

Abstract

Transient receptor potential ankyrin 1 (TRPA1) antagonists have generated broad interest in the pharmaceutical industry for the treatment of both pain and asthma. Over the past decade, multiple antagonist classes have been reported in the literature with a wide range of structural diversity. Our own work has focused on the development of proline sulfonamide and hypoxanthine-based antagonists, two antagonist classes with distinct physicochemical properties and pharmacokinetic (PK) trends. Late in our discovery program, cryogenic electron microscopy (cryoEM) studies revealed two different antagonist binding sites: a membrane-exposed proline sulfonamide transmembrane site and an intracellular hypoxanthine site near the membrane interface. A retrospective look at the discovery program reveals how the different binding sites, and their location relative to the cell membrane, influenced the optimization trajectories and overall drug profiles of each antagonist class.
Keywords: TRPA1 antagonists, cryogenic electron microscopy, antagonist binding sites, membrane, membrane partitioning
Recent advances in the preparation, stabilization, and visualization of membrane proteins via X-ray crystallography and cryogenic electron microscopy (cryoEM) have resulted in an increasing number of published transmembrane protein structures with bound small molecule modulators.1−3 These structures have revealed diverse small molecule binding sites located either extracellularly (Figure 1A), intracellularly (Figure 1B), or within the transmembrane region (Figure 1C) of integral membrane proteins. With a growing number of small molecule binding sites found within the lipid bilayer,4,5 there is an increasing awareness that drug properties are dependent on the membrane exposure at a ligand binding site.6,7 While free drug in the aqueous compartment may directly interact with binding sites in the extracellular or cytosolic space, intramembrane binding pockets require a ligand to first partition into the lipid membrane prior to interacting with a target site.8 This microkinetic model of ligand binding suggests that observed potency is dependent upon both intrinsic affinity for a target protein and the ability of a ligand to associate with the membrane.9,10
Figure 1.
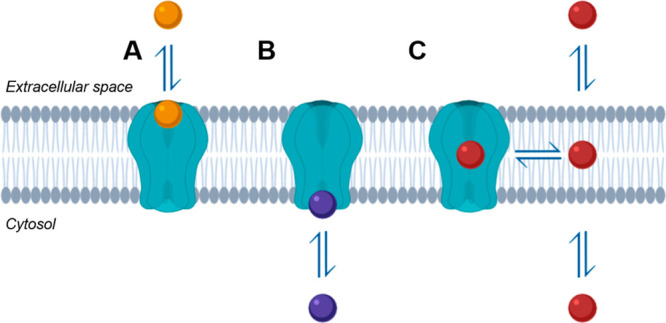
Cartoon representation of transmembrane target drug binding via (A) extracellular space, (B) intracellular space or cytosol, and (C) phospholipid membrane. Drug binding sites within the lipid membrane require ligand partitioning to the lipid membrane.
Physicochemical properties necessarily impact both the membrane partitioning event and the observed pharmacokinetics (PK) of drugs targeting membrane-associated proteins.11 Historically, structure-based analyses of medicinal chemistry campaigns at these transmembrane binding sites are lacking. We therefore sought to examine how two different binding sites impact drug properties and optimization trajectories for an ion channel target.
Transient receptor potential ankyrin 1 (TRPA1) is a cation-selective calcium-permeable ion channel that acts as a sensor for exogenous irritants and endogenous pro-inflammatory signals.12 During the course of our work toward the discovery of TRPA1 antagonists for the treatment of pain and asthma, we developed two structurally distinct classes of antagonists.13−15 While the program was not enabled by structure-based design methods, we were aware that each series possessed divergent properties and that they likely shared non-overlapping binding sites by evaluating species-specific potency trends using rat–human TRPA1 chimeras.16,17 Late in the discovery phase, cryoEM structures of representative antagonists from each class were elucidated, including the proline sulfonamide GDC-0334 (1) and fluorophenyl hypoxanthine 2 (Figure 2), confirming predictions that these antagonists modulate TRPA1 activity at discrete binding sites.14,15
Figure 2.

Human TRPA1 cryoEM maps and binding site details for (A) GDC-0334 (1) at a membrane-exposed intrahelical site, and (B) hypoxanthine 2, at an intracellular binding site. Human TRPA1 cryoEM maps are depicted as isosurface renderings, with the TRPA1 protein colored in white with bound TRPA1 antagonists in yellow. Lipid bilayer boundaries are depicted as gray lines. Binding site details are depicted as transparent surface and cartoon renderings with TRPA1 antagonists shown as spheres.
The structure of GDC-0334 (1) bound to TRPA1 reveals a membrane-exposed intrahelical binding site deep within the lipid bilayer (Figure 2A).15 While the proline sulfonamide portion of the molecule is bound to a largely hydrophobic protein site, the highly fluorinated biaryl is exposed to the lipid environment, making minimal contact with the protein. This finding came as a surprise to the discovery team, as structure–activity relationships (SARs) developed on the biaryl region of the molecule were not driven by specific protein interactions as had been assumed. Instead, potency derived from the biaryl moiety is likely based on ligand–lipid interactions in the TRPA1-bound state and the ability of the ligand to partition in the lipid bilayer, where the biaryl may actually function as a lipophilic tail, or anchor. The U-shaped binding conformation of GDC-0334 (1) had been predicted from conformational analyses and also suggests that small molecule conformation may play a role in both shielding ligand polarity and facilitating partitioning in the membrane.13
The structure of TRPA1 bound to hypoxanthine 2, on the other hand, reveals a very different binding site environment. The hypoxanthine antagonist makes more complete contact with the protein at a generally more polar intracellular site. While this site lacks direct exposure to the lipid bilayer, it is positioned near the interface of the cytosol and plasma membrane inner leaflet.14
Differences in the character of the binding sites, and their exposure (or lack thereof) to the lipid membrane, affected the observed physicochemical property trends of potent compounds within each class (Figure 3). This led to very specific sets of challenges toward the optimization of potency and properties of each series. We found that roughly 80% of proline sulfonamide antagonists with a cellular IC50 < 10 nM possess a log D7.4 greater than 3, suggesting that high lipophilicity is required for potency, reflective of the lipid-exposed intramembrane binding site. Meanwhile, roughly 75% of the proline sulfonamides studied suffer from poor kinetic solubility (<10 μM), consistent with the hypothesis that enhanced membrane partitioning is required for potency yet is negatively correlated with aqueous solubility. The proline sulfonamide class of TRPA1 antagonists also caused the activation of the human pregnane X receptor (hPXR activation at 10 μM of antagonist), presenting potential cytochrome P450 (CYP) induction liabilities.18 It is conceivable that this xenobiotic detoxification mechanism is triggered by a large drug depot within the cellular and nuclear membranes due to the high lipophilicity of this class of antagonists.
Figure 3.

Comparison of selected properties within the proline sulfonamide and hypoxanthine series. Data shown only for compounds with hTRPA1 IC50 < 10 nM in a CHO cell line-based FLIPR assay. Compound counts listed in gray above each pie chart.
The more polar character of the intracellular hypoxanthine binding site and the lack of membrane exposure allow for the optimization of intrinsic receptor affinity without consideration of ligand–lipid interactions. As a result, antagonists in the hypoxanthine series are more soluble, occupy a broader range of log D7.4 values, and maintain high passive permeability (Figure 3), resulting in favorable oral drug profiles with wider property flexibility.
Detailed accounts describing the ligand-based discovery of both the proline sulfonamide and hypoxanthine class of antagonists have been previously reported, enabling retrospective analysis of decision-making in light of new structural information.13,14,19 Lead compounds from each series, representative of the optimization trajectory within the two drug classes, are shown in Table 1 alongside potency, physicochemical properties, and in vitro and in vivo DMPK data.
Table 1. Compound Profiles, Rat PK, and Rat AITC Target Engagement for TRPA1 Antagonists.

Data for the corresponding prodrug.13
PPB was determined in rat with 4 h compound incubation time (compared to standard 24 h due to compound instability in rat plasma).
Within the proline sulfonamide series, a significant effort was focused on optimization of the biaryl moiety, as SAR around the proline sulfonamide region of the molecule was steep. Two observations guided SAR optimization within the biaryl region (3 → 4 → 5 → 1): proper placement of fluorine atoms had a positive impact on potency, while nitrogen heterocycles (e.g., pyridines and pyrimidines) were tolerated and increased lipophilic ligand efficiency (LLE). Of note, the terminal trifluoromethylpyrimidine was particularly effective at improving potency while increasing aqueous solubility and reducing unbound clearance (CLu). Knowing that this group is fully exposed to the lipid bilayer when bound to TRPA1 (see Figure 2A), it is likely that the trifluoromethyl group effectively “hides” or “shields” the polar nitrogens of the pyrimidine from the lipid environment. In support of this hypothesis, despite an increase in the topographical polar surface area (TPSA) between pyridine 3 and pyrimidine 4, the experimental polar surface area (EPSA) remains constant between analogs.20 In the end, the highly fluorinated development candidate GDC-0334 (1) was identified with high in vitro and in vivo potency (see Table 1).15
The hypoxanthine-based series of antagonists bound at the intracellular site possessed very different SAR trends. In an effort to improve the potency within this antagonist class, the team sought to rigidify the linker region between the oxadiazole/oxadiazolone and the chlorophenyl substituent (6 → 7 → 8). While this strategy led to improved potency, it also increased the measured log D7.4 and reduced kinetic solubility. The introduction of a tetrahydrofuran ring (→ 9) broke this trend, improving in vitro potency and solubility while lowering lipophilicity. The ability to optimize for low lipophilicity compounds within this series is likely a result of the more polar character of the intracellular antagonist binding site compared to the phospholipid exposed binding site of the proline sulfonamide series.14
The TRPA1 antagonist program offers a unique case study given the identification of compounds 1 and 9 as leads from two series occupying very different physicochemical property space. We first compared the activities of 1 and 9 using both cellular and in vivo measures of potency. While in vitro TRPA1 potency was measured in a CHO cell line-based fluorometric imaging plate reader (FLIPR) Ca2+ assay, in vivo TRPA1 potency was assessed in an allyl isothiocyanate (AITC) rat pain model.13−15 In the experiment, the TRPA1 antagonist is orally administered and pain behaviors, such as flinching and licking, are monitored following AITC injection in the paw of a rat at the Tmax. Dose-dependent reductions in pain behavior are then monitored, allowing correlation of drug exposures with measured pain responses across animals. Surprisingly, compound 9 was less potent than GDC-0334 (1) in vivo, despite comparable in vitro potency against the rat TRPA1 channel. This in vitro/in vivo potency discrepancy was observed across lead compounds and is more pronounced in lower lipophilicity analogs, such as compound 6 (see data in Table 1).14
We hypothesize that the cellular assay does not accurately reflect the complex tissue environment where TRPA1 channels reside in vivo and that lower lipophilicity compounds may possess a decreased ability to access the membrane-bound TRPA1 channel within the target tissues.21 In support of this hypothesis, we noticed that larger in vivo potency shifts correlated with lower measured log D7.4. While log D7.4 is a common measure of bulk hydrophobicity, it does not accurately model interactions with zwitterionic phospholipids (i.e., phospholipophilicity) which may impact target access for TRPA1 antagonists bound at the membrane.22 We thought that unbound volume of distribution (Vss,u = Vss/fu), an in vivo measure of drug distribution in the body, might be a better reflection of the overall tissue (i.e., membrane) binding.21,23,24 Using rat PK data, we found that molecules with a higher Vss,u possess a reduced potency shift in vivo (see data Table 1). While both log D7.4 and Vss,u show similar trends, we found Vss,u to be a useful measure of in vivo lipophilicity that helped reconcile PKPD disconnects observed in the program. Of note, we anticipate these trends will be applicable across species, since differences in tissue binding are not significant between species.21
Despite observed potency differences, dose optimization is often the ultimate goal of a drug discovery campaign and is impacted by both in vivo potency and exposure.25 It has been shown that these parameters may offset each other when viewed across wide ranges of lipophilicity, resulting in similar efficacious doses.21 Again, GDC-0334 (1) and hypoxanthine 9 are valuable tools to explore this concept given their different potency and PK profiles. Notably, despite roughly 10-fold differences in unbound clearance (CLu) and in vivo potency, both 1 and 9 are able to block pain responses in the rat AITC experiment at similar doses (3–10 mg/kg).14,15
We next compared the optimization trajectories for each series using plots of cellular potency versus lipophilicity looking for binding site-specific trends that might inform future drug discovery programs targeting membrane-bound proteins (Figure 4).26 LLE plots (pIC50versus log D7.4) for the proline sulfonamides (Figure 4A) and the hypoxanthines (Figure 4B) reveal very different trends between series. At the beginning of the proline sulfonamide optimization campaign (Figure 4A, 3 → 4 → 5), improvements in TRPA1 potency were achieved without increases in measured log D7.4, thus increasing LLE between leads. The optimization trajectory ends with GDC-0334 (1), which is among the more efficient compounds within the series. Its position in the upper right quadrant of the plot (high potency, high lipophilicity), however, is somewhat unusual. Among the most potent compounds identified within the series, the in vivo potency of GDC-0334 (1) was a distinguishing characteristic that resulted in its selection as a development candidate.15 In the end, the membrane-exposed intrahelical site favors highly lipophilic molecules and ultimately directs the overall trajectory of the series optimization toward a higher lipophilicity space.
Figure 4.

Optimization trajectories based on lipophilic ligand efficiency (LLE) for (A) the proline sulfonamide series and (B) the hypoxanthine series. Optimization trajectories based on membrane ligand efficiency (MLE) for (C) the proline sulfonamide series and (D) the hypoxanthine series.
In comparison, initial potency improvements within the intracellular hypoxanthine-based series were obtained through added lipophilicity (Figure 4B, 6 → 7 → 8), yet concluded with the highly efficient hypoxanthine 9 (high potency, low lipophilicity). Unlike the membrane-exposed proline sulfonamide series, lipophilicity could be varied widely while maintaining potency at the intracellular antagonist site, allowing for more diverse exploration of property space. The ability to explore multiple antagonist series with different binding sites ultimately enabled the discovery team to explore a wide range of chemical space to interrogate safety and efficacy profiles in vivo.
Intrigued by an in vivo measure of lipophilicity that incorporates drug phospholipophilicity, we wondered whether evaluating optimization trajectories using Vss,u might offer a valuable perspective into membrane protein drug design. We therefore explored a novel “membrane ligand efficiency” metric (MLE, eq 1) using rat Vss,u as a measure of lipophilicity, replacing the octanol:water partition coefficient in the common LLE metric (LLE = pIC50 – log D7.4), wherein
| 1 |
During a drug discovery program targeting a membrane-bound protein, MLE can be used, in conjunction with LLE, to provide useful insights into the origins of target potency. While it may not be desirable to optimize MLE, it may prove useful in distinguishing potency driven by membrane partitioning (i.e., ligand–lipid interactions) or intrinsic target potency (i.e., ligand–protein interactions).10
Applied to the TRPA1 program, MLE plots (pIC50versus log(Rat Vss,u)) for the proline sulfonamides (Figure 4C) and the hypoxanthines (Figure 4D) show overall trajectories similar to those using LLE (Figure 4A,B, respectively). However, closer analysis of the lead trajectory within the proline sulfonamide series reveals a noteworthy relationship among leads. Despite large successive increases in potency between compounds 4, 5, and 1, MLE values between these leads are unchanged; this is visualized by the constant MLE trajectory line at approximately 6 (Figure 4C, 4 → 5 → 1). We hypothesize that the MLE-neutral trajectory observed during the optimization of the fluorinated biaryl ring reflects the lipid-exposed environment of this functional group in the protein-bound state (see Figure 2A). Thus, all fluorine-derived potency within this region of the molecule is likely due to enhanced ligand–lipid interactions within the membrane, not improved ligand–protein interactions.
As expected, the MLE optimization trajectory for the intracellular series (Figure 4D) is similar to that observed using LLE (Figure 4B), as the hypoxanthine cryoEM structure (see Figure 2B) suggests ligand–lipid interactions are not necessary for high potency, resulting in the discovery of the highly efficient tetrahydrofuran 9. Despite this, efforts to rigidify the chlorophenyl linker between 6, 7, and 8 unexpectedly increased the Vss,u (i.e., membrane partitioning) of these analogs, contributing to increases in observed in vivo potencies and smaller in vitro/ in vivo potency discrepancies (see Table 1). This suggests that hypoxanthine potency may still be influenced by increased membrane partitioning through local accumulation of the ligand near the membrane-proximal intracellular site, though lipophilicity is not required for high potency, as demonstrated by hypoxanthine 9. While MLE may be a useful tool for teams working at lipid-exposed intramembrane binding sites to differentiate protein- versus membrane-derived potency improvements,10 additional target examples are required to fully evaluate its utility.
We next assessed the impact of the binding site environment on PK optimization within each antagonist class by comparing plots of rat CLuversusVss,u (Figure 5). The proline sulfonamide series (Figure 5A) occupied a higher CLu and Vss,u space compared to the intracellular hypoxanthine series (Figure 5B), consistent with the higher lipophilicity required to target the intramembrane binding site. Interestingly, the proline sulfonamide series is enriched in molecules possessing in vivo half-lives > 6 h. This is likely the result of the team’s simultaneous optimization of both potency and in vitro metabolic stability in a high lipophilicity space. Not surprisingly, fluorine emerged as a common tactic to increase in vivo potency (viaVss,u) while improving metabolic stability (via CLu) at the membrane-exposed binding site, providing a source of metabolically inert lipophilicity.21,23,24 This series also suffers from poor rodent oral bioavailability which impaired preclinical lead evaluation and progression.13 In contrast, shorter half-lives (<2 h) were common in the intracellular hypoxanthine series, where low CLu resulted in high free drug exposures that could offset lower in vivo potency in a low lipophilicity space (see above discussion). Notably, the improved kinetic solubility within this series also translated to improved oral bioavailability.14
Figure 5.

Rat CLu (mL/min/kg) versus rat Vss,u (L/kg) for (A) the proline sulfonamide series and (B) the hypoxanthine series shaded by rat t1/2 (<2, 2–6, >6 h). Lead compounds within each series are denoted by colored stars.
Lessons learned on the Genentech TRPA1 program reveal a link between binding site environment, drug properties, and medicinal chemistry optimization. Newly revealed lipid-exposed intramembrane binding sites suggest that the lipid bilayer likely serves as a relevant target biophase that critically determines many aspects of a drug profile. Within this context, ligand–lipid interactions must be considered during the optimization of both observed potency and in vivo PK when targeting intramembrane sites. We show that membrane-exposed sites may influence optimization trajectories for a given series such that advanced knowledge of the binding site environment may enable better decision-making during the discovery phase. The TRPA1 program highlights the added value of interrogating multiple series and binding sites during a discovery program, enabling more diverse property exploration and increasing chances of success to identify potent and safe drug candidates. Because structure-based techniques are increasingly available to elucidate small molecules bound to membrane proteins, discovery teams will be better positioned to leverage these learnings toward more rational compound design.
Acknowledgments
We thank Alexis Rohou and Lionel Rougé (Genentech Structural Biology) for cryoEM structures and help with image preparation. We thank chemistry team members at Pharmaron Beijing and Paraza Pharma for their contributions to compound design and synthesis. We thank Huy Nguyen and Newton Wu (Genentech Discovery Chemistry—Analytical) for EPSA and physicochemical property measurements. We also thank Justin Ly and the Genentech DMPK and in vivo studies groups for study support.
Glossary
Abbreviations
- AITC
allylisothiocyanate
- AUC
area under the curve
- CHO
Chinese hamster ovary
- CryoEM
cryogenic electron microscopy
- CYP
cytochrome P450
- DMPK
drug metabolism and pharmacokinetics
- EPSA
experimental polar surface area
- FLIPR
fluorometric imaging plate reader
- hPXR
human pregnane X receptor
- LLE
lipophilic ligand efficiency
- LM
liver microsome
- MDCK
Madin–Darby canine kidney cells
- MLE
membrane ligand efficiency
- PK
pharmacokinetic
- PPB
plasma protein binding
- SAR
structure–activity relationship
- TRPA1
transient receptor potential cation channel, subfamily A, member 1
- TPSA
topographical polar surface area
The authors declare no competing financial interest.
References
- Cheng Y. Membrane protein structural biology in the era of single particle cryo-EM. Curr. Opin. Struct. Biol. 2018, 52, 58–63. 10.1016/j.sbi.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisshammer R. New approaches towards the understanding of integral membrane proteins: A structural perspective on G protein-coupled receptors. Protein Sci. 2017, 26, 1493–1504. 10.1002/pro.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishin A.; Gusach A.; Luginina A.; Marin E.; Borshchevskiy V.; Cherezov V. An outlook on using serial femtosecond crystallography in drug discovery. Expert Opin. Drug Discovery 2019, 14, 933–945. 10.1080/17460441.2019.1626822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A.; Yano J.; Hirozane Y.; Kefala G.; Gruswitz F.; Snell G.; Lane W.; Ivetac A.; Aertgeerts K.; Nguyen J.; Jennings A.; Okada K. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 2014, 513, 124–127. 10.1038/nature13494. [DOI] [PubMed] [Google Scholar]
- Liu F.; Zhang Z.; Levit A.; Levring J.; Touhara K. K.; Shoichet B. K.; Chen J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. 10.1126/science.aaw7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szlenk C. T.; Gc J. B.; Natesan S. Does the lipid bilayer orchestrate access and binding of ligands to transmembrane orthosteric/allosteric sites of G protein-coupled receptors?. Mol. Pharmacol. 2019, 96, 527–541. 10.1124/mol.118.115113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Yu Z.; Xiao W.; Lu S.; Zhang J. Allosteric binding sites at the receptor-lipid bilayer interface: novel targets for GPCR drug discovery. Drug Discovery Today 2021, 26, 690–703. 10.1016/j.drudis.2020.12.001. [DOI] [PubMed] [Google Scholar]
- Vauquelin G.; Packeu A. Ligands, their receptors and··· plasma membranes. Mol. Cell. Endocrinol. 2009, 311, 1–10. 10.1016/j.mce.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason R. P.; Rhodes D. G.; Herbette L. G. Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interaction with biological membranes. J. Med. Chem. 1991, 34, 869–877. 10.1021/jm00107a001. [DOI] [PubMed] [Google Scholar]
- Dickson C. J.; Hornak V.; Velez-Vega C.; McKay D. J. J.; Reilly J.; Sandham D. A.; Shaw D.; Fairhurst R. A.; Charlton S. J.; Sykes D. A.; Pearlstein R. A.; Duca J. S. Uncoupling the structure-activity relationships of β2 Adrenergic receptor ligands from membrane binding. J. Med. Chem. 2016, 59, 5780–5789. 10.1021/acs.jmedchem.6b00358. [DOI] [PubMed] [Google Scholar]
- Herbette L. G. Membrane pathways for drug/ion channel interactions: Molecular basis for pharmacokinetic properties. Drug Dev. Res. 1994, 33, 214–224. 10.1002/ddr.430330305. [DOI] [Google Scholar]
- Talavera K.; Startek J. B.; Alvarez-Collazo J.; Boonen B.; Alpizar Y. A.; Sanchez A.; Naert R.; Nilius B. Mammalian transient receptor potential TRPA1 channels: from structure to disease. Physiol. Rev. 2020, 100, 725–803. 10.1152/physrev.00005.2019. [DOI] [PubMed] [Google Scholar]
- Chen H.; Volgraf M.; Do S.; Kolesnikov A.; Shore D. G.; Verma V. A.; Villemure E.; Wang L.; Chen Y.; Hu B.; Lu A.-J.; Wu G.; Xu X.; Yuen P.-w.; Zhang Y.; Erickson S. D.; Dahl M.; Brotherton-Pleiss C.; Tay S.; Ly J. Q.; Murray L. J.; Chen J.; Amm D.; Lange W.; Hackos D. H.; Reese R. M.; Shields S. D.; Lyssikatos J. P.; Safina B. S.; Estrada A. A. Discovery of a potent (4R,5S)-4- fluoro-5-methylproline sulfonamide transient receptor potential ankyrin 1 antagonist and its methylene phosphate prodrug guided by molecular modeling. J. Med. Chem. 2018, 61, 3641–3659. 10.1021/acs.jmedchem.8b00117. [DOI] [PubMed] [Google Scholar]
- Terrett J. A.; Chen H.; Shore D. G.; Villemure E.; Larouche-Gauthier R.; Déry M.; Beaumier F.; Constantineau-Forget L.; Grand-Maître C.; Lépissier L.; Ciblat S.; Sturino C.; Chen Y.; Hu B.; Lu A.; Wang Y.; Cridland A. P.; Ward S. I.; Hackos D. H.; Reese R. M.; Shields S. D.; Chen J.; Balestrini A.; Riol-Blanco L.; Lee W. P.; Liu J.; Suto E.; Wu X.; Zhang J.; Ly J. Q.; La H.; Johnson K.; Baumgardner M.; Chou K.-J.; Rohou A.; Rougé L.; Safina B. S.; Magnuson S.; Volgraf M. Tetrahydrofuran-based transient receptor potential ankyrin 1 (TRPA1) antagonists: ligand-based discovery, activity in a rodent asthma model, and mechanism-of-action via cryogenic electron microscopy. J. Med. Chem. 2021, 64, 3843–3869. 10.1021/acs.jmedchem.0c02023. [DOI] [PubMed] [Google Scholar]
- Balestrini A.; Joseph V.; Dourado M.; Reese R. M.; Shields S. D.; Rougé L.; Bravo D. D.; Chernov-Rogan T.; Austin C. D.; Chen H.; Wang L.; Villemure E.; Shore D. G.; Verma V. A.; Hu B.; Chen Y.; Leong L.; Bjornson C.; Hotzel K.; Gogineni A.; Lee W. P.; Suto E.; Wu X.; Liu J.; Zhang J.; Gandham V.; Wang J.; Payandeh J.; Ciferri C.; Estevez A.; Arthur C. P.; Kortmann J.; Wong R. L.; Heredia J. E.; Doerr J.; Jung M.; Vander Heiden J. A.; Roose-Girma M.; Tam L.; Barck K. H.; Carano R. A. D.; Ding H. T.; Brillantes B.; Tam C.; Yang X.; Gao S. S.; Ly J. Q.; Liu L.; Chen L.; Liederer B. M.; Lin J. H.; Magnuson S.; Chen J.; Hackos D. H.; Elstrott J.; Rohou A.; Safina B. S.; Volgraf M.; Bauer R. N.; Riol-Blanco L. A TRPA1 inhibitor suppresses neurogenic inflammation and airway contraction for asthma treatment. J. Exp. Med. 2021, 218, e20201637. 10.1084/jem.20201637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Zhang X.-F.; Kort M. E.; Huth J. R.; Sun C.; Miesbauer L. J.; Cassar S. C.; Neelands T.; Scott V. E.; Moreland R. B.; Reilly R. M.; Hajduk P. J.; Kym P. R.; Hutchins C. W.; Faltynek C. R. Molecular determinants of species-specific sctivation or blockade of TRPA1 channels. J. Neurosci. 2008, 28, 5063–5071. 10.1523/JNEUROSCI.0047-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernov-Rogan T.; Gianti E.; Liu C.; Villemure E.; Cridland A. P.; Hu X.; Ballini E.; Lange W.; Deisemann H.; Li T.; Ward S. I.; Hackos D. H.; Magnuson S.; Safina B.; Klein M. L.; Volgraf M.; Carnevale V.; Chen J. TRPA1 modulation by piperidine carboxamides suggests an evolutionarily conserved binding site and gating mechanism. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 26008–26019. 10.1073/pnas.1913929116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S. S.; Sakamuru S.; Huang R.; Moeller T. A.; Shinn P.; VanLeer D.; Auld D. S.; Austin C. P.; Xia M. Identification of clinically used drugs that activate pregnane X receptors. Drug Metab. Dispos. 2011, 39, 151–159. 10.1124/dmd.110.035105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel L. B.; Olivieri P. R.; Boezio A. A.; Deak H. L.; Emkey R.; Graceffa R. F.; Gunaydin H.; Guzman-Perez A.; Lee J. H.; Teffera Y.; Wang W.; Youngblood B. D.; Yu V. L.; Zhang M.; Gavva N. R.; Lehto S. G.; Geuns-Meyer S. Optimization of a novel quinazolinone-based series of transient receptor potential A1 (TRPA1) antagonists demonstrating potent in vivo activity. J. Med. Chem. 2016, 59, 2794–2809. 10.1021/acs.jmedchem.6b00039. [DOI] [PubMed] [Google Scholar]
- Goetz G. H.; Philippe L.; Shapiro M. J. EPSA: a novel supercritical fluid chromatography technique enabling the design of permeable cyclic peptides. ACS Med. Chem. Lett. 2014, 5, 1167–1172. 10.1021/ml500239m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. R.; Madeira M.; Wood H. B.; Geissler W. M.; Raab C. E.; Martin I. J. Integrating the impact of lipophilicity on potency and pharmacokinetic parameters enables the use of diverse chemical space during small molecule drug optimization. J. Med. Chem. 2020, 63, 12156–12170. 10.1021/acs.jmedchem.9b01813. [DOI] [PubMed] [Google Scholar]
- Tsopelas F.; Giaginis C.; Tsantili-Kakoulidou A. Lipophilicity and biomimetic properties to support drug discovery. Expert Opin. Drug Discovery 2017, 12, 885–896. 10.1080/17460441.2017.1344210. [DOI] [PubMed] [Google Scholar]
- Broccatelli F.; Aliagas I.; Zheng H. Why decreasing lipophilicity alone is often not a reliable strategy for extending IV half-life. ACS Med. Chem. Lett. 2018, 9, 522–527. 10.1021/acsmedchemlett.8b00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin H.; Altman M. D.; Ellis J. M.; Fuller P.; Johnson S. A.; Lahue B.; Lapointe B. Strategy for extending half-life in drug design and its significance. ACS Med. Chem. Lett. 2018, 9, 528–533. 10.1021/acsmedchemlett.8b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer T. S.; Smith D.; Beaumont K.; Di L. Dose predictions for drug design. J. Med. Chem. 2020, 63, 6423–6435. 10.1021/acs.jmedchem.9b01365. [DOI] [PubMed] [Google Scholar]
- Young R. J.; Leeson P. D. Mapping the efficiency and physicochemical trajectories of successful optimizations. J. Med. Chem. 2018, 61, 6421–6467. 10.1021/acs.jmedchem.8b00180. [DOI] [PubMed] [Google Scholar]