

Abstract

Bromodomain containing proteins and the acetyl-lysine binding bromodomains contained therein are increasingly attractive targets for the development of novel epigenetic therapeutics. To help validate this target class and unravel the complex associated biology, there has been a concerted effort to develop selective small molecule bromodomain inhibitors. Herein we describe the structure-based efforts and multiple challenges encountered in optimizing a naphthyridone template into selective TAF1(2) bromodomain inhibitors which, while unsuitable as chemical probes themselves, show promise for the future development of small molecules to interrogate TAF1(2) biology. Key to this work was the introduction and modulation of the basicity of a pendant amine which had a substantial impact on not only bromodomain selectivity but also cellular target engagement.
Keywords: TAF1, TAF1L, Bromodomain, Epigenetics
The 61 human bromodomains are composed of 48 typical bromodomains where two conserved amino acid residues, asparagine and tyrosine, form a direct and a water-mediated hydrogen-bond interaction to an acetyl-lysine (KAc) residue.1 This interaction drives the recruitment of cellular transcriptional machinery to a specific epigenetic histone mark and regulates gene expression.2 These epigenetic reader modules have emerged as a new exciting class of therapeutic targets for a variety of diseases including immune disorders, metabolic disease, and oncology.3,4 The majority of research to date has been focused on the dual bromodomain containing bromodomain and extra terminal (BET) family of proteins, and a number of molecules targeting the BET proteins are currently undergoing oncology clinical trials.5 Driven by the profound and fundamental biology associated with the BET proteins, interest in the 53 non-BET bromodomain containing proteins (BCPs) has increased. To help understand the role these bromodomains play in modulating healthy and disease states, a number of small molecule inhibitors and chemical probes have been developed and disclosed.6 Used in conjunction with other techniques such as gene knockdown or knockout, small molecule chemical probes can provide insight into the role of individual protein domains present in multidomain proteins such as BCPs.7,8 To give increased confidence in phenotype assignment, access to and use of multiple well characterized inhibitors of different chemotypes is beneficial for robust target validation.9,10 Although there are a significant number of reported small molecule inhibitors and probes for non-BET bromodomain targets, several lack the sufficient breadth of chemical equity for robust target validation.
TAF1 and the highly related TAF1L are multidomain proteins, each of which contains two kinase domains, a histone acetyl transferase domain and two bromodomains termed TAF1(1)/TAF1L(1) (N-terminal bromodomain) and TAF1(2)/TAF1L(2) (C-terminal bromodomain). TAF1 and TAF1L are associated factors of the TATA-binding protein (TBP), a subunit of transcription factor IID (TFIID), which plays a critical role in RNA polymerase transcription. TFIID acts as a scaffold for the assembly of the preinitiation complex formed prior to transcription, coordinates the alignment of RNA polymerase with DNA, and helps facilitate binding to DNA.11,12 Dysregulation of TAF1 has been associated with a number of diseases across oncology and neurology: overexpression of TAF1 has been shown to increase androgen receptor activity several fold, resulting in the progression of prostate cancer, and it has been proposed that TAF1 mutation may play a role in colorectal and gastric cancers.13,14 TAF1 bromodomain mutations have also been shown to contribute to the phenotypes displayed across multiple neurodegenerative X-linked syndromes, including intellectual disability and facial dysmorphology.15
While the involvement of TAF1 within such disease has been established, the role of the TAF1 bromodomains is less clear. To this end, in 2014 Bradner and co-workers reported the first submicromolar TAF1/TAF1L bromodomain small molecule inhibitor, UMB-32 (1) with comparable activity against BRD4 (Figure 1).16
Figure 1.
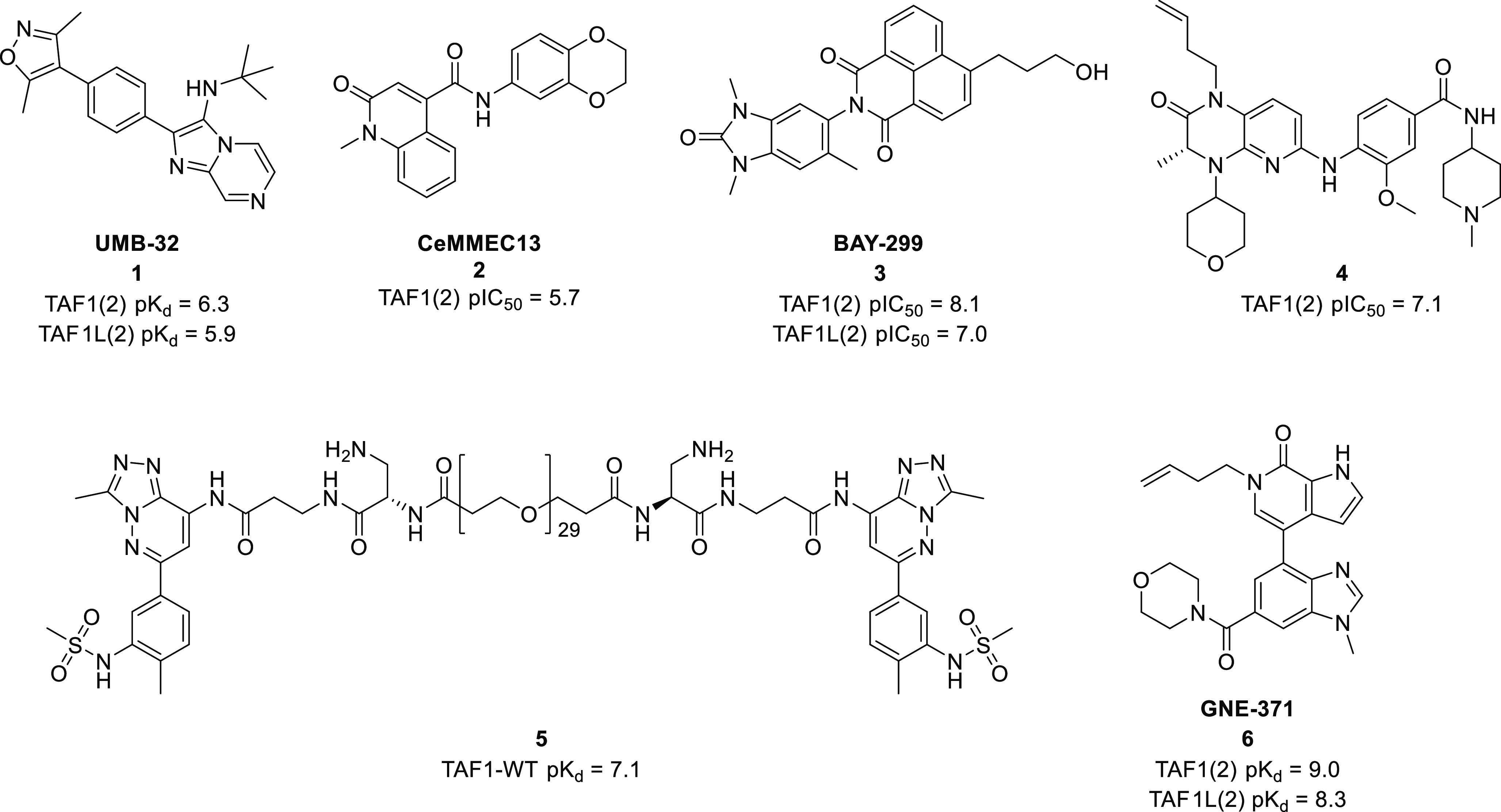
Disclosed TAF1/TAF1L inhibitors and their reported bromodomain affinities.
Since this demonstration of the small molecule tractability of TAF1(2)/TAF1L(2), there have been several reports in the patent and peer-reviewed literature of inhibitors. CeMMEC13 (2) was disclosed by Sdelci and co-workers as a TAF1 inhibitor,17 and researchers from Bayer developed BAY-299 (3), a potent inhibitor of both TAF1(2)/TAF1L(2) and BRPF2.18 Compound 4 was developed from polo-like kinase 1 inhibitor BI-2536 and demonstrates >120-fold selectivity for TAF1(2) over BRD4(1).19 Bifunctional molecule 5 was designed to bind to both bromodomains of TAF1 bivalently and demonstrated significant affinity gain over monovalent ligand binding.20 Additionally, a bivalent molecule designed to inhibit the BET bromodomains was also reported to inhibit TAF1(2), although it is unclear whether the TAF1 inhibition is bivalent.21 The selective and potent TAF1(2)/TAF1L(2) chemical probe GNE-371 (6) was recently developed by Genentech and Constellation Pharmaceuticals.22 The 1-butenyl group in GNE-371 functions as the KAc methyl mimetic and takes advantage of the differential stability of conserved bromodomain water networks to induce selectivity at TAF1(2) through rearrangement and stabilization of the waters in the KAc binding site.23 To aid TAF1 target validation efforts, we sought to develop a structurally diverse, potent and selective small molecule inhibitor of the second bromodomain of these targets. Selectivity is an important consideration when linking a biological phenotype to small molecule inhibition.7 Due to the profound biological phenotype associated with BET family bromodomain inhibition,2 a minimum target of >100-fold selectivity was set, together with >30-fold over other non-BET bromodomains. Herein, we report the structure guided optimization of a naphthyridone template into potent and selective biochemical inhibitors of TAF1(2) that while unsuitable as chemical probes, represent leads for the future development of TAF1(2) inhibitors.
TAF1 and TAF1L both contain tandem bromodomains with differing levels of homology between the four domains (Figure 2a). The N-terminal bromodomains in both proteins only differ by a single amino acid, and the C-terminal bromodomains are 97% identical. In contrast, the C- and N-terminal bromodomains within TAF1 and TAF1L show significant differences to each other (37–39% identity). In particular, a change in gatekeeper residues from leucine in TAF1(1)/TAF1L(1) to tyrosine in TAF1(2)/TAF1L(2) creates a fundamentally different binding pocket and impacts access to the lipophilic WPF shelf.24 Interestingly, there are no commercial assays for TAF1(1) or TAF1L(1), so it is unclear if the molecules described in the course of this work are selective over the first bromodomain or not and the majority of published reports also focus on the C-terminal bromodomains of these proteins. Due to the high homology between TAF1(2) and TAF1L(2) it was not expected that selectivity between the bromodomains would be achievable and as such, was not sought or monitored.
Figure 2.

(a) Table comparing percent of sequence identity between TAF1(1, 2) and TAF1L(1, 2); (b) X-ray crystal structure of 18 (yellow) bound to human TAF1(2) (gray) (pdb: 7p4s); (c) X-ray crystal structure of 18 (yellow) bound to human TAF1(2) (gray) with a protein surface (gray) (pdb: 7p4s).
To identify TAF1(2) inhibitors, a screen of molecules from the GSK compound collection known to or predicted to contain acetyl lysine mimetics was carried out against TAF1(2) using a time-resolved fluorescence resonance energy transfer (TR-FRET) competition assay. A TR-FRET assay against the N-terminal bromodomain of BRD4, termed BRD4(1), was also run as an antitarget as a representative member of the BET family. While several TAF1(2) active compounds were identified, the majority were also at least as potent against BRD4(1). Naphthyridone 7 stood out among the hit compounds with 8-fold bias over BRD4(1) and a TAF1(2) ligand efficiency (LE) of 0.31 (Table 1). Compound 7 had been accessed during a previous research effort that resulted in a high quality ATAD2 chemical probe and the template was also used to develop a tool for the first bromodomain of the BET bromodomains.25−28 While activity against TAF1(2) with this template had been identified previously,26,28 compound 7 demonstrated a 10-fold selectivity over ATAD2. Compound 7 was highly polar (ChromLogDpH7.4: 0.3) which translated into good aqueous solubility, excellent LLE (6.5) and poor (<3 nm/s) passive permeability as measured by an artificial membrane permeability assay (AMP).
Table 1. Profiling of Compounds 7–18a.


LE = (1.37 × pIC50)/heavy atom count. n.d. = not determined. For statistics, see Supporting Information, Table S1.
Removing the primary carbamate to reduce the polarity was tolerated at TAF1(2), together with a concomitant increase in LE, although no measurable increase in passive permeability was observed (compound 8). Capping the piperidine NH with a methyl group with compound 9 to reduce the number of hydrogen bond donor also had little effect on the bromodomain activity profile or the low passive permeability. Modeling of compound 9 into apo TAF1(2) (pdb: 3uv4) indicated that the piperidine ring may offer a vector for further interactions to boost potency, in particular with Asp1539 (TAF1(2) numbering, vide infra).29 A pendant alcohol was broadly tolerated with compound 10, whereas primary amine 11 gave a far improved profile indicating for the first time that high levels of selectivity over BRD4(1) and ATAD2 while retaining TAF1(2) activity was possible from the naphthyridone template. Homologation of the primary amine to compound 12 gave a similar profile to compound 11, although a minor improvement in passive permeability was observed.
The 5-position pyridine ring was a preferred group on the naphthyridone template for ATAD2 activity due to its displacement of a water atom and subsequent hydrogen bonding interaction with Asp1014 (ATAD2 numbering) by the ring nitrogen.25 In order to modulate the persistent ATAD2 activity of this template, replacement of this pyridine ring to remove this interaction was prioritised. Pyridyl replacement with a benzene ring with 13 was not well tolerated at TAF1(2) with a complete erosion of selectivity over BRD4(1) observed. Similarly, a cyclohexyl ring and a tetrahydropyran (compounds 14 and 15) decreased the activity against TAF1(2), BRD4(1) and ATAD2 indicating that sp3 substitution is not well tolerated in the naphthyridone 5-position. However, restoring some sp2 hybridization with dihydropyran 16 gave a boost in TAF1(2) potency without a concomitant increase in BRD4(1) or ATAD2 potency resulting in a promising bromodomain selectivity profile. This finding confirmed the design hypotheses that the 5-position pyridine ring was at least partly responsible for persistent ATAD2 potency observed on the template to date. Highlighting the importance of the amine moiety, primary carboxamide 17 ablated activity at TAF1(2).
Initial attempts to obtain crystallography of naphthyridone compounds in complex with TAF1(2) were unsuccessful; however, a structure of related compound 18 bound to human TAF1(2) (pIC50: 7.5) was ultimately achieved (Figure 2b and c). As expected from the binding pose of analogues with ATAD2,25 the carbonyl group functions as the KAc mimetic, forming the canonical direct hydrogen bond to Asn1583 and a water mediated interaction with Tyr1540. Additional hydrogen bond interactions between the naphthyridone NH and the 8-position NH to Asn1583 completes the tridentate interaction to the conserved residue. The methyl group of the naphthyridone core acts as the KAc methyl mimetic, protruding toward the conserved water molecules in the binding site. Tyr1589, the gatekeeper residue, has rotated 18° compared to the published apo structure (pdb: 3uv4) to make a face-to-face interaction with the bicyclic core.29 Confirming the design hypothesis, the flexible primary amine sits on the protein surface and makes a salt bridge to Asp1539 which has shifted 0.7 Å from the apo structure to make this interaction. BRD4(1) and ATAD2 all contain an acidic group in the same three-dimensional space as Asp1539 in TAF1(2), so it is unclear as to why the presence of the primary amine is only beneficial for TAF1(2) potency. The 5-position pyridine sits with a 60° dihedral angle against the WPF stack at the edge of the ZA channel and drives substantial reorganization of both Trp1526 and Phe1536 compared to the apo structure to accommodate. Whereas the pyridine nitrogen points into bulk solvent, the pendant alcohol makes a direct hydrogen bonding interaction with the backbone NH of Asn1533 and through-water interactions to Pro1531, His1530, and Pro1527. It should be noted that these hydrogen bonding interactions between the pendant alcohol and the protein contributes little to TAF1(2) potency as seen by comparing the similar activity of matched molecular pair 12 and 18 (Table 1). The narrow nature of the channel that the 5-position substituent protrudes into helps explain the drop of potency upon substitution with a sp3 carbon: presumably the fully saturated ring system is not tolerated due to an increased dihedral angle which drives steric clashes with the protein (Figure 2c).
Naphthyridone 16 showed a promising bromodomain selectivity profile with encouraging solubility. However, the measured passive permeability was low (<5 nm/s), indicating that bromodomain target engagement in a cellular system would be unlikely.
To prioritize new design hypotheses, a regression analysis of 30 calculated physicochemical descriptors of the naphthyridone compounds in the GSK database (570 compounds) was carried out. This study revealed a dependence of permeability on the pKa of the most basic unit, with passive permeability through an artificial membrane being >100 nm/s more likely for molecules with pKa <9.5. Consistent with this finding was the low permeability observed with 16 which demonstrated predicted pKa’s: 9.9 (primary amine) and 7.7 (piperidine). To probe this hypothesis, a series of molecules were designed targeting reduction of the basicity of both basic centers, profiled and only those with a predicted pKa <9.5 taken forward into synthesis.
To speed up data generation across multiple bromodomains, a switch was made to utilize BROMOscan assays (pKi values) as opposed to the internal TR-FRET assays (pIC50 values) that had been used to guide the chemistry to this point.30 Due to the presence of the 5-position dihydropyran which had a substantial impact on ATAD2 potency, activity against this bromodomain was no longer routinely measured. As has been noted previously, the BROMOscan potency was routinely higher than the corresponding TR-FRET data.28 To further investigate this, an analysis of unrelated compounds in the GSK collection with data in both assay formats against TAF1(2), BRD4(1) and BRD4(2) was carried out (Figures S2–S4, Supporting Information). These analyses highlighted not only the strong correlation between the two assay formats for the three bromodomain targets (R2 = 0.76–0.92), but also provided a predictive equation to translate between them. For example, BROMOscan TAF1(2) pKi = 0.39 + (1.17 × TR-FRET TAF1(2) pIC50) and BROMOscan BRD4(1) pKi = 0.56 + (1.01 × TR-FRET BRD4(1) pIC50).
Addition of fluorine to both restrict conformation and reduce the pKa of basic groups in a predictable manner is a commonly used method in medicinal chemistry.31 To modulate the basicities of both amines simultaneously, substitution of the propylamine 2-carbon of 16 was targeted (Table 2). Monofluorination with racemic 19 was tolerated at TAF1(2), albeit without improved permeability. Double fluorination to give compound 20 did show the desired levels of passive permeability (220 nm/s), unfortunately at the expense of TAF1(2) potency. Using the BROMOscan to TR-FRET conversion equation (Figures S2 and S3, Supporting Information), predicted TAF1(2) TR-FRET pIC50 = 5.9 and BRD4(1) pIC50 <4.7, highlighting that the desired TAF1(2) potency and 100-fold selectivity had likely not been achieved. Carboxamide 21 showed high levels of TAF1(2) potency together with a drop of selectivity over BRD4(1). The low passive permeability is presumably related to the remaining basic amine. Adding one fluorine to give racemic α-fluoro 22 gave the most potent compound of the series at TAF1(2), but despite a pKa of 7.8, poor passive permeability was still observed. To complete the set of compounds, α-difluoro 23 was made and to our surprise nanomolar activity at TAF1(2) was still achieved as judged by BROMOscan. Excellent selectivity over BRD4(1) was retained, and measurable levels of permeability were observed in the passive permeability assay. Although the design hypothesis that reducing the predicted pKa would drive passive permeability was ultimately correct, the level by which the basicity needed to be reduced had been underestimated. The data indicated that for this set of compounds, meaningful passive permeability was only observed with a predicted pKa ≤7.3.
Table 2. Profiling of Compounds 16 and 19–24a.


LE = (1.37 × pKi)/heavy atom count. For statistics, see Supporting Information Table S2.
CAD solubility.
The predicted pKa values matched the most basic measured values well; for example, compound 19 measured pKa = 8.6, compound 22 measured pKa = 7.5, and compound 23 measured pKa = 6.4.
Restricted docking of matched molecular pair 20 and 23 into the crystal structure of TAF1(2) from the complex with 18 was carried out to give insight into the difference in activity at TAF1(2) (Figure 3). As constrained by the docking protocol, both molecules maintained the tridentate hydrogen-bonding interaction with Asn1683 and the through-water interaction with Tyr1540 observed with the crystal structure of compound 18 bound to TAF1(2) (Figure 2b). The dihydropyran is accommodated in the cleft between Trp1526 and Phe1536 with a predicted 62° dihedral angle, like that observed with compound 18 bound to TAF1(2) (Figure 3c and d). The pendant amine for both molecules maintained the interaction with Asp1539, however the presence, or not, of the amide has a drastic impact on the orientation of the fluoro groups (Figure 3c and d). When the amide is present, a conformation where one fluorine sits anti to the carbonyl group is adopted, resulting in the fluorine groups protruding away from the protein surface.32 Presumably, this flip of the fluorine groups predicted for compound 23 relieves an unfavorable interaction with the protein surface that impacts the TAF1(2) activity of compound 20.
Figure 3.

Docking of (a) compound 20 (green) into TAF1(2) (gray); (b) compound 23 (yellow) into TAF1(2) (gray); (c) compound 20 (green) into TAF1(2) (gray) viewed from an alternative angle; (d) compound 23 (yellow) into TAF1(2) (gray) view from an alternative angle using Maestro (Schrodinger Inc.) 2018. pdb: 7p4s was used for all dockings.
The knowledge built around the template binding to TAF1(2) demonstrated that the naphthyridone methyl group functioned as the KAc methyl mimetic (Figure 2b). It has been previously demonstrated that TAF1(2) is able to recognize not just methyl acetyl lysine, but also atypical crotonyl and butyryl substituted acetyl lysines.33 Genentech and Constellation Pharmaceuticals have elegantly utilized this facet of the TAF1(2) bromodomain with the 1-butenyl group as an atypical acetyl lysine methyl mimetic on a pyrrolopyridinone scaffold to induce TAF1(2) selectivity through the rearrangement and stabilization of the KAc binding site water molecules.22,23 This approach was also successfully applied to a dihydropyridopyrazine scaffold in the development of a dual BET, TAF1(2) inhibitor 4.19 Overlaying the published structure of GNE-371 bound to TAF1(2) with 18 bound to TAF1(2) indicated that the methyl groups occupied approximately the same space, even if the angle of the C–C bond was slightly different (Supporting Information Figure S1). Accordingly, compound 24 bearing a 1-butenyl acetyl lysine methyl mimetic was accessed. Despite the successful translatability of the atypical acetyl lysine methyl mimetic approach for BRD7/9 bromodomain inhibitors across multiple chemotypes,34 with the naphthyridone template, an unexpected >200-fold drop in TAF1(2) potency was observed, making it challenging to understand whether selectivity against other bromodomains had been improved (compare matched molecular pair 23 and 24). As the angle of the methyl groups is not the same across GNE-371 and compound 18 bound to TAF1(2), the incorporation of the 1-butenyl chain presumably forces the naphthyridone template to shift in the TAF1(2) binding site which results in a less complementary fit and concomitant drop in affinity.
Despite this disappointing result with the 1-butenyl group, further characterization of compound 23 was undertaken to determine the broader bromodomain selectivity profile. Single shot screening at 10 μM in the DiscoverX BROMOscan assay (Figure 4) was followed up by 11-point dose–response curves of selected bromodomains where substantial activity was observed (Table 3). Importantly, considering the compound genesis, activity against ATAD2 remained low (%I = 24% at 10 μM; see Supporting Information Table S3 for more details).
Figure 4.

BROMOscan profiling at 10 μM for compound 23.
Table 3. Full Curve BROMOscan Profiling of Compound 23a.
| bromodomain | compd 23 pKi |
|---|---|
| TAF1(2) | 9.1 |
| BRD4(1) | <5.3 (>×6310) |
| BRD4(2) | 6.1 (×1000) |
| BAZ2B | 6.0 (×1260) |
| BRD9 | 7.4 (×50) |
| CECR2 | 6.0 (×1260) |
| BRPF1 | 5.3 (×6310) |
TAF1(2) fold selectivity is shown in parentheses after the pKi value.
The single shot profile was borne out when moving to the full curve assays with compound 23 showing ≥1000-fold selectivity over BRD4 and >50-fold selectivity over the broader bromodomains tested. Prediction of TR-FRET potency from the BROMOscan data indicates TAF1(2) pIC50 = 7.4, BRD4(1) pIC50 <4.7, and BRD4(2) pIC50 = 5.3, indicating that the predefined potency and selectvity criteria had been met in both assay formats. Although not measured, it is expected that 23 is active against TAF1L(2). 23 was also profiled in a human microsomal clearance assay which showed a moderate apparent intrinsic clearance with Clint,app = 20.4 mL/min/kg. With 23 meeting the predefined bromodomain selectivity criteria together with encouraging lipophilicity, solubility, and permeability, attention turned to the demonstration of TAF1(2) cellular target engagement. A nanoBRET assay was generated in HEK-293 cells measuring the displacement of NanoLuc-tagged TAF1(2) from Halo-tagged histone H4 (Promega).18,35 However, 23 appeared inactive at the concentrations tested with a TAF1(2) nanoBRET pIC50 <5. In contrast, 20 displayed a TAF1(2) nanoBRET pIC50 of 5.5, despite being 63-fold less potent against TAF1(2) in a biochemical binding assay compared with 23 (Table 2). The improved passive permeability of 20 appears to be critical to driving measurable activity in the nanoBRET assay (compound 20 AMP, 220 nm/s; compound 23 AMP, 67 nm/s). Active compound efflux may also be a factor but attempts to unravel this have thus far been unsuccessful.
The detailed synthetic routes to compounds 7–24 are described in the Supporting Information, and the preparation of compound 23 is shown in Scheme 1. Previously described 25(25) was reacted under Buchwald amination conditions followed by chemoselective bromination with NBS to give flexible intermediate 26. Suzuki–Miyaura coupling with the commercially available dihydropyran pinacol boronate was followed by global deprotection to give piperidine 27. Amide coupling and then acid mediated Boc deprotection proceeded in a 10% yield over the two steps to provide 23.
Scheme 1. Synthesis of Compound 23.

Reagents and conditions: (a) 1-Boc-4-aminopiperidine, Pd2(dba)3, BrettPhos, NaOtBu, THF, 60 °C, 4 h, 72%; (b) NBS, CHCl3, rt, 1.5 h, 99%; (c) DHP-Bpin, Pd(OAc)2, CataCXium A, K2CO3, 1,4-dioxane/H2O, microwave 100 °C, 1 h, 86%; (d) TFA, reflux, 3 h, 95%. (e) HATU, DIPEA, DMF, rt, 5 h, 26%; (f) 4 M HCl in 1,4-dioxane, rt, 2 h, 40%.
In summary, the optimization of unselective naphthyridone 7 into potent and TAF1(2) selective 23 has been achieved via crystallography guided structure-based drug design to target Asp1539 with a pendant amine. This salt bridge interaction with Asp1539 drove selectivity over BRD4(1) and ATAD2, with selectivity further enhanced by the introduction of a semisaturated dihydropyran. Careful modulation of the pKa was critical to drive permeability with cellular target engagement demonstrated only with the less selective, yet more permeable 20. Although none of the molecules described meet the criteria of a chemical probe and should not be used as such, compound 23 represents an early lead for the future development of small and/or bifunctional molecules to interrogate the biological function of TAF1(2).
Acknowledgments
M.A.C and N.H.T. are grateful to GlaxoSmithKline R&D, Stevenage, and the University of Strathclyde for Ph.D. studentships, and we thank the EPSRC for funding via Prosperity Partnership EP/S035990/1. We also thank Sean Lynn for assistance with NMR and Tony Cook for high resolution mass spectrometry.
Glossary
Abbreviations
- AMP
artificial membrane permeability
- ATAD2
ATPase family, AAA domain containing 2A
- ATAD2B
ATPase family, AAA domain containing 2B
- BAF
BRG1/BRM-associated factor
- BAZ2A
bromodomain adjacent to zinc finger domain 2A
- BAZ2B
bromodomain adjacent to zinc finger domain 2B
- BCP
bromodomain containing protein
- BD
bromodomain
- BET
bromodomain and extra terminal domain
- BRD1
bromodomain containing protein 1
- BRD2
bromodomain containing protein 2
- BRD3
bromodomain containing protein 3
- BRD4
bromodomain containing protein 4
- BRD7
bromodomain containing protein 7
- BRD8
bromodomain containing protein 8
- BRD9
bromodomain containing protein 9
- BRDT
bromodomain containing protein, testis-specific
- BRPF1
bromodomain and PHD finger-containing protein 1
- BRPF3
bromodomain and PHD finger-containing protein 3
- CAD
charged aerosol detection
- CECR2
cat eye syndrome chromosome region candidate 2
- CLND
chemiluminescent nitrogen detection
- CREBBP
CREB binding protein
- EP300
E1A-associated protein p300
- FALZ
bromodomain PHD finger transcription factor
- GCN5L2
general control nondepressible 5
- KAc
acetylated lysine
- LE
ligand efficiency
- PBAF
polybromo-associated BAF
- PBRM1
polybromo 1
- PCAF
P300/CREBBP associated factor
- PEPPSI
pyridine-enhanced precatalyst preparation stabilization and initiation
- PHD
plant homeodomain
- pIC50
–log10(IC50)
- SMARCA2
SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily A, member 2
- SMARCA4
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4
- TRIM24
tripartite motif containing 24
- TRIM33
tripartite motif containing 33
- WDR9
WD repeat-containing protein 9
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00294.
Additional text describing all biochemical methods and results, all chemistry experimental procedures, BROMOscan data for 23, docking methods, overlay of GNE-371 and 18 bound to TAF1(2), comparison between TR-FRET/BROMOscan data for TAF1(2) BRD4(1) and BRD4(2), and X-ray data collection and refinement statistics (PDF)
Author Present Address
§ MSD, Francis Crick Institute, 1 Midland Road, London, NW1 1AT, UK
Author Present Address
∥ The University of Manchester, Oxford Road, Manchester M13 9PL, UK
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): All authors except M.A.C and N.C.O.T. are current or former employees of GlaxoSmithKline.
Notes
Coordinates have been deposited with the Protein Data Bank under accession code 7p4s (TAF1(2)/18 complex). Authors will release atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Filippakopoulos P.; Knapp S. Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Smith S. G.; Zhou M.-M. The Bromodomain: A New Target in Emerging Epigenetic Medicine. ACS Chem. Biol. 2016, 11, 598–608. 10.1021/acschembio.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as Therapeutic Targets. Expert Rev. Mol. Med. 2011, 13, e29. 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran A. G.; Conery A. R.; Sims R. J. III Bromodomains: A New Target Class for Drug Development. Nat. Rev. Drug Discovery 2019, 18, 609–628. 10.1038/s41573-019-0030-7. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Ma S. Disrupting Acetyl-Lysine Interactions: Recent Advance in the Development of BET Inhibitors. Curr. Drug Targets 2018, 19, 1148–1165. 10.2174/1389450119666171129165427. [DOI] [PubMed] [Google Scholar]
- Clegg M. A.; Tomkinson N. C. O.; Prinjha R. K.; Humphreys P. G. Advancements in the Development of Non-BET Bromodomain Chemical Probes. ChemMedChem 2019, 14, 362–385. 10.1002/cmdc.201800738. [DOI] [PubMed] [Google Scholar]
- Bunnage M. E.; Piatnitski Chekler E. L.; Jones L. H. Target Validation Using Chemical Probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- Schiedel M.; Moroglu M.; Ascough D. M. H.; Chamberlain A. E. R.; Kamps J. J. A. G.; Sekirnik A. R.; Conway S. J. Chemical Epigenetics: The Impact of Chemical and Chemical Biology Techniques on Bromodomain Target Validation. Angew. Chem., Int. Ed. 2019, 58, 17930–17952. 10.1002/anie.201812164. [DOI] [PubMed] [Google Scholar]
- Blagg J.; Workman P. Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology. Cancer Cell 2017, 32, 9–25. 10.1016/j.ccell.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Schapira M. The Promise and Peril of Chemical Probe Negative Controls. ACS Chem. Biol. 2021, 16, 579–585. 10.1021/acschembio.1c00036. [DOI] [PubMed] [Google Scholar]
- Verrijzer C. P.; Tjian R. TAFs Mediate Transcriptional Activation and Promoter Selectivity. Trends Biochem. Sci. 1996, 21, 338–342. 10.1016/0968-0004(96)10044-X. [DOI] [PubMed] [Google Scholar]
- Patel A. B.; Louder R. K.; Greber B. J.; Grünberg S.; Luo J.; Fang J.; Liu Y.; Ranish J.; Hahn S.; Nogales E. Structure of Human TFIID and Mechanism of TBP Loading Onto Promoter DNA. Science 2018, 362, eaau8872. 10.1126/science.aau8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli P.; Wafa L. A.; Cheng H.; Zoubeidi A.; Fazli L.; Gleave M.; Snoek R.; Rennie P. S. TAF1 Differentially Enhances Androgen Receptor Transcriptional Activity via Its N-Terminal Kinase and Ubiquitin-Activating and -Conjugating Domains. Mol. Endocrinol. 2010, 24, 696–708. 10.1210/me.2009-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H. R.; An C. H.; Yoo N. J.; Lee S. H. Frameshift Mutations in the Mononucleotide Repeats of TAF1 and TAF1L Genes in Gastric and Colorectal Cancers with Regional Heterogeneity. Pathol. Oncol. Res. 2017, 23, 125–130. 10.1007/s12253-016-0107-0. [DOI] [PubMed] [Google Scholar]
- O’Rawe J. A.; Wu Y.; Dörfel M. J.; Rope A. F.; Au P. Y. B.; Parboosingh J. S.; Moon S.; Kousi M.; Kosma K.; Smith C. S.; Tzetis M.; Schuette J. L.; Hufnagel R. B.; Prada C. E.; Martinez F.; Orellana C.; Crain J.; Caro-Llopis A.; Oltra S.; Monfort S.; Jiménez-Barrón L. T.; Swensen J.; Ellingwood S.; Smith R.; Fang H.; Ospina S.; Stegmann S.; Hollander N. D.; Mittelman D.; Highnam G.; Robison R.; Yang E.; Faivre L.; Roubertie A.; Rivière J.-B.; Monaghan K. G.; Wang K.; Davis E. E.; Katsanis N.; Kalscheuer V. M.; Wang E. H.; Metcalfe K.; Kleefstra T.; Innes A. M.; Kitsiou-Tzeli S.; Rosello M.; Keegan C. E.; Lyon G. J. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am. J. Hum. Genet. 2015, 97, 922–932. 10.1016/j.ajhg.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown M. R.; Shaw D. L.; Fu H.; Liu S.; Xu X.; Marineau J. J.; Huang Y.; Zhang X.; Buckley D. L.; Kadam A.; Zhang Z.; Blacklow S. C.; Qi J.; Zhang W.; Bradner J. E. Biased Multicomponent Reactions to Develop Novel Bromodomain Inhibitors. J. Med. Chem. 2014, 57, 9019–9027. 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdelci S.; Lardeau C.-H.; Tallant C.; Klepsch F.; Klaiber B.; Bennett J.; Rathert P.; Schuster M.; Penz T.; Fedorov O.; Superti-Furga G.; Bock C.; Zuber J.; Huber K. V. M.; Knapp S.; Müller S.; Kubicek S. Mapping the chemical chromatin reactivation landscape identifies BRD4-TAF1 cross-talk. Nat. Chem. Biol. 2016, 12, 504–510. 10.1038/nchembio.2080. [DOI] [PubMed] [Google Scholar]
- Bouché L.; Christ C. D.; Siegel S.; Fernández-Montalván A. E.; Holton S. J.; Fedorov O.; ter Laak A.; Sugawara T.; Stöckigt D.; Tallant C.; Bennett J.; Monteiro O.; Díaz-Sáez L.; Siejka P.; Meier J.; Pütter V.; Weiske J.; Müller S.; Huber K. V. M.; Hartung I. V.; Haendler B. Benzoisoquinolinediones as Potent and Selective Inhibitors of BRPF2 and TAF1/TAF1L Bromodomains. J. Med. Chem. 2017, 60, 4002–4022. 10.1021/acs.jmedchem.7b00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard D.; Buckley D. L.; Seo H.-S.; Ferguson F. M.; Dhe-Paganon S.; Bradner J. E.; Gray N. S. Dual Inhibition of TAF1 and BET Bromodomains from the BI-2536 Kinase Inhibitor Scaffold. ACS Med. Chem. Lett. 2019, 10, 1443–1449. 10.1021/acsmedchemlett.9b00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh J. L.; Watts B.; Stuckey J.; Norris-Drouin J. L.; Cholensky S. H.; Dickson B. M.; An Y.; Mathea S.; Salah E.; Knapp S.; Khan A.; Adams A. T.; Strahl B. D.; Sagum C. A.; Bedford M. T.; James L. I.; Kireev D. B.; Frye S. V. Quantitative Characterization of Bivalent Probes for a Dual Bromodomain Protein, Transcription Initiation Factor TFIID Subunit 1. Biochemistry 2018, 57, 2140–2149. 10.1021/acs.biochem.8b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; Chen D.-Q.; Sun L.; Huan X.-J.; Bao X.-B.; Tian C.-Q.; Hu J.; Lv K.-K.; Wang Y.-Q.; Xiong B.; Miao Z. H. Novel Bivalent BET Inhibitor N2817 Exhibits Potent Anticancer Activity and Inhibits TAF1. Biochem. Pharmacol. 2021, 185, 114435. 10.1016/j.bcp.2021.114435. [DOI] [PubMed] [Google Scholar]
- Wang S.; Tsui V.; Crawford T. D.; Audia J. E.; Burdick D. J.; Beresini M. H.; Côté A.; Cummings R.; Duplessis M.; Flynn E. M.; Hewitt M. C.; Huang H.-R.; Jayaram H.; Jiang Y.; Joshi S.; Murray J.; Nasveschuk C. G.; Pardo E.; Poy F.; Romero F. A.; Tang Y.; Taylor A. M.; Wang J.; Xu Z.; Zawadzke L. E.; Zhu X.; Albrecht B. K.; Magnuson S. R.; Bellon S.; Cochran A. G. GNE-371, a Potent and Selective Chemical Probe for the Second Bromodomains of Human Transcription-Initiation-Factor TFIID Subunit 1 and Transcription-Initiation-Factor TFIID Subunit 1-Like. J. Med. Chem. 2018, 61, 9301–9315. 10.1021/acs.jmedchem.8b01225. [DOI] [PubMed] [Google Scholar]
- Crawford T. D.; Tsui V.; Flynn E. M.; Wang S.; Taylor A. M.; Côte A.; Audia J. E.; Beresini M. H.; Burdick D. J.; Cummings R.; Dakin L. A.; Duplessis M.; Good A. C.; Hewitt M. C.; Huang H.-R.; Jayaram H.; Kiefer J. R.; Jiang Y.; Murray J.; Nasveschuk C. G.; Pardo E.; Poy F.; Romero F. A.; Tang Y.; Wang J.; Xu Z.; Zawadzke L. E.; Zhu X.; Albrecht B. K.; Magnuson S. R.; Bellon S.; Cochran A. G. Diving into the Water: Inducible Binding Conformations for BRD4, TAF1(2), BRD9, and CECR2 Bromodomains. J. Med. Chem. 2016, 59, 5391–5402. 10.1021/acs.jmedchem.6b00264. [DOI] [PubMed] [Google Scholar]
- Chung C.-W.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J.-M. G.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clément C. A.; Boullay A.-B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med. Chem. 2011, 54, 3827–3838. 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- Demont E. H.; Chung C.-W.; Furze R. C.; Grandi P.; Michon A.-M.; Wellaway C.; Barrett N.; Bridges A. M.; Craggs P. D.; Diallo H.; Dixon D. P.; Douault C.; Emmons A. J.; Jones E. J.; Karamshi B. V.; Locke K.; Mitchell D. J.; Mouzon B. H.; Prinjha R. K.; Roberts A. D.; Sheppard R. J.; Watson R. J.; Bamborough P. Fragment-Based Discovery of Low-Micromolar ATAD2 Bromodomain Inhibitors. J. Med. Chem. 2015, 58, 5649–5673. 10.1021/acs.jmedchem.5b00772. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Chung C.-W.; Furze R. C.; Grandi P.; Michon A.-M.; Sheppard R. J.; Barnett H.; Diallo H.; Dixon D. P.; Douault C.; Jones E. J.; Karamshi B.; Mitchell D. J.; Prinjha R. K.; Rau C.; Watson R. J.; Werner T.; Demont E. H. Structure-ased Optimization of Naphthyridones into Potent ATAD2 Bromodomain Inhibitors. J. Med. Chem. 2015, 58, 6151–6178. 10.1021/acs.jmedchem.5b00773. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Chung C.-W.; Demont E. H.; Furze R. C.; Bannister A. J.; Che K. H.; Diallo H.; Douault C.; Grandi P.; Kouzarides T.; Michon A. M.; Mitchell D. J.; Prinjha R. K.; Rau C.; Robson S.; Sheppard R. J.; Upton R.; Watson R. J. A Chemical Probe for the ATAD2 Bromodomain. Angew. Chem., Int. Ed. 2016, 55, 11382–11386. 10.1002/anie.201603928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R. J.; Bamborough P.; Barnett H.; Chung C.-W.; Davis R.; Gordon L.; Grandi P.; Petretich M.; Phillipou A.; Prinjha R. K.; Rioja I.; Soden P.; Werner T.; Demont E. H. GSK789: A Selective Inhibitor of the First Bromodomains (BD1) of the Bromo and Extra Terminal Domain (BET) Proteins. J. Med. Chem. 2020, 63, 9045–9069. 10.1021/acs.jmedchem.0c00614. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone Recognition and Large-scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROMOscan recombinant protein binding assays were carried out at DiscoverX, http://www.discoverx.com.
- Morgenthaler M.; Schweizer E.; Hoffmann-Röder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Müller K. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem 2007, 2, 1100–1115. 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- Briggs C. R.S.; O’Hagan D.; Howard J. A.K.; Yufit D. S. The C-F bond as a tool in the conformational control of amides. J. Fluorine Chem. 2003, 119, 9–13. 10.1016/S0022-1139(02)00243-9. [DOI] [Google Scholar]
- Flynn M.; Huang O.; Poy F.; Oppikofer M.; Bellon S.; Tang Y.; Cochran A. G. A Subset of Human Bromodomains Recognizes Butyryllysine and Crotonyllysine Histonepeptide Modifications. Structure 2015, 23, 1801–1814. 10.1016/j.str.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Clegg M. A.; Bamborough P.; Chung C.-W.; Craggs P. D.; Gordon L.; Grandi P.; Leveridge M.; Lindon M.; Liwicki G. M.; Michon A.-M.; Molnar J.; Rioja I.; Soden P. E.; Theodoulou N. H.; Werner T.; Tomkinson N. C. O.; Prinjha R. K.; Humphreys P. G. Application of Atypical Acetyl-lysine Methyl Mimetics in the Development of Selective Inhibitors of the Bromodomain-Containing Protein 7 (BRD7)/Bromodomain-Containing Protein 9 (BRD9) Bromodomains. J. Med. Chem. 2020, 63, 5816–5840. 10.1021/acs.jmedchem.0c00075. [DOI] [PubMed] [Google Scholar]
- Machleidt T.; Woodroofe C. C.; Schwinn M. K.; Mendez J.; Robers M. B.; Zimmerman K.; Otto P.; Daniels D. L.; Kirkland T. A.; Wood K. V. NanoBRET-a novel BRET platform for the analysis of protein-protein interactions. ACS Chem. Biol. 2015, 10, 1797–1804. 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.