

Abstract

Herein, we report the SAR leading to the discovery of VU6028418, a potent M4 mAChR antagonist with high subtype-selectivity and attractive DMPK properties in vitro and in vivo across multiple species. VU6028418 was subsequently evaluated as a preclinical candidate for the treatment of dystonia and other movement disorders. During the characterization of VU6028418, a novel use of deuterium incorporation as a means to modulate CYP inhibition was also discovered.
Keywords: muscarinic acetylcholine receptor M4, dystonia, deuterium, cytochrome P450, SAR
The muscarinic acetylcholine receptors (mAChRs) have been established as promising drug targets for a variety of disorders affecting both the central nervous system (CNS) and periphery, including Alzheimer’s disease (AD), schizophrenia, Parkinson’s disease (PD), ischemic stroke, chronic obstructive pulmonary disorder (COPD), dystonia, and others.1,2 Although the literature contains a wealth of information describing the role of these receptors in biological processes, their structures, and their ligands (both agonists and antagonists), few mAChR modulators have been disclosed that are both truly subtype-selective and possess favorable pharmacokinetic (PK) properties.
The mAChR family of receptors is separated into five subtypes (M1–M5), which belong to the Class A rhodopsin-like G protein-coupled receptor (GPCR) superfamily.3 As is the case with many Class A GPCRs, the structures of the five mAChR subtypes reveal a high degree of sequence homology in the orthosteric pocket.4 This high level of similarity has challenged the development of subtype-selective orthosteric antagonists; the majority of mAChR drug discovery programs describe ligands that are only modestly selective or behave as pan-subtype agonists or antagonists,5−8 although examples of subtype-selective series are known.9,10
The M4 mAChR has recently emerged as a promising therapeutic target for debilitating movement disorders including PD and dystonia. M4 is highly expressed in the striatum, and a complex interplay between dopaminergic and cholinergic input has long been understood to regulate extrapyramidal motor control.11 M4 knockout (KO) studies in mice have suggested that the anticataleptic effects of muscarinic antagonists such as scopolamine are dependent on acute blockade of M4 signaling.12 Additionally, M4 KO mice are characterized by an increase in basal locomotor activity and increased locomotor responses compared to wild type.13 These studies, and the fact that the M4 receptor has been specifically implicated in the modulation of striatal dopamine release,14 suggest that development of M4 antagonists represents a promising therapeutic approach to movement disorders.15
In a previous report,16 we described the discovery and characterization of novel M4 antagonist compounds derived from PCS1055 (1).17 These compounds, VU6013720 (2), VU6021625 (3), and VU6021302 (4), represent a major advance in mAChR tool compound development in terms of both receptor subtype selectivity and druggability (Figure 1). Specifically, VU6021625 was found to be highly potent at both human and rat M4 with minimal inhibition of human acetylcholinesterase (hAChE): h/rM4 IC50 = 0.44 and 57 nM, respectively; hAChE IC50 >10 μM (Eurofins Panlabs).16 Here, we report the subsequent structure–activity relationship (SAR) studies on these compounds, which culminated in the discovery of VU6028418 (8i), a highly selective M4 antagonist evaluated as a preclinical candidate for the treatment of movement disorders.
Figure 1.

Structures of selected M4 antagonist compounds.
Our synthetic chemistry effort was carried out as described in the previous report.16 Analogues of compounds 2–4 were synthesized as described in Scheme 1. Substituted [3.3.0]cyclopentylpyrrolidine 5, synthesized as previously described,18,19 was coupled to 3,6-dichloropyridazine under nucleophilic aromatic substitution (SNAr) conditions to give chloropyridazine 6, which then underwent a facile Boc-deprotection/reductive amination sequence to give tetrahydropyran (THP) 7. Compound 7 was then substituted with the appropriate boronic acid or pinacol ester under Suzuki–Miyaura conditions to give substituted pyridazines 8a–i. The third generation BrettPhos palladacycle20 proved to be particularly useful in realizing these challenging cross couplings.
Scheme 1. Synthesis of Compounds 8a–i.

Reagents and conditions: (a) 3,6-dichloropyridazine, DIPEA, tBuOH, microwave, 150 °C, 65%; (b) HCl, 1,4-dioxane, rt; (c) tetrahydro-2H-pyran-4-carbaldehyde; NaBH(OAc)3, DCM, THF, rt, 89% over two steps; (d) boronic acid or pinacol ester, K2CO3, BrettPhos-Pd-G3, 1,4-dioxane, H2O, 100 °C, 11–47%.
Selected SAR is shown in Table 1. Early in our SAR campaign on 2, it was found that the eastern THP motif consistently provided robust hM4 potency and subtype selectivity (relative to hM2) as well as low predicted hepatic microsomal clearance compared to benzylic and non-heteroatom-containing aliphatic substituents (predicted human hepatic clearance (hClhep) generally <10 mL/min/kg for THP compounds). The THP group was therefore held constant throughout our subsequent synthetic efforts, and the majority of our SAR was focused on replacements to the western halogenated phenyl ring. Additionally, substitution and/or replacement of the aminopyridazine (with respect to both the pyridazine ring and capping/replacement of the free NH) was found to consistently give a decrease in M4 potency, and we avoided these types of modifications as the program progressed.
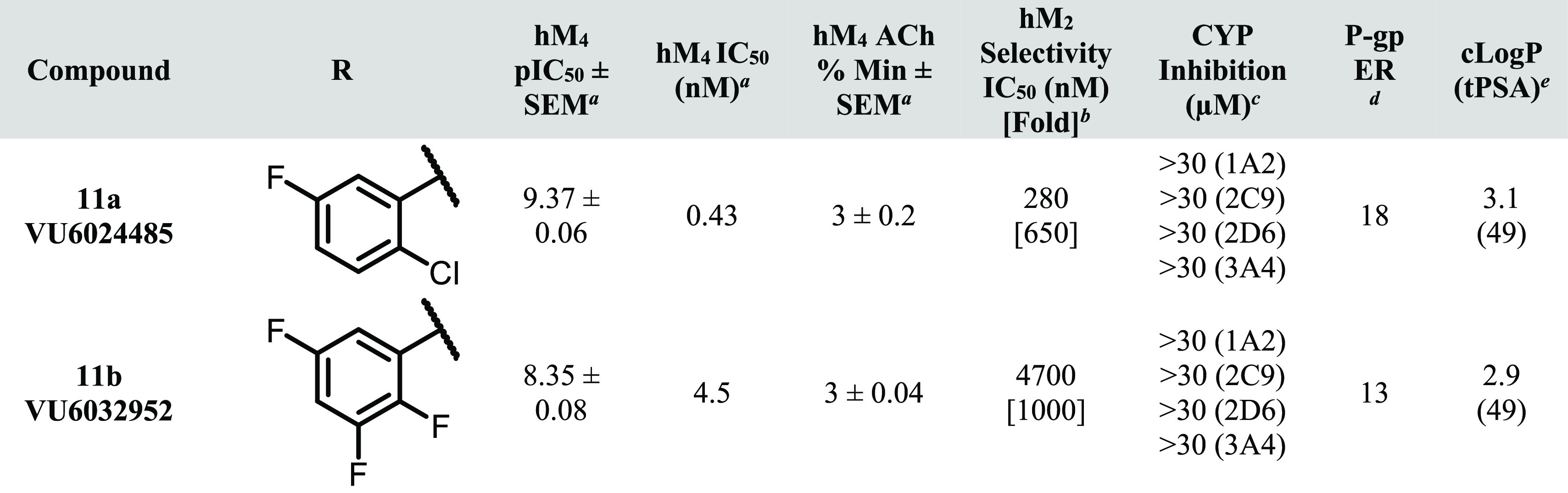
Table 1. hM4 Potency, Selectivity, CYP Inhibition, and P-gp Efflux for 8a–i.


Calcium mobilization assays with hM4. IC50 values for hM4 represent means from at least three (n = 3) independent experiments performed in triplicate.
Calcium mobilization assays with hM2/Gqi5-CHO cells performed in the presence of an EC80 fixed concentration of acetylcholine. IC50 values for hM2 represent one or two independent experiments performed in triplicate.
Cytochrome P450 cocktail inhibition assay in human liver microsomes (see the SI for further details).
BBB penetration potential using Madin Darby canine kidney cells with overexpression of MDR1 gene (encoding for P-gp) in cell monolayers; efflux ratio (ER) is defined as Papp(B-to-A)/Papp(A-to-B) (see the SI for further details).
Lilly Laboratories cell porcine kidney 1-MDR1 cell line was used for comparison.
Calculated using ChemDraw Professional version 20.1.
As shown in Table 1, a variety of substitutions on the western phenyl ring provided excellent hM4 potency (<10 nM) and subtype selectivity relative to hM2 (>1000-fold). Although there are reports in the literature of mAChR antagonists with modest subtype-selectivity,8,9 to our knowledge, these analogues represent the most selective orthosteric mAChR antagonist ligands for the M4 receptor subtype. Notably, potencies in this series generally displayed a disconnect between human and rat, although in most cases rM4 IC50’s were <1 μM (data not shown). Although consistent within this series, the rationale for the decrease in rat potency is not fully understood at present; we are hopeful that future crystallography and/or molecular pharmacology experiments will further clarify this discrepancy.
At the onset of our SAR campaign, two major areas of improvement were identified for 2: (1) CYP inhibition and (2) P-gp efflux. It was hypothesized that the suboptimal CYP inhibition profile of 2 (VU6013720) could be improved by a general decrease in clogP, particularly with respect to the western halogenated phenyl group. Toward this end, compounds with increased polar surface area (PSA) and/or decreased lipophilic character (as exemplified by analogues 8a–d) were synthesized. Compound 8a, in which the 2-chloro, 5-fluoro substituents are deleted to give an unsubstituted phenyl ring, displayed a marked reduction in CYP inhibition (particularly with respect to 1A2 and 3A4), although P-gp efflux remained comparable to 2. An increase in PSA (i.e., 3-pyridyl analogue 8b, acetylaminophenyl 8c and benzolactam 8d) provided no erosion in the robust hM4 potency of 2 and 8a, although the high P-gp efflux displayed by these analogues precluded their further characterization in the CYP inhibition assay.
Because the introduction of polarity on the western biaryl system proved detrimental to P-gp efflux, it was reasoned that an exhaustive exploration of halogenated aromatic groups could potentially provide a reduction of CYP inhibition while maintaining or decreasing the P-gp efflux ratio (ER). As exemplified by fluorinated analogues 8e–i, the pattern of halogen substitution appears to play a crucial role in the overall CYP inhibition profile. Replacement of the 2-chloro substituent with fluorine (2,5-difluoro analogue 8h) reduced CYP inhibition relative to 2 across three isoforms while also modestly decreasing P-gp efflux. Unfortunately, potent 2C9 inhibition persisted with this and many of the other analogues tested in this series. 2,3,5-Trifluoro analogue 8i was found to improve the CYP inhibition profile relative to 2 even further (2C9 IC50 = 9.8 μM; >30 μM across all other isoforms tested), with decreased P-gp efflux relative to 2 (8.9 vs 14). In an LLC-PK1-MDR1 cell line, 8i did not appear to be a substrate for P-gp efflux (ER = 0.93). Other than species differences (Madin-Darby canine kidney vs porcine kidney), the observed differences in P-gp ER are likely a result of differences in surface-level cell expression between these cell lines. This result highlights the importance of testing for P-gp activity across multiple cell types followed by rigorous in vivo verification. This type of (relative) 2,3,5-trifluorophenyl substitution pattern is well-known in drug discovery literature, and the ability of this arrangement to confer favorable properties is perhaps best exemplified by the diabetes mellitus type 2 medication sitagliptin (Januvia).21,22
Although it is well established that orthosteric mAChR ligands generally require the presence of a basic amine for activity,4,23 we were interested in chemistry aimed at modulating the basicity of the pyrrolidine nitrogen in an attempt to further decrease CYP inhibition and/or P-gp ER. Installation of a methyl group on the THP pendant either α or β to the amine generally resulted in a loss of subtype-selectivity, and β-fluoro THP analogues (relative to the amine) gave analogues with high predicted hClhep values (data not shown). Conversely, although such a modification is likely neutral with respect to modulating basicity, it was found that an α-gem-deutero analogue of 2 (11a) not only maintained the robust potency and subtype selectivity of 2, but completely reversed the unfavorable CYP inhibition profile (>30 μM across all isoforms tested, Table 2). Although the incorporation of deuterium has been used to beneficial effect across a number of different parameters and programs in medicinal chemistry, particularly to slow CYP-mediated oxidative metabolism through the kinetic isotope effect,24 to the best of our knowledge, this is the first report in the literature describing this type of dramatic effect on CYP inhibition reversal. Although this trend was found to hold true for the 2,3,5-trifluoro analogue 8i as well (gem-deutero analogue 11b), this modification consistently provided a slight increase in P-gp ER relative to the CH2 counterparts.
Table 2. hM4 Potency, M2 Selectivity, CYP Inhibition, and P-gp Efflux for Compounds 11a and 11b.

Calcium mobilization assays with (a) hM4. IC50 values for hM4 represent means from at least three (n = 3) independent experiments performed in triplicate.
hM2/Gqi5-CHO cells performed in the presence of an EC80 fixed concentration of acetylcholine. IC50 values for hM2 represent one independent experiment performed in triplicate.
Cytochrome P450 cocktail inhibition assay in human liver microsomes (see SI for further details).
BBB penetration potential using MDR1-MDCK cell monolayers; ER is defined as Papp(B-to-A)/Papp(A-to-B) (see SI for further details).
Calculated using ChemDraw Professional version 20.1.
gem-Deutero analogues of this type were synthesized as shown in Scheme 2. Boc deprotection/amide coupling on substituted pyridazines 9a and 9b (synthesized by the Suzuki–Miyaura chemistry described in Scheme 1; see the Supporting Information for more details) gave amides 10a and 10b, and subsequent reduction with lithium aluminum deuteride (LAD) gave gem-deutero analogues 11a and 11b.
Scheme 2. Synthesis of Compounds 11a and 11b.

Reagents and conditions: (a) HCl, 1,4-dioxane, rt; (b) tetrahydro-2H-pyran-4-carboxylic acid, HATU, DIPEA, DMF, rt, 88% (10a) and 78% (10b) over two steps; (c) LiAlD4, THF, rt, 25% (10a) and 14% (10b).
Based on its potent and selective M4 mAChR activity (see Figure 2 for full mAChR selectivity), potency across species (rat M4 IC50 = 145 nM), CYP inhibition (>9 μM across all isoforms tested), P-gp ER (<10), and favorable physicochemical properties (melting point = 201–203 °C, kinetic solubility = 85.1 μM (pH = 6.8)), compound 8i was selected at this stage for further profiling.
Figure 2.
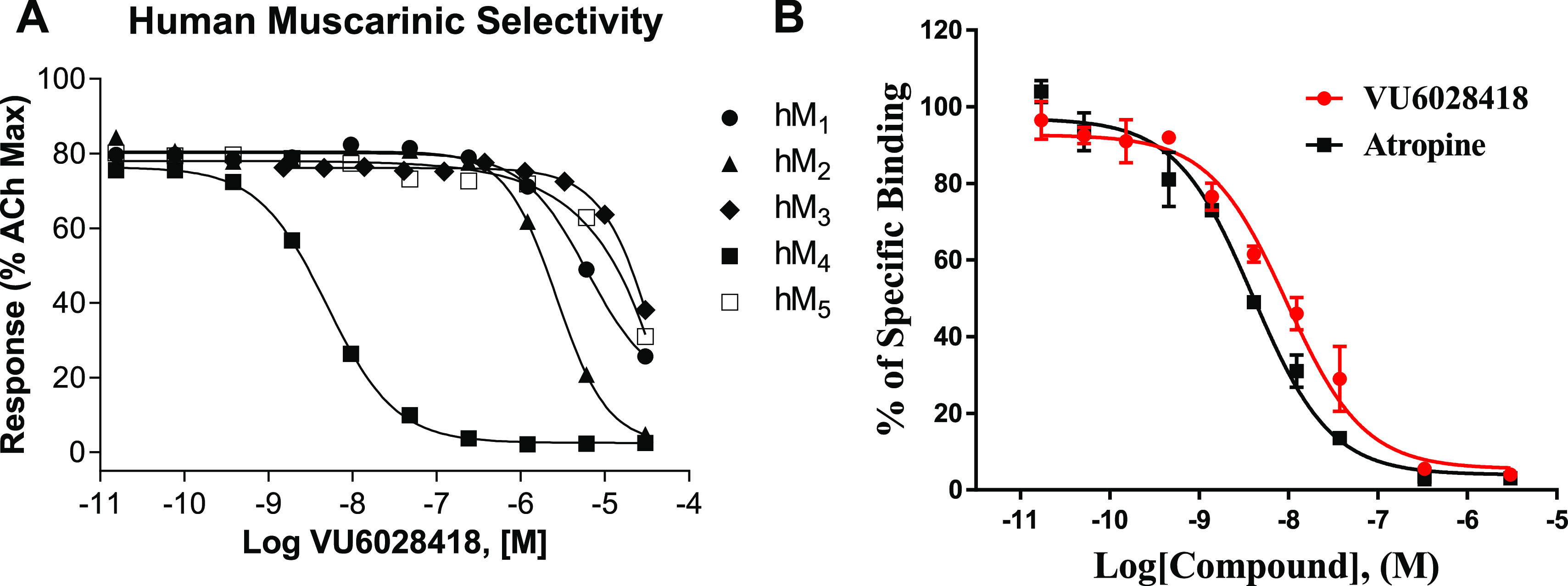
VU6028418 (8i) mAChR selectivity and radioligand displacement. (A) VU6028418 is highly selective for hM4 versus all other human muscarinic subtypes (hM4 IC50 = 4.1 nM, hM2 IC50 = 3.5 μM, hM1,3,5 IC50 >10 μM). VU6028418 was applied to cells before the addition of an EC80 concentration of acetylcholine and the response was measured via calcium mobilization. Data represent one independent determination performed in triplicate using CHO cells stably transfected with the specified mAChR. (B) VU6028418 fully competes with the equilibrium of [3H]NMS in a concentration-dependent manner (Ki = 3.2 nM). Data represent one independent determination performed in triplicate using membranes harvested from CHO cells stably expressing human M4. Competition displacement data from both VU6028418 and atropine fitted to a one-site model well.
The in vivo PK parameters for 8i (VU6028418) are listed in Table 3. 8i displayed high oral bioavailability (≥100% rat and mouse, 86% dog; suspension doses). Clearance from plasma was low in rat and mouse (6.1 and 17 mL/min/kg respectively) and conversely high in dog (43 mL/min/kg). Vss was large in all species tested, and elimination t1/2 was generally quite long (>13 h). Distribution to brain on a total and unbound concentration basis (Kp, Kp,uu) was high (Table 3), while unbound distribution to CSF (Kp,u) was moderate (rat). Consistent with the observed low clearance profile, low turnover was observed in metabolite ID experiments (cryopreserved hepatocytes), with >80% of the parent remaining across all tested species after 4 h (see the Supporting Information for additional details and results).
Table 3. In Vivo PK Parameters for VU6028418 (8i).
| parameter | rat (SD)a | mouse (CD-1)a | dog (beagle)a |
|---|---|---|---|
| dose (mg/kg) iv/po | 1/10 | 1/3 | 1/3 |
| CLp (mL/min/kg) | 6.1 | 17 | 43 |
| Vss (L/kg) | 6.7 | 10.6 | 8.5 |
| elimination t1/2 (h) | 13 | NC | 15 |
| Cmax (ng/mL) po | 17 000 | 181 | 70 |
| Tmax (h) po | 1.5 | 6.67 | 17 |
| AUCo-inf (ng/mL·h) po | 30 000 | NC | 1100 |
| F (%) po | ≥100 | ≥100 | 86 |
| total brain/total plasma (Kp) | 6.4 | ND | ND |
| unbound brain/unbound plasma (Kp,uu) | 0.61 | ND | ND |
| CSF/plasma unbound (Kp,u) | 0.24 | ND | ND |
Values represent means from two to three animals. ND = not determined. NC = not calculated; there was insufficient data to define the elimination phase (i.e., Cmax was one of the last three time points).
In a rat single PO dose escalation PK study, 8i demonstrated a linear increase in mean AUC0-last by dose between 1, 3, 10, and 30 mg/kg with a sublinear increase between 30, 100, and 300 mg/kg. Importantly, no adverse events were observed for any of the dose groups in this study (see the Supporting Information for additional details and results). Based on the in vivo data from rat and dog as well as in vitro intrinsic clearance data from rat, dog, and human (hepatic microsomes; data not shown), preliminary human PK predictions (Table 4) were generated. Consistent with the rat and dog data, low clearance, high volume of distribution, and long t1/2 were predicted in human.
Table 4. Predicted Human PK Parameters for VU6028418 (8i).
IVIVE approach with empirical correction factors using mean rat and dog in vivo data.
Scaling of unbound Vss using mean rat and dog in vivo data.
Based on the encouraging in vivo PK profile for 8i, the CYP inhibition and toxicity profiles for this compound were further evaluated. Across seven isoforms (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5), essentially no reversible inhibition was observed; weak time-dependent signals were observed for 2C9, 2C19, and 2D6 (IC50’s >100 μM). Phenotyping experiments revealed 8i to possess a predominantly 3A4 metabolism phenotype (∼90%), with smaller contributions from 2D6 (∼2.0%) and 2J2 (∼8.3%). However, induction experiments showed induction of 1A2F, 2B6, and 3A4 in hepatocytes from one of the three donors examined; EC50 and Emax values were ∼1.9–3.6 uM and ∼2.1–3.8x respectively (see the Supporting Information for further details regarding all CYP450 experiments). 8i was negative in a Mini Ames test (4 strains ± S9).
VU6028418 (8i) demonstrated a robust, dose-dependent response in a rat model of haloperidol-induced catalepsy (MED = 1 mg/kg, Figure 3) after oral administration (F3,36 = 8.7; p < 0.001).25 Vehicle-treated rats demonstrated a mean latency to withdraw of 43.4 ± 4.3 s. The administration of 0.3 mg/kg of VU6028418 did not significantly alter the mean latency to withdraw (32 ± 5.2 s; p > 0.05 compared to vehicle treated controls). Treatment with either 1 or 3 mg/kg of VU6028418 significantly reduced mean latency to withdraw in these animals (21.3 ± 4.6 for 1 mg/kg; p < 0.01; 15.1 ± 2.1 for 3 mg/kg p < 0.001). VU6028418 significantly reversed the cataleptic behavior when compared to vehicle treated control animals (F3,36 = 8.7 p < 0.001; Figure 3B). The administration of 0.3 mg/kg of VU6028418 reversed cataleptic behavior by 26.2 ± 12.0% (p > 0.05), 1 mg/kg reversed the catalepsy by 50.9 ± 10.7% (p < 0.01), and 3 mg/kg reversed catalepsy by 65.2 ± 4.9% (p < 0.001). Post study (1 h) at 1 mg/kg, brain/plasma Kp and Kp,uu were determined to be 3.4 and 0.32 respectively, with a mean Cbrain,unbound of 12.5 ng/g.
Figure 3.

Dose dependent effect of VU6028418 in a rat model of haloperidol-induced catalepsy. (A) VU6028418 dose-dependently reduced the mean latency to withdraw in haloperidol treated animals. (B) VU6028418 significantly reversed cataleptic behavior. n = 10 for all groups, **p < 0.01, ***p < 0.001.
An ancillary pharmacology screen (Eurofins Panlabs)26 of 8i revealed 101% inhibition of the σ1 receptor at 10 μM, and 61% hERG inhibition at the same concentration (see the Supporting Information for the full ancillary pharmacology profile). In follow up dose response assays for these respective targets, 8i was found to possess a Ki of 16.9 nM (σ1 radioligand binding) and an IC50 of 431 nM (hERG patch clamp). 8i was also found to have a strong inhibitory effect on hERG tail current (94 ± 1% inhibition, n = 3) in subsequent electrophysiology studies (HEK, tail current recorded at −40 mV after depolarizing step to +30 mV, data not shown). In the [3H]dofetilide binding assay, VU6028418 was found to have an IC50 of 1.1 μM (Ki = 790 nM, Eurofins Panlabs).26 Despite the strong hERG inhibition displayed by this compound, 8i was not found to strongly inhibit any other cardiac ion channels tested (all ion channel inhibitions ≤42% at 10 μM (patch clamp), see the Supporting Information for additional details).27 This result is perhaps not entirely surprising given the chemotype of these analogues; the presence of a basic amine flanked by relatively lipophilic groups inherently biases the series toward hERG activity.28
Although the potent hERG inhibition precluded VU6028418 (8i) from further development, the M4 antagonists described herein represent some of the most potent and selective tool compounds yet discovered for the mAChR family. Because of its excellent subtype selectivity, PK, and toxicity profile, VU6028418 (8i) should find extensive use in the probing of mAChR biology toward a better understanding of dystonia and other movement disorders. Although it is difficult to speculate on the origins of the robust M4 selectivity observed within this series in the absence of a cocrystal structure, we are hopeful that future crystallography experiments will clarify this encouraging trend. Additionally, the observation that a gem-deuterium modification can bias chemotypes away from CYP inhibition should be of broad utility to medicinal chemists. Experiments in our laboratory aimed at understanding the hERG SAR of this series are ongoing and will be reported in due course.
Acknowledgments
The authors would like to thank the following funding sources for their generous support of this work: Ancora Innovation, LLC, and DoD Grant W81XWH-19-1-0355 (C.W.L., P.J.C.). C.W.L. and A.M.B. thank the William K. Warren Family and Foundation for funding the William K. Warren, Jr. Chair in Medicine and support of our programs. We also thank Dr. Roman Lazarenko and Dr. Jerod Denton for assistance with hERG electrophysiology experiments, and Dr. Chris Presley for assistance with HRMS.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00363.
Full experimental procedures and characterization for new compounds, experimental details for all assays (PDF)
Author Contributions
M.S., T.R.C., K.A.B., L.A.B., C.H., J.L.E., D.W.E., and A.M.B. performed synthetic chemistry and scaled up key compounds. A.L.R., L.P., A.Q., and C.M.N. performed and analyzed molecular pharmacology and radioligand binding experiments. J.W.D. and J.M.R. performed and analyzed behavioral pharmacology experiments. J.C.O., K.J.W., S.C., and T.M.B. performed and analyzed DMPK experiments. P.J.C., C.W.L., A.M.B., C.M.N., and J.M.R. oversaw experimental design, and A.M.B. wrote the manuscript with input from all authors.
The authors declare the following competing financial interest(s): C.M.N., J.W.D., C.W.L., P.J.C., T.M.B., J.M.R., T.R.C., L.A.B., C.H., M.S., J.L.E., D.W.E., and A.M.B. are inventors on applications for composition of matter patents that protect several series of M4 antagonists.
Supplementary Material
References
- Wess J.; Eglen R. M.; Gautam D. Muscarinic Acetylcholine Receptors: Mutant Mice Provide New Insights for Drug Development. Nat. Rev. Drug Discovery 2007, 6, 721–733. 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Kruse A. C.; Kobilka B. K.; Gautam D.; Sexton P. M.; Christopoulos A.; Wess J. Muscarinic Acetylcholine Receptors: Novel Opportunities for Drug Development. Nat. Rev. Drug Discovery 2014, 13, 549–560. 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A.; Schrage R.; Mohr K. Allosteric Modulators Targeting CNS Muscarinic Receptors. Neuropharmacology 2018, 136, 427–437. 10.1016/j.neuropharm.2017.09.024. [DOI] [PubMed] [Google Scholar]
- Vuckovic Z.; Gentry P. R.; Berizzi A. E.; Hirata K.; Varghese S.; Thompson G.; van der Westhuizen E. T.; Burger W. A. C.; Rahmani R.; Valant C.; Langmead C. J.; Lindsley C. W.; Baell J. B.; Tobin A. B.; Sexton P. M.; Christopoulos A.; Thal D. M. Crystal Structure of the M5Muscarinic Acetylcholine Receptor. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 26001–26007. 10.1073/pnas.1914446116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. M.; Weiner R. L.; Luscombe V. B.; Cho H. P.; Niswender C. M.; Engers D. W.; Bridges T. M.; Conn P. J.; Lindsley C. W. Synthesis and Evaluation of 4,6-Disubstituted Pyrimidines as CNS Penetrant Pan-Muscarinic Antagonists with a Novel Chemotype. Bioorg. Med. Chem. Lett. 2017, 27, 2479–2483. 10.1016/j.bmcl.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. M.; Weiner R. L.; Luscombe V. B.; Ajmera S.; Cho H. P.; Chang S.; Zhan X.; Rodriguez A. L.; Niswender C. M.; Engers D. W.; Bridges T. M.; Conn P. J.; Lindsley C. W. Discovery and Optimization of 3-(4-Aryl/heteroarylsulfonyl) piperazin-1-yl)-6-(piperidin-1-yl) Pyridazines as Novel, CNS Penetrant Pan-Muscarinic Antagonists. Bioorg. Med. Chem. Lett. 2017, 27, 3576–3581. 10.1016/j.bmcl.2017.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapecchi S.; Nesi M.; Matucci R.; Bellucci C.; Buccioni M.; Dei S.; Guandalini L.; Manetti D.; Martelli C.; Martini E.; Marucci G.; Orlandi F.; Novella Romanelli M.; Teodori E.; Cirilli R. Synthesis, Affinity Profile and Functional Activity of Potent Chiral Muscarinic Antagonists with a Pyrrolidinylfuran Structure. J. Med. Chem. 2010, 53, 201–207. 10.1021/jm901048j. [DOI] [PubMed] [Google Scholar]
- Böhme T. M.; Augelli-Szafran C. E.; Hallak H.; Pugsley T.; Serpa K.; Schwarz R. D. Synthesis and Pharmacology of Benzoxazines as Highly Selective Antagonists at M4 Muscarinic Receptors. J. Med. Chem. 2002, 45, 3094–3102. 10.1021/jm011116o. [DOI] [PubMed] [Google Scholar]
- Liu H.; Hofmann J.; Fish I.; Schaake B.; Eitel K.; Bartuschat A.; Kaindl J.; Rampp H.; Banerjee A.; Hübner H.; Clark M. J.; Vincent S. G.; Fisher J. T.; Heinrich M. R.; Hirata K.; Liu X.; Sunahara R. K.; Shoichet B. K.; Kobilka B. K.; Gmeiner P. Structure-Guided Deveopment of Selective M3Muscarinic Acetylcholine Receptor Antagonists. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 12046–12050. 10.1073/pnas.1813988115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.; Lachapelle E. A.; Kablaoui N. M.; Webb D.; Popiolek M.; Grimwood S.; Kozak R.; O’Connor R. E.; Lazzaro J. T.; Butler C. R.; Zhang L. Discovery of Selective M4Muscarinic Acetylcholine Receptor Agonists with Novel Carbamate Isosteres. ACS Med. Chem. Lett. 2019, 10, 941–948. 10.1021/acsmedchemlett.9b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle M. S.; Conn P. J. Roles of the M4 Acetylcholine Receptor in the Basal Ganglia and the Treatment of Movement Disorders. Mov. Disord. 2019, 34, 1089–1099. 10.1002/mds.27740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa H.; Taketo M. M.; Matsui M. Loss of Anti-Cataleptic Effect of Scopolamine in Mice Lacking Muscarinic Acetylcholine Receptor Subtype 4. Eur. J. Pharmacol. 2003, 468, 15–19. 10.1016/S0014-2999(03)01642-X. [DOI] [PubMed] [Google Scholar]
- Gomeza J.; Zhang L.; Kostenis E.; Felder C.; Bymaster F.; Brodkin J.; Shannon H.; Xia B.; Deng C.; Wess J. Enhancement of D1 Dopamine Receptor-Mediated Locomotor Stimulation in M4 Muscarinic Acetylcholine Receptor Knockout Mice. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 10483–10488. 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster D. J.; Wilson J. M.; Remke D. H.; Mahmood M. S.; Uddin J. M.; Wess J.; Patel S.; Marnett L. J.; Niswender C. M.; Jones C. K.; Xiang Z.; Lindsley C. W.; Rook J. M.; Conn P. J. Antipsychotic-like Effects of M4 Positive Allosteric Modulators Are Mediated by CB2 Receptor-Dependent Inhibition of Dopamine Release. Neuron 2016, 91, 1244–1252. 10.1016/j.neuron.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs A. M.; Roman K. M.; Campbell S. A.; Pisani A.; Hess E. J.; Bonsi P. The Neurobiological Basis for Novel Experimental Therapeutics in Dystonia. Neurobiol. Dis. 2019, 130, 104526. 10.1016/j.nbd.2019.104526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle M. S.; Bender A. M.; Dickerson J. W.; Foster D. J.; Qi A.; Cho H. P.; Donsante Y.; Peng W.; Bryant Z.; Stillwell K. J.; Bridges T. M.; Chang S.; Watson K. J.; O’Neill J. C.; Engers J. L.; Peng L.; Rodriguez A. L.; Niswender C. M.; Lindsley C. W.; Hess E. J.; Conn P. J.; Rook J. M. Discovery of the First Selective M4Muscarinic Acetylcholine Receptor Antagonsits with In Vivo Anti-Parkinsonian and Anti-Dystonic Efficacy. ACS Pharmacol. Transl. Sci. 2021, 10.1021/acsptsci.0c00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croy C. H.; Chan W. Y.; Castetter A. M.; Watt M. L.; Quets A. T.; Felder C. C. Characterization of PCS1055, a Novel Muscarinic M4 Receptor Antagonist. Eur. J. Pharmacol. 2016, 782, 70–76. 10.1016/j.ejphar.2016.04.022. [DOI] [PubMed] [Google Scholar]
- Cho T. P.; Long Y. F.; Gang L. Z.; Yang W.; Jun L. H.; Yuan S. G.; Hong F. J.; Lin W.; Liang G. D.; Lei Z.; Jing L. J.; Shen G. A.; Hong S. G.; Dan W.; Ying F.; Ke Y. P.; Ying L.; Jun F.; Tai M. X. Synthesis and Biological Evaluation of Azobicyclo[3.3.0]octane Derivatives as Dipeptidyl Peptidase 4 Inhibitors for the Treatment of Type 2 Diabetes. Bioorg. Med. Chem. Lett. 2010, 20, 3565–3568. 10.1016/j.bmcl.2010.04.120. [DOI] [PubMed] [Google Scholar]
- Lemoine R. C.; Petersen A. C.; Setti L.; Baldinger T.; Wanner J.; Jekle A.; Heilek G.; deRosier A.; Ji C.; Rotstein D. M. Evaluation of 3-Amino-8-azabicyclo[3.2.1]octane Replacement in the CCR5 Antagonist Maraviroc. Bioorg. Med. Chem. Lett. 2010, 20, 1674–1676. 10.1016/j.bmcl.2010.01.080. [DOI] [PubMed] [Google Scholar]
- Ruiz-Castillo P.; Buchwald S. L. Applications of Palladium-Catalyzed C-N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Wang L.; Beconi M.; Eiermann G. J.; Fisher M. H.; He H.; Hickey G. J.; Kowalchick J. E.; Leiting B.; Lyons K.; Marsilio F.; McCann M. E.; Patel R. A.; Petrov A.; Scapin G.; Patel S. B.; Roy R. S.; Wu J. K.; Wyvratt M. J.; Zhang B. B.; Zhu L.; Thornberry N. A.; Weber A. E. (2R)-4-Oxo-4-[3-(Trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin- 7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: A Potent, Orally Active Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. J. Med. Chem. 2005, 48, 141–151. 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- Gordeev M. F.; Yuan Z. Y. New Potent Antibacterial Oxazolidinone (MRX-I) with an Improved Class Safety Profile. J. Med. Chem. 2014, 57, 4487–4497. 10.1021/jm401931e. [DOI] [PubMed] [Google Scholar]
- Thal D. M.; Sun B.; Feng D.; Nawaratne V.; Leach K.; Felder C. C.; Bures M. G.; Evans D. A.; Weis W. I.; Bachhawat P.; Kobilka T. S.; Sexton P. M.; Kobilka B. K.; Christopoulos A. Crystal Structures of the M1 and M4Muscarinic Acetylcholine Receptors. Nature 2016, 531, 335–340. 10.1038/nature17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirali T.; Serafini M.; Cargnin S.; Genazzani A. A. Applications of Deuterium in Medicinal Chemistry. J. Med. Chem. 2019, 62, 5276–5297. 10.1021/acs.jmedchem.8b01808. [DOI] [PubMed] [Google Scholar]
- Panarese J. D.; Engers D. W.; Wu Y.; Bronson J. J.; Macor J. E.; Chun A.; Rodriguez A. L.; Felts A. S.; Engers J. L.; Loch M. T.; Emmitte K. A.; Castelhano A. L.; Kates M. J.; Nader M. A.; Jones C. K.; Blobaum A. L.; Conn P. J.; Niswender C. M.; Hopkins C. R.; Lindsley C. W. Discovery of VU2957 (Valiglurax): An mGlu4 Positive Allosteric Modulator Evaluated as a Preclinical Candidate for the Treatment of Parkinson’s Disease. ACS Med. Chem. Lett. 2019, 10, 255–260. 10.1021/acsmedchemlett.8b00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.eurofinsdiscoveryservices.com.
- https://www.criver.com/products-services/safety-assessment/safety-pharmacology/vitro-assays/chantest-cardiac-channel-panel-savetytm-assessment?region=3601.
- Kratz J. M.; Schuster D.; Edtbauer M.; Saxena P.; Mair C. E.; Kirchebner J.; Matuszczak B.; Baburin I.; Hering S.; Rollinger J. M. Experimentally Validated hERG Pharmacophore Models as Cardiotoxicity Prediction Tools. J. Chem. Inf. Model. 2014, 54, 2887–2901. 10.1021/ci5001955. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.