

Abstract
Mitogen-activated protein kinases (MAPKs) play critical roles in the central nervous system immune responses through glial function, which are regulated with relative selectivity (or preference) by MAPK phosphatases (MKP). Phosphorylated extracellular signal-regulated protein kinase (p-ERK) is preferentially dephosphorylated by MKP-3, which display little activity over p-p38 and p–c-Jun NH2-terminal kinases (p-JNK). It has been proposed that these substrate preferences may vary depending on tissue or functional cellular processes. Since astrocytes display a prominent activity of JNK > ERK under stressed or reactive phenotype, we hypothesize that MKP-3 possess a similar or differential substrate preference in astrocytes for JNK and ERK (ERK = JNK or JNK > ERK). We generated transient expression of MKP-3 by transfecting a specific cDNA in primary rat neonatal brain cortex astrocytes. Cells were stimulated with lipopolysaccharide (LPS), and MAPKs and downstream pro-inflammatory products were measured by Western blot and ELISA analyses. MKP-3 expression in primary astrocytes reduced LPS-induced p-ERK and p-p38 by ~50%, and p-JNK by ~75%, and moderately reduced nitrite oxide (NO), while completely blocked Interleukin (IL)-6 and tumor necrosis factor alpha (TNFα). We confirmed MKP-3 specific activity by developing a BV-2 microglia cell line stably overexpressing MKP-3 and using a specific siRNA against MKP-3. Our data demonstrate MKP-3 has differential substrate preference in astrocytes compared to other cells types, since it preferentially dephosphorylated p-JNK over p-ERK. Our results indicate also that astrocytic immune functions can be modulated by MKP-3 induction, a strategy that could be beneficial in neurological conditions in which astrocytes play a pathophysiological role, i.e. persistent pain.
Keywords: MKP-3, Phosphatase, Cytokines, Glia, Astrocytes, Microglia
1. Introduction
Mitogen-activated protein kinase (MAPK) phosphatases (MKPs) are the major deactivators (by dephosphorylation) of extracellular signal-regulated protein kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 kinase [4,8,20]. The major role of MKPs is to limit inflammatory processes and promote homeostasis [8]. MKPs are a group of dual specificity proteins phosphatases [8] that display a relative degree of substrate specificity. For example, MKP-1 mainly dephosphorylates p-p38 and p-JNK over p-ERK [20], and MKP-3 has been identified as a selective ERK pathway negative regulator [12,14]. However, these MKP substrate preferences may vary depending on the cell type or the physiological or pathophysiological condition in which a particular MKP is expressed [20]. Since the major inducers of MKP expression and enzymatic activity are the activation of their own substrates [8], the cellular availability of specific MAPKs may also determine the preference of MKPs. Thus, MKPs may modulate different cellular responses in different tissues or cell types.
In rodents, central MAPKs are activated following peripheral nerve injury and induce a stressed or reactive (pro-inflammatory) phenotype in glial cells. Central JNK is activated almost exclusively in astrocytes following peripheral nerve injury [5,24]. Central p-ERK is expressed in a sequential manner, in neurons (h), then in microglia (days), and then in astrocytes (weeks) following peripheral nerve injury [23]. In contrast, p-p38 is mostly expressed in microglia following peripheral skin or nerve injury [10,11], and in some cases in astrocytes [1]. Thus, reactive astrocytes in the central nervous system display a preferential activity of JNK and ERK. This particular nature of astrocytes may determine differential substrate preferences for MKP-3. We propose to test this in vitro, and hypothesize that MKP-3 modulate these MAPK in a differential fashion with preference for JNK, JNK > ERK.
2. Materials and methods
2.1. Cloning of the rat MKP-3 gene
Total RNA was extracted from spinal tissue of 250 g Sprague Dawley rats (Harlan, Indianapolis, IN) using RNA easy columns (Qiagen, Basel, Switzerland) according to the manufacturer’s protocol. Total cDNA was synthesized from 1 μg of total RNA using a quanti-tect reverse transcription kit (Qiagen, Basel, Switzerland), following the manufacturer’s protocol. The rat MKP-3 cloning was started by DNA amplification (PCR) from cDNA sample using two specifically design primers: forward (5’TGCTAGGCACCCCGCCTTCT3’) and reverse (5’TGACACCTGGGGCCACACAC3’) homologous to rat MKP-3 gene (Genbank accession number NM_053883). PCR was performed as follows: 95 °C for 5 min followed by 30 cycles at 95 °C for 15 s, 61 °C for 1 min, 72 °C for 1 min 40 s followed by 72 °C for 10 min. The PCR product (1.2 kb DNA fragment) was checked on agarose gel, eluted, and ligated into pcDNA3.1 expression vector (Invitrogen, San Diego, CA) using the TA cloning technology. Competent Escherichia coli cells were transformed, and the expression vector containing the rat MKP-3 insert was isolated and sequenced on both strands using the Big Dye terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) vector to confirm rat MKP-3 identity.
2.2. Cell culture, transfection, and lipopolysaccharide (LPS) treatment
The procedures used in these studies were approved by the Dartmouth College Institutional Animal Care and Use Committee. Highly purified primary astrocytes cultures were prepared as P2–P3 Harlan Sprague Dawley pups (Indianapolis, IN) that were killed by decapitation. The cerebral cortices were removed and meninges dissected away; cortical tissue was minced with a sterile scalpel blade and digested with Trypsin/EDTA 1< (Mediatech, Herndon, VA) for 15 min at 37 °C. The tissue was allowed to settle, and the supernatant was discarded. Dulbecco’s modified Eagle’s medium (DMEM, Mediatech) – supplemented with 10% charcoal-stripped fetal bovine serum (FBS, Hyclone, Logan, UT), 1.1% GlutaMax (Gibco-Invitrogen, Carlsbad, CA), and 1% penicillin/streptomycin (P/S, 100 U/ml penicillin, 100 μg/ml streptomycin, Mediatech) containing 2000 units DNase (Sigma, St. Louis, MO) – was added to the tissue on ice. The tissue was triturated several times, clumps were allowed to settle, and the supernatant was removed to a sterile 50 ml conical tube on ice between triturations. Triturations were then repeated until no tissue clumps were observed. The final volume was diluted to 25 ml with media and centrifuged at 310 × g for 15 min. The supernatant was discarded and the cells re-suspended in media. A small aliquot of cells was stained for trypan blue exclusion, and cells were plated at 1 × 106 cells per 75 cm2 flask. Cultures were maintained at 37 °C and 5% CO2. Media was changed every three to four days. After eight days in vitro (DIV 8), the flasks were confluent with astrocytes and microglia. Flasks were lightly shaken by hand for 1 min, and the media containing microglia was removed. The remaining adherent cells cultures were found to be >95% astrocytes (GFAP positive cells) and were used for further experiments. Cells were challenged with 1 μg/ml of LPS (0111:B4 serotype, Sigma, St. Louis, MO) in complete media for 1, 2, 6, 12, 24 and 48 h. Following LPS treatment, supernatants were removed and stored at −80 °C until further used.
2.3. Quantitative real time (RT)-PCR and Western blot analyses
mRNA level was determined as described previously [17]. Briefly, 1 μg of total RNA from each sample was reverse transcribed into cDNA as described above. Quantitative RT-PCR was performed with the Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, Foster city, CA) using the following conditions: 1 cycle of 50 °C for 2 min, 1 cycle of 95 °C for 10 min, then 40 cycles at 95 °C for 15 s, 60 °C for 1 min. All samples were run in duplicate using Taqman gene expression assays (Applied Biosystems, Foster City, CA) with rat MKP-3 (Rn00518185 m1) predesigned and preformulated primer and probe. Expression of MKP-3 in primary astrocytes and BV-2 cells was, respectively, normalized to a predesigned and preformulated rat (Rn01775763 g1) or mouse (Mm99999915 g1) control gene GAPDH (Applied Biosystems, Foster City, CA). The difference in MKP-3 mRNA expression between treatments was analyzed using the comparative CT method as described previously [17]. Total RNA for each sample was also included during each run as a negative control.
MAP Kinases (MAPKs) were evaluated at 1 and 2 h following LPS stimulation in BV-2 and primary astrocytes cells. Total proteins were extracted from cells using 1× Laemmli buffer (Bio-Rad, Hercules, CA) containing 5% 2-β Mercaptoethanol (Sigma, St. Louis, MO). Proteins were separated on a 10% SDS polyacrylamide gel (Bio-Rad, Hercules, CA) and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA). Immunoblotting was performed as described previously [17] using mouse anti-rat MKP-3 monoclonal antibody (1:500, Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-phospho-ERK 44/42 (Phospho-MAP Kinase 1:500, Cell Signaling, Danvers, MA), mouse anti-phospho-p38 (1:500, Cell Signaling, Danvers, MA), or mouse anti-phospho-JNK (1:500, Cell Signaling, Danvers, MA). Blots were incubated with goat HRP-conjugated secondary antibody (1:3000, Pierce, Rockford, IL), treated with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Rockford, IL), and imaged with Syngene G-box (Synoptics, Frederick, MD). Membranes were stripped and re-probed with rabbit anti-ERK 44/42 (Total MAP Kinase 1:500, Cell Signaling, Danvers, MA), rabbit anti-p38 (total MAP kinase 1:500, Cell Signaling, Danvers, MA), rabbit anti-JNK (1:500, Cell Signaling, Danvers, MA), or mouse anti-β-actin antibody (1:3000, Abcam, Cambridge, MA). Quantification of protein expression was performed using Syngene Tools software (Synoptics, Frederick, MD) by measuring the band densities in relation to the loading controls (total MAPK and beta actin).
2.4. Measurement of nitric oxide (NO) production and ELISA assays
Nitric oxide produced in culture supernatants of BV-2 and primary astrocytes cells following LPS treatment were quantified according to the Griess assay (Promega, Madison, WI) following the manufacturer protocol. Cytokine release (IL-6, and TNF-α) in the same supernatants was assessed using R&D Systems DuoSet ELISA (R&D Systems, Minneapolis, MN) kits as suggested by the manufacturer.
2.5. Statistical analysis
All in vitro experiments were completed at least three times. Data are expressed as mean ± s.e.m. Statistical analyses were completed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA). One-way ANOVA and Bonferroni post-test were used for MKP-3 mRNA (Figs. 1A and 2D and Supplement Fig. 1A), protein (Fig. 1C) quantifications and NO and cytokines accumulation after siRNA and LPS treatment (Fig. 2E–G). Two-way ANOVA and Bonferroni post-test were used for NO, cytokines and MAPKs accumulation after LPS treatment (Fig. 2A–C, Supplement Fig. 1G–I and Supplement Fig. 2A–C). Unpaired t-test was used for MAPKs accumulation in primary astrocyes (Fig. 1G–I) and MKP-3 protein quantification in BV2 (Supplement Fig. 1C). Significance was determined at a level of P < 0.05.
Fig. 1.
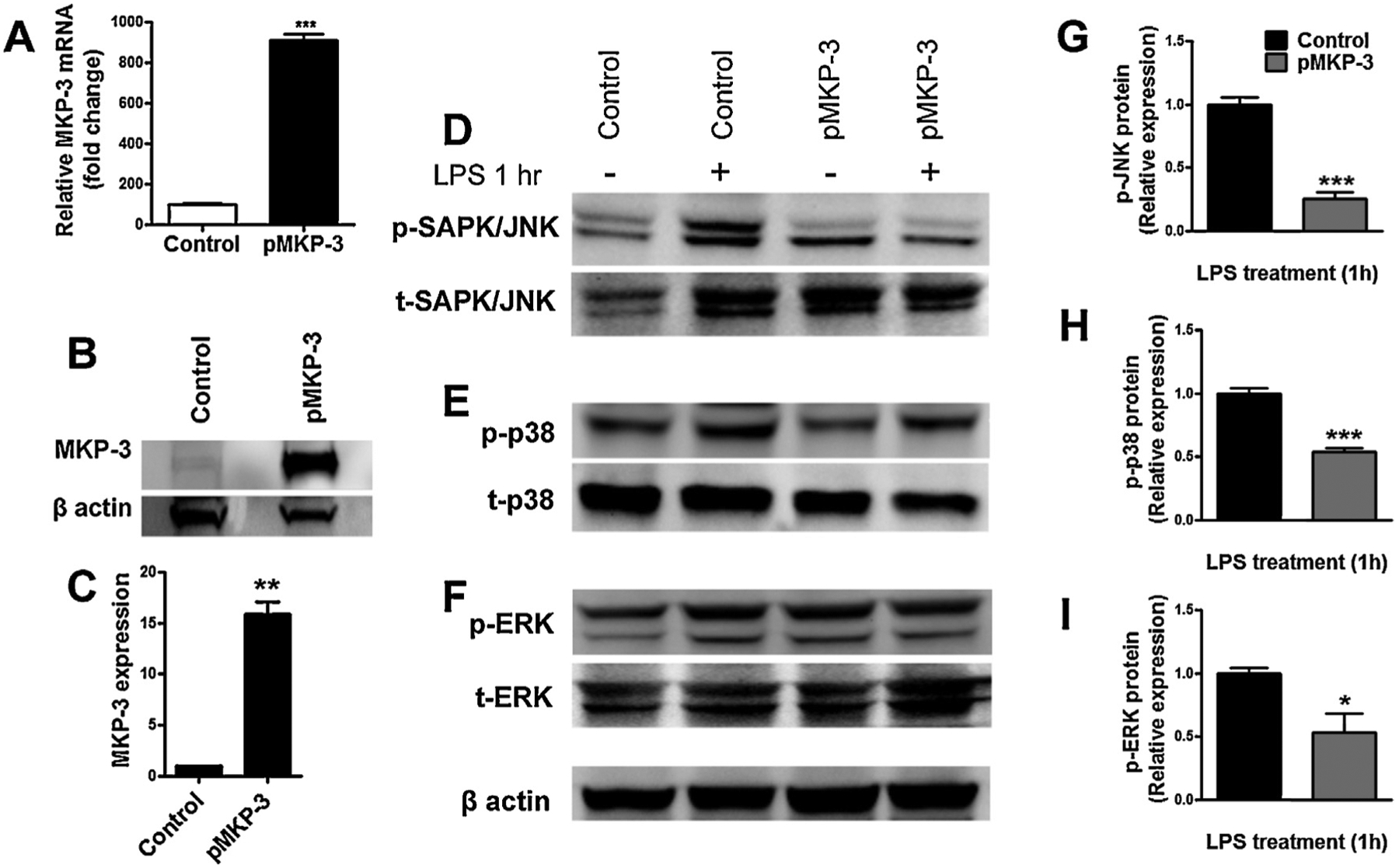
In vitro characterization of MKP-3 and MAPKs in primary astrocytes cells transiently expressing MKP-3. MKP-3 mRNA levels determined by qRT-PCR (A), representative Western blot (B) and quantification of MKP-3 protein (C) in normal primary astrocytes (control) and primary astrocytes transiently overexpressing MKP-3 (pMKP-3). Representative Western blots of p-JNK (D), p-p38 (E), and p-ERK (F), and quantification of p-JNK (G), p-p38 (H), and p-ERK (I) protein in normal primary astrocytes (control) and primary astrocytes transiently overexpressing MKP-3 (pMKP-3), following 1 h incubation in medium (control alone) or lipopolysaccharide (LPS) stimulation (1 μg/ml). Results are expressed as mean ± s.e.m. of three experiments in triplicate. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. control primary astrocytes.
Fig. 2.
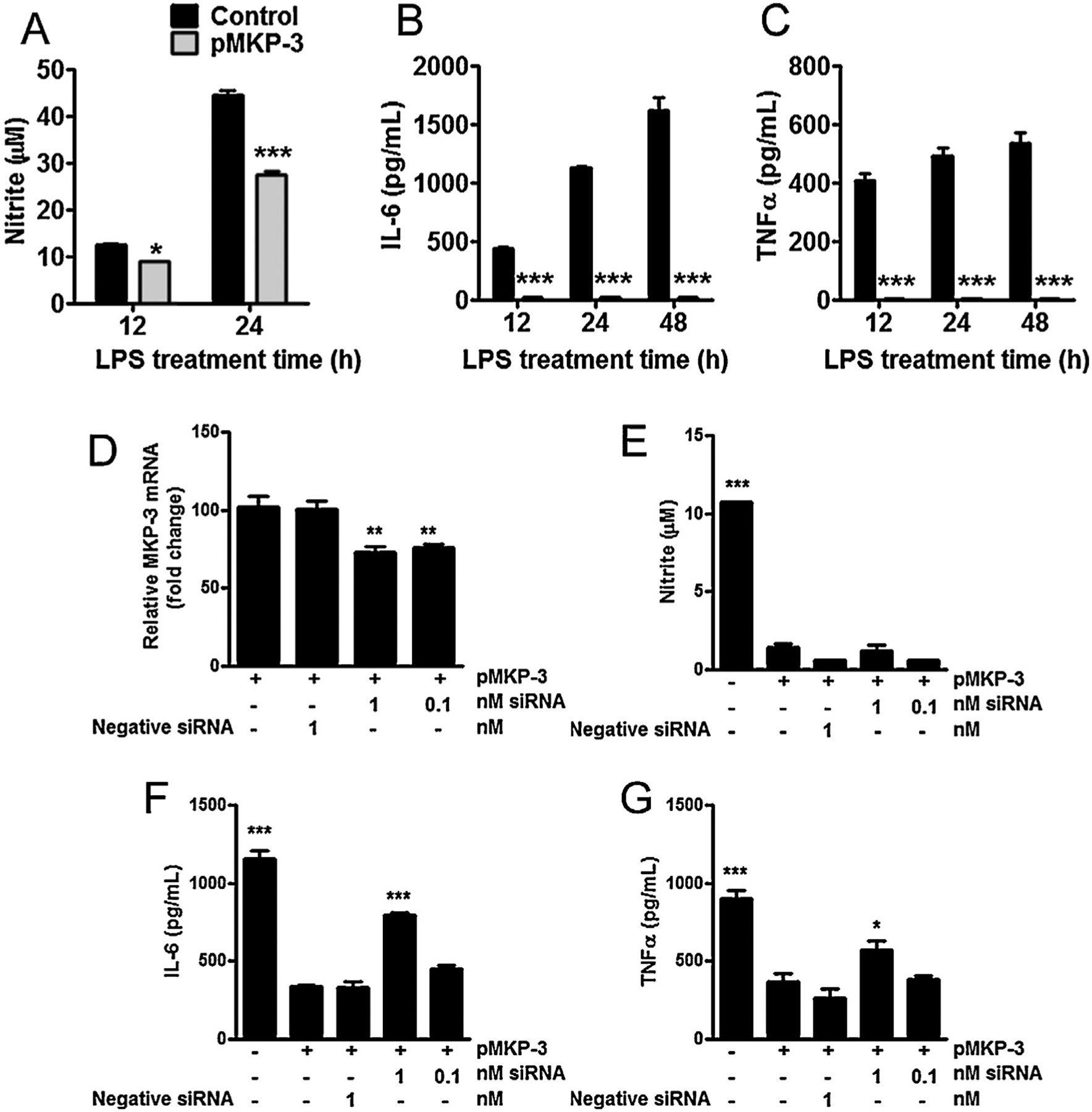
Overexpression of MKP-3 reduces inflammatory effectors in primary astrocytes cells and reversal effects of MKP-3 mRNA knockdown in BV-2 microglia cells. (A) Nitrite oxide (measured as nitrite), (B) IL-6 and (C) TNFα concentration in cell culture supernatant in control (normal) and transiently expressing MKP-3 primary astrocytes cells challenged with LPS (1 μg/ml) for 12, 24 or 48 h. MKP-3 mRNA levels (D) in BV-2 cells stably expressing MKP-3 after 24 h transfection with MKP-3 siRNA compared to negative control siRNA and vehicle treated cells. Nitrite oxide (measured as nitrite) (E), IL-6 (F), and TNFα (G) cytokines release in control (normal) and stably expressing MKP-3 BV-2 cells transfected 24 h with a negative siRNA or a siRNA against MKP-3 and challenged with LPS (1 μg/ml) for 24 h. Results are expressed as mean ± s.e.m. of three experiments in duplicate. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. control primary astrocytes or BV-2 cells stably expressing MKP-3.
3. Results
3.1. In vitro over expression of MKP-3 in primary astrocytes cells
The rat MKP-3 cDNA was cloned from rat spinal cord homologous to the cloned MKP-3 (NM 053883). qRT-PCR and Western blot experiments showed an increase in both MKP-3 mRNA and protein in primary astrocyte cells (Fig. 1A–C) compared to non-transfected primary astrocytes. Then, we studied the functional/activity capabilities of MKP-3 overexpression by measuring MAPK phosphorylation. No differences in p-JNK, p-p38 and p-ERK expression were found between non-transfected astrocytes and MKP-3 transfected astrocytes (Fig. 1D–F). However, when challenged with LPS, non-transfected primary astrocytes cells, show a significant increase in p-JNK, p-p38 and p-ERK (Fig. 1 D–F). Over expression of MKP-3 in LPS-stimulated primary astrocytes induced a significant decrease in p-JNK (75 ± 0.05% of control), p-ERK (50 ± 0.15% of control), p-p38 (50 ± 0.03% of control) 1 h after the stimulation (D–I). Additionally, we measured the functional/activity capabilities (downstream effects) of this MAPK inhibition. Over-expressing MKP-3 astrocytes stimulated with LPS for 12, 24, and 48 h significantly reduced NO accumulation (Fig. 2A) and completely reduced the accumulation of IL-6 and TNFα when compared to control cells (Fig. 2B and C). We then tried to inhibit MKP-3 expression by double astrocytic transfections, MKP-3 cDNA and MKP-3 siRNA; however this manipulation induced cell death. Therefore, we tested the specificity of MKP-3 activity using BV-2 microglia cells stably expressing MKP-3, as we have previously done for other phosphatase family member, MKP-1 [17] (See Supplemental material for methods).
3.2. In vitro over expression and knockdown of MKP-3 in the BV-2 microglia cell line
Through qRT-PCR and Western blot analyses we observed an increase in both MKP-3 mRNA and protein in BV-2 microglia stably expressing MKP-3 (Supplement Fig. 1A–C) when compared to normal BV-2 microglia. In non-transfected BV2 microglia cells, LPS challenge result in an increase in p-JNK and p-p38 as observed with primary astrocytes cells but not in p-ERK. BV-2 microglia stably expressing MKP-3 and challenged with LPS (1 μg/ml, 1 and 2 h) showed a reduction of p-JNK and p-p38 levels (~30% of control; Supplement Fig. 1D–E and G–H). Conversely, no significant change in p-ERK was observed (Supplement Fig. 1F and I). The release of NO, IL-6, and TNFα was significantly decreased in BV-2 stably expressing MKP-3 when compared to control cells (Supplement Fig. 2A–C). This data confirmed the similar functionality of MKP-3 in BV-2 microglia to primary astrocytes cells (substrate preference JNK > ERK). We therefore conducted silencing studies using BV-2 microglia stably expressing MKP-3 and a stealth siRNA sequence targeting MKP-3 mRNA to test its specific activity.
Analysis of MKP-3 mRNA accumulation using qRT-PCR in BV-2 microglia treated with 1 or 0.1 nM siRNA showed a significant decrease in MKP-3 mRNA compared to BV-2 microglia exposed to negative control siRNA or vehicle after 24 h of treatment (Fig. 2D). Then, as we have done previously for MKP-1 [17], we measured the effects of this siRNA in the final pro-inflammatory effectors affected by MKP-3 induction. MKP-3 mRNA knockdown using 1 nM siRNA, but not 0.1 nM, resulted in an increase in IL-6 and TNFα (but not NO) release following LPS treatment in BV-2 cells stably expressing MKP-3 (Fig. 2E–G). The lack of effect of our MKP-3 siRNA on NO production (controlled by inducible nitric oxide synthase) suggests that NO production is marginally dependent on MKP-3 signaling. An alternative explanation for this finding could be the partial knockdown of MKP-3 by our siRNA.
4. Discussion
The main findings of our studies are (1) MKP-3 preferentially reduces p-JNK over p-ERK and p-p38 in primary astrocytes; (2) This MAPK modulation pattern in primary astrocytes significantly reduced NO and completely abolished IL-6 and TNF accumulation; and (3) These effects are specifically induced by MKP-3 since block-age of MKP-3 mRNA expression reversed its action on MAPKs and pro-inflammatory mediators in BV-2 microglia cells.
Our current data demonstrate that MKP-3 preserves its enzymatic activity on MAPKs in both microglia and astrocytes when compared to peripheral cells [22]. Interestingly, we observed a reduced p-ERK preference of MKP-3 in primary astrocytes and in BV-2 microglia cells when compared to MKP-3 actions on p-JNK. This finding contrasts with early studies reporting a natural preference of MKP-3 for p-ERK in other cells types [12,14]. Accordingly, the overexpression of MKP-3 in astrocytes did induce a 30% reduction in p-ERK accumulation, that was however considerably lower than its effects in p-JNK (~70%) and p-p-38 (~50%). Interestingly, these preferential effects of MKP-3 on p-JNK over p-ERK and p-p38 are associated with the fact that astrocytes preferentially express p-JNK over p-ERK and p-p38 [9,24], a condition that may explain our findings. It is known that the activation of MKP substrates play a key role in specific MKP expression [8], and this may determine the substrate preference of phosphatases in particular cell types or physiological or pathophysiological conditions [20]. This postulation had not been tested in brain-derived cells such as astrocytes. Our data strongly suggest that the MAPK activity profile of astrocytes (JNK > ERK) determines the differential substrate preference of MKP-3, JNK > ERK. Our findings using BV-2 microglia cells also support this assumption. It is known that following LPS stimulation, immortalized BV-2 microglial cells express lower levels of p-ERK than primary microglia [7]. This may, explain the reason why MKP-3 did not show a substrate preference for ERK in this cell line, but rather a JNK and p38 preference. Thus, our data with BV-2 cells provide a scenario in which cellular p-ERK activity, the natural preferential target for MKP-3, is not prominent, as it is the case for primary astrocytes. Accordingly, we have previously observed in primary microglial cells that p-ERK is primarily regulated by MKP-3 but in conjunction with another family member MKP-1 overexpression using pharmacological approaches [16].
The phosphorylation of MAPKs in glial cells promotes the efficient production of pro-algesic factors such as cytokines or nitric oxide [9]. Therefore, the dephosphorylation of MAPKs by MKPs directly affects the production of these effectors [17]. Thus, our results not only confirmed the functional outcome of MKP-3 induction directly on its targets (MAPKs), but also over downstream factors that depend upon MAPK activity. The current observed effects of MKP-3/JNK > ERK pathways suggests that MAPKs (mainly JNK) are pivotal in the production of IL-6 and TNF cytokines in reactive primary astrocytes. Our results also show the differential immune capabilities of astrocytes compared to microglia [16] and how MAPKs and potentially MKP regulation are pivotal in the differential biology of these brain-derived cells.
One of the limitations of our studies is the unpractical co-transfection of MKP-3 cDNA and its corresponding siRNA in primary astrocytes cells. Contrary to single transfections attempt, co-transfection of primary astrocytes significantly produced astrocytic cell death. Our own and previous other studies have shown co-transfections to be detrimental to primary microglia and other primary cells [6,15,17]. These outcomes lead us to use the immortalized BV-2 microglia cell line and developed a line that is stably overexpressing MKP-3 as previously done with MKP-1 [17]. The use of this central nervous derived BV-2 microglia cell line seems more appropriate than the use of cells of peripheral origins such as HELA and CHO cells to assess the role of MKPs in brain derived cells. We recognize that the BV-2 system does not completely mimic our astrocyte system. For example, we observed some differences in the extent of the effects of MKP-3 over MAPKs between BV-2 and primary astrocytes cells. However, the lower level of negative modulation of MAPKs (and downstream effectors) in BV-2 cells compared to primary astrocytes is explained by the lower extent of MKP-3 expression (mRNA and protein) of our BV-2 cell line (~2.5-fold) in comparison to its expression in primary astrocytes (>15-fold). Nevertheless, we consider that the MKP-3/MAPK/pro-inflammatory factor signaling pattern displayed by our BV-2 cell line was sufficient to test the specificity of our MKP-3 induction in astrocytes (the primary goal for these experiments). Since reduction of MKP-3 mRNA expression in BV-2 microglia cells (using siRNA) significantly reverse the effects on JNK and p38 activation, and downstream pro-inflammatory factor production/release, our data strongly suggest that our results using primary astrocytes are due specifically to MKP-3 gene induction.
5. Conclusions
We demonstrate that MKP-3 in brain-derived astrocytes possess a differential MAPK/target preference, most likely due to the particular MAPK profile/biology of astrocytes under reactive states. Our studies add to our knowledge in astrocyte biological functions and phosphatase activity under brain-derived cellular reactive conditions. In a more translational aspect, the relevance of our studies might be interpreted on the basis that the induction of MKPs [16,17], the inhibition of MAPKs [9], or the specific blockade of TNF [19], IL-6 [3], or NO [13], reduces pain behaviors in animal models. Likewise, similar effects have seen in pain patients using an IL-6 antibody [18] or p38 inhibitors [2,21]. In these lines, our data indicate that the induction of MKP-3 could be beneficial to the treatment of pain such as postoperative or neuropathic pain conditions in which MAPKs and glial cells play a role.
Supplementary Material
Acknowledgments
The authors would like to acknowledge The Rita Allen Foundation and American Pain Society 2011 Pain grant (EAR-S), and The Hitchcock Foundation Award 2011-2012 (EAR-S). We thank Beth Wilkerson, Presbyterian College School of Pharmacy, for edits and revision.
Abbreviations:
- MKP
mitogen-activated protein kinase-phosphatase
- PCR
poly-merase chain reaction
- cDNA
complementary DNA
- LPS
lipopolysaccharide
- ERK
extracellular signal-regulated kinase
- p-ERK
phospho-extracellular signal-regulated kinase
- t-ERK
total-extracellular signal-regulated kinase
- JNK
c-Jun N terminal kinase
- p-JNK
phospho-c-Jun N terminal kinase
- t-JNK
total-c-Jun N terminal kinase
- p-p38
phospho-p38
- Il
interleukin
- TNF
tumor necrosis factor
- NO
nitrite oxide
- siRNA
small interfering RNA
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
Footnotes
Competing interests
The authors declare that they have no competing interests.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.neulet.2014.05.039.
References
- [1].Alkaitis MS, Solorzano C, Landry RP, Piomelli D, DeLeo JA, Romero-Sandoval EA, Evidence for a role of endocannabinoids, astrocytes and p38 phosphorylation in the resolution of postoperative pain, PLoS One 5 (2010) e10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Anand P, Shenoy R, Palmer JE, Baines AJ, Lai RY, Robertson J, Bird N, Ostenfeld T, Chizh BA, Clinical trial of the p38 MAP kinase inhibitor dilmapi-mod in neuropathic pain following nerve injury, Eur. J. Pain 15 (2011) 1040–1048. [DOI] [PubMed] [Google Scholar]
- [3].Arruda JL, Sweitzer S, Rutkowski MD, DeLeo JA, Intrathecal anti-IL-6 antibody and IgG attenuates peripheral nerve injury-induced mechanical allodynia in the rat: possible immune modulation in neuropathic pain, Brain Res. 879 (2000) 216–225. [DOI] [PubMed] [Google Scholar]
- [4].Ducruet AP, Vogt A, Wipf P, Lazo JS, Dual specificity protein phosphatases: therapeutic targets for cancer and Alzheimer’s disease, Annu. Rev. Pharmacol. Toxicol 45 (2005) 725–750. [DOI] [PubMed] [Google Scholar]
- [5].Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR, JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain, J. Neurosci 29 (2009) 4096–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hamm A, Krott N, Breibach I, Blindt R, Bosserhoff AK, Efficient transfection method for primary cells, Tissue Eng. 8 (2002) 235–245. [DOI] [PubMed] [Google Scholar]
- [7].Horvath RJ, Nutile-McMenemy N, Alkaitis MS, Deleo JA, Differential migration LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures, J. Neurochem 107 (2) (2008) 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jeffrey KL, Camps M, Rommel C, Mackay CR, Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses, Nat. Rev. Drug Discovery 6 (2007) 391–403. [DOI] [PubMed] [Google Scholar]
- [9].Ji RR, Gereau R.W.t., Malcangio M, Strichartz GR, MAP kinase and pain, Brain Res. Rev 60 (2009) 135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ji RR, Suter MR, p38 MAPK, microglial signaling, and neuropathic pain, Mol. Pain 3 (2007) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jin SX, Zhuang ZY, Woolf CJ, Ji RR, p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain, J. Neurosci 23 (2003) 4017–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kawakami Y, Rodriguez-Leon J, Koth CM, Buscher D, Itoh T, Raya A, Ng JK, Esteban CR, Takahashi S, Henrique D, Schwarz MF, Asahara H, Izpisua Belmonte JC, MKP3 mediates the cellular response to FGF8 signalling in the ver-tebrate limb, Nat. Cell Biol 5 (2003) 513–519. [DOI] [PubMed] [Google Scholar]
- [13].Kim KH, Kim JI, Han JA, Choe MA, Ahn JH, Upregulation of neuronal nitric oxide synthase in the periphery promotes pain hypersensitivity after peripheral nerve injury, Neuroscience 190 (2011) 367–378. [DOI] [PubMed] [Google Scholar]
- [14].Kim Y, Rice AE, Denu JM, Intramolecular dephosphorylation of ERK by MKP3, Biochemistry 42 (2003) 15197–15207. [DOI] [PubMed] [Google Scholar]
- [15].Kizjakina K, Bryson JM, Grandinetti G, Reineke TM, Cationic glycopolymers for the delivery of pDNA to human dermal fibroblasts and rat mesenchymal stem cells, Biomaterials 33 (2012) 1851–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Landry RP, Martinez E, DeLeo JA, Romero-Sandoval EA, Spinal cannabinoid receptor type 2 agonist reduces mechanical allodynia and induces mitogen-activated protein kinase phosphatases in a rat model of neuropathic pain, J. Pain 13 (2012) 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ndong C, Landry RP, Deleo JA, Romero-Sandoval EA, Mitogen activated protein kinase phosphatase-1 prevents the development of tactile sensitivity in a rodent model of neuropathic pain, Mol. Pain 8 (2012) 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohtori S, Miyagi M, Eguchi Y, Inoue G, Orita S, Ochiai N, Kishida S, Kuniyoshi K, Nakamura J, Aoki Y, Ishikawa T, Arai G, Kamoda H, Suzuki M, Takaso M, Furuya T, Kubota G, Sakuma Y, Oikawa Y, Toyone T, Takahashi K, Efficacy of epidural administration of anti-interleukin-6 receptor antibody onto spinal nerve for treatment of sciatica, Eur. Spine J 21 (2012) 2079–2084 (official publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research Society; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raghavendra V, Rutkowski MD, DeLeo JA, The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats, J. Neurosci 22 (2002) 9980–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Theodosiou A, Ashworth A, MAP kinase phosphatases, Genome Biol. 3 (2002) (REVIEWS3009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tong SE, Daniels SE, Black P, Chang S, Protter A, Desjardins PJ, Novel p38alpha mitogen-activated protein kinase inhibitor shows analgesic efficacy in acute postsurgical dental pain, J. Clin. Pharmacol 52 (2012) 717–728. [DOI] [PubMed] [Google Scholar]
- [22].Wong VC, Chen H, Ko JM, Chan KW, Chan YP, Law S, Chua D, Kwong DL, Lung HL, Srivastava G, Tang JC, Tsao SW, Zabarovsky ER, Stanbridge EJ, Lung ML, Tumor suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell invasion and epithelial–mesenchymal transition (EMT)-associated phenotype, Int. J. Cancer 130 (2012) 83–95. [DOI] [PubMed] [Google Scholar]
- [23].Zhuang ZY, Gerner P, Woolf CJ, Ji RR, ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model, Pain 114 (2005) 149–159. [DOI] [PubMed] [Google Scholar]
- [24].Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decos-terd I, Ji RR, A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance, J. Neurosci 26 (2006) 3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.