

Abstract
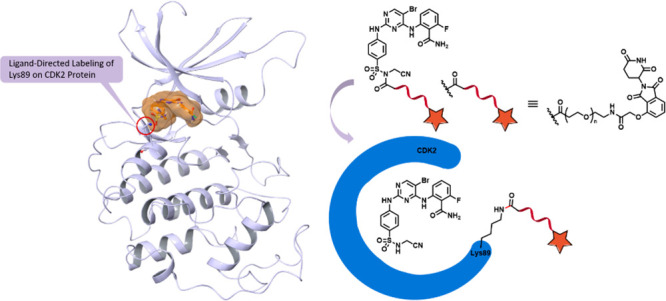
Ligand-directed bioconjugation strategies have been used for selective protein labeling in live cells or tissue samples in applications such as live-cell imaging. Here we hypothesized that a similar strategy could be used for targeted protein degradation. To test this possibility, we developed a series of CDK2-targeting N-acyl-N-alkylsulfonamide (NASA)-containing acylation probes. The probes featured three components: a CDK2 homing ligand, a CRL4CRBN E3 ligase recruiting ligand, and a NASA functionality. We determined that upon target binding, NASA-mediated reaction resulted in selective functionalization of Lys89 on purified or native CDK2. However, we were unable to observe CDK2 degradation, which is in contrast to the efficient degradation achieved by the use of a structurally similar reversible bivalent degrader. Our analysis suggests that the lack of degradation is due to the failure to form a productive CDK2:CRBN complex. Therefore, although this work demonstrates that NASA chemistry can be used for protein labeling, whether this strategy could enable efficient protein degradation remains an open question.
Keywords: Covalent degrader, CDK2, ligand-directed, N-acyl-N-alkylsulfonamide, chemical modification
Targeted protein degradation is a rapidly developing research field focusing on the discovery and development of agents that can induce selective and rapid degradation of a protein target.1−3 The most common approach in this space has been the use of small molecules that induce ternary complex formation between the target of interest and an E3 ubiquitin ligase, resulting in ubiquitination of the target and its subsequent proteasome-mediated degradation. Degradation-based targeting strategies have garnered intense interest because of several key advantages over the traditional inhibitor-based approaches, such as demonstrating potential in cases where a ligand binds directly to the target but has difficulty affecting or blocking the function of the protein. Unlike “molecular glues” (or monovalent degraders) that currently lack rational development strategies for new targets,4,5 proteolysis-targeting chimeras (PROTACs), which are bivalent degraders that include two ligands, one to recruit the target and the other to recruit the E3 ligase, are amenable to rational design, as evidenced by rapidly growing list of targets for which PROTACs have been developed.6 Most PROTACs developed to date are degraders that bind to the target and the E3 ligase reversibly. An orthogonal strategy to access proteins that are difficult to target through reversible binding alone is to equip the small-molecule ligand with an electrophilic warhead to form a covalent bond with a nucleophilic side-chain residue (e.g., cysteine, serine, threonine, lysine, or tyrosine) found in the target protein.7 One of the key advantages of covalent binders is their prolonged duration of action. Recently, several PROTACs featuring either target recruiting ligands with reactive warheads (e.g., HaloTag7 fusion proteins,8,9 ERK1/2,10 BTK,11 and KRAS12) or E3 ligase recruiting ligands with reactive warheads (e.g., RNF4,13 DCAF16,14 RNF114,15 and KEAP116) have been reported. However, the majority of the reported PROTACs degrade targets traditionally considered as ligandable, leaving the space of “unligandable” targets poorly explored.
Over the past decade, a range of bio-orthogonal reactions that enable chemo- and regioselective covalent labeling of proteins have been developed to enable selective chemical modification of proteins in their native environment.17,18 These strategies deliver different types of probes that can be used to study protein structure, function, dynamics, and localization. Recently, ligand-directed N-acyl-N-alkylsulfonamide (NASA) chemistry was shown to target nucleophilic lysine residues located in the proximity of the ligand binding site.19 In this strategy, the ligand serves as a homing device to recognize and bind a protein target of interest and deliver a payload to the proximal lysine via NASA-mediated acylation, as shown for heat shock protein 90 (HSP90)19 and human double minute 2 (HDM2).20 Because this reaction is rapid and irreversible, a possible advantage of this strategy is the ability to covalently label a protein of interest even in the absence of a tightly bound homing ligand. Building on this, degrader molecules that use a NASA delivery approach to covalently tag a protein target with a recruitment ligand for an E3 ligase could potentially expand the scope of degradable proteome, especially for unligandable proteins that have protein–protein interactions with their known ligandable partner proteins. Here, as a proof of concept, we integrated the NASA approach into the development of degraders and developed the initial series of efficient NASA-directed cyclin-dependent kinase 2 (CDK2) acylation probes. We demonstrated that the probes effectively labeled Lys89 on CDK2. Notably, we successfully acylated CDK2 with the degrader compound TMX-3054 and examined the consequences of this modification in living cells. In summary, TMX-3054 can efficiently engage native CDK2 with high site selectivity, but it failed to induce productive binary complex formation between acylated CDK2 and cereblon (CRBN), potentially explaining why we failed to observe effective degradation of CDK2 in OVCAR8 cells.
Design and Characterization of NASA-Based Acylation Probes
To identify a suitable proof-of-concept target, we queried kinome-wide sequence alignments and crystal structures of human protein kinases. One of our selection criteria was the presence of a solvent-exposed lysine residue located near the ATP-binding pocket. On this basis, more than 30 kinases met our criteria (Table S1), among which Lys89 on CDK2 emerged as a top hit particularly because it was previously targeted by the covalent CDK2 inhibitor NU6300.21 Moreover, we recently described the non-covalent picomolar CDK2 inhibitor TMX-3013,22 which contains a sulfonamide functionality suitable for installation of a NASA warhead. Thus, we used TMX-3013 as the CDK2 homing ligand and introduced the NASA warhead at the 4-position of the phenyl ring, resulting in the generation of TMX-3063, which displayed potent inhibitory activity against CDK2/cyclin A with an IC50 value of 2.2 nM as measured using a commercially available CDK2 biochemical assay (Figure 1A,B and Table S2). To examine our hypothesis that TMX-3063 would perform ligand-directed acetylation of the ε-amino group of Lys89, we first used liquid chromatography–mass spectrometry (LC–MS) to analyze the recombinant CDK2 protein incubated with a 2-fold molar excess of TMX-3063 for 1 h at room temperature. As expected, a mass shift consistent with the stoichiometric acetylation of CDK2 was observed (Figure 1C). To determine the site of modification, the labeled protein was digested with trypsin, and the peptides were analyzed by capillary electrophoresis–mass spectrometry (CE–MS). A database search revealed exclusive modification of Lys89 (Figure 1D). Taken together, our mass-spectrometry-based analyses confirmed that TMX-3063 is a NASA-based acylation reagent that acetylates Lys89 on CDK2.
Figure 1.
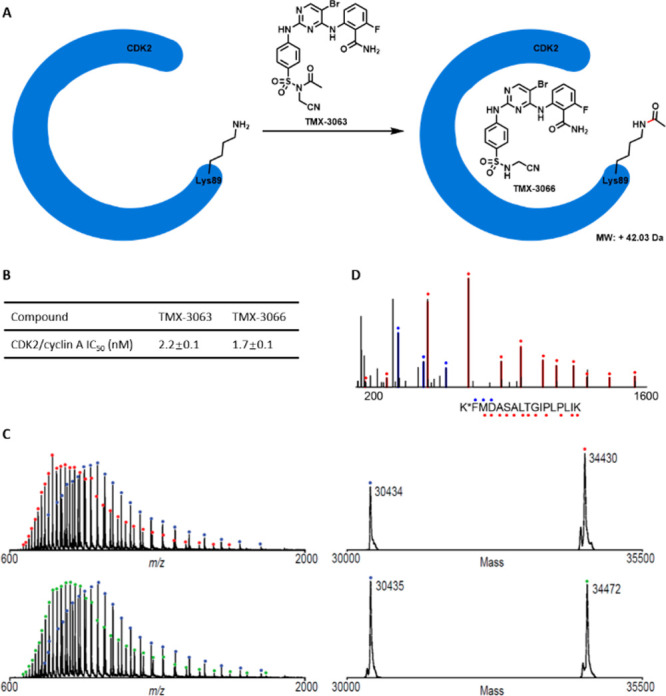
Mass-spectrometry-based analyses demonstrated that TMX-3063 reacted with CDK2 protein at Lys89. (A) Schematic illustration of covalent modification of CDK2 by TMX-3063. (B) CDK2 inhibitory activities of TMX-3063 and TMX-3066. (C) Mass spectra (left) and zero-charge mass spectra (right) of CDK2 protein treated with DMSO (top) or a 2-fold molar excess of TMX-3063 (bottom) for 1 h at room temperature. The observed mass shift of 42 Da is consistent with covalent addition of a single molecule of TMX-3063 (with loss of the TMX-3066 fragment). (D) CE–MS/MS spectrum of chymotryptic CDK2 peptide (residues 89 to 104) with Lys89 modified by TMX-3063. Ions of type b and y are indicated with blue and red glyphs, respectively. K* denotes TMX-3063-modified lysine.
Next, to determine whether TMX-3063 engages CDK2 in a cellular environment, we performed competitive pull-down studies. To enable this experiment, we synthesized TMX-3052 (Figure 2A), a NASA-linked, desthiobiotin-conjugated compound that retains potency as a CDK2 inhibitor. To investigate cellular target engagement, we treated OVCAR8 cells with DMSO or TMX-3063 at a concentration of 1 μM for 4 h, and the cell lysates were then incubated with desthiobiotin-conjugated TMX-3052 (1 μM) for 16 h at 4 °C. Protein bound by TMX-3052 was affinity-purified using streptavidin beads and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. As shown in Figure 2B, the desthiobiotinylated CDK2 band was clearly observed regardless of the denaturing condition (lanes 2 and 3), consistent with covalent modification of CDK2. Pretreatment of the cells with TMX-3063 resulted in protection of CDK2 from pull-down by the desthiobiotin compound (lanes 4 and 5). Taken together, these results confirmed cellular target engagement of CDK2 by TMX-3063. Similar experiments revealed that TMX-3063 can also engage CDK1 and CDK5, which also possess a lysine analogous to Lys89 of CDK2 (Figure S1).
Figure 2.

TMX-3063 labels CDK2 in cells. (A) Chemical structure of TMX-3052 and its CDK2/cyclin A inhibitory activity. (B) OVCAR8 cells were pretreated with either DMSO or TMX-3063 (1 μM) for 4 h followed by cell lysis, and the cell lysates were then incubated with TMX-3052 (1 μM) for 16 h. The protein was denatured with 4 mM urea + 1% SDS to eliminate the reversible binding.
Design and Evaluation of NASA-Based Covalent Probes for CDK2 Degradation
Prior to this study, we and others have successfully developed PROTAC-based CDK2 degraders, including TMX-2172, which incorporates CRBN-recruiting ligands.22,23 We designed a series of NASA-directed degrader molecules based on TMX-3063. As shown in Figure 3, four CRBN-recruiting molecules with PEG linkers of various lengths (TMX-3054, TMX-3166, TMX-3164, and TMX-3185) were synthesized. Biochemical activity tests confirmed that all of these compounds were potent CDK2 inhibitors with IC50 values below 20 nM (Table S2). Next, to determine whether these molecules could also form a covalent bond with Lys89 of recombinant CDK2 protein, we performed MS labeling studies using TMX-3054 as a representative. A mass shift consistent with ca. 75% modification of CDK2 protein was observed after incubation for 2 h at room temperature, and subsequent CE–MS analysis of chymotryptic peptides confirmed the site of modification (Figure 4A,B). To visualize the CRBN-recruiting ligand-tagged structure, recombinant CDK2 was cocrystallized in complex with TMX-3054, and a structure was determined to a resolution of 3.08 Å. We observed electron density in the ATP-binding pocket consistent with the chemical structure of TMX-3066, which is the expected product following reaction of TMX-3054 with Lys89 of CDK2. We were unable to observe any electron density for the CRBN-recruiting tag on Lys89, presumably because it was solvent-exposed and disordered (Figure 4C).
Figure 3.
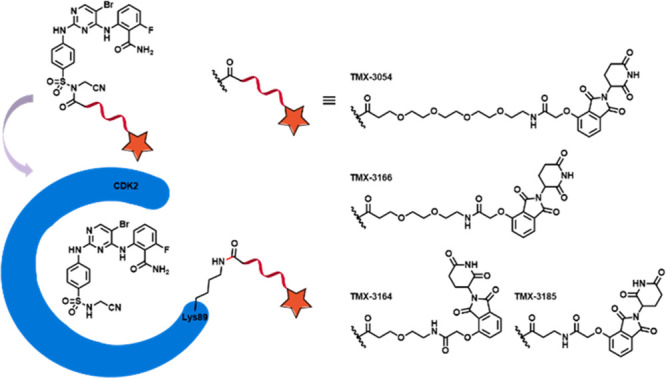
Schematic illustration of ligand-directed delivery of a CRBN-recruiting tag onto Lys89 of CDK2.
Figure 4.
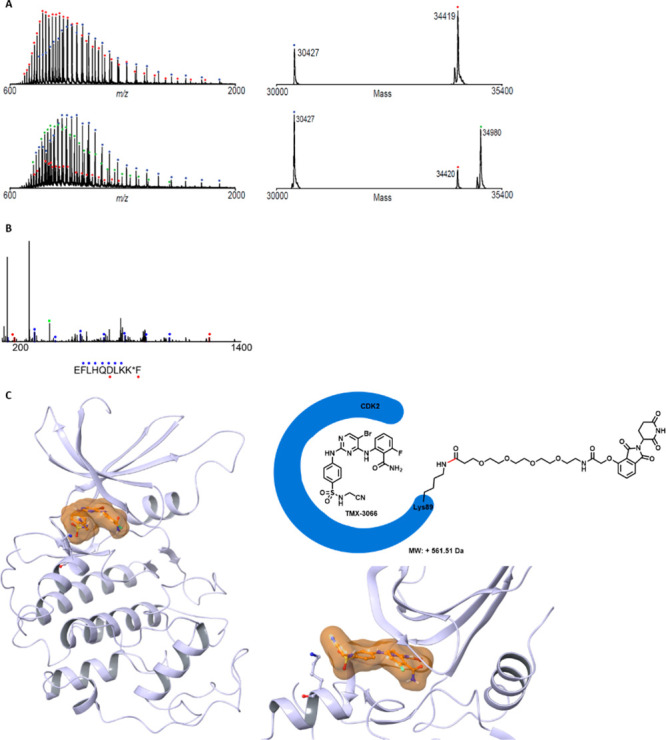
Mass spectrometry analysis and an X-ray crystallography study revealed that TMX-3054 reacted with CDK2 protein at Lys89. (A) Mass spectra (left) and zero-charge mass spectra (right) of CDK2 protein treated with DMSO (top) or a 10-fold molar excess of TMX-3054 (bottom) for 2 h at room temperature. The observed mass shift of 561 Da is consistent with covalent addition of a single molecule of TMX-3054 (with loss of the TMX-3066 fragment). (B) CE–MS/MS spectrum of chymotryptic CDK2 peptide (residues 81 to 90) with Lys89 modified by TMX-3054. Ions of type b and y are indicated with blue and red glyphs, respectively. K* denotes TMX-3054-modified lysine. (C) Cocrystal structure of CDK2 (3.08 Å) in complex with TMX-3054 (PDB ID 7MKX). The electron density of the released bound ligand TMX-3066 is colored in clay. The electron density of Lys89 (shown as gray sticks) was present, but the linked CRBN-recruiting moiety was not and was presumed to be completely solvent-exposed and disordered.
To investigate whether any of the NASA-based compounds could degrade CDK2, we generated the CDK2 HiBit assay, which enabled quantitative measurement of CDK2 protein levels in living cells.24 As shown in Figure 5A, none of the CRBN-recruiting-tag molecules resulted in significant reduction of the HiBit signals in the concentration range of 0.1–10 μM. In contrast, our previously developed reversible bivalent degrader TMX-2172 significantly reduced the signal, indicating efficient degradation of HiBit–CDK2. To examine whether the lack of degradation in the presence of covalently attached CRBN-recruiting tag was due to the inability to bind CRBN or the failure to form a productive binary complex, we generated a NanoBRET assay. Surprisingly, none of the NASA-based molecules produced a fluorescence signal following incubation of cells at concentrations up to 30 μM (Figure 5B). This was cross-validated by a negative result from immunoblot analysis of native CDK2 in OVCAR8 cells after 6 h treatment with the compounds (Figure S2). As shown in Figure 6A, no CDK2 degradation was observed upon treatment with TMX-3054 in the concentration range of 10 nM–10 μM. To rule out the possibility of poor cell permeability, a cellular target pull-down study was performed. Similar to the results for TMX-3063, as shown in lanes 4 and 5 of Figure 6B, the biotinylated CDK2 band was significantly attenuated after 4 h treatment of OVCAR8 cells with TMX-3054 at a concentration of 1 μM, indicating efficient target engagement on CDK2 (the cellular engagement data for TMX-3054 with CDK5 and CDK1 are available in Figure S1). Taken together, these results indicated that CDK2 is fully modified with a CRBN-recruiting tag on Lys89 via NASA-based molecules. However, the modified CDK2 is unable to bind CRBN and is therefore not targeted for degradation.
Figure 5.
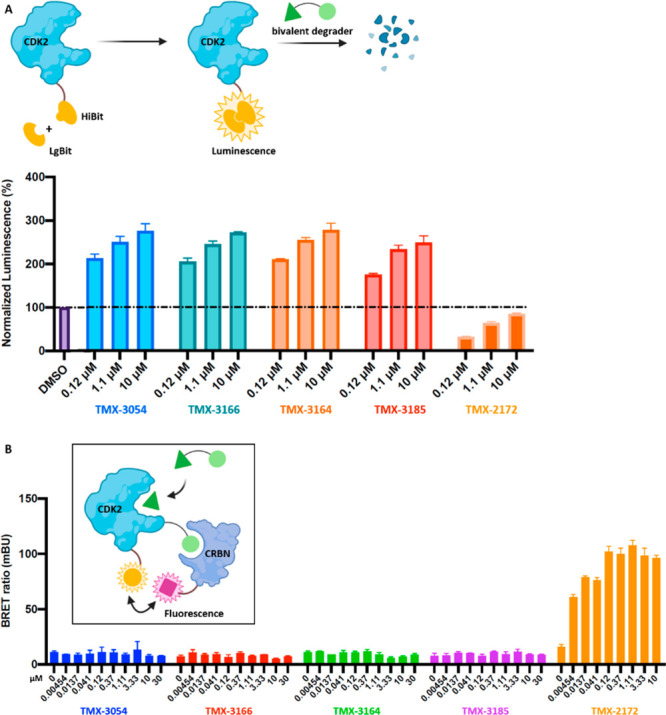
HiBiT (top) and NanoBRET (bottom) target engagement with degraders. (A) HiBiT, which complements with LgBiT to form the luminescence, was inserted via CRISPR/Cas9 to the C-terminus of CDK2 in HEK293T cells. Following treatment with degraders, protein degradation was monitored by the loss of luminescence. (B) In HEK293T cells, NanoLuc–CDK2 was used as the energy donor and HaloTag–CRBN was used as the energy acceptor. Following treatment with degraders, the CDK2:CRBN complex formation was monitored by the NanoBRET ratio (acceptor emission at 618 nm/donor emission at 460 nm).
Figure 6.

Immunoblot analysis of CDK2 in OVCAR8 cells. (A) OVCAR8 cells were treated with TMX-3054 for 6 h in the concentration range of 10 nM–10 μM. (B) OVCAR8 cells were pretreated with either DMSO or TMX-3054 (1 μM) for 4 h followed by cell lysis, and then the cell lysates were incubated with TMX-3052 (1 μM) for 16 h.
Targeted protein degradation is one of the most rapidly growing areas of research within chemical biology and drug discovery. In addition to reversible bivalent degraders that are commonly pursued, covalent bivalent degraders provide an alternative way to access proteins that are difficult to target through reversible binding alone. Although successful examples of covalent bivalent degraders either covalently binding target proteins or covalently binding E3 ligases have been reported, degrading proteins that lack a small-molecule binding pocket are considered challenging. Here we explored the possibility of using proximity-driven, ligand-directed reaction to label a target protein with an E3 recruiting ligand as a simple degradation tag. With CDK2 as a model system, our probes successfully labeled the native CDK2 with a CRBN-recruiting tag on Lys89. However, we did not observe CDK2 degradation under the conditions tested, and our follow-up analysis suggests that the likely reason for this is the failure of productive CDK2:CRBN complex formation. Thus, our work demonstrates that NASA-based chemistry can be an effective approach for installing an E3 ligase recruiting tag on the target of interest. However, our work also shows that the presence of the recruiting tag is insufficient to ensure target degradation and that additional factors, such as the exact location and orientation of the tag as well as the target selection, appear to play a major role, similar to what has been seen for reversible bivalent degraders. Collectively, our results indicate that further work is needed to establish the most effective way to rapidly optimize NASA-based acylation chemistry toward achieving degradation within and beyond kinase family of proteins.
Acknowledgments
We thank Dr. Milka Kostic (Twitter: @MilkaKostic) for assistance with critical reading and scientific editing of the manuscript. This work was supported in part by the National Institutes of Health (R01CA218278 and P01CA154303 awarded to N.S.G. and R01CA233800 awarded to J.A.M.).
Glossary
Abbreviations
- CDK2
cyclin-dependent kinase 2
- NASA
N-acyl-N-alkylsulfonamide
- CRBN
cereblon
- CE–MS
capillary electrophoresis–mass spectrometry
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00285.
Biological activities, assay protocols, synthetic procedures, and compound characterization data (PDF)
Author Contributions
# M.T., J.J., and S.B.F. contributed equally. M.T., T.Z., and N.S.G. generated the concept. M.T. designed and synthesized compounds, analyzed and interpreted data, and prepared the manuscript. S.B.F. and J.A.M. carried out mass spectrometry analyses. H.-S.S., J.H.B., and S.D.-P. expressed and purified protein constructs and performed cocrystallization. J.J. conducted immunoblot analysis and biochemical assays. K.A.D. and E.S.F. performed quantitative proteomics. J.A.M. and N.S.G. provided strategic direction and coordinated manuscript preparation. All of the authors discussed the results and commented on the manuscript. All of the authors approved the final version of the manuscript.
The authors declare the following competing financial interest(s): N.S.G. is a founder, science advisory board (SAB) member, and equity holder in Syros, Jengu, Inception, C4, B2S, Larkspur (board member), and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Arbella, Epiphanes, Deerfield, and Sanofi. J.A.M. serves on the SAB of 908 Devices and has received sponsored research support from AstraZeneca and Vertex. E.S.F. is a founder, SAB member, and equity holder in Civetta Therapeutics, Jengu Therapeutics (board member), and Neomorph Inc., an equity holder in C4 Therapeutics, and a consultant to Novartis, Sanofi, EcoR1 Capital, and Deerfield. The Fischer lab receives or has received research funding from Astellas, Novartis, Voronoi, Ajax, and Deerfield. S.D.-P. receives or has received research funding from the Linde Family Foundation, Taiho Pharmaceuticals, and the Doris Duke Charitable Foundation.
Supplementary Material
References
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18 (12), 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Chamberlain P. P.; Hamann L. G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15 (10), 937–944. 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- Kostic M.; Jones L. H. Critical Assessment of Targeted Protein Degradation as a Research Tool and Pharmacological Modality. Trends Pharmacol. Sci. 2020, 41 (5), 305–317. 10.1016/j.tips.2020.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S. L. The Rise of Molecular Glues. Cell 2021, 184 (1), 3–9. 10.1016/j.cell.2020.12.020. [DOI] [PubMed] [Google Scholar]
- Hanan E. J.; Liang J.; Wang X.; Blake R. A.; Blaquiere N.; Staben S. T. Monomeric Targeted Protein Degraders. J. Med. Chem. 2020, 63 (20), 11330–11361. 10.1021/acs.jmedchem.0c00093. [DOI] [PubMed] [Google Scholar]
- PROTAC database: http://cadd.zju.edu.cn/protacdb/.
- Zhang T.; Hatcher J. M.; Teng M.; Gray N. S.; Kostic M. Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation. Cell Chem. Biol. 2019, 26 (11), 1486–1500. 10.1016/j.chembiol.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Raina K.; Darricarrere N.; Hines J.; Gustafson J. L.; Smith I. E.; Miah A. H.; Harling J. D.; Crews C. M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10 (8), 1831–1837. 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovell H.; Testa A.; Maniaci C.; Zhou H.; Prescott A. R.; Macartney T.; Ciulli A.; Alessi D. R. Rapid and Reversible Knockdown of Endogenously Tagged Endosomal Proteins via an Optimized HaloPROTAC Degrader. ACS Chem. Biol. 2019, 14 (5), 882–892. 10.1021/acschembio.8b01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebraud H.; Wright D. J.; Johnson C. N.; Heightman T. D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2 (12), 927–934. 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon R.; Shraga A.; Gehrtz P.; Livnah E.; Shorer Y.; Gurwicz N.; Avram L.; Unger T.; Aharoni H.; Albeck S.; Brandis A.; Shulman Z.; Katz B. Z.; Herishanu Y.; London N. Efficient targeted degradation via reversible and irreversible covalent PROTACs. J. Am. Chem. Soc. 2020, 142 (27), 11734–11742. 10.1021/jacs.9b13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M. J.; Chu L.; Nalawansha D. A.; Li K.; Crews C. M. Targeted Degradation of Oncogenic KRASG12C by VHL-recruiting PROTACs. ACS Cent. Sci. 2020, 6 (8), 1367–1375. 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C. C.; Kleinman J. I.; Brittain S. M.; Lee P. S.; Chung C. Y. S.; Kim K.; Petri Y.; Thomas J. R.; Tallarico J. A.; McKenna J. M.; Schirle M.; Nomura D. K. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14 (11), 2430–2440. 10.1021/acschembio.8b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Crowley V. M.; Wucherpfennig T. G.; Dix M. M.; Cravatt B. F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15 (7), 737–746. 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradlin J. N.; Hu X.; Ward C. C.; Brittain S. M.; Jones M. D.; Ou L.; To M.; Proudfoot A.; Ornelas E.; Woldegiorgis M.; Olzmann J. A.; Bussiere D. E.; Thomas J. R.; Tallarico J. A.; McKenna J. M.; Schirle M.; Maimone T. J.; Nomura D. K. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019, 15 (7), 747–755. 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong B.; Luo M.; Xie Y.; Spradlin J. N.; Tallarico J. A.; McKenna J. M.; Schirle M.; Maimone T. J.; Nomura D. K. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci. Rep. 2020, 10 (1), 15543. 10.1038/s41598-020-72491-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 2009, 48 (38), 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer C. D.; Davis B. G. Selective chemical protein modification. Nat. Commun. 2014, 5, 4740. 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- Tamura T.; Ueda T.; Goto T.; Tsukidate T.; Shapira Y.; Nishikawa Y.; Fujisawa A.; Hamachi I. Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide. Nat. Commun. 2018, 9, 1870. 10.1038/s41467-018-04343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T.; Tamura T.; Kawano M.; Shiono K.; Hobor F.; Wilson A. J.; Hamachi I. Enhanced Suppression of a Protein-Protein Interaction in Cells Using Small-Molecule Covalent Inhibitors Based on an N-Acyl-N-alkyl Sulfonamide Warhead. J. Am. Chem. Soc. 2021, 143 (12), 4766–4774. 10.1021/jacs.1c00703. [DOI] [PubMed] [Google Scholar]
- Anscombe E.; Meschini E.; Mora-Vidal R.; Martin M. P.; Staunton D.; Geitmann M.; Danielson U. H.; Stanley W. A.; Wang L. Z.; Reuillon T.; Golding B. T.; Cano C.; Newell D. R.; Noble M. E.; Wedge S. R.; Endicott J. A.; Griffin R. J. Identification and Characterization of an Irreversible Inhibitor of CDK2. Chem. Biol. 2015, 22 (9), 1159–1164. 10.1016/j.chembiol.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M.; Jiang J.; He Z.; Kwiatkowski N. P.; Donovan K. A.; Mills C. E.; Victor C.; Hatcher J. M.; Fischer E. S.; Sorger P. K.; Zhang T.; Gray N. S. Development of CDK2 and CDK5 Dual Degrader TMX-2172. Angew. Chem., Int. Ed. 2020, 59 (33), 13865–13870. 10.1002/anie.202004087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F.; Chen L.; Cao C.; Yu J.; Luo X.; Zhou P.; Zhao L.; Du W.; Cheng J.; Xie Y.; Chen Y. Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur. J. Med. Chem. 2020, 187, 111952. 10.1016/j.ejmech.2019.111952. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.