to the training process as discussed below.
In situations where there is limited access to data, a common strategy, is to use a real-world data validation set so the NN weights are saved according to the correct target distribution. We examine the effect of replacing the synthetic validation set with a 413-image hand-drawn validation set, varying the size of the synthetic training set from 50 000 to 500 000 (Fig. S8†). Using a hand-drawn validation set has little impact on the hand-drawn recognition accuracy in comparison to using a synthetic validation set since the number of images available is so limited.
We now incorporate hand-drawn data into the training set so that it can directly impact the weight optimization during training, allowing the NN to learn from the target data, rather than only determine if the weights should be saved. The number of remaining images of hand-drawn hydrocarbon structures in our dataset after the removal of the test set is 413, which must be distributed between the training set and validation set. We assign 213 images to the training set and 200 images to the validation set. A dataset of 500 000 images is chosen since it reached the highest accuracies in our synthetic data experiments.
We trained the image-to-SMILES network with varying ratios of augmented and degraded real-world hand-drawn and synthetic data, and tested the weights on the 200 image hand-drawn test set. Due to the very limited hand-drawn hydrocarbon data, we augmented and degraded the images to produce the number needed in the training set to satisfy each given ratio. For example, to generate a training set of 50% hand-drawn and 50% synthetic images (250 000 images each), each hand-drawn image was augmented ∼1173 times using the augment molecule pipeline (Fig. 5b, excluding the final translation step). Although this introduces a large number of repeated SMILES and similar images, the small amount of hand-drawn data makes this necessary to ensure that the information is not overridden by the large amount of synthetic data. Once the molecules have been augmented and degraded, the synthetic and hand-drawn data are randomly shuffled together for training.
We investigate ratios of 0 : 100, 10 : 90, 50 : 50, 90 : 10 and 100 : 0 synthetic : hand-drawn data. From Fig. 6a, it can be seen that using entirely hand-drawn data results in an out-of-sample accuracy of 0% due to the network overfitting to the very narrow distribution of hand-drawn training data. Adding synthetic data allows the NN to be exposed to many more molecules and image types, and hence leads to a rapid increase in test set accuracy up to 90 : 10 synthetic : hand-drawn data. Removing the final 10% of hand-drawn hydrocarbon molecules from the training set (equivalent to the 500 000 image training run presented in Fig. S8†), however, leads to a decrease in the hydrocarbon recognition accuracy from 62% to 56%. Therefore, the results suggest that two opposing effects are at play: (i) including target data in the training set allows the weights to be optimized for the target application and (ii) including only a narrow or sparse distribution of target data leads to overfitting. As a result, including a small portion of target data, specifically 10% hand-drawn molecules, yields the highest recognition accuracy.
Fig. 6. Recognition accuracy of the hand-drawn hydrocarbon test set of trained neural network with different training/validation datasets. (a) Results of training with varying ratios of augmented and degraded hand-drawn hydrocarbon to synthetic data training sets (500 000 image total) and hand-drawn hydrocarbons validation set. (b) The effect of fine tuning is investigated by restarting the weights from training with a 500 000-image synthetic dataset used for both training and validation, and a 500 000-image synthetic dataset used for training with a hand-drawn validation set. The weights are restarted with a training set consisting of 90% synthetic data and 10% augmented and degraded hand-drawn data, and a validation set of hand-drawn hydrocarbons.

In all the experiments discussed so far, the image-to-SMILES network has been trained from scratch, i.e., the weights are randomly initialized. When applying deep learning to tasks with limited available data, training the network with a large dataset before restarting the weights with a similar dataset has been shown to increase NN accuracy.50 This approach is termed fine-tuning due to the NN weights being tuned from a related task to better suit the desired datatype. We apply fine-tuning to our problem by first training with synthetic data and then restarting the NN weights with training data that includes real-life images of hand-drawn hydrocarbon structures. We fine-tune two trained NNs, both of which use 500 000 image synthetic training datasets but that differ in their validation data: the first uses a synthetic validation set (pre-training results shown in Fig. S7b†) and the second uses a hand-drawn validation set (pre-training results shown in Fig. S8†). The two trained NNs are restarted with a training set made up of 90% synthetic data and 10% hand-drawn data – the optimal ratio according to Fig. 6a. The results from the two fine-tuning runs (Fig. 6b) show that pre-training with synthetic data before incorporating hand-drawn data into the training set improves the molecule recognition accuracy. The network reaches 67.5% accuracy after pre-training with a hand-drawn validation set, in comparison to the best NN trained from scratch which was 61.5% accurate.
Ensemble learning
Instead of relying on a single model to predict a desired output, combining several models can result in improved performance, a process called ensemble learning.51 There are several ways in which ensemble models can operate, such as boosting, bagging and random forests.52 Here, we form an ensemble model comprised of a committee of trained NNs, where each NN casts a single vote according to their prediction. The predictions can then be ordered from most to least votes and the prediction that has the most votes is output. The number of agreeing votes for a prediction can give insight into the confidence of the ensemble model. If all of the NNs predict the same output, there is a high probability the prediction is accurate. However, if the NNs disagree, there is higher uncertainty in the prediction.
We build an ensemble model comprised of trained NNs from previous experiments that achieve at least 50% accuracy on the hand-drawn test set (5 out of the 17 trained NNs). The out-of-sample hand-drawn hydrocarbon recognition accuracy for the ensemble model is shown in Fig. 7a, comparing the three predictions that have the most votes with the reference SMILES label. The ensemble model achieves an accuracy of 76% on the hand-drawn test set for the top prediction and 85.5% if the top three predictions are considered. By forming a committee of NNs, we see a significant improvement in accuracy in comparison to the constituent NNs (the highest of which obtained 67.5% on out-of-sample hand-drawn data).
Fig. 7. (a) The out-of-sample hand-drawn hydrocarbon recognition accuracy of the highest N ranked predictions of an ensemble model made up of trained NNs with over 50% recognition accuracy on out-of-sample images of hand-drawn hydrocarbon molecules. (b) The out-of-sample hand-drawn hydrocarbon recognition accuracy of the ensemble model when the top prediction has a given number of agreeing votes, V (blue) and the percentage occurrence of a given number of agreeing votes for the top prediction (red). The accuracy is attributed to the confidence of the model when there are V votes for the top SMILES prediction.

The agreement between the models that make up the committee offers insight into the certainty of the prediction. Fig. 7b shows the increase of recognition accuracy as the number of votes for the top-ranked prediction, V, rises. Here, we assign the accuracy of the ensemble model when there are V agreeing votes to its confidence value. When all the models disagree (V = 1) the model has low out-of-sample accuracy, equating to a low confidence value of the model. When more models agree, the prediction tends to have a higher accuracy. All of the models agreeing (V = 5) translates to a confidence value of 98% in the predicted hydrocarbon.
In addition to knowing the confidence of the model's prediction, it is useful to know how often it achieves this confidence: if the model was 100% confident when all the votes agreed but this only occurred 1% of the time its use would be limited. We therefore investigate the portion of times that a confidence value occurs in the ensemble model's test set predictions (Fig. 7b). It can be seen that the percentage of times that V votes occurs increases with the number of votes – there are few instances where all the NNs disagree (V = 1), and by far the most common occurrence is all NNs agreeing (V = 5).
The importance of knowing the uncertainty of a model's prediction should not be underestimated. In many cases, it is more important to achieve a lower accuracy but be able to predict when the model will fail, than to achieve a higher accuracy without insight into when it will fail. For example, in the case of autonomous vehicles, a model that is able to determine when it will fail and prompt a human to take over controls would be far safer than a model that failed less but was unable to forecast failure. In the case of hand-drawn molecule recognition, the software could, for example, prompt the user to take a second photograph if the uncertainty of the model was high. It may also offer insight into if an erroneous molecule was input by the user, as this would likely cause confusion and result in disagreement between committee members. A potential feature for the ChemPix app is to show the top three predictions if the uncertainty of the first prediction is high; the user could then select the correct molecule from the three if it appears. This data could be continuously collected and fed back into the NN to iteratively re-train and improve its performance as more data is collected.
Of course, both the accuracy and confidence of the NN output should be optimized. Here, our ensemble model recognizes the correct molecule with 89% confidence in over 70% of cases and with near 100% confidence in over 50% of cases. This is a promising result for deploying this technology to real-world applications.
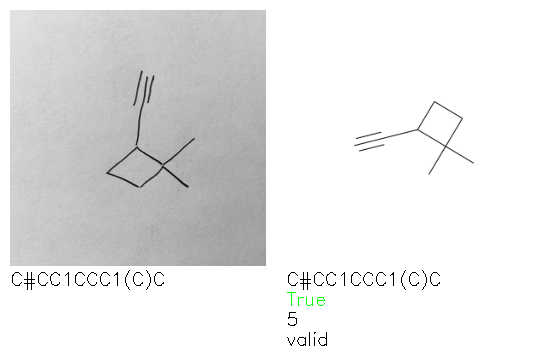
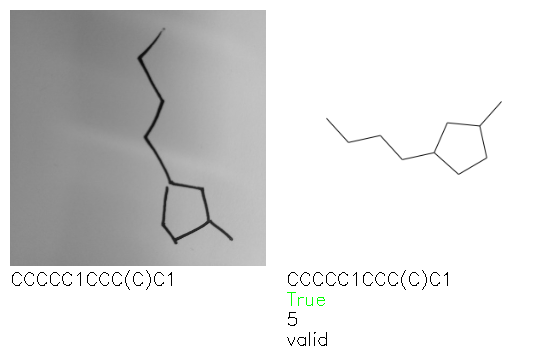
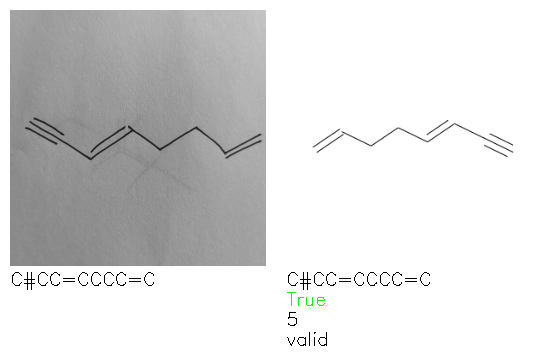
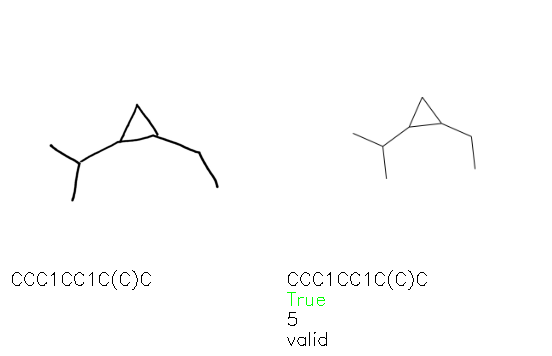
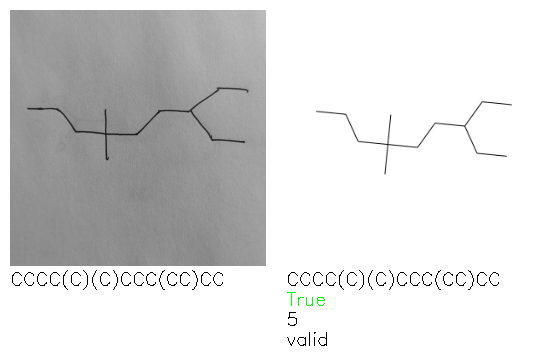
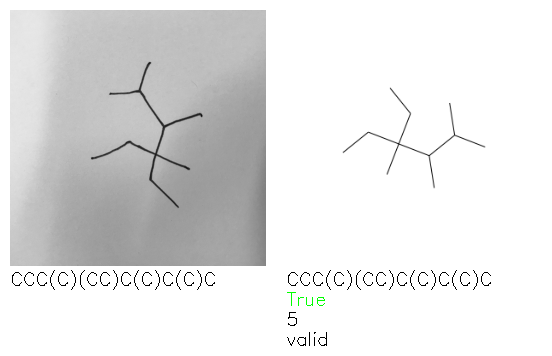
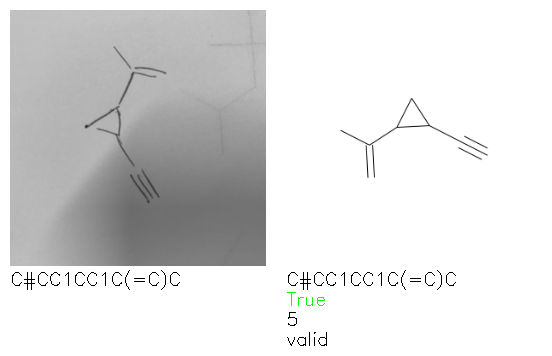
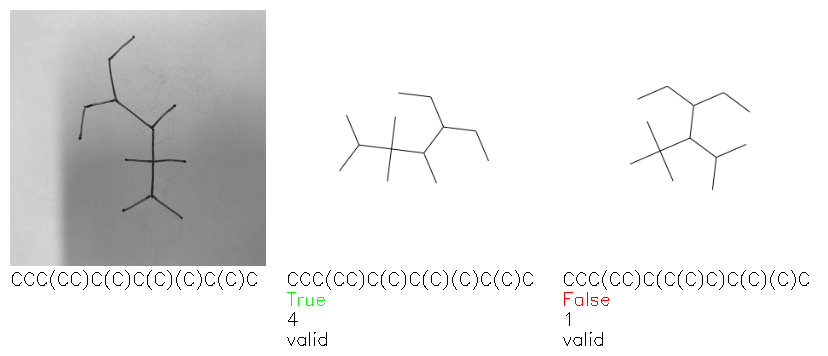
A selection of examples from the hand-drawn test set and their respective predictions from the ensemble are highlighted in Fig. 8. The model is able to recognise a wide variety of hydrocarbons with different sized rings and chain lengths. The network confidently recognizes hydrocarbons drawn on a variety of textured materials, including a napkin, whiteboard and paper. The network is able to determine the molecular structure despite dark shadows and bright spots in the photograph, as well as molecules drawn with a range of pen and pencil types. Wavy lines and “unnatural” bond angles are generally handled well.
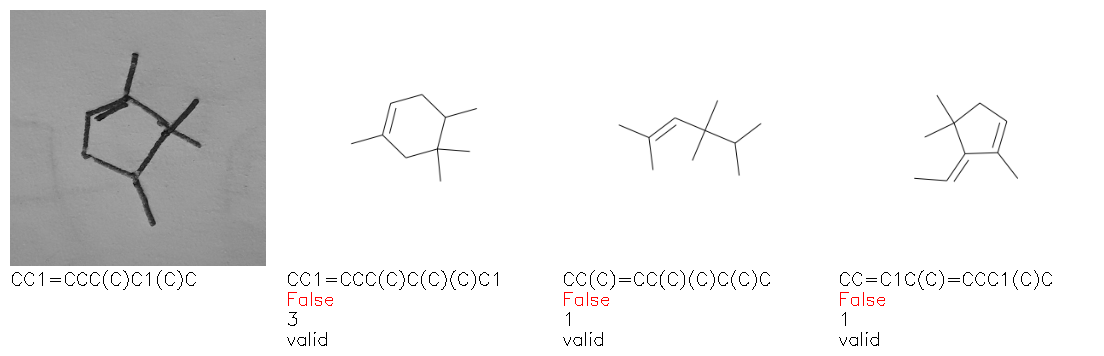
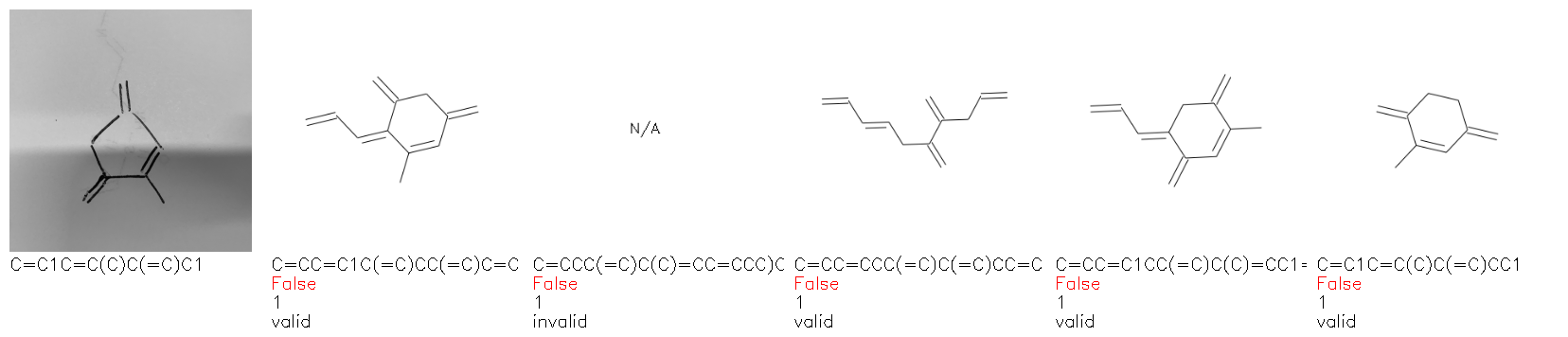
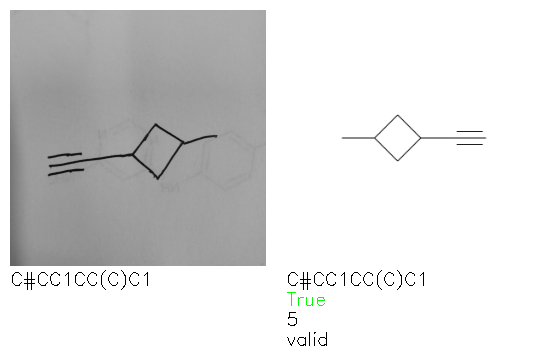
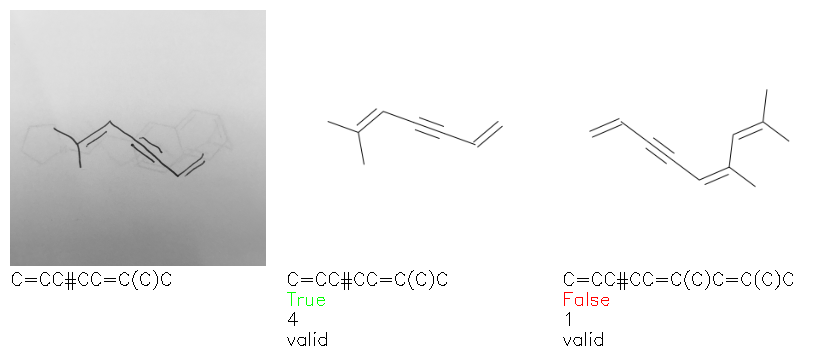
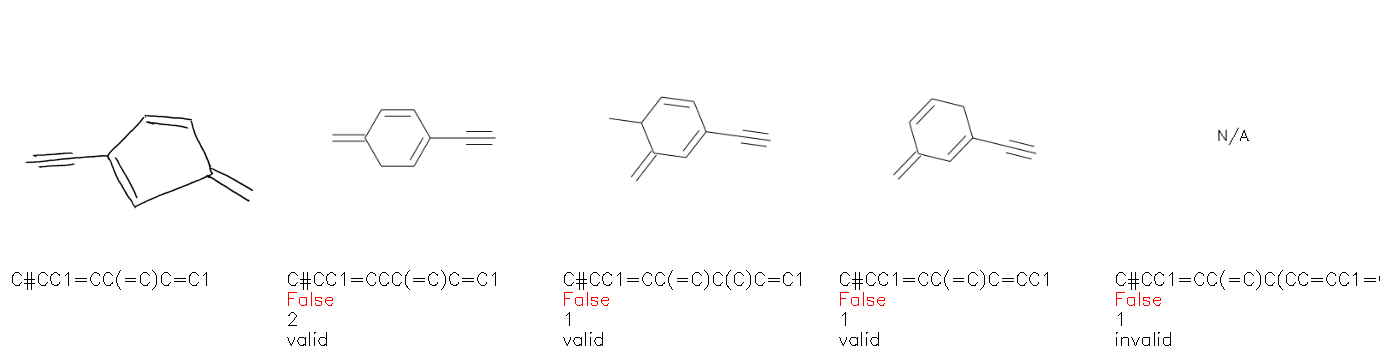
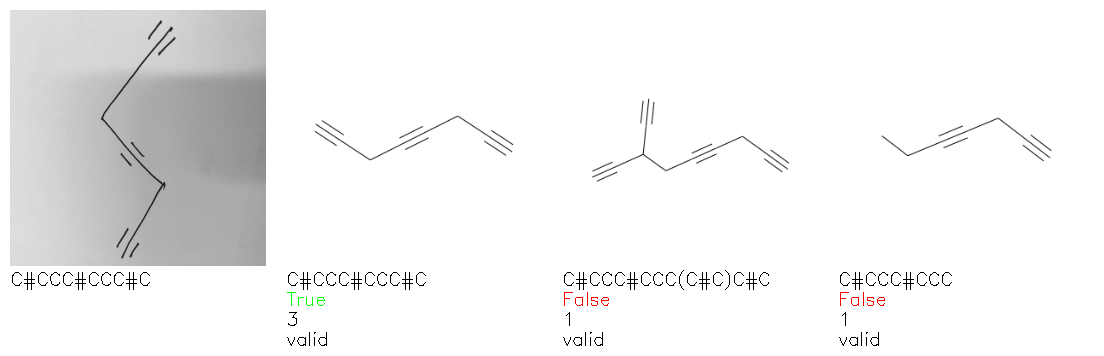
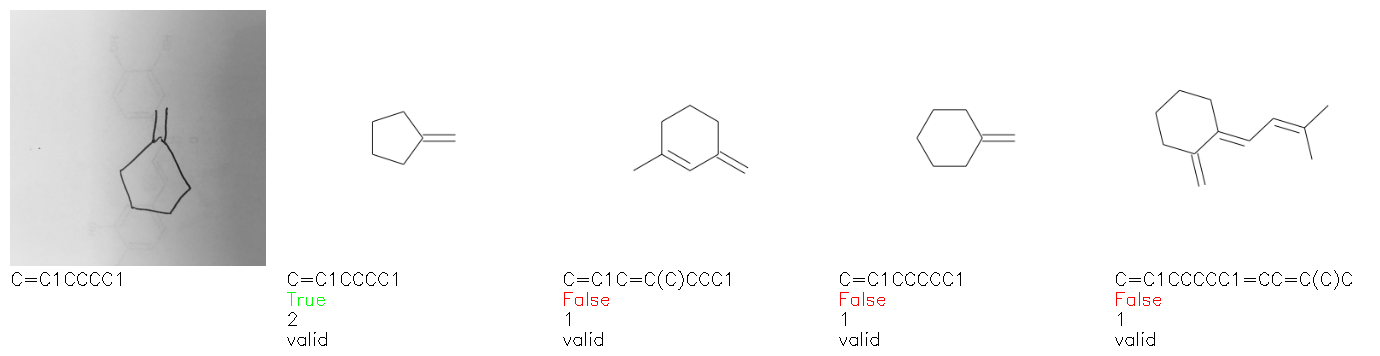
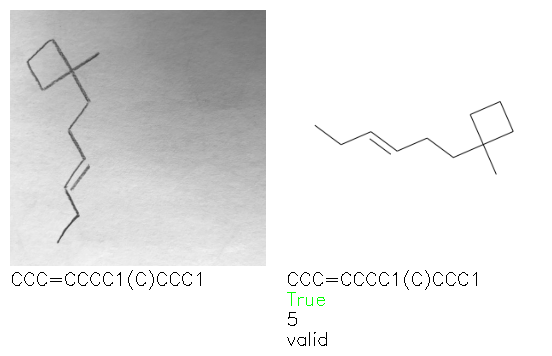
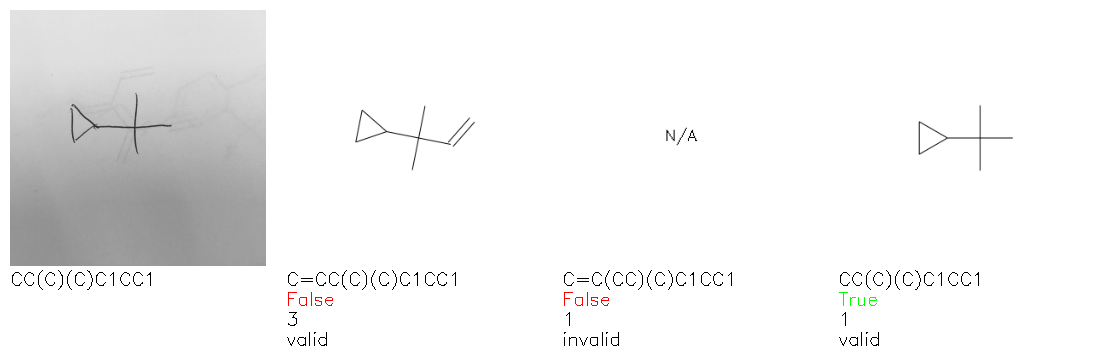
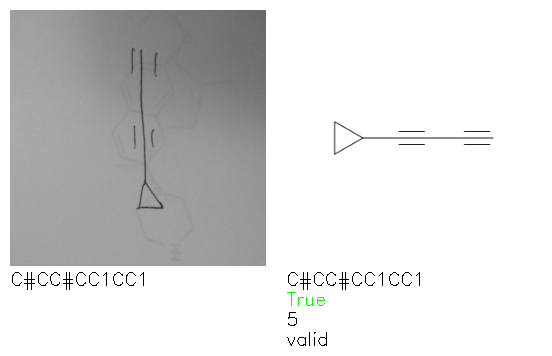
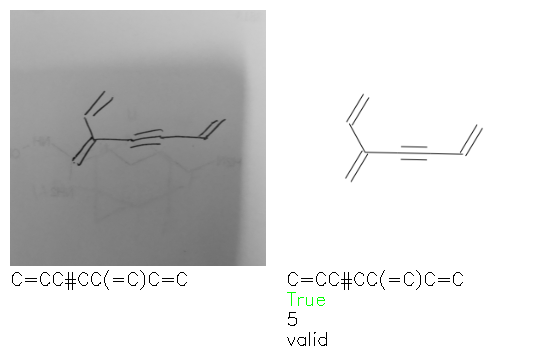
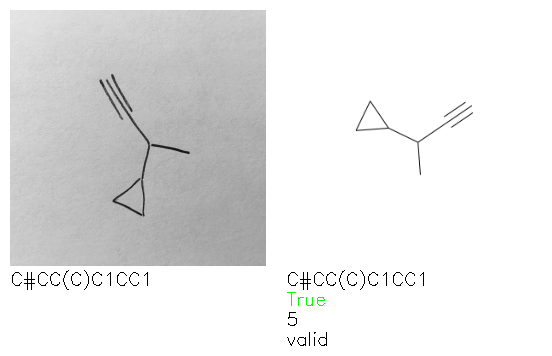
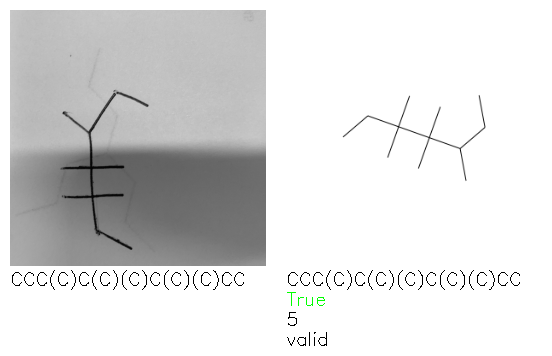
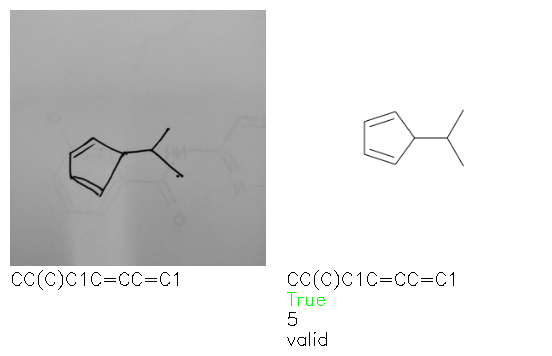
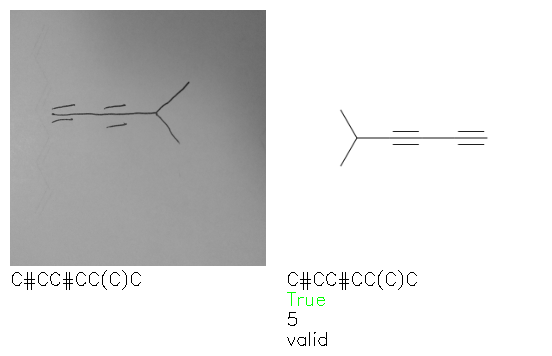
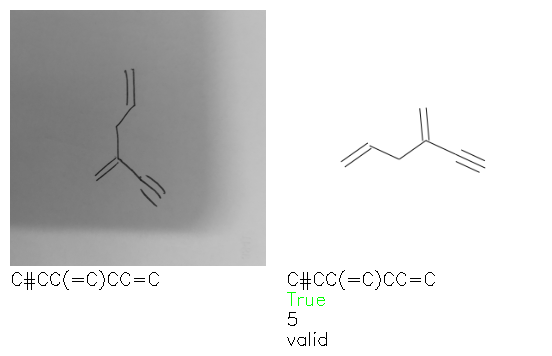
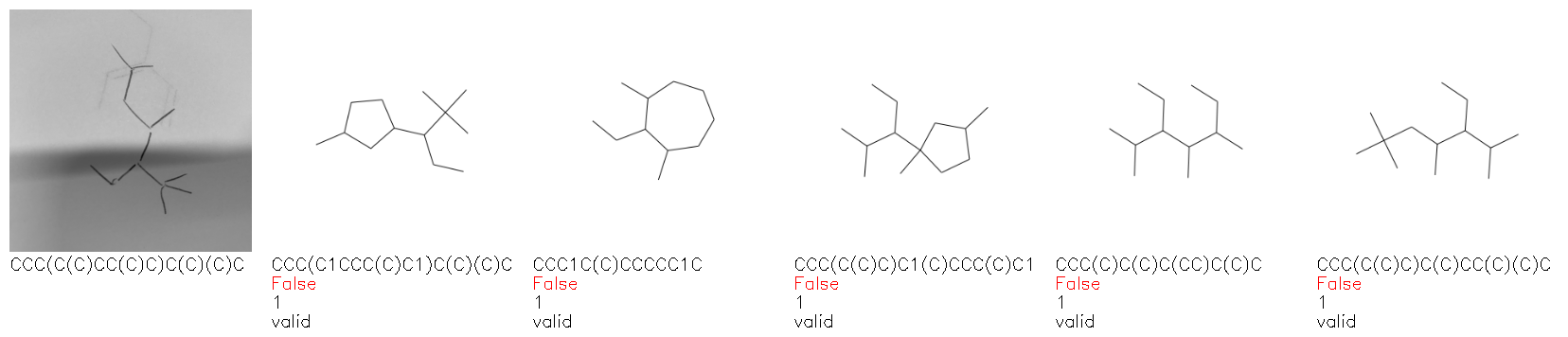
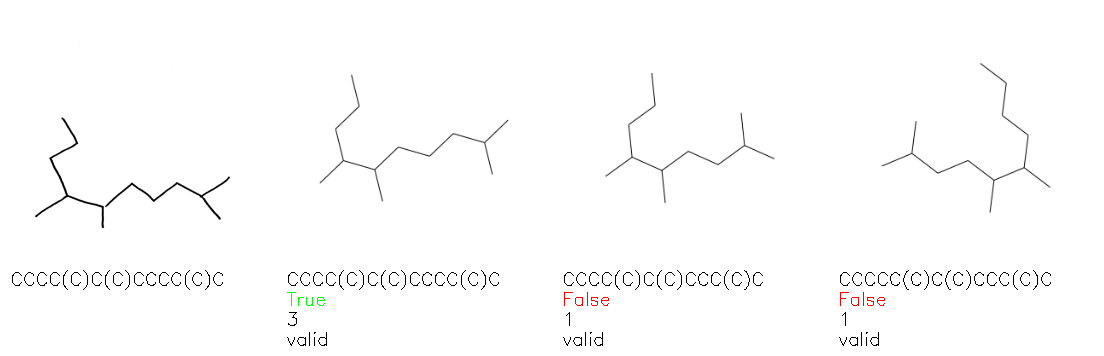
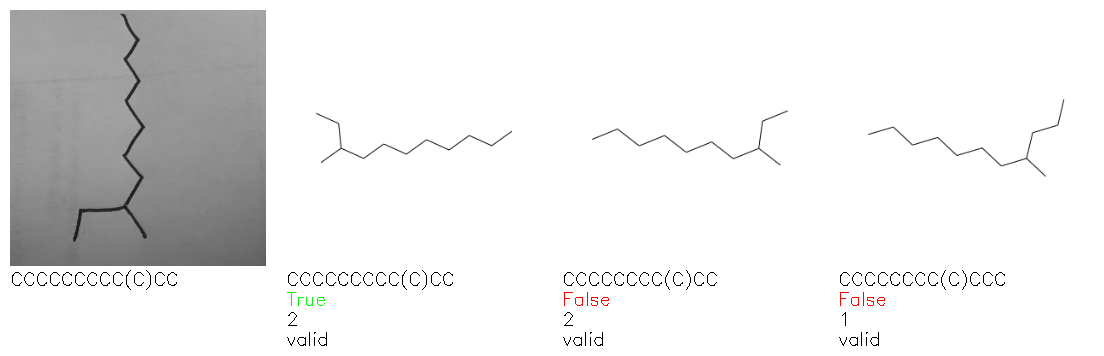
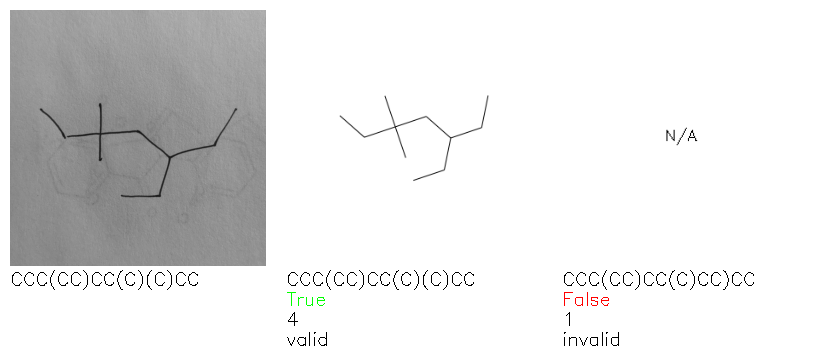
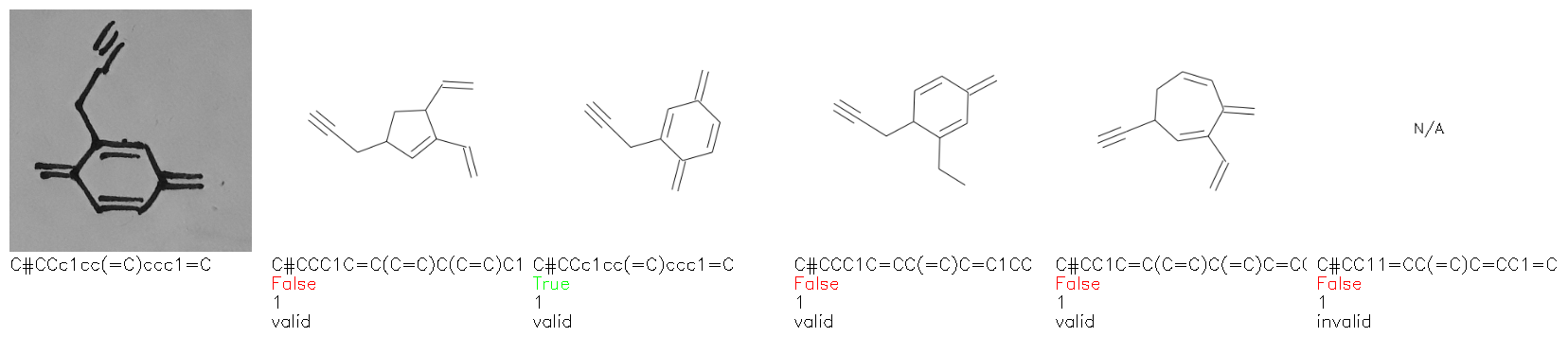
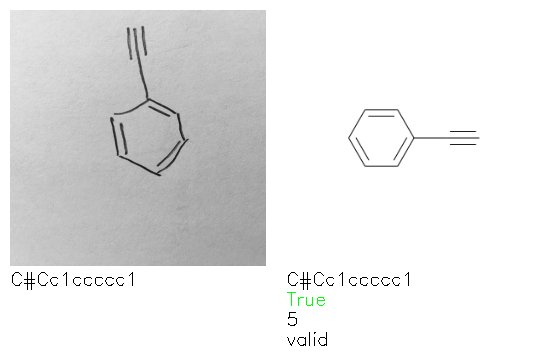
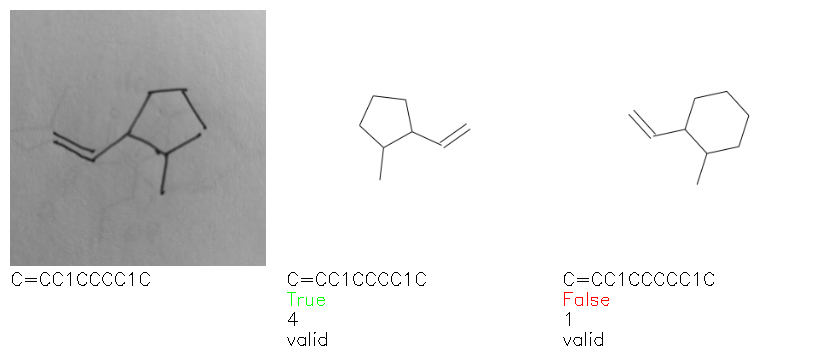
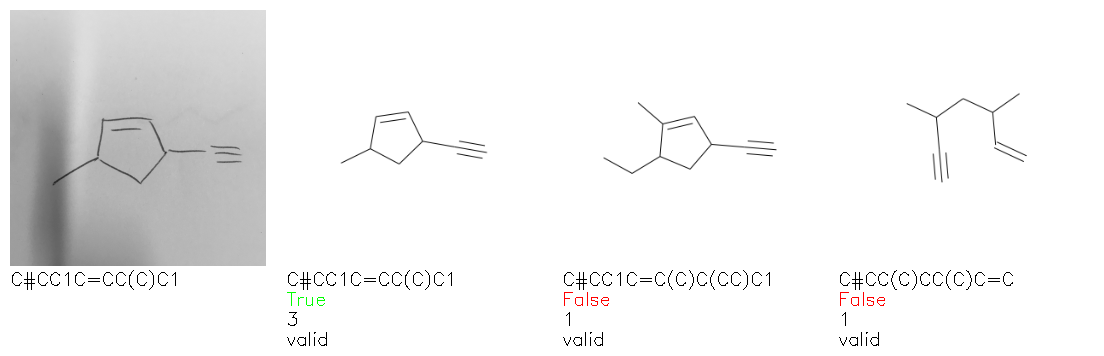
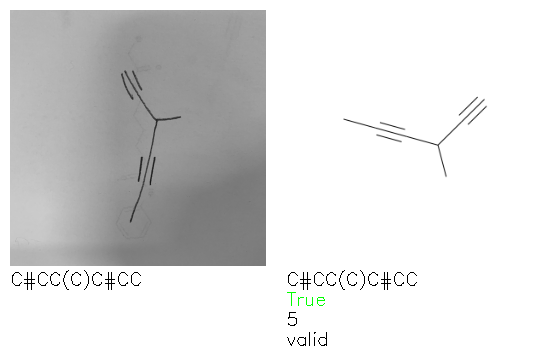
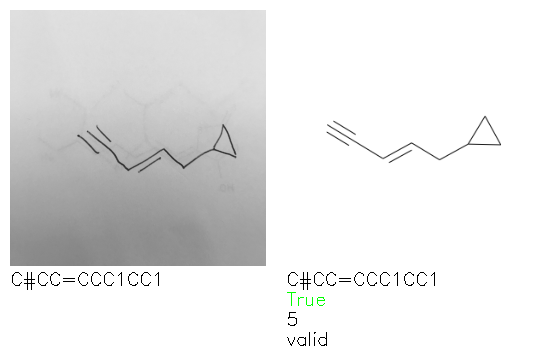
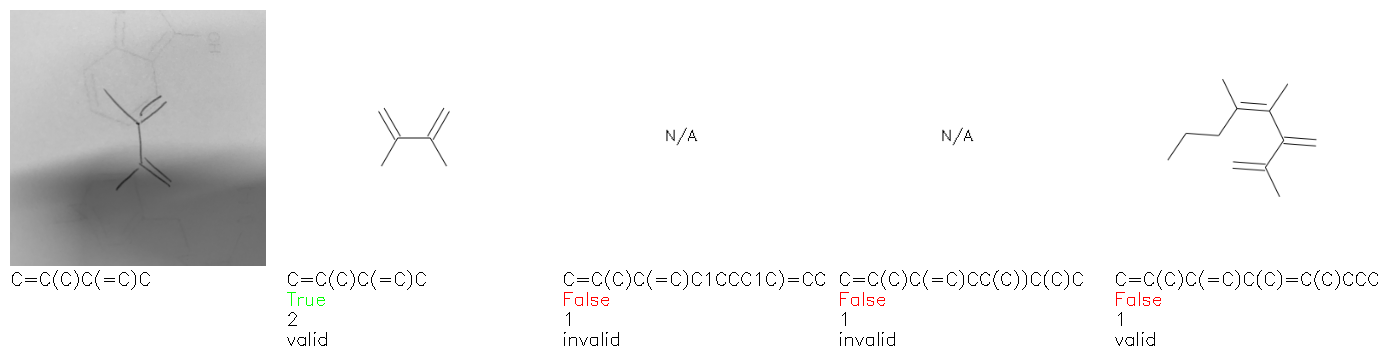
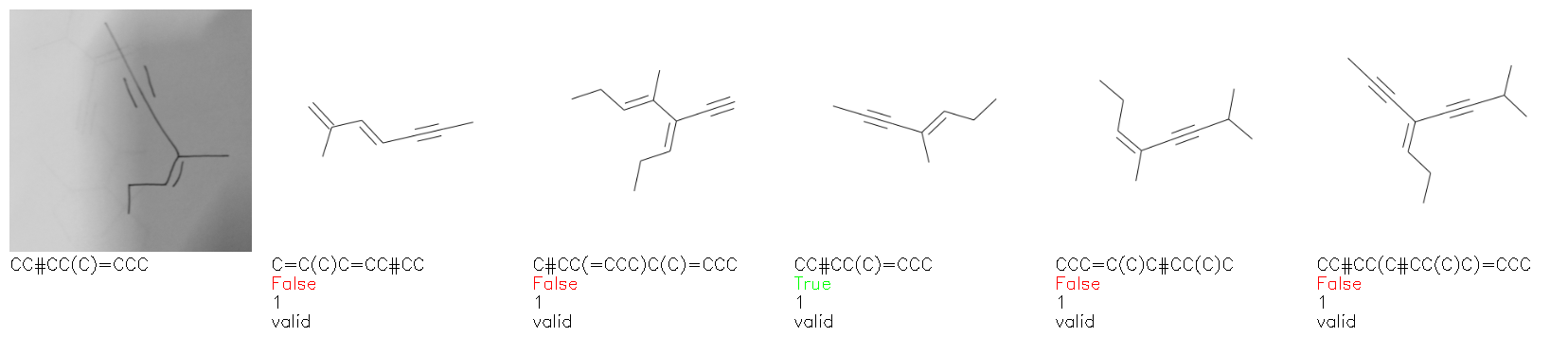
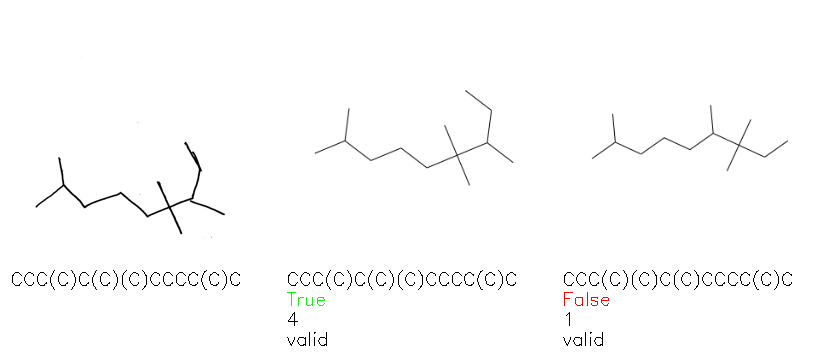
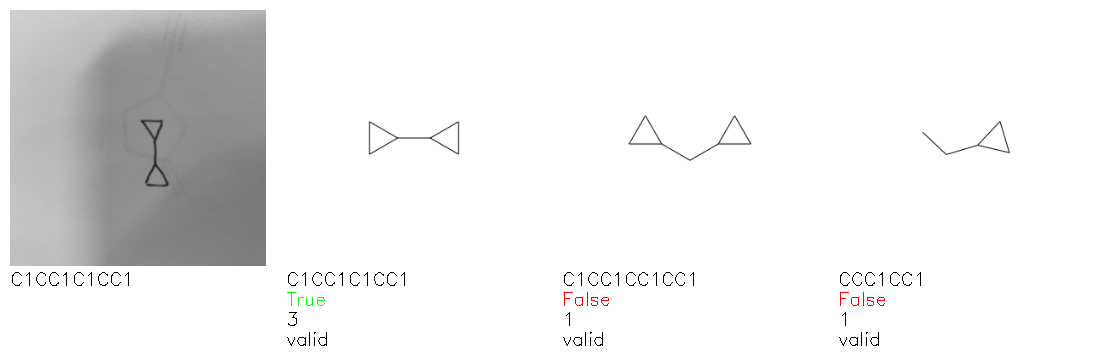
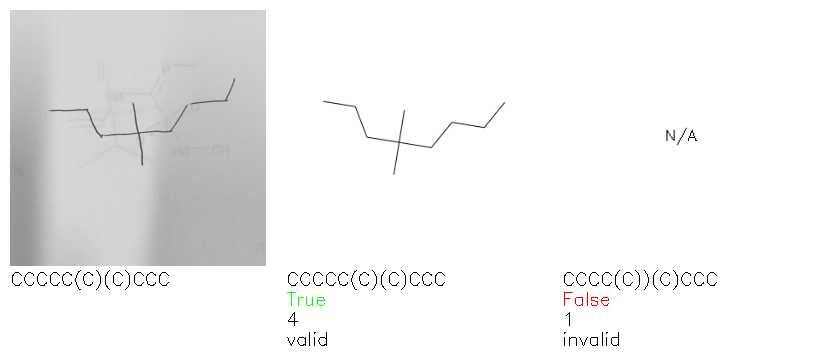
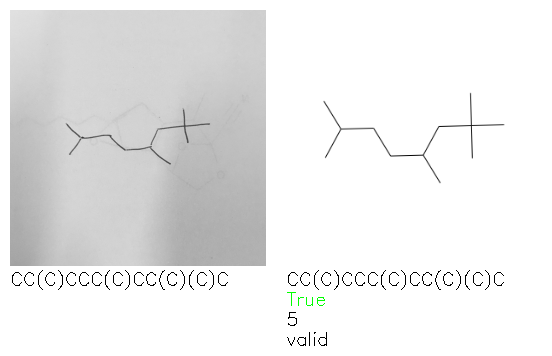
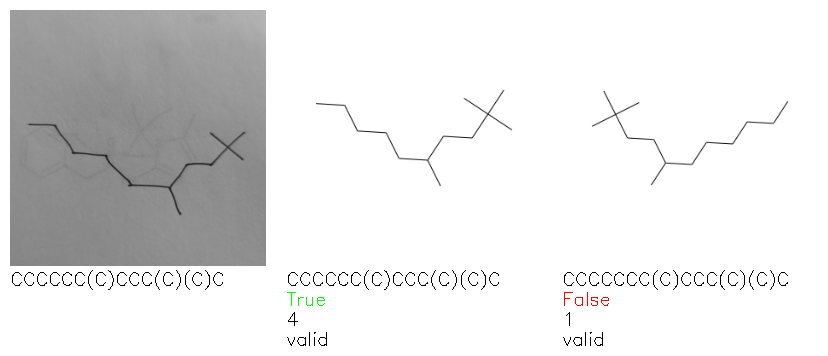
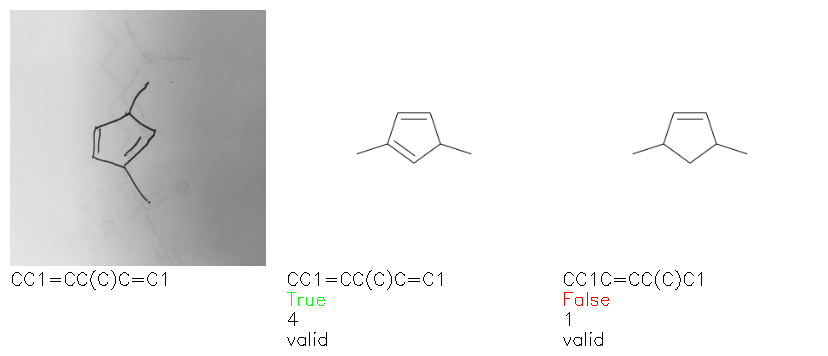
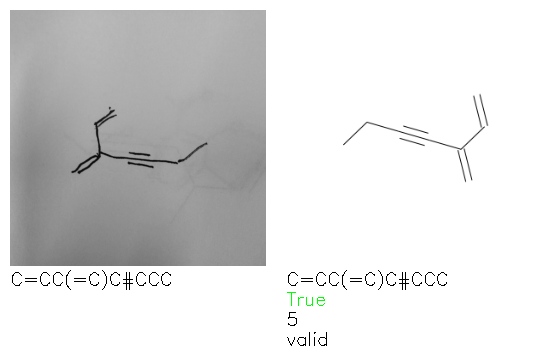
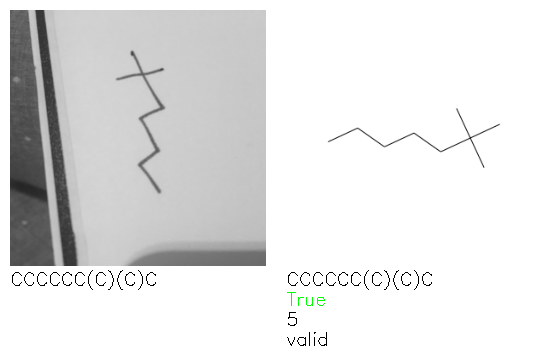
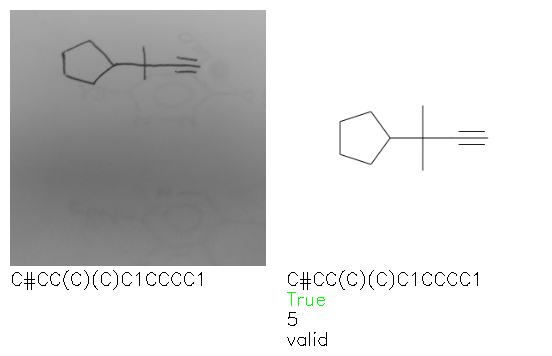
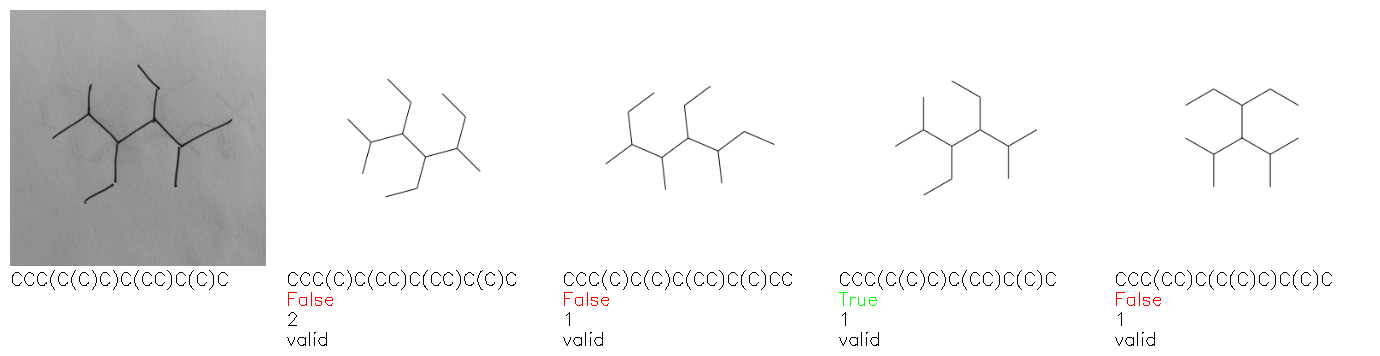
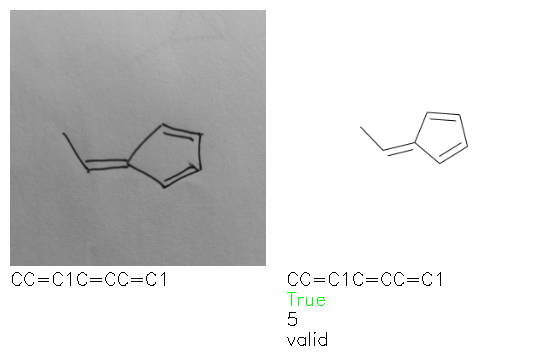
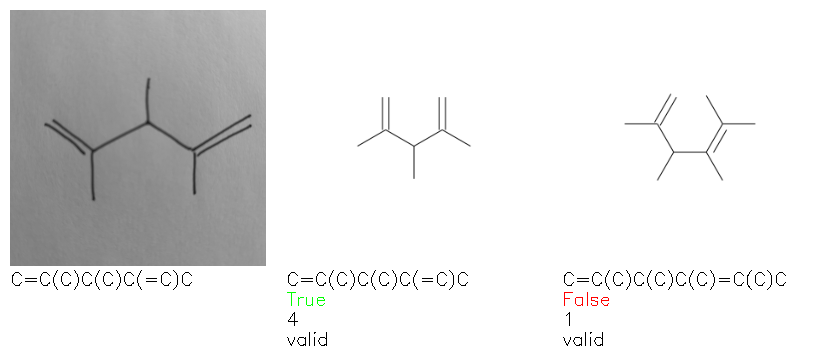
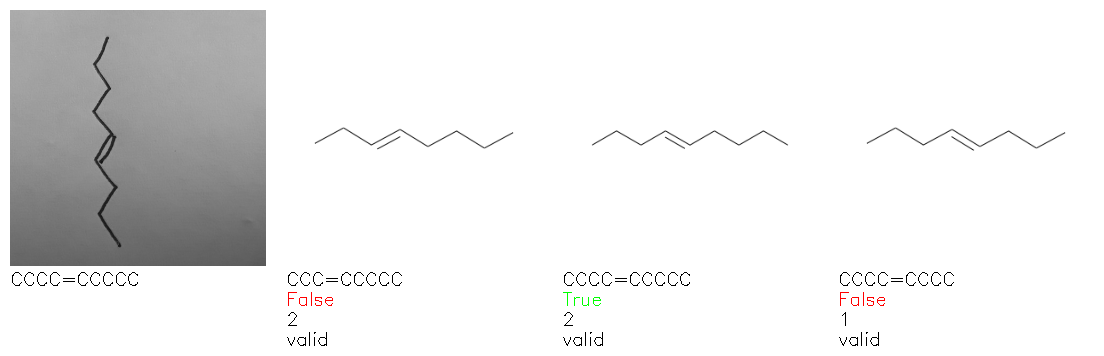
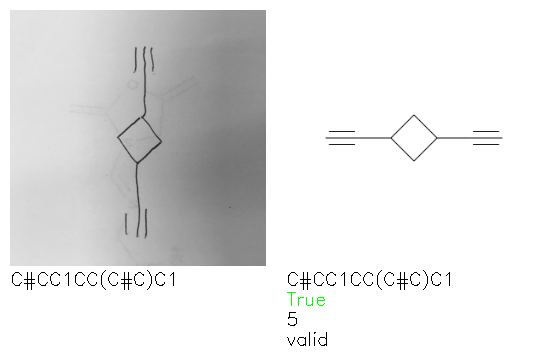
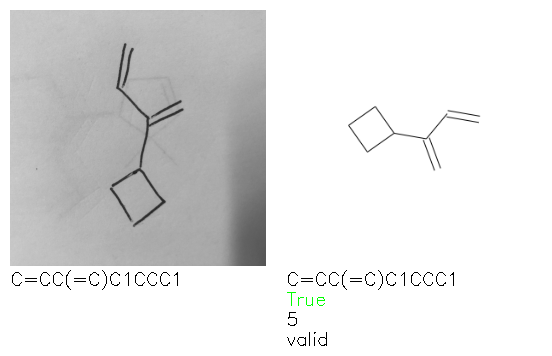
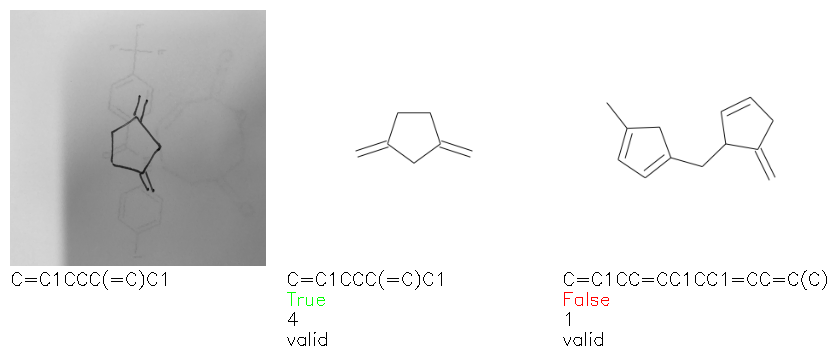
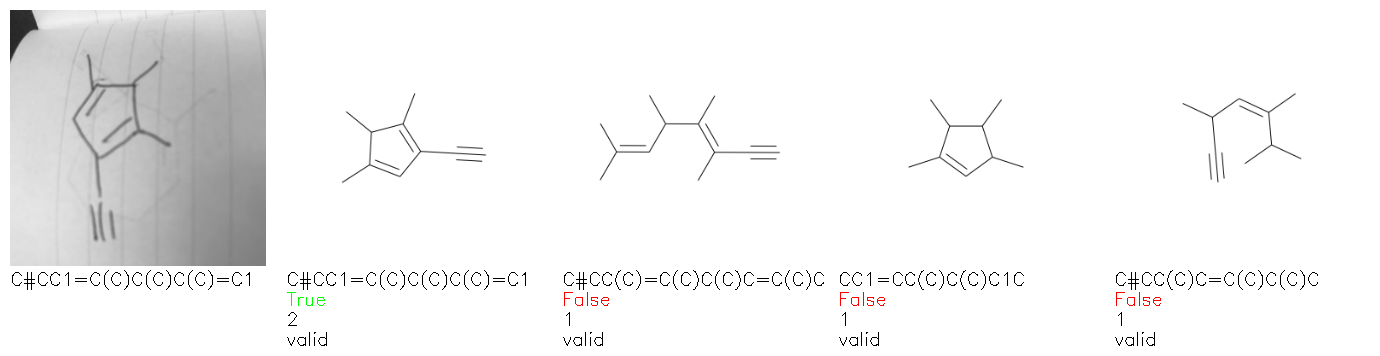
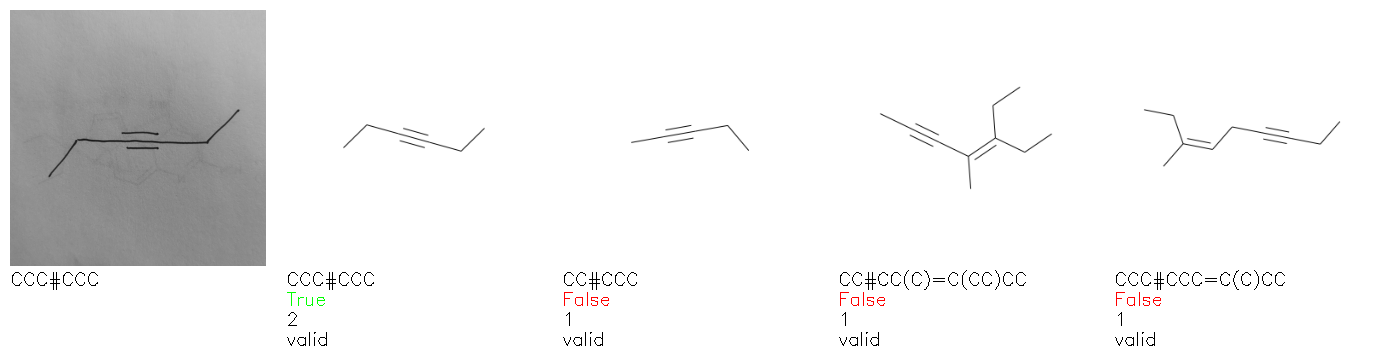
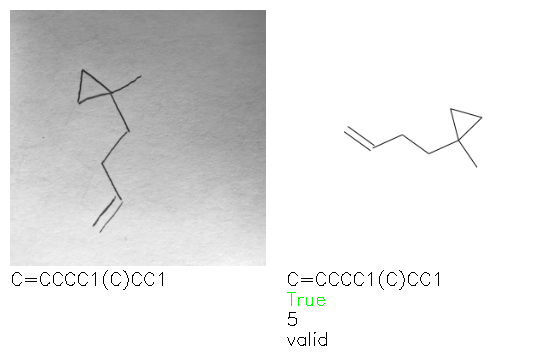
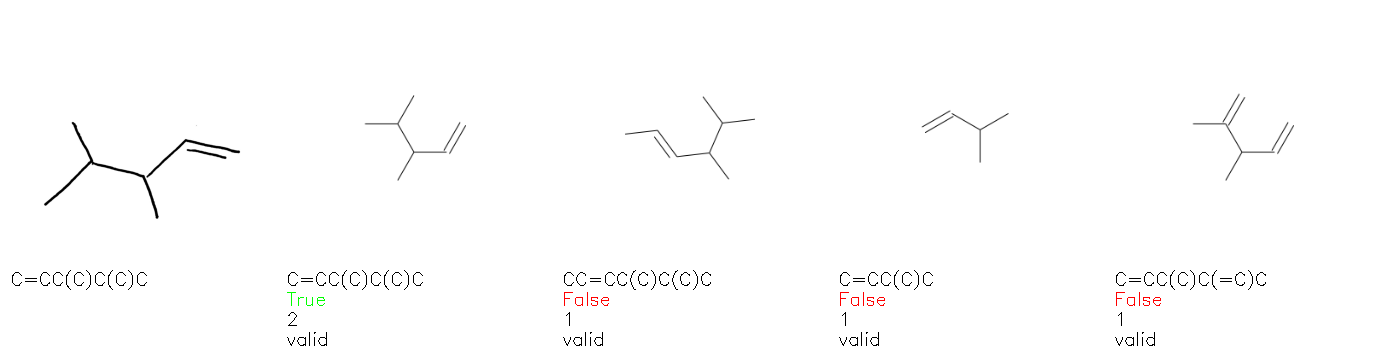
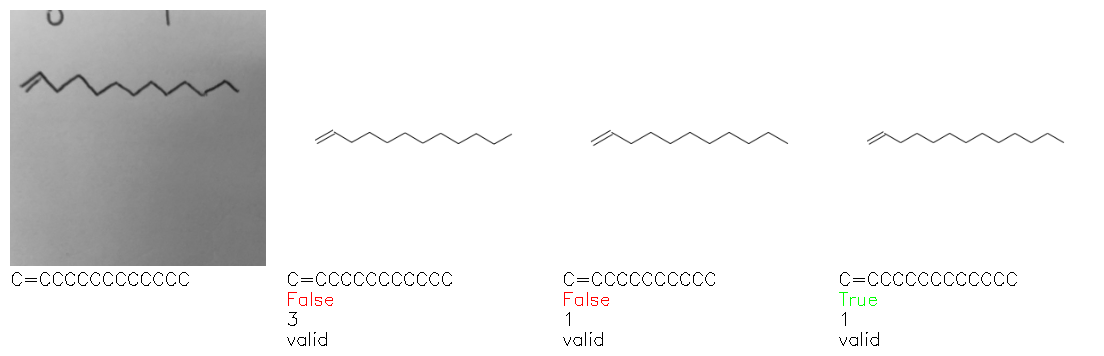
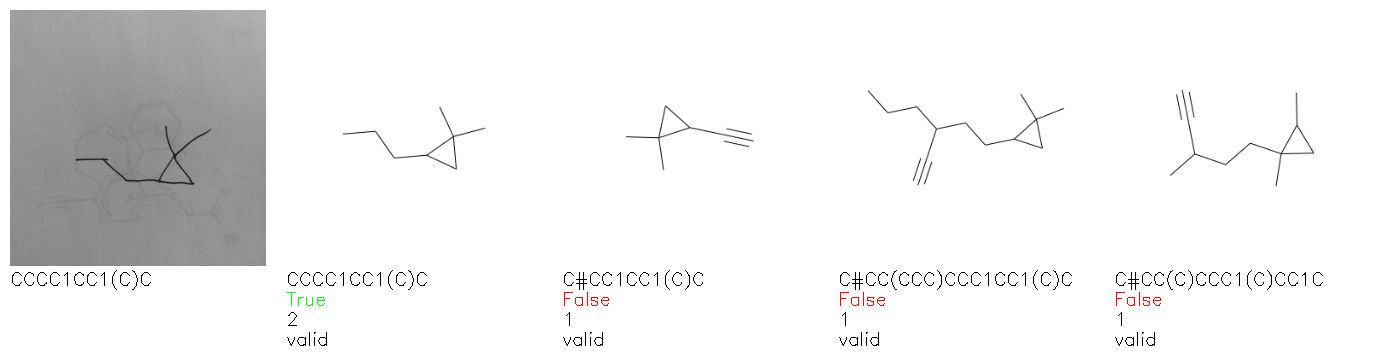
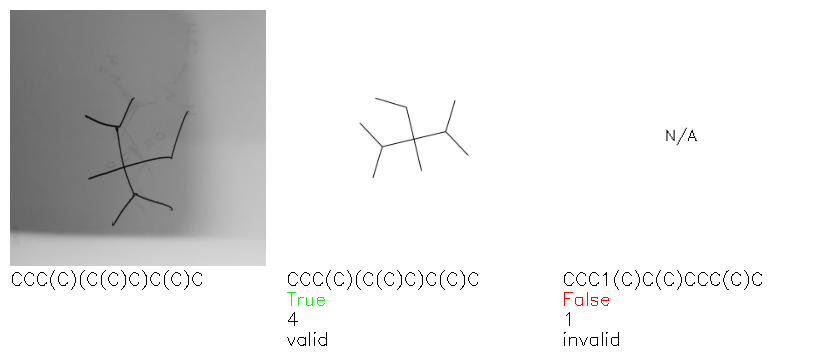
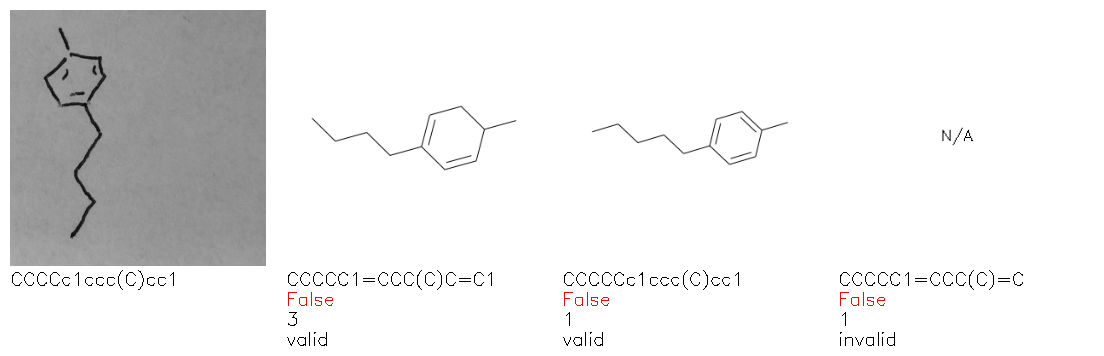
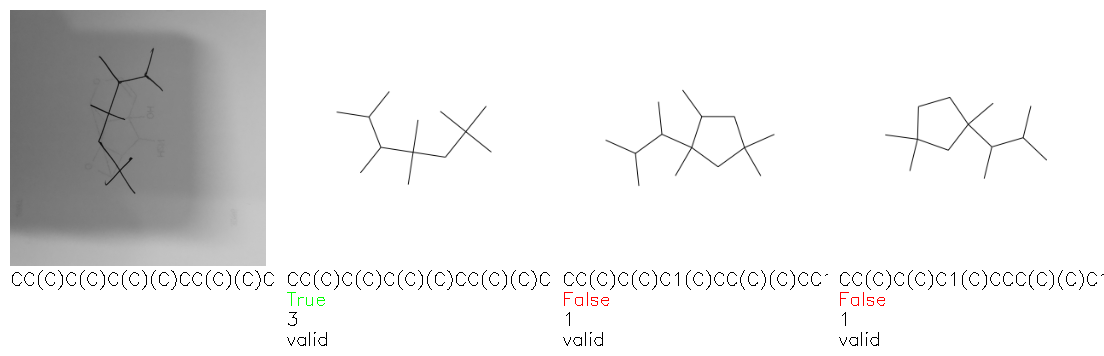
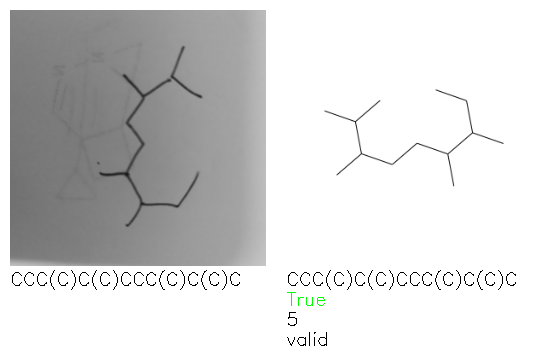
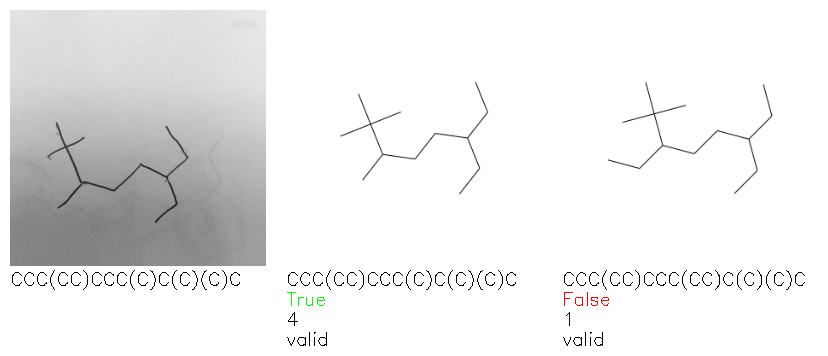
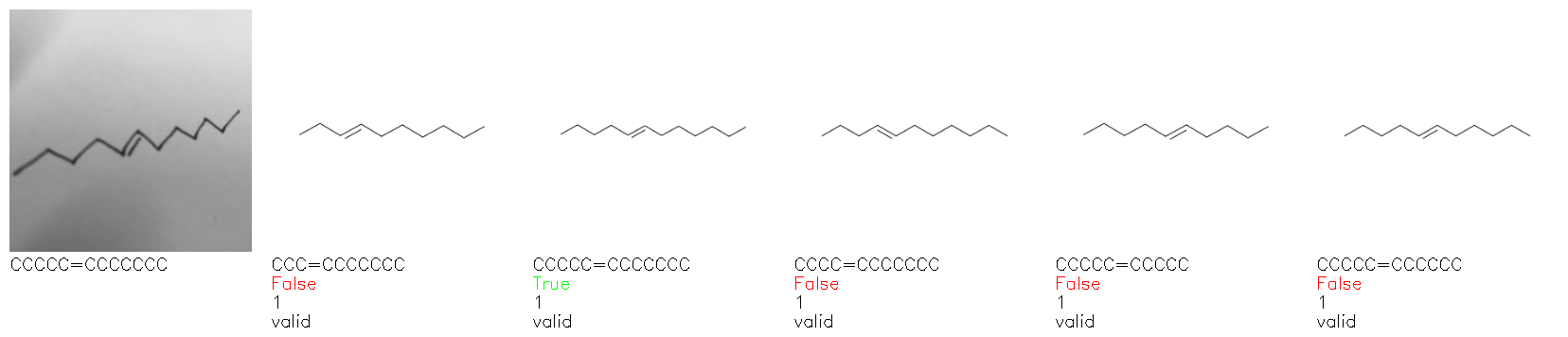
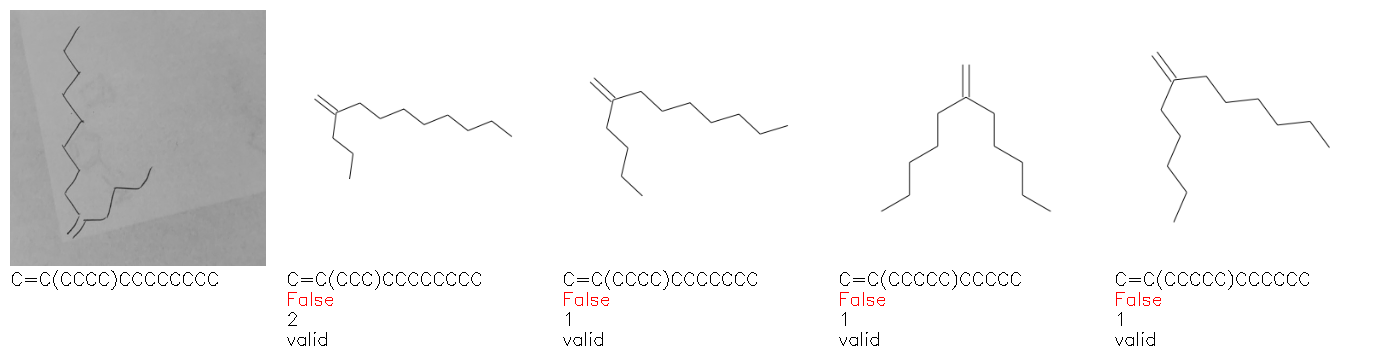
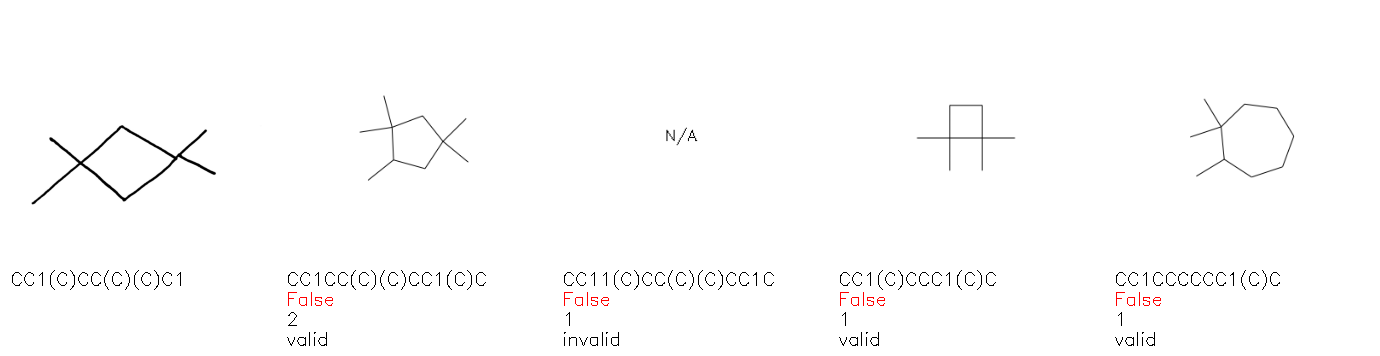
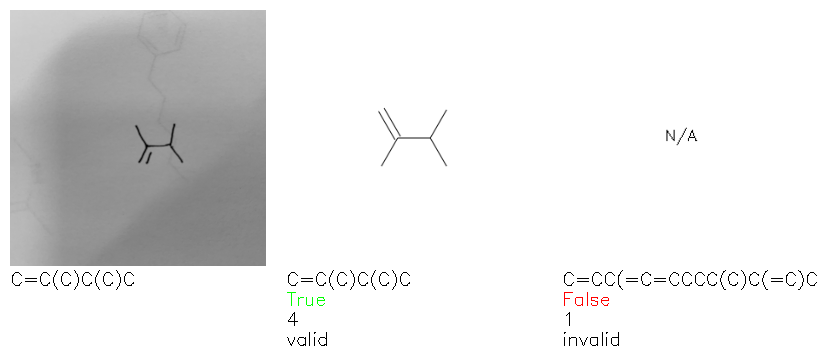
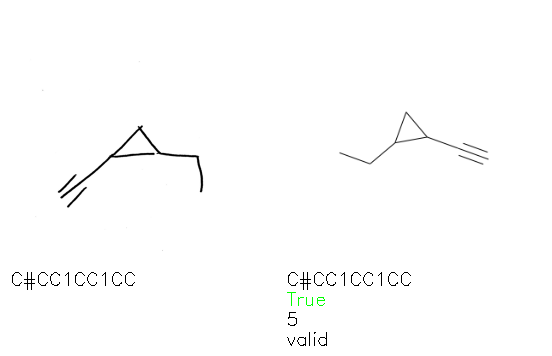
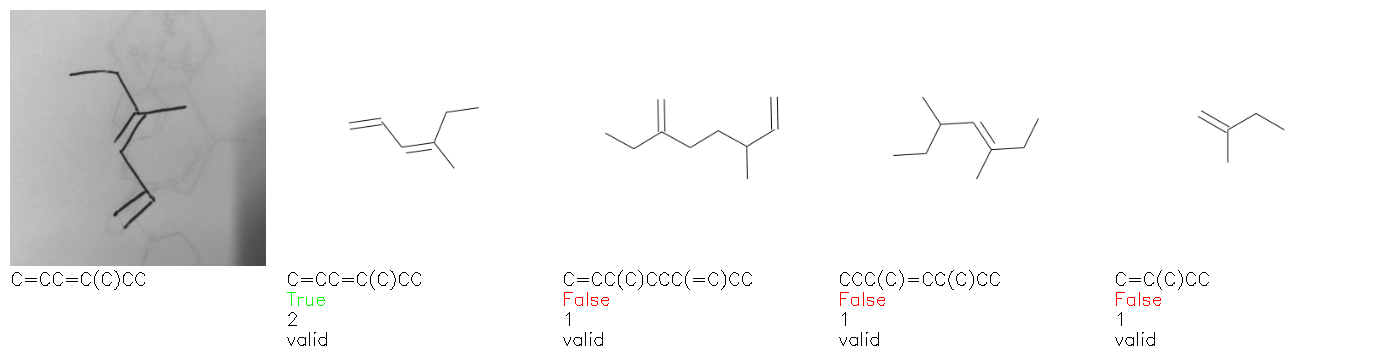
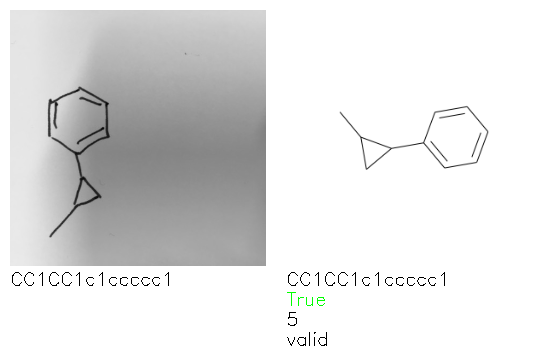
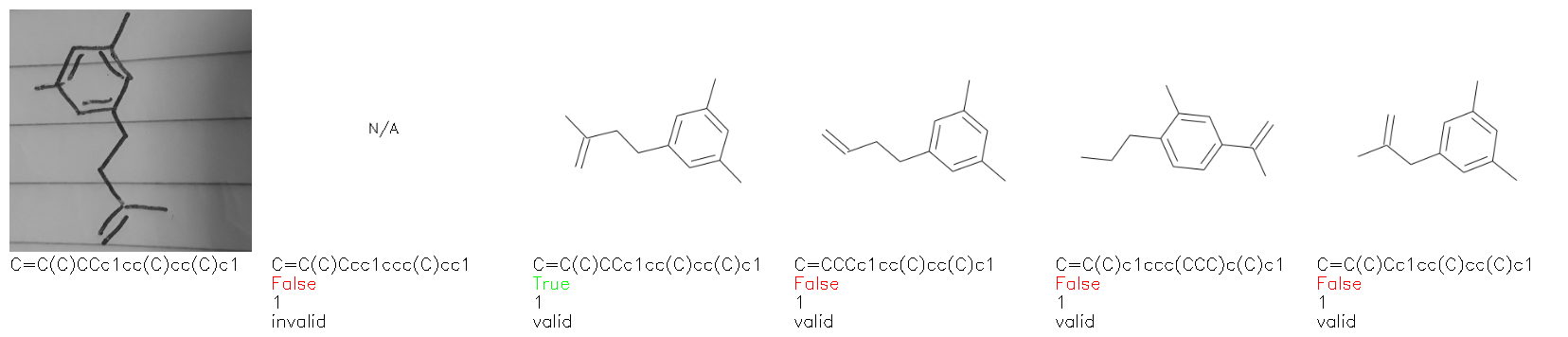
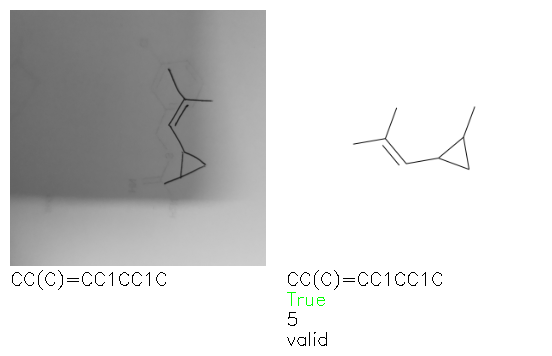
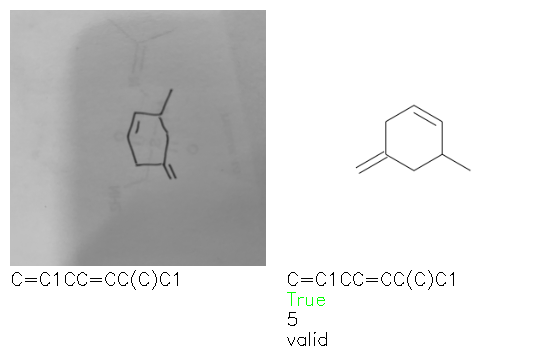
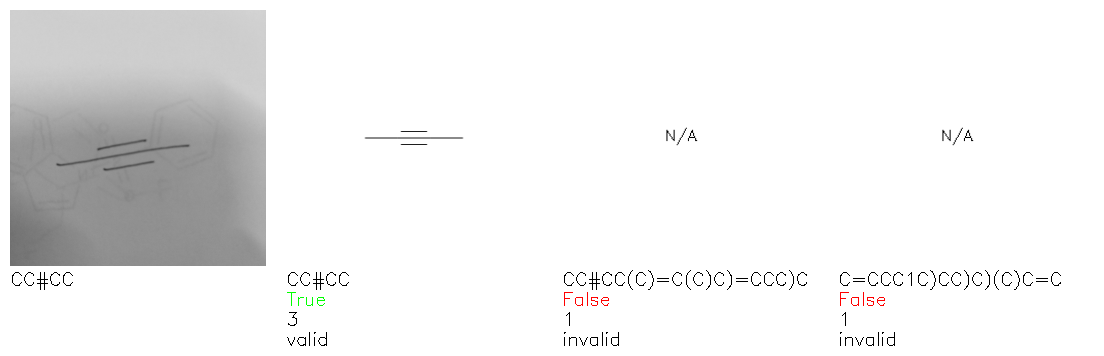
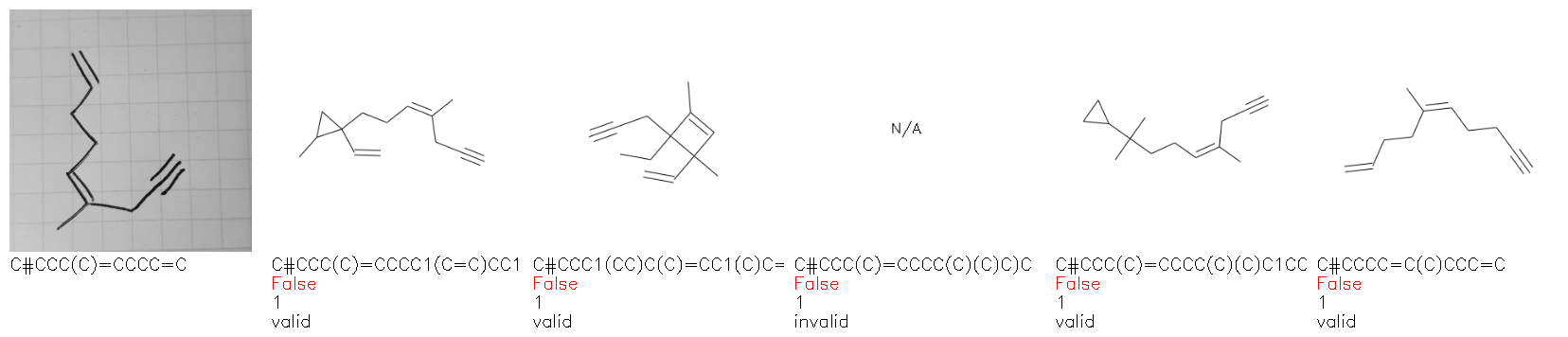
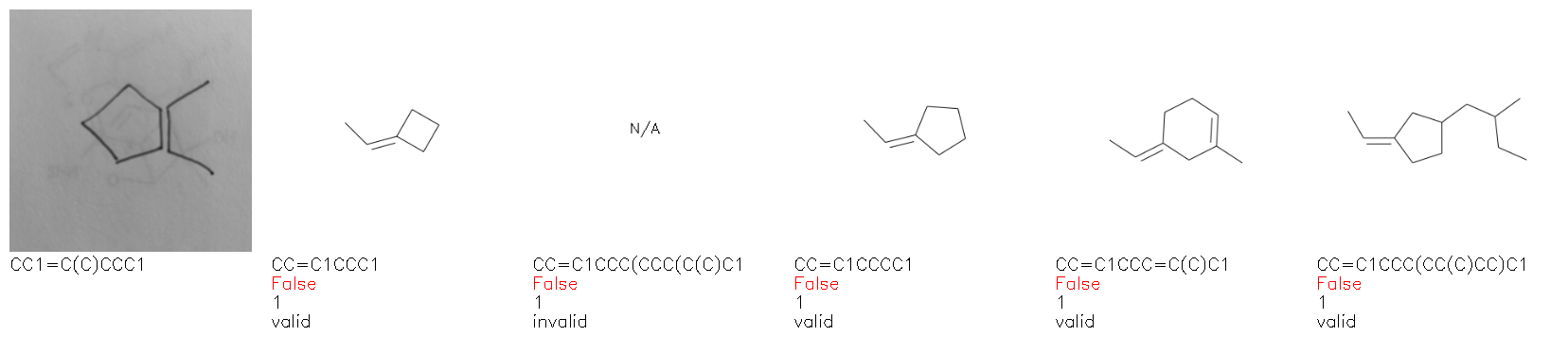
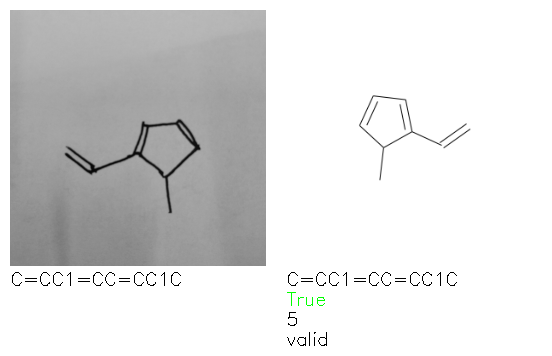
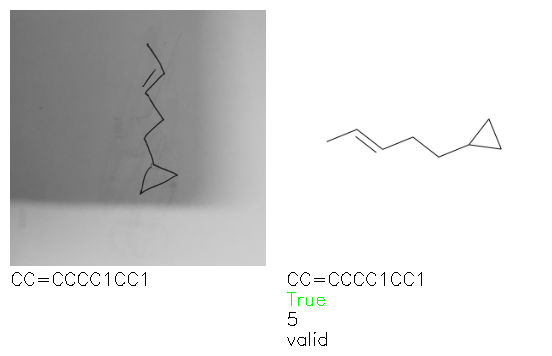
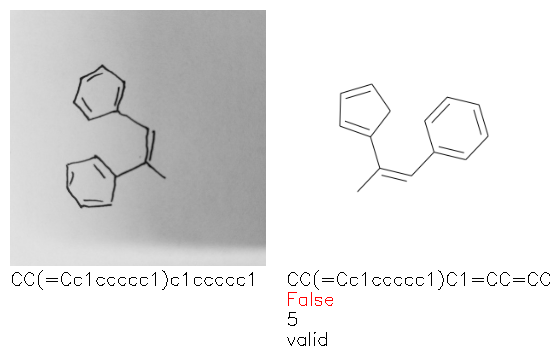
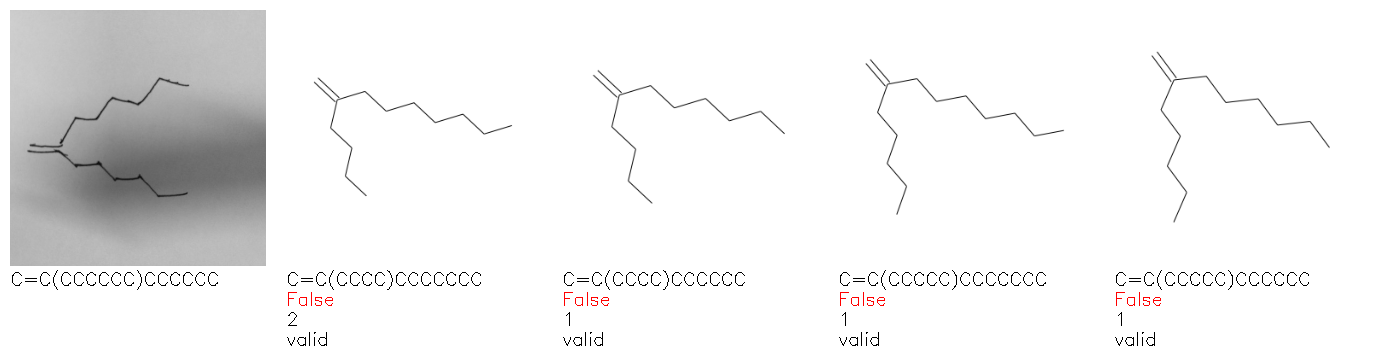
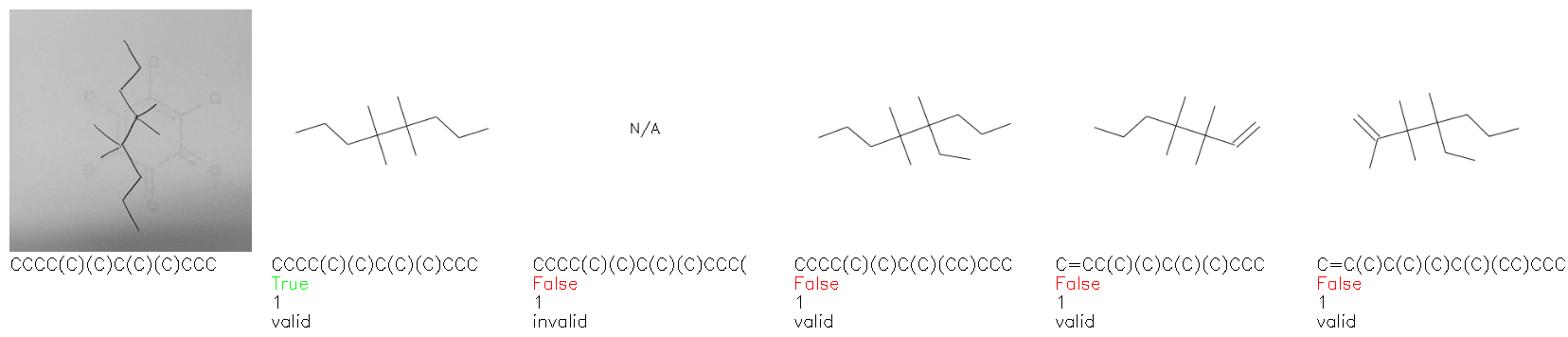
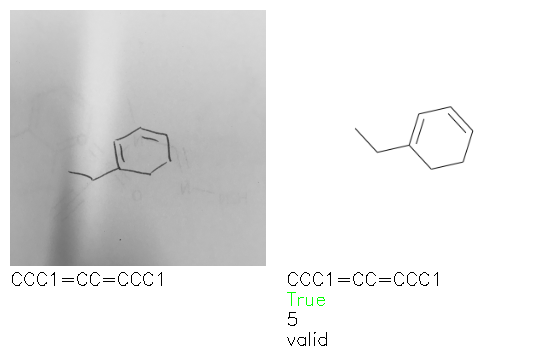
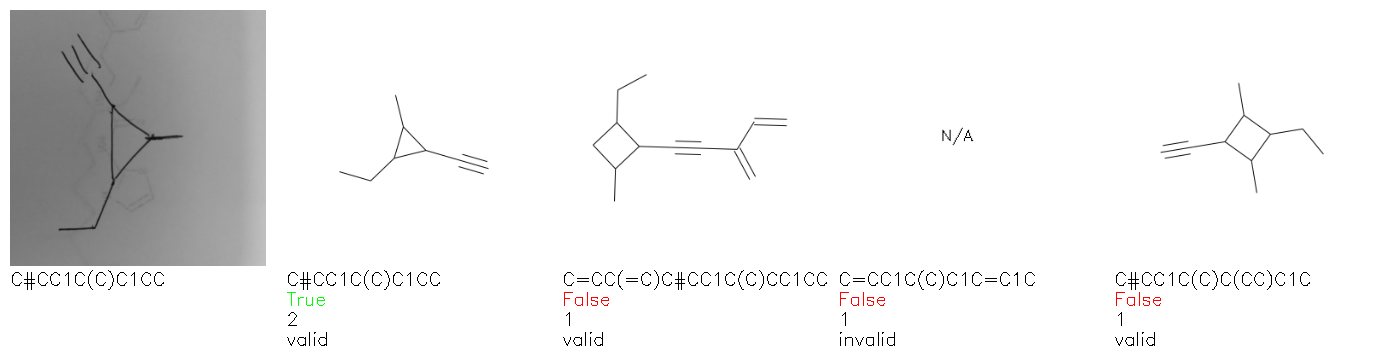
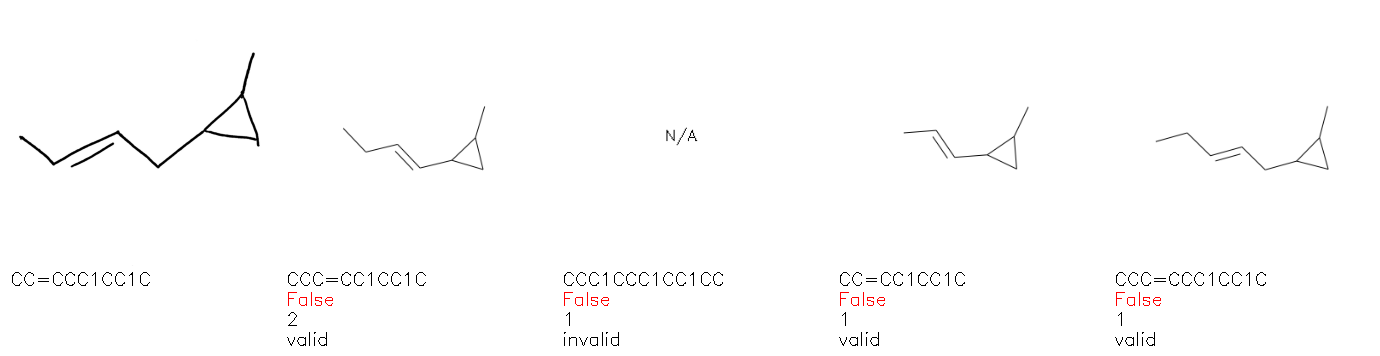
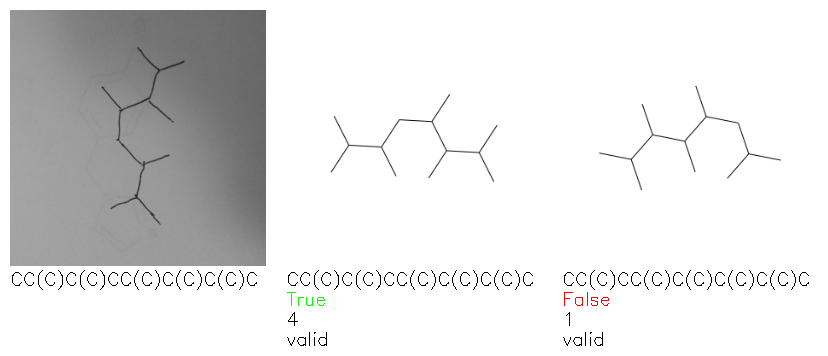
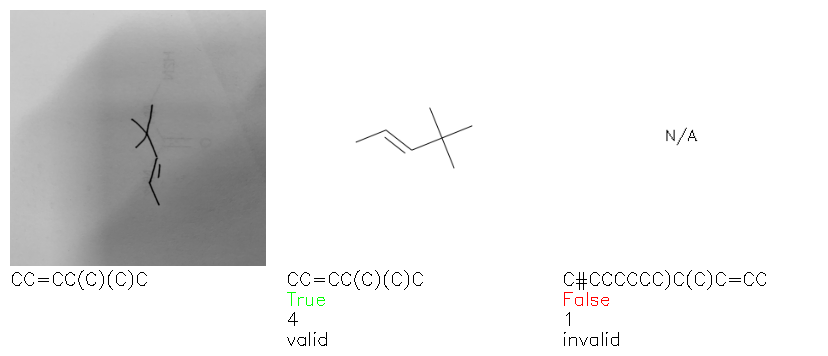
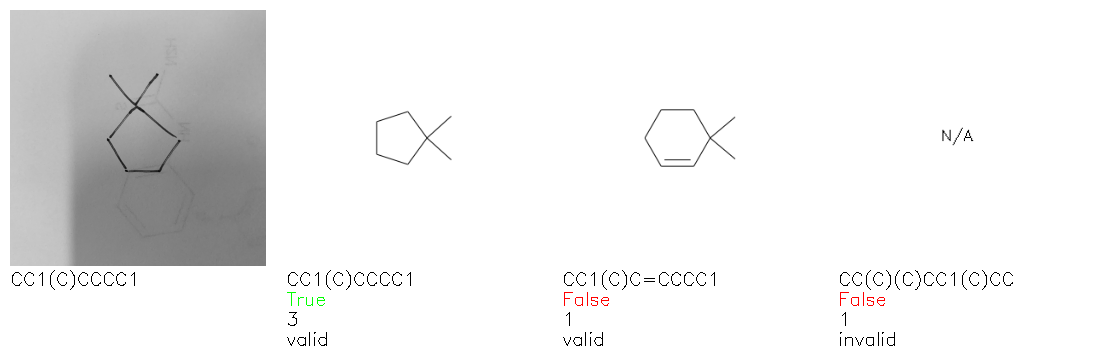
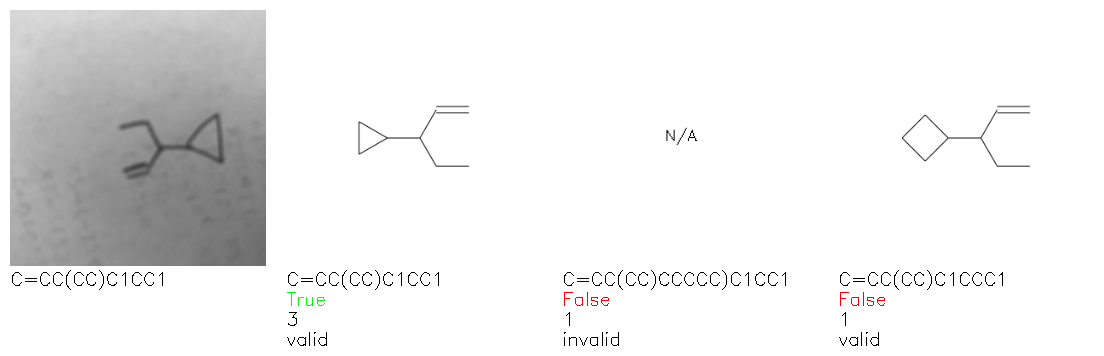
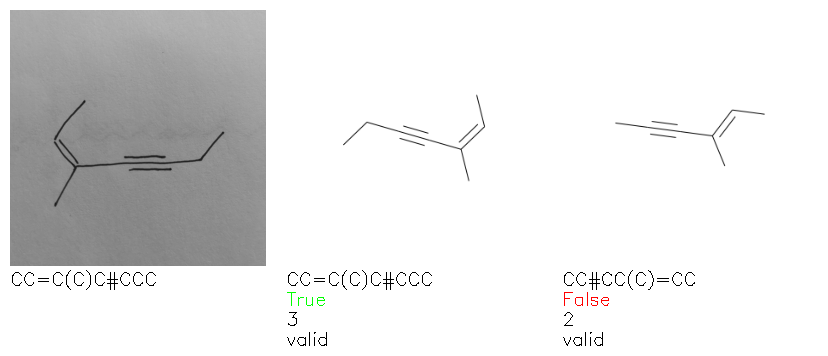
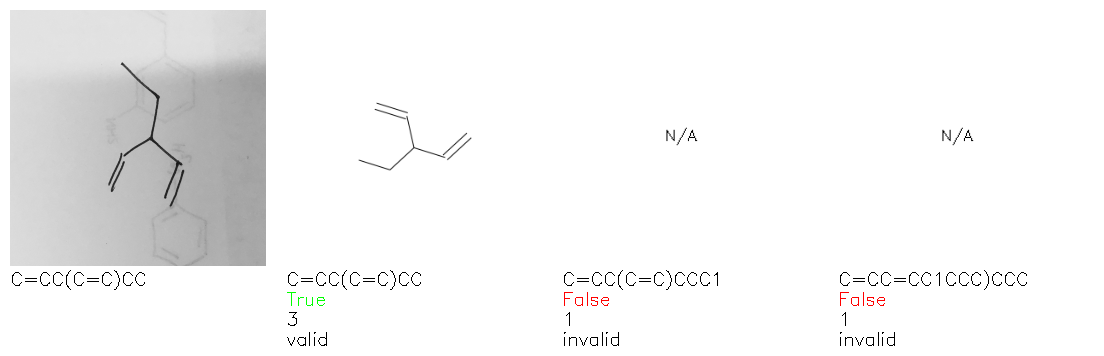
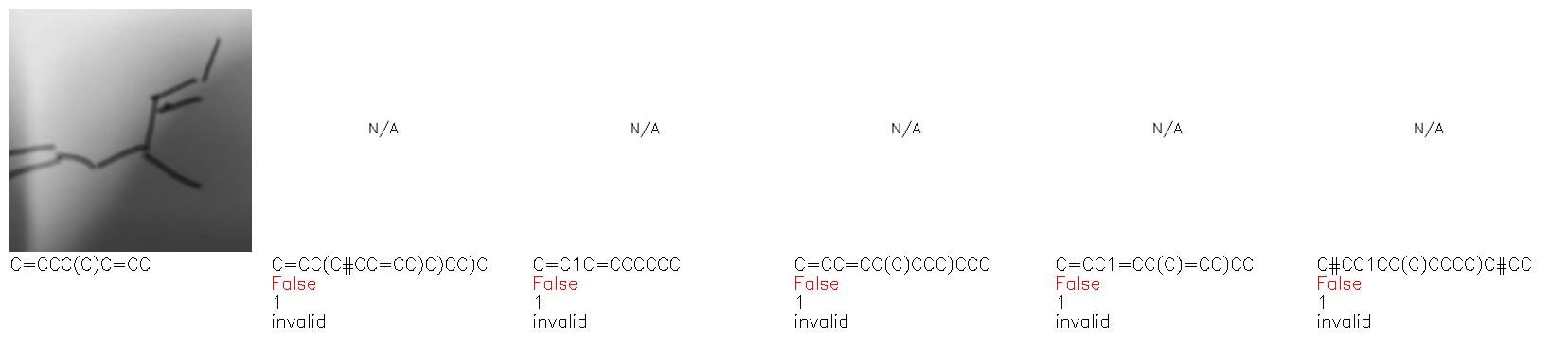
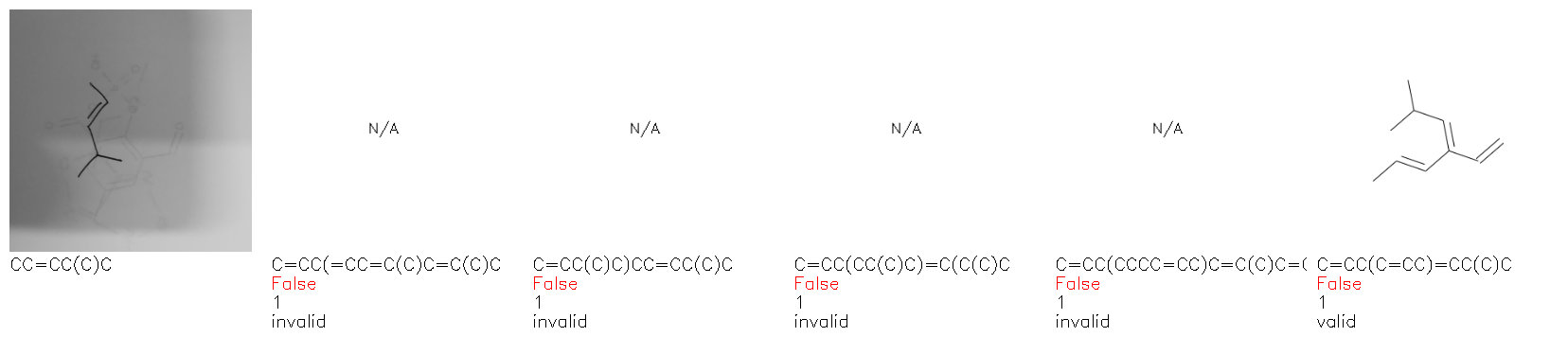
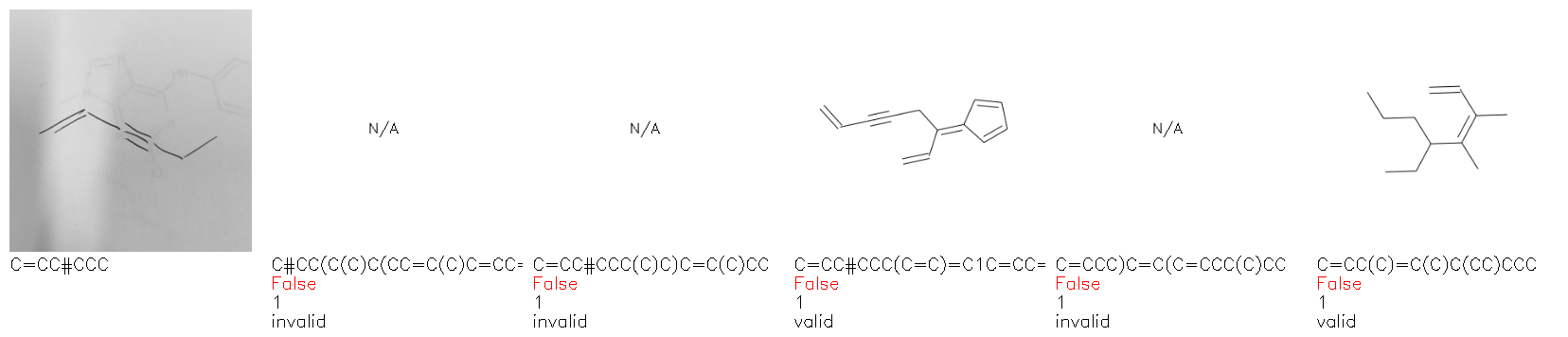
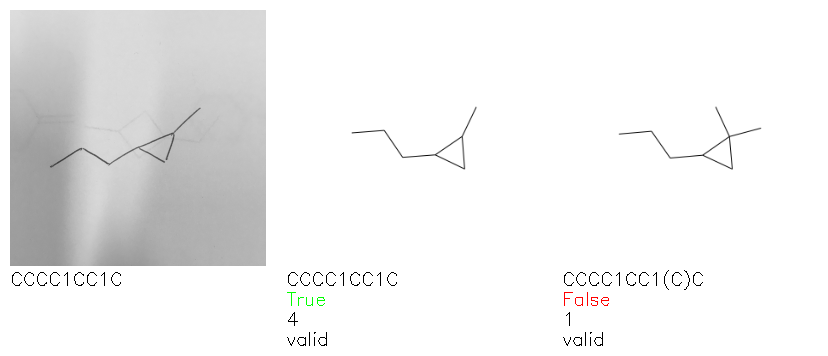
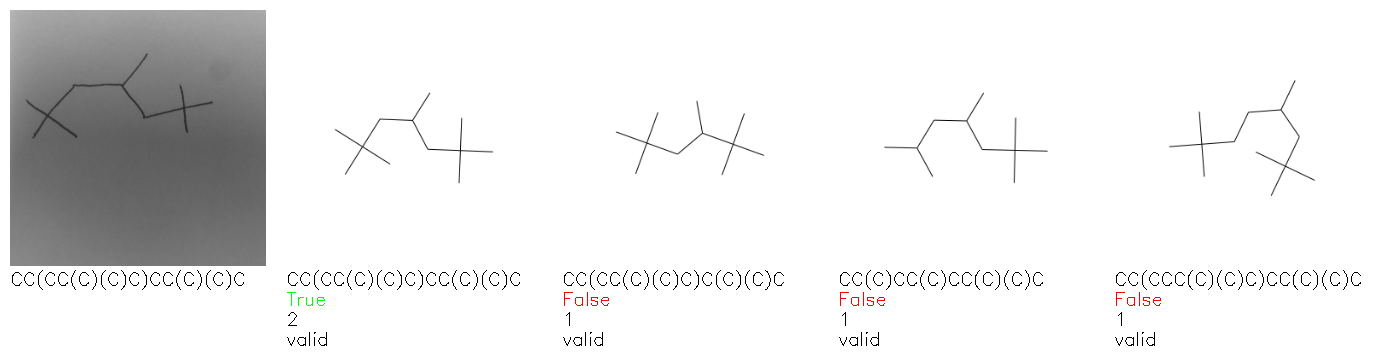
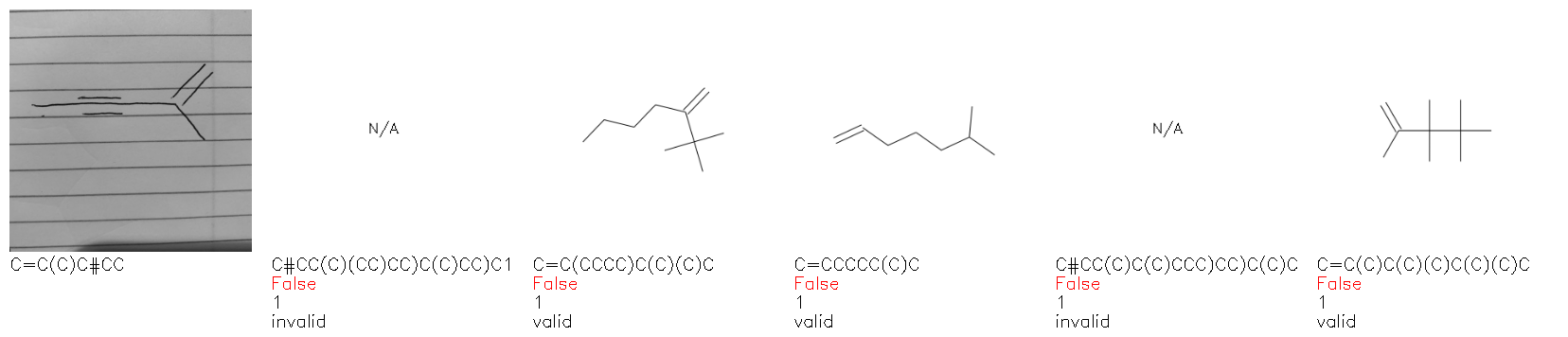
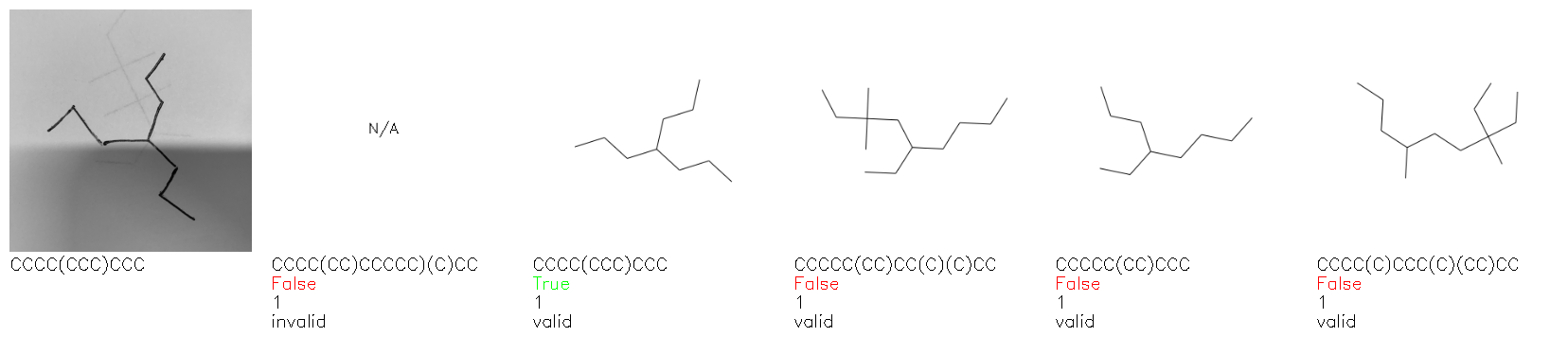
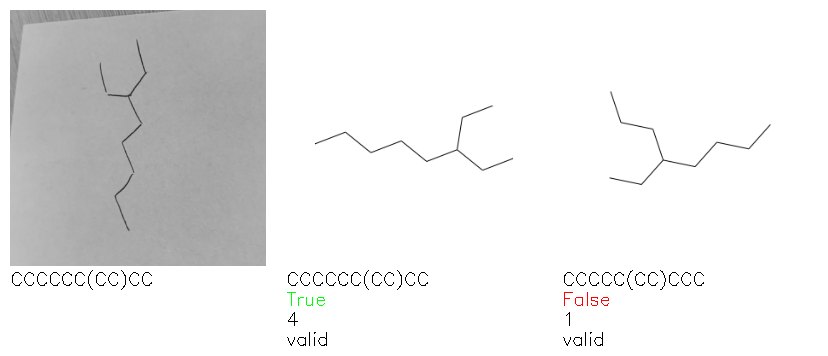
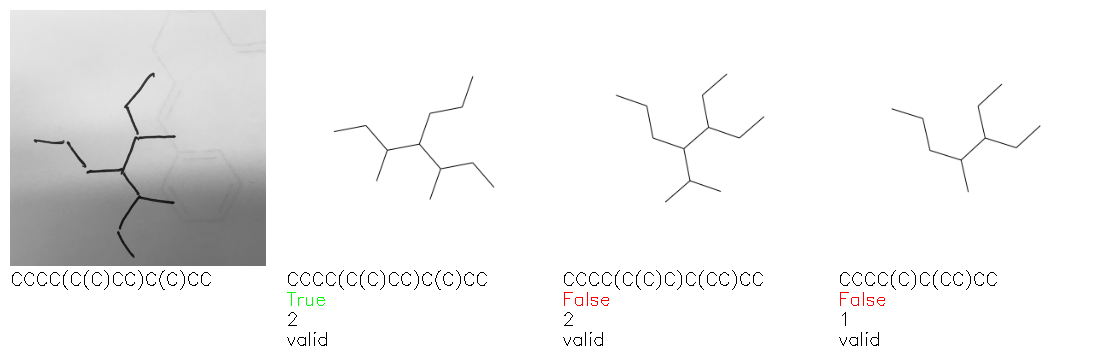
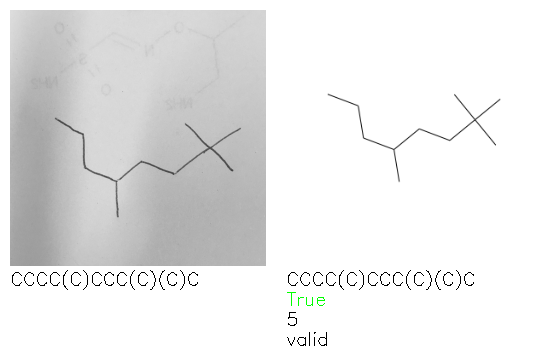
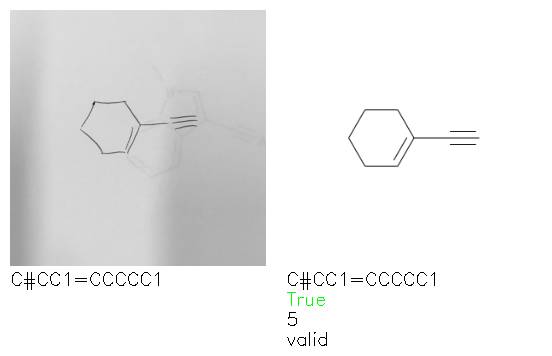
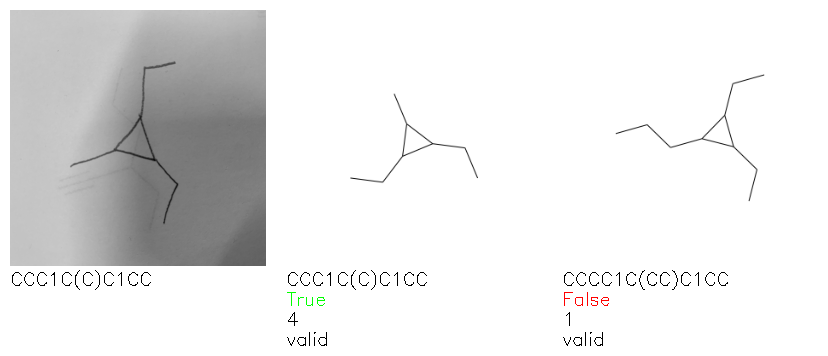
Fig. 8. Representative examples of cyclic (top) and acyclic (bottom) molecules from the hand-drawn hydrocarbon test set and their corresponding predictions from the ensemble. The input image is presented next to the predictions; the number of votes for each predicted molecule, V, is shown, along with if the molecule was recognised correctly. Predictions of invalid SMILES strings are shown as N/A. Hydrocarbons that are recognised correctly by one of the models predictions are outlined in green, and those that fail to be predicted correctly are outlined in red.

As far as we can determine, there is not a clear pattern between molecules that are predicted correctly and incorrectly, however we notice some features that make the recognition more challenging. Molecules drawn on lined and squared paper can increase the difficulty in comparison to those drawn on plain paper. The networks also struggle more when benzene rings are drawn in the resonance hybrid style (with a circle) in comparison to the Kekulé structure. This is likely due to the RDKit generated training imaged being exclusively Kekulé. As discussed previously, incorrectly predicted structures generally have disagreeing committee members. A rare case in which all the committee members agree on an incorrect prediction is shown in Fig. 8: α-Methyl-cis-stilbene is wrongly identified since two bonds are mistaken for one, resulting in a structure that is very close to correct. It is common for wrong predictions to contain only a minor mistake such as this. We also note that a large portion of the images in the hand-drawn dataset consists of molecules that are drawn on paper with chemical structures drawn on the opposite side of the page. In many cases, these structures bleed through the page, confusing the network. Lastly, we note that the model currently does not handle conjoined rings due to limitations of RDKit's image generation, which depicts bridges differently from the standard chemistry drawing style. This could be addressed by applying a different chemical structure renderer and/or collecting more hand-drawn structure data. The full test set with the corresponding reference and predicted SMILES can be found in the ESI.†
Fig. 9a highlights the SMILES error for predictions of invalid molecules. The largest portion of errors corresponds to unclosed or unopened parenthesis, with the next most prominent error being rings left unclosed or the closure duplicated. This gives insight into the somewhat lower accuracy of branched molecules and rings. Lastly, a small portion of errors correspond to carbons with a valence greater than four, syntactical errors (e.g. a SMILES string ending in “=”), and aromatic carbons outside of a ring. Invalid SMILES predictions are quite rare (6.5% of the total predictions), and tend to be correlated with challenging images where the model has low confidence of the prediction.
Fig. 9. (a) Proportion of errors associated with invalid SMILES predictions. (b) Recognition accuracy of highest ranked ensemble prediction for subsets of the hand-drawn hydrocarbon test set. From left-to-right: all hydrocarbons (vocab: “Cc=#()1”), acyclic hydrocarbons only (vocab: “C=#()”), cyclic hydrocarbons only (vocab: “Cc=#()1”, must contain “1”), unbranched hydrocarbons only (vocab:“Cc=#1”), invalid predicted SMILES removed from predictions.

To gain insight into if the model more or less accurately recognizes certain types of molecules, we compute the accuracy of the ensemble's first prediction for subsets of the test set, including acyclic, cyclic and unbranched hydrocarbons (Fig. 9b). The recognition accuracy is seen to be relatively consistent between the different groups of molecules, however, molecules without rings are correctly recognised slightly more often than those with rings, and un-branched molecules (those without “()” in their SMILES string) are more accurate still. We also investigate the effect of removing all invalid SMILES from the predictions, which leads to an insignificant change in accuracy.
Conclusions
In this work, we demonstrate how deep learning can be used to develop an offline hand-drawn hydrocarbon structure recognition tool. We curated a large synthetic dataset and a small hand-drawn dataset and explored how to best leverage the two to maximize molecule recognition accuracy. The datasets were used to train an image-to-SMILES neural network to extract the molecule from a photographed hand-drawn hydrocarbon structure. Training with synthetic data only leads to only 50% recognition accuracy on real-life hand-drawn hydrocarbons. Replacing a small fraction of the training set with augmented hand-drawn images and applying fine-tuning leads to an improvement of hand-drawn hydrocarbon recognition accuracy to nearly 70%. The trained data-driven models were combined with ensemble learning to achieve superior accuracy to the constituent models and gain information on when the model would fail. The final model achieved an accuracy of 76%, and the top three predictions included the exactly correct molecule over 85% of the time.
Extending the hydrocarbon recognition results presented in this paper to the recognition of all molecules offers an obvious extension, however, variation in hand-drawn font style and letter location provides a significant challenge. A hybrid rule-based and data-driven workflow offers one strategy to overcome these barriers. For example, a functional group detector network and hydrocarbon backbone recognition network, such as that presented in this study, could be combined with a rule-based model to produce the complete molecular structure. We also plan to explore neural style transfer to enhance the quality of the synthetic data.53
The chemical structure recognition software developed in this work has many interesting use cases, such as connecting it to a user interface to be used as a phone or tablet application. A wide range of chemistry software could then be connected to the backend such as theoretical chemistry packages, lab notebooks and analytical tools. It would be particularly useful for software that currently requires knowledge of coding, command line scripting, and specialized input file format and so is inaccessible to large sections of the chemistry community. Connection to ChemVox6 voice control and TeraChem Cloud54 electronic structure service offers one example of a potentially powerful integrated tool. Since drawing a chemical structure by hand is a familiar task for all chemists, this app would lower the barrier of accessing such software. As a result, these currently unattainable tools could be readily incorporated into laboratories and classrooms to catalyse advances in both chemical research and education.
Data availability
Data and source code can be found at https://github.com/mtzgroup/ChemPixCH.
Author contributions
HW, KT, and TJM conceptualized the project and contributed to methodology. HW wrote the original draft. All authors contributed to reviewing and editing of the manuscript. HW led the investigation and implemented the models. AW, BC, and AB contributed to data generation and acquisition. TJM supervised the project and contributed to funding acquisition, resources, and project administration.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by the Office of Naval Research (N00014-18-1-2659).
Electronic supplementary information (ESI) available: Details of image processing, neural network training, and example image predictions. Link to code to generate data and run training experiments. See DOI: 10.1039/d1sc02957f
References
- Rosenblatt F. The perceptron: a probabilistic model for information storage and organization in the brain. Psychol. Rev. 1958;65:386. doi: 10.1037/h0042519. [DOI] [PubMed] [Google Scholar]
- Goodfellow I., Bengio Y., Courville A. and Bengio Y., Deep learning, MIT Press, Cambridge, 2016 [Google Scholar]
- Noé F. Tkatchenko A. Müller K.-R. Clementi C. Machine learning for molecular simulation. Annu. Rev. Phys. Chem. 2020;71:361–390. doi: 10.1146/annurev-physchem-042018-052331. [DOI] [PubMed] [Google Scholar]
- Rupp M. Tkatchenko A. Müller K.-R. Von Lilienfeld O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012;108:058301. doi: 10.1103/PhysRevLett.108.058301. [DOI] [PubMed] [Google Scholar]
- Behler J. Perspective: Machine learning potentials for atomistic simulations. J. Chem. Phys. 2016;145:170901. doi: 10.1063/1.4966192. [DOI] [PubMed] [Google Scholar]
- Raucci U. Valentini A. Pieri E. Weir H. Seritan S. Martinez T. J. Voice-controlled quantum chemistry. Nat. Comput. Sci. 2021;1:42–45. doi: 10.1038/s43588-020-00012-9. [DOI] [PubMed] [Google Scholar]
- Bluche T., Louradour J. and Messina R., Scan, attend and read: End-to-end handwritten paragraph recognition with mdlstm attention, Proceedings of 14th IAPR International Conference on Document Analysis and Recognition, ICDAR, 2017, pp. 1050–1055 [Google Scholar]
- Michael J., Labahn R., Grüning T. and Zöllner J., Evaluating sequence-to-sequence models for handwritten text recognition, Proceedings of International Conference on Document Analysis and Recognition (ICDAR), 2019, pp. 1286–1293 [Google Scholar]
- Graves A. and Schmidhuber J., Offline handwriting recognition with multidimensional recurrent neural networks, Proceedings of Advances in Neural Information Processing Systems, 2009, pp. 545–552 [Google Scholar]
- Ingle R. R., Fujii Y., Deselaers T., Baccash J. and Popat A. C., A scalable handwritten text recognition system, Proceedings of 2019 International Conference on Document Analysis and Recognition ICDAR, 2019, pp. 17–24 [Google Scholar]
- Plamondon R. Srihari S. N. Online and off-line handwriting recognition: a comprehensive survey. IEEE Trans Pattern Anal Mach Intell. 2000;22:63–84. [Google Scholar]
- Rozas R. Fernandez H. Automatic processing of graphics for image databases in science. J. Chem. Inf. Comput. Sci. 1990;30:7–12. [Google Scholar]
- Contreras M. L. Allendes C. Alvarez L. T. Rozas R. Computational perception and recognition of digitized molecular structures. J. Chem. Inf. Comput. Sci. 1990;30:302–307. [Google Scholar]
- McDaniel J. R. Balmuth J. R. Kekule: OCR-optical chemical (structure) recognition. J. Chem. Inf. Comput. Sci. 1992;32:373–378. [Google Scholar]
- Casey R., Boyer S., Healey P., Miller A., Oudot B. and Zilles K., Optical recognition of chemical graphics, Proceedings of 2nd International Conference on Document Analysis and Recognition (ICDAR'93), 1993, pp. 627–631 [Google Scholar]
- Ibison P. Jacquot M. Kam F. Neville A. Simpson R. W. Tonnelier C. Venczel T. Johnson A. P. Chemical literature data extraction: the CLiDE Project. J. Chem. Inf. Comput. Sci. 1993;33:338–344. [Google Scholar]
- Rajan K. Brinkhaus H. O. Zielesny A. Steinbeck C. A review of optical chemical structure recognition tools. J. Cheminf. 2020;12:1–13. doi: 10.1186/s13321-020-00465-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkoutos G. V. Rzepa H. Clark R. M. Adjei O. Johal H. Chemical machine vision: automated extraction of chemical metadata from raster images. J. Chem. Inf. Comput. Sci. 2003;43:1342–1355. doi: 10.1021/ci034017n. [DOI] [PubMed] [Google Scholar]
- Rosania G. R. Crippen G. Woolf P. Shedden K. A cheminformatic toolkit for mining biomedical knowledge. Pharm. Res. 2007;24:1791–1802. doi: 10.1007/s11095-007-9285-5. [DOI] [PubMed] [Google Scholar]
- Algorri M.-E., Zimmermann M., Friedrich C. M., Akle S. and Hofmann-Apitius M., Reconstruction of chemical molecules from images, Proceedings of 29th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 2007, pp. 4609–4612 [DOI] [PubMed] [Google Scholar]
- Valko A. T. Johnson A. P. CLiDE Pro: the latest generation of CLiDE, a tool for optical chemical structure recognition. J. Chem. Inf. Model. 2009;49:780–787. doi: 10.1021/ci800449t. [DOI] [PubMed] [Google Scholar]
- Filippov I. V. Nicklaus M. C. Optical structure recognition software to recover chemical information: OSRA, an open source solution. J. Chem. Inf. Model. 2009;49:740–743. doi: 10.1021/ci800067r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. Rosania G. R. Shedden K. A. Nguyen M. Lyu N. Saitou K. Automated extraction of chemical structure information from digital raster images. Chem. Cent. J. 2009;3:4. doi: 10.1186/1752-153X-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Saitou K. and Rosania G., Image-based automated chemical database annotation with ensemble of machine-vision classifiers, Proceedings of 2010 IEEE International Conference on Automation Science and Engineering, 2010, pp. 168–173 [Google Scholar]
- Sadawi N. M., Sexton A. P. and Sorge V., Chemical structure recognition: a rule-based approach, Proceedings of Document Recognition and Retrieval XIX, 2012, p. 82970E [Google Scholar]
- Tharatipyakul A., Numnark S., Wichadakul D. and Ingsriswang S., ChemEx: information extraction system for chemical data curation, Proceedings of BMC Bioinformatics, 2012, vol. S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasconi P. Gabbrielli F. Lippi M. Marinai S. Markov logic networks for optical chemical structure recognition. J. Chem. Inf. Model. 2014;54:2380–2390. doi: 10.1021/ci5002197. [DOI] [PubMed] [Google Scholar]
- Beard E. J. Cole J. M. ChemSchematicResolver: A Toolkit to Decode 2D Chemical Diagrams with Labels and R-Groups into Annotated Chemical Named Entities. J. Chem. Inf. Model. 2020;60:2059–2072. doi: 10.1021/acs.jcim.0c00042. [DOI] [PubMed] [Google Scholar]
- Staker J. Marshall K. Abel R. McQuaw C. M. Molecular Structure Extraction from Documents Using Deep Learning. J. Chem. Inf. Model. 2019;59:1017–1029. doi: 10.1021/acs.jcim.8b00669. [DOI] [PubMed] [Google Scholar]
- Oldenhof M., Arany A., Moreau Y. and Simm J., ChemGrapher: Optical Graph Recognition of Chemical Compounds by Deep Learning, arXiv preprint arXiv:2002.09914, 2020 [DOI] [PubMed]
- Rajan K. Zielesny A. Steinbeck C. DECIMER: towards deep learning for chemical image recognition. J. Cheminf. 2020;12:1–9. doi: 10.1186/s13321-020-00469-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang T. Y. and Davis R., Recognition of hand drawn chemical diagrams, Proceedings of AAAI, 2007, pp. 846–851 [Google Scholar]
- Ramel J.-Y., Boissier G. and Emptoz H., Automatic reading of handwritten chemical formulas from a structural representation of the image, Proceedings of Fifth International Conference on Document Analysis and Recognition. ICDAR'99, 1999, pp. 83–86 [Google Scholar]
- VISIONARCANUM, InkToMolecule online, https://visionarcanum.com/ink2mol/, accessed May 1, 2021 [Google Scholar]
- Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988;28:31–36. [Google Scholar]
- Hirschberg J. Manning C. D. Advances in natural language processing. Science. 2015;349:261–266. doi: 10.1126/science.aaa8685. [DOI] [PubMed] [Google Scholar]
- Vinyals O., Toshev A., Bengio S. and Erhan D., Show and tell: A neural image caption generator, Proceedings of IEEE Conference on Computer Vision and Pattern Recognition, 2015, pp. 3156–3164 [Google Scholar]
- Xu K., Ba J., Kiros R., Cho K., Courville A., Salakhudinov R., Zemel R. and Bengio Y., Show, attend and tell: Neural image caption generation with visual attention, Proceedings of International conference on machine learning, 2015, pp. 2048–2057 [Google Scholar]
- LeCun Y. Bengio Y. Hinton G. Deep learning. Nature. 2015;521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Krizhevsky A. Sutskever I. Hinton G. E. Imagenet classification with deep convolutional neural networks. Commun. ACM. 2017;60:84–90. [Google Scholar]
- Hochreiter S. Schmidhuber J. Long short-term memory. Neural Comput. 1997;9:1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- Deng Y., Kanervisto A., Ling J. and Rush A. M., Image-to-markup generation with coarse-to-fine attention, Proceedings of International Conference on Machine Learning, 2017, pp. 980–989 [Google Scholar]
- Fink T. Bruggesser H. Reymond J. L. Virtual exploration of the small-molecule chemical universe below 160 daltons. Angew. Chem. Int. Ed. 2005;44:1504–1508. doi: 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- Fink T. Reymond J.-L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model. 2007;47:342–353. doi: 10.1021/ci600423u. [DOI] [PubMed] [Google Scholar]
- Blum L. C. Reymond J.-L. 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 2009;131:8732–8733. doi: 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- Bradski G., The OpenCV Library, Dr Dobb's Journal of Software Tools, 2000, 120, pp. 122–125 [Google Scholar]
- Weir H., ChemPixCH, 2021, https://github.com/mtzgroup/ChemPixCH
- Wang Q., Gao J., Lin W. and Yuan Y., Learning from synthetic data for crowd counting in the wild, Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, 2019, pp. 8198–8207 [Google Scholar]
- Kuznichov D., Zvirin A., Honen Y. and Kimmel R., Data augmentation for leaf segmentation and counting tasks in rosette plants, Proceedings of IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops, 2019 [Google Scholar]
- Tajbakhsh N. Shin J. Y. Gurudu S. R. Hurst R. T. Kendall C. B. Gotway M. B. Liang J. Convolutional neural networks for medical image analysis: Full training or fine tuning? IEEE Trans. Med. Imaging. 2016;35:1299–1312. doi: 10.1109/TMI.2016.2535302. [DOI] [PubMed] [Google Scholar]
- Bishop C. M., Neural networks for pattern recognition, Oxford University Press, 1995 [Google Scholar]
- Polikar R., Ensemble learning in Ensemble machine learning, Springer, 2012, pp. 1–34 [Google Scholar]
- Gatys L. A., Ecker A. S. and Bethge M., Image style transfer using convolutional neural networks, Proceedings of IEEE Conference on Computer Vision and Pattern Recognition, 2016, pp. 2414–2423 [Google Scholar]
- Seritan S. Thompson K. Martínez T. J. TeraChem Cloud: A high-performance computing service for scalable distributed GPU-accelerated electronic structure calculations. J. Chem. Inf. Model. 2020;60:2126–2137. doi: 10.1021/acs.jcim.9b01152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and source code can be found at https://github.com/mtzgroup/ChemPixCH.