

Abstract
Soluble adenylyl cyclase (sAC) has gained attention as a potential therapeutic target given the role of this enzyme in intracellular signaling. We describe successful efforts to design improved sAC inhibitors amenable for in vivo interrogation of sAC inhibition to assess its potential therapeutic applications. This work culminated in the identification of TDI-10229 (12), which displays nanomolar inhibition of sAC in both biochemical and cellular assays and exhibits mouse pharmacokinetic properties sufficient to warrant its use as an in vivo tool compound.
Keywords: sAC, Soluble adenylyl cyclase, ADCY10, Inhibitor, Structure-based drug design
Cyclic AMP (cAMP), a critical second messenger, is involved in a broad range of physiological processes. cAMP is produced from ATP by adenylyl cyclases (ACs) and degraded by catabolizing phosphodiesterases (PDEs). Presently, there are two known distinct classes of adenylyl cyclases in mammals: bicarbonate-regulated soluble adenylyl cyclase (sAC, ADCY10) and G protein-regulated transmembrane adenylyl cyclases (tmACs; ADCY1–9). While PDEs are established drug targets, at this juncture, there exist no therapeutic agents targeting sAC.
In contrast to tmACs, which respond to signals originating in other surrounding cells, sAC functions as an environmental sensor: it is directly regulated by bicarbonate and serves as a physiological CO2/HCO3–/pHi sensor.1 sAC is localized in intracellular microdomains and is found distributed through the cytoplasm and in cellular organelles, including inside the nucleus and the mitochondrial matrix.2 It regulates a diverse array of biological functions, including sperm activation and motility, intraocular pressure, ciliary beat frequency in airways, luminal pH in the epididymis, the mitochondrial electron transport chain, and glucose-stimulated insulin release from β cells of the pancreas.1,3
While sAC is ubiquitously expressed, two independently derived sAC knockout (KO) mouse strains show male-specific sterility with only mild other phenotypes (e.g., elevated intraocular pressure).3−6 Importantly, in humans, loss of sAC function appears similarly benign: two adult males bearing an ADCY10 frameshift variant were sterile but otherwise healthy.7
Based on its apparent functions, potent and selective inhibitors of sAC potentially provide a mechanism for therapeutic intervention in multiple disease states, including for hypotony or as an on-demand, nonhormonal contraceptive. While tool compounds, including LRE1 (1)2 and KH7 (1b),8 are known (Figure 1), their properties (weak potencies, low selectivities, poor pharmacokinetic profiles, etc.) render them unsuitable for careful scrutiny of sAC driven pharmacology in cellular or animal models.9 While additional sAC inhibitors have been described by pharma (primarily via patent disclosures),10,11 limited data exists regarding their profiles.
Figure 1.

Structures of LRE1 and KH7.
This work describes our efforts to identify compounds, typified by TDI-10229, that combine significantly improved intrinsic potency with the ability to sustain systemic exposure above cell-based IC50 values to enable investigations of sAC-mediated biology in preclinical species.
First generation sAC inhibitors, such as KH7, suffered from general cell toxicity and potential non-sAC mediated effects on ATP/cAMP levels that are likely to confound their utility as tool molecules.2 The more recently discovered LRE12 binds in the bicarbonate binding site of sAC, preventing its enzymatic function. LRE1 represented a significant improvement in overall profile relative to KH7 and permitted more extensive interrogation of sAC biology in vitro. However, its relatively modest potency in biochemical and cell-based assays suggests that high micromolar levels would be required in vivo to elicit sAC-driven pharmacology in translational experiments. LRE1 also showed low oral bioavailability (<5%) in mice, a liability exacerbated by rapid mouse and human microsomal clearance (>768 and 360 μL/(min/mg), respectively), further limiting in vivo options.
More potent sAC inhibitors with appropriate systemic PK to afford sustained sAC inhibition in vivo would represent a significant advance in enabling interrogation of sAC pharmacology in vivo. As a low molecular weight (280.8 g/mol) lead, LRE1 represented an auspicious entry point for medicinal chemistry efforts focused on discovering superior sAC inhibitors. Along with improved potency, microsomal stability and pharmacokinetics, we also wished to evolve the LRE1 lead toward more drug-like molecules, lacking the cyclopropylamine and monosubstituted thiophene moieties, which represent potential metabolic liabilities.12,13
We leveraged significant structural biology data throughout this project. In the first instance, utilization of free-energy perturbation (FEP) calculations with FEP+14 based on the X-ray crystal structure of LRE1 bound to sAC2 led to the design of 2 through scaffold hopping (Table 1). Compound 2 bears a methylpyrazole in lieu of the less desirable cyclopropylamino moiety and yields a 10-fold improvement in potency over LRE1. Medicinal chemistry efforts next focused on modifications around the conserved aminochloropyrimidine headgroup. Unfortunately, this headgroup proved to have extremely narrow SAR, as predicted by FEP+, with very small changes, such as chloro (6) to methyl (10), leading to a dramatic loss in activity. We then moved on to replacement of the undesirable thiophene ring. It was noted that various aromatic and heteroaromatic moieties were well tolerated on the 4-position methylene of the pyrazole (3–8), whereas direct attachment of the thiophene ring to the pyrazole (9) was not. Of particular note is that substituted phenyl rings, such as the fluorophenyl in compound 6, yielded similar potency to the thiophene compound 2.
Table 1. 4-Substituted Pyrazole Analogs.
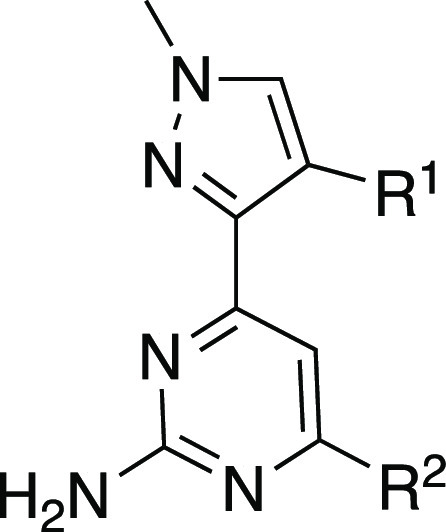

Compounds were tested at minimum in triplicate.
Addition of a methyl group at the 5-position of the pyrazole was also noted to lead to a small improvement in potency as shown in Table 2 (IC50 of 140 nM in 2 vs IC50 of 74 nM in 11). Gratifyingly, the equivalent methyl substitution on the phenyl compound 7 also improved potency and yielded compound 12 with an IC50 of 195 nM. Alternative ring systems were also found to retain potency in this context (13–15), while others abrogated affinity (16 and 17). Next, we investigated the effect of substitution at the 5-position of the pyrazole ring (Table 2). Substitution at R2 on the central pyrazole was found to be sensitive to functionality. For example, the methyl ester substituted analog 18 exhibited higher potency, while other closely related analogs displayed reduced potency (e.g., acid 19 or methyl amide 20). Furthermore, substitution of the R2 methyl group with hydroxy (21) or dimethylamino (22) resulted in reduced potency versus methyl (12). To analyze the binding details of our improved inhibitors and to aid in further lead optimization, we then obtained crystallographic data with 12 bound to sAC (see Figure 2; Supplementary Table 1; Supplementary Figure 1).
Table 2. 5-Substituted Pyrazole Analogs.
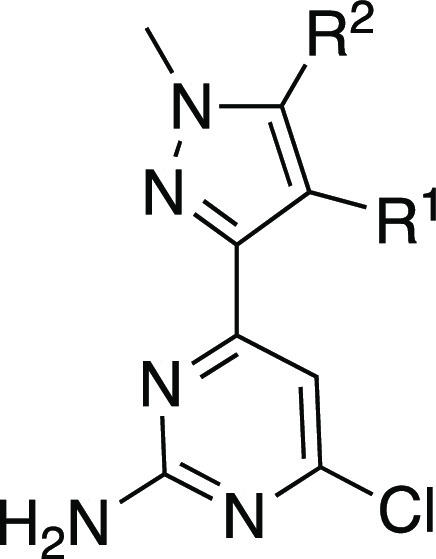
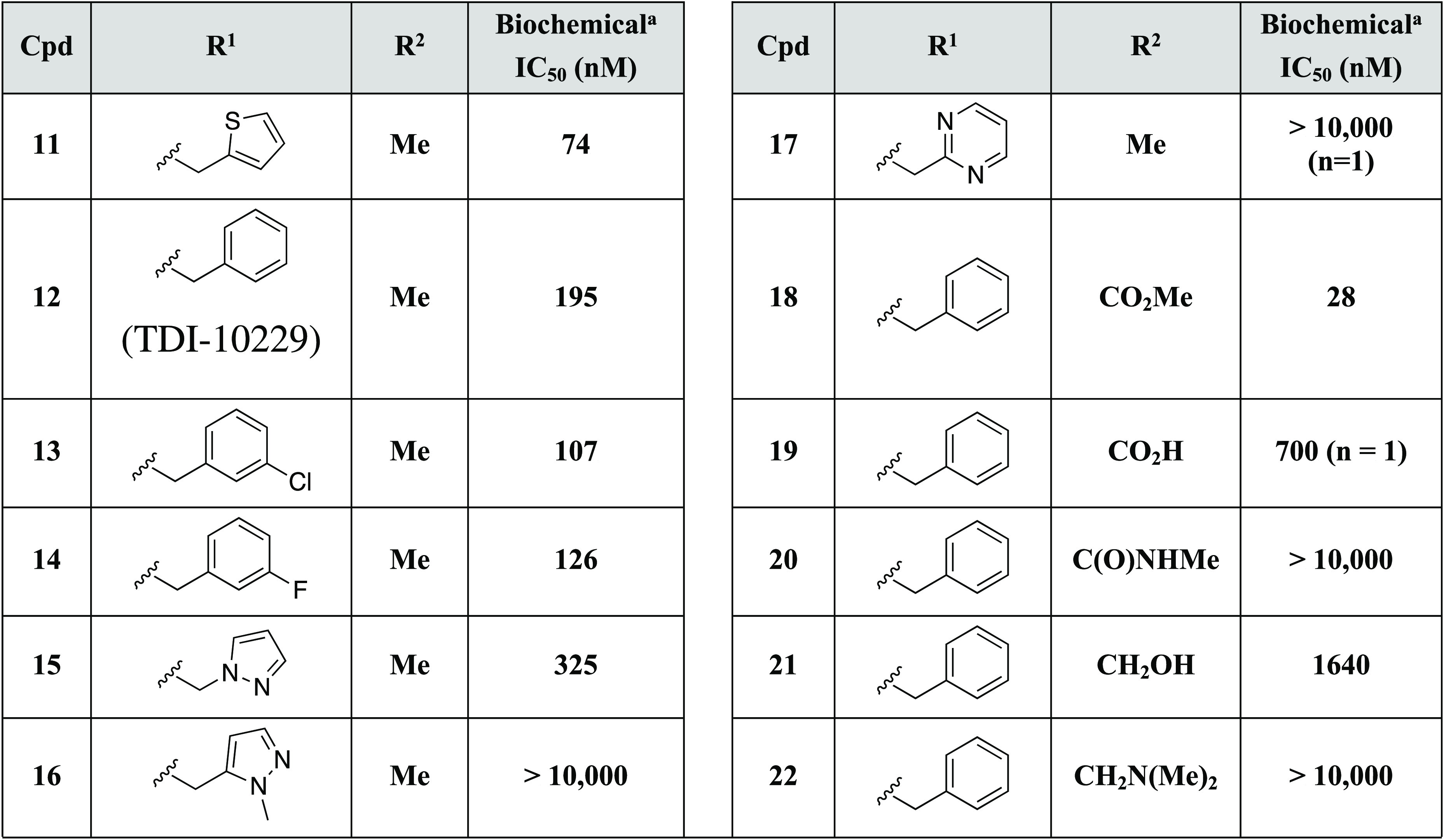
Compounds were tested at minimum in triplicate unless otherwise noted.
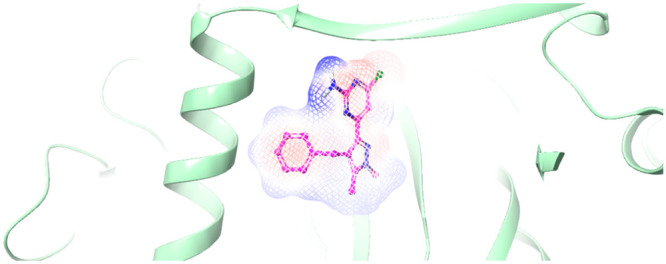
Figure 2.
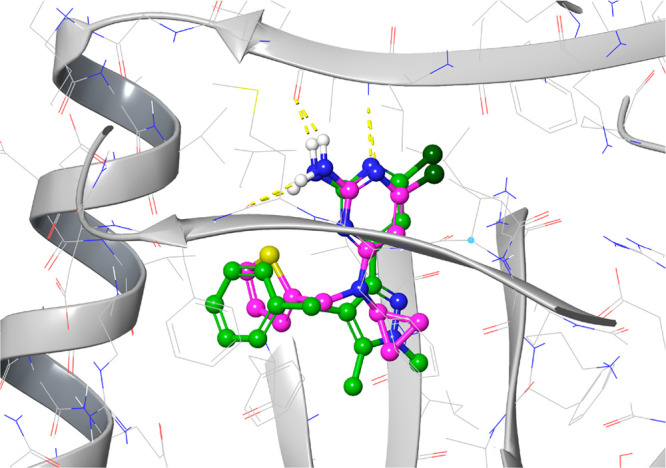
Overlay of crystal structures of sAC complexes with compound 12 and LRE1. The crystal structure of sAC in complex with 12 is displayed as a gray cartoon with gray wires and overlaid with the ligand from the crystal structure of sAC in complex with LRE1. Compound 12 is displayed as atom-colored balls and sticks with green carbons. LRE1 is displayed as atom-colored balls and sticks with magenta carbons.
Figure 2 shows the conserved overlap of the two aminochloropyrimidine headgroups. Importantly, the key hydrogen-bonding contacts made by the aminochloropyrimidine headgroup of LRE1 are maintained in 12. Compared to LRE1, a very subtle rotation of the headgroup of 12 allows it to position its central pyrazole group in the area occupied by LRE1’s central amine and additional cyclopropane substituent. The larger pyrazole moiety fills this area more completely and its additional interactions likely contribute to the higher affinities of 2 and 12. The phenyl group of 12 is also oriented similarly to LRE1’s thiophene, albeit with a slightly rotated ring plane. The rotation and larger ring of 12 are accommodated by a minor rearrangement of the interacting Arg176, which previously was observed to adopt multiple different rotamers.2 The protein conformations are otherwise very similar between the two structures.
With sAC inhibitors displaying improved intrinsic potency relative to LRE1 in hand, their activity was evaluated in a cellular context. Given that many compounds exhibited good permeability (parallel artificial membrane permeability assay, PAMPA), it was gratifying to see much improved inhibition of sAC in a cellular assay (Table 3).
Table 3. Cell Activity and Selected Parameters.
| compd | cell IC50a (nM) | PAMPA (nm/s) | HPLC LogD (pH = 7.4) | kinetic solubility (μg/mL) |
|---|---|---|---|---|
| 2 | 196 | 258 | 2.5 | 6.3 |
| 3 | 225 | 257 | 2.8 | 7.5 |
| 11 | 102 | 280 | 2.7 | 0.38 |
| 12 | 92 | 274 | 2.9 | 1.3 |
Human 4-4 cell data: compounds were tested at minimum in triplicate.
Of the new analogs generated, TDI-10229 (12) was deemed most promising overall in terms of activity coupled with drug-like qualities and was therefore selected for additional characterization. Mouse pharmacokinetic studies of TDI-10229 indicated that this compound, with its 59% bioavailability, is well suited for interrogation of sAC inhibition in an in vivo context at reasonable dosages with coverage up to 50-fold over the cell IC50 based on free drug concentrations (mouse fu = 0.1, Table 4). Furthermore, TDI-10229 was not cytotoxic at 20 μM (200-fold above its IC50 for sAC in cells), it had a low propensity to form reactive intermediates (glutathione trapping study),16 and it did not show appreciable activity against a panel of 310 kinases and 46 other well-known drug targets (GPCRs, ion channels, and nuclear receptors) [Supporting Information S1]. TDI-10229 also displayed high selectivity for sAC versus the closely related tmAC subtypes.15
Table 4. Mouse Pharmacokinetic Properties of TDI-10229 (12).
| dosage | AUC (μM·h) | Cmax (μM) | MRTa (h) |
|---|---|---|---|
| 20 mg/kg (po) | 94 | 15.5 | 3.95 |
| 20 mg/kg (ip) | 72 | 45 | 2.82 |
Mean residence time.
To summarize, in order to identify an inhibitor suitable for exploration of sAC-driven pharmacology in vivo, a set of sAC inhibitors derived from LRE1 were designed and synthesized, efforts facilitated by structural biology input coupled to computational drug design tools. From the novel analogs synthesized, TDI-10229 displayed suitable potency, selectivity, and drug-like properties to facilitate in vivo studies of sAC pharmacology in preclinical animal models of disease, including its utility for hypotony or as an on-demand, nonhormonal contraceptive.3,9,15
Acknowledgments
We thank the BESSY beamline staff for excellent technical support and Dr. Tanweer Khan for generating 13C NMR data. The authors gratefully acknowledge the support to the project generously provided by the Tri-Institutional Therapeutics Discovery Institute (TDI), a 501(c)(3) organization. TDI receives financial support from Takeda Pharmaceutical Company, TDI’s parent institutes (Memorial Sloan Kettering Cancer Center, The Rockefeller University, and Weill Cornell Medicine) and from a generous contribution from Mr. Lewis Sanders and other philanthropic sources.
Glossary
Abbreviations
- AC
adenylyl cyclase
- sAC
soluble adenylyl cyclase
- tmAC
transmembrane adenylyl cyclase
- po
per os
- ip
intraperitoneal
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00273.
Synthesis and characterization for compounds 2–22, biology assay methods, ADMET assay methods, mouse pharmacokinetic studies, crystal structure determination, diffraction and refinement statistics, crystal structure of hsAC-cat in complex with TDI-10229 (12), and supplementary references (PDF)
Accession Codes
Coordinates and structure factors for the sAC/TDI-10229 crystal structure have been deposited with the worldwide PDB (www.pdb.org) under accession ID 7OVD.
Author Contributions
‡ D.J.H. and P.T.M. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by NIH Grants P50 HD100549 and R01 HD088571 (to J.B. and L.R.L.), DFG grant STE1701/11 (C.S.), F31 AG069501 (T.R.), T32 GM073546-13 (J.F.), and Male Contraceptive Initiative (M.B.).
The authors declare no competing financial interest.
This paper was published ASAP on July 14, 2021, with errors in the Supporting Information. The corrected version was reposted on July 20, 2021.
Supplementary Material
References
- Rossetti T.; Jackvony S.; Buck J.; Levin L. R. Bicarbonate, carbon dioxide and pH sensing via mammalian bicarbonate-regulated soluble adenylyl cyclase. Interface Focus 2021, 11, 20200034. 10.1098/rsfs.2020.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Espiritu L.; Kleinboelting S.; Navarrete F. A.; Alvau A.; Visconti P. E.; Valsecchi F.; Starkov A.; Manfredi G.; Buck H.; Adura C.; Zippin J. H.; van den Heuvel J.; Glickman J. F.; Steegborn C.; Levin L. R.; Buck J. Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat. Chem. Biol. 2016, 12, 838–844. 10.1038/nchembio.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins S. V.; Steegborn C.; Levin L. R.; Buck J. Pharmacological modulation of the CO2/HCO3-/pH-, calcium-, and ATP-sensing soluble adenylyl cyclase. Pharmacol. Ther. 2018, 190, 173–186. 10.1016/j.pharmthera.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G.; Jaiswal B. S.; Xie F.; Krajnc-Franken M. A. M.; Robben T. J. A. A.; Strik A. M.; Kuil C.; Philipsen R. L. A.; van Duin M.; Conti M.; Gossen J. A. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 2993–2998. 10.1073/pnas.0400050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess K. C.; Jones B. H.; Marquez B.; Chen Y.; Ord T. S.; Kamenetsky M.; Miyamoto C.; Zippin J. H.; Kopf G. S.; Suarez S. S.; Levin L. R.; Williams C. J.; Buck J.; Moss S. B. The ″soluble″ adenylyl cyclase in sperm mediates multiple signaling events required for fertilization. Dev. Cell 2005, 9, 249–259. 10.1016/j.devcel.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F.; Garcia M. A.; Carlson A. E.; Schuh S. M.; Babcock D. F.; Jaiswal B. S.; Gossen J. A.; Esposito G.; van Duin M.; Conti M. Soluble adenylyl cyclase (sAC) is indispensable for sperm function and fertilization. Dev. Biol. 2006, 296, 353–362. 10.1016/j.ydbio.2006.05.038. [DOI] [PubMed] [Google Scholar]
- Akbari A.; Pipitone G. B.; Anvar Z.; Jaafarinia M.; Ferrari M.; Carrera P.; Totonchi M. ADCY10 frameshift variant leading to severe recessive asthenozoospermia and segregating with absorptive hypercalciuria. Hum. Reprod. 2019, 34, 1155–1164. 10.1093/humrep/dez048. [DOI] [PubMed] [Google Scholar]
- Steegborn C. Structure, mechanism, and regulation of soluble adenylyl cyclases - similarities and differences to transmembrane adenylyl cyclases. Biochim. Biophys. Acta, Mol. Basis Dis. 2014, 1842, 2535–2547. 10.1016/j.bbadis.2014.08.012. [DOI] [PubMed] [Google Scholar]
- Balbach M.; Fushimi M.; Huggins D. J.; Steegborn C.; Meinke P. T.; Levin L. R.; Buck J. Optimization of lead compounds into on-demand, nonhormonal contraceptives: leveraging a public-private drug discovery institute collaboration. Biol. Reprod. 2020, 103, 176–182. 10.1093/biolre/ioaa052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saalau-Bethell S. M.; Berdini V.; Cleasby A.; Congreve M.; Coyle J. E.; Lock V.; Murray C. W.; O’Brien M. A.; Rich S. J.; Sambrook T.; Vinkovic M.; Yon J. R.; Jhoti H. Crystal structure of human soluble adenylate cyclase reveals a distinct, highly flexible allosteric bicarbonate binding pocket. ChemMedChem 2014, 9, 823–32. 10.1002/cmdc.201300480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchmann B.; Harter M.; Cancho-Grande Y.; Kosemund D.; Schirok H.; Nguyen D.; Fritsch M.. Azaindoles as Inhibitors of Soluble Adenylate Cyclase. WO 2009/030725 A2, 2009.
- Macmillan D. The use of Derek Nexus to facilitate decision-making in chemical safety assessment. Application of non-animal approaches for decision-making in chemical safety assessment, December 10–11, 2018, NC3Rs, London, UK. https://www.lhasalimited.org/publications/the-use-of-derek-nexus-to-facilitate-decision-making-in-chemical-safety-assessment/5183.
- Ellison C. M.; Madden J. C.; Judson P.; Cronin M. T. D. Using In Silico Tools in a Weight of Evidence Approach to Aid Toxicological Assessment. Mol. Inf. 2010, 29, 97–110. 10.1002/minf.200900006. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wu Y.; Deng Y.; Kim B.; Pierce L.; Krilov G.; Lupyan D.; Robinson S.; Dahlgren M. K.; Greenwood J.; Romero D. L.; Masse C.; Knight J. L.; Steinbrecher T.; Beuming T.; Damm W.; Harder E.; Sherman W.; Brewer M.; Wester R.; Murcko M.; Frye L.; Farid R.; Lin T.; Mobley D. L.; Jorgensen W. L.; Berne B. J.; Friesner R. A.; Abel R. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- Balbach M.; Ghanem L.; Rossetti T.; Kaur N.; Ritagliati C.; Ferreira J.; Krapf D.; Molina L. P.; Santi C. M.; Hansen J. N.; Wachten D.; Fushimi M.; Meinke P. T.; Buck J.; Levin L. R.. Soluble adenylyl cyclase inhibition prevents human sperm functions essential for fertilization. Mol. Hum. Reprod. 2021, submitted for publication [DOI] [PMC free article] [PubMed]; https://www.biorxiv.org/content/10.1101/2021.04.27.441671v1.article-info.
- Sameshima T.; Miyahisa I.; Yamasaki S.; Gotou M.; Kobayashi T.; Sakamoto J. High-Throughput Quantitative Intrinsic Thiol Reactivity Evaluation Using a Fluorescence-Based Competitive Endpoint Assay. SLAS Discov 2017, 22, 1168–1174. 10.1177/2472555217704654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.