

Abstract
Purpose:
Colorectal cancer (CRC) patients with peritoneal metastases (CRPM) have limited treatment options and the lowest CRC survival rates. We aimed to determine whether organoid testing could help guide precision treatment for CRPM patients, as the clinical utility of prospective, functional drug screening including non-standard agents is unknown.
Experimental Design:
CRPM organoids (peritonoids) isolated from patients underwent parallel next-generation sequencing and medium-throughput drug panel testing ex vivo to identify specific drug sensitivities for each patient. We measured the utility of such a service including: success of peritonoid generation, time to cultivate peritonoids, reproducibility of the medium-throughput drug testing, and documented changes to clinical therapy as a result of the testing.
Results:
Peritonoids were successfully generated and validated from 68% (19/28) of patients undergoing standard care. Genomic and drug profiling was completed within 8 weeks and a formal report ranking drug sensitivities was provided to the medical oncology team upon failure of standard care treatment. This resulted in a treatment change for 2 patients, one of whom had a partial response despite previously progressing on multiple rounds of standard care chemotherapy. The barrier to implementing this technology in Australia is the need for drug access and funding for off-label indications.
Conclusions:
Our approach is feasible, reproducible and can guide novel therapeutic choices in this poor prognosis cohort, where new treatment options are urgently needed. This platform is relevant to many solid organ malignancies.
Keywords: Organoids, precision medicine, colorectal cancer, drug screening, genomics
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer -related mortality worldwide(1). The peritoneum is a common site for metastases(2), but confers the worst survival rates among metastatic CRC (mCRC) patients. Patients with unresectable colorectal peritoneal metastases (CRPM) have a median survival of 12–16 months and five-year survival of less than 5% (3,4). CRPM responds poorly to modern chemotherapy regimens compared to other sites of CRC metastasis (5). While approximately 60% of CRC liver metastases respond to modern systemic therapy, less than a third of CRPM demonstrate any response (6,7). With only a handful of systemic therapy options available, patients with CRPM rapidly exhaust treatment options.
The advent of cytoreductive surgery (CRS) with hyperthermic intraperitoneal chemotherapy (HIPEC) has offered carefully selected patients with CRPM a favourable 27 to 41 months median survival, with a 23 to 42% five-year survival (8,9). However, despite successful CRS and HIPEC, up to 80% of patients recur within two years (10–12). Treatment after disease recurrence centres on systemic chemotherapy, but has shown limited efficacy. Novel drug delivery methods such as pressurised aerosolised chemotherapy (PIPAC) show promise as a feasible palliative surgical option in patients with recurrent or unresectable CRPM, but need further evaluation in clinical trials(13,14). There is an urgent need to explore new modalities and means of selecting treatment for patients with CRPM. Precision medicine is both a current challenge and opportunity facing the oncology community. The fundamental question becomes how to rationally assign drug treatments, not by cancer site, or even pathological subtype, but based on the unique molecular biology of each cancer and each patient. Genomics has successfully guided choice of targeted therapies across multiple cancer types (15–17), but clinical outcomes led purely by cancer genomics have been disappointing (18). Previous analyses by targeted next-generation sequencing of >1000 patients with metastatic cancer, found that only 11% could be matched with an on-indication, FDA-approved drug and an additional 9% could be matched when off-label use of targeted FDA-approved drugs were considered (19). Genomic sequencing is a static measure evaluating alterations in the tumor, and fails to provide any functional assessment of tumor responses to drugs. Functional screening using patient-derived tumor cells can bridge this gap to provide information on drug sensitivity specific to the patient, even in the absence of actionable biomarkers.
Organoid culture has emerged as a promising pre-clinical model of disease (20). This culture system enables long-term propagation of cells from percutaneous biopsies or operative specimens with good success rates, that represent the genetics (21), inter-patient variation and epithelial cell types of the original sample (22,23). Recent co-clinical trials using gastrointestinal cancer patient -derived organoids have reported that organoid drug responses ex vivo mimic patient responses in the clinic (24–27). While only containing small numbers of patients, these studies and others (28–31) validate the use of tumor-derived organoids for functional testing of patient specific therapies.
In this multicentre prospective study, we aimed to firstly establish an ex vivo organoid-based platform to integrate functional drug sensitivity testing with genomic profiling to identify suitable therapeutic options in patients with CRPM. We subsequently utilise this platform to evaluate responses to standard chemotherapeutics and guide choice of novel therapy options for patients that fail standard treatment.
Materials and methods
Study design
APOLLO is a multicentre Australian study designed to measure the utility of an organoid-based platform integrating genomics and functional testing to guide treatment choice for CRPM patients that have exhausted standard care. The objectives were to assess: success rate of peritonoid generation; time to cultivate peritonoids; reproducibility of high-throughput drug testing; plus document changes to, and outcomes of, clinical therapy as a result of the testing.
All participants gave informed written consent and research was conducted in accordance with the Declaration of Helsinki, the NHMRC Statement on Ethical Conduct in Human Research and institutional approvals (PMCC 15/76 and HREC/16/SAC/344 SSA/17/TQEH/291). Patients with microsatellite stable (MSS) CRPM undergoing surgical or percutaneous intervention were eligible for participation. Patients were excluded if they had microsatellite high (MSI-H) cancers as this group of patients could enter immune checkpoint inhibitor trials. Tissue was received from operative specimens at time of staging laparoscopy, CRS and HIPEC, or percutaneous biopsies. In all operative specimens, a minimum of two tissue samples from different sites was received and pooled. Organoid cultures were derived from peritoneal deposits (termed peritonoids) or synchronously resected primary colorectal cancer (termed tumoroids).
Patient-derived organoid establishment and passaging
Tumour samples were first minced and enzymatically digested in Organoid digestion media (DMEM [Gibco] containing Collagenase IV 67.5U/mL [CLS-4 Worthington], Dispase 0.23U/mL [Gibco, Massachusetts, USA], Hyaluronidase 8–20U/mL, DNase Type I 50 Kunitz units/mL, 100 U/mL Pen and 100 μg/mL Strep [all Sigma Aldrich, St. Louis, MO, USA]) in a water bath at 37°C for 30–60 minutes. Peritonoids were cultured in low (5–6%) oxygen conditions in CRC media containing advanced DMEM/F12, 10 mM Hepes, 1x Glutamax, 10 mg/L gentamicin, 1x antibiotic-antimycotic, 2x B27 [all ThermoFisher], 500 nM A83–01 [Tocris], 50 ng/ml hEGF, 1 nM [Leu15]-Gastrin 1 human, 1 mM N-Acetyl-L-cysteine, 5 μM SB202190, 10 μM SB431542 and 10 μM Y27632 [all Sigma Aldrich, St. Louis, MO, USA]. CRC media was changed twice weekly, with growth monitored until passaging was required. Peritonoids were passaged upon reaching 100–200μm in diameter by digestion with TrypLE [Gibco, Massachusetts, USA] at 37°C followed by tituration with a pipette. Cells were replated in matrigel, with the general aim to expand the number of wells by at least double at each passage. In our experience, it was noticeable within 36–48 hours if organoids were starting to grow. Peritonoids that did not grow within 14 days were discarded. Peritonoid cultures were documented by short tandem repeat (STR) DNA profiling to represent the source peritoneal tumour and were ambient shipped overnight between collaborating institutes in Australia and Seattle, USA, with minimal loss of viability.
In vivo tumorigenicity assays
Peritonoids (1× 105 cells suspended in 1:1 matrigel/PBS) were subcutaneously injected into a single flank of NOD.Cg-PrkdcscidIl2rgtm1Wjl/Szj (NSG) mice. Tumor growth was noted for all lines examined and monitored twice weekly for up to 63 days using calipers.
Immunohistochemistry
Formalin fixed, paraffin embedded organoid and tissue sections (4μm) were dewaxed and stained using haematoxylin and eosin. For immunohistochemical staining, slides were subjected to either sodium citrate or EDTA antigen retrieval at 125°C for 3 minutes followed by 90°C for 10 seconds. This was followed by incubation with the following primary antibodies: CDX2 (1:500 #EPR2764Y Millipore), CK20 (1:200 #M7019 DAKO/Agilent) or anti-human mitochondria (1:500 #MAB1273 Millipore). Biotin conjugated secondary antibodies and ABC reagent (Vector laboratories) were used and developed with DAB solution (DAKO). Slides were counterstained using Meyer’s haematoxylin.
National Association of Testing Authorities (NATA) accredited next-generation sequencing
Whole exome sequencing (WES) libraries were prepared using the NEBNext DNA (New England Biolabs) or KAPA DNA Kits (Roche) using DNA from matched peripheral blood mononuclear cells (normal germline), peritonoids and tumoroids. Exonic fragments were captured using the Roche SeqCap EZ Exome v3.0 kit (Roche) and 75-bp paired-end sequencing was performed on a HiSeq2500 and a NextSeq 500 (Illumina). Analysis of NGS data can be found in Supplementary Methods.
SEngine Precision Medicine drug testing
Peritonoids were assessed for purity and viability following ex vivo expansion in media containing DMEM/F12, 50ng/mL rhEGF [both Corning CellGro] with 100ug/mL Primocin (Invivogen), 10mM HEPES, 1x B27,1x GlutaMax [all Gibco], 1x N2 [Thermo Fisher], 100ng/mL Noggin, 100ng/mL Wnt-3a [both R&D Systems], 500ng/mL R-Spondin-1 [PeproTech], 1mM Nicotinamide [Sigma Aldrich] and 10μM Y-27632 [Selleckchem]. Drug tests were conducted on peritonoids from passage 4 to 8. Eight hundred cells per well were seeded into 384 well assay plates containing 50uL media supplemented with 5% Matrigel [Corning] for high throughput screening as described. A broadly targeted 87 drug pan-cancer focused small molecule library (first 5 samples, Table S1A) was acoustically administered (Labcyte Echo) as single agents using contactless, nanovolume liquid transfers to create a 3-log, 6-dose drug curve; drug concentrations ranged from 33pM to 200μM, depending on individual drug properties. Dose ranges for targeted agents were designed to capture previously reported Cmax values (serum level) and the asymptotic response range. Dose ranges for chemotherapies were derived from clinical dosage guidelines, one log above and two below an assumed 1.8M2 average body surface area. This library was further refined to create a 35-drug CRC-focused panel (Table S1B) to better reflect clinically available options for the subsequent patient samples. Peritonoids and tumoroids were challenged with drugs for 6 days, following which, relative viability was determined by whole-well ATP quantification using Cell-Titer-Glo 2.0 (Promega) and normalized to vehicle-only controls (maximal DMSO concentration used was 0.2%). Additional drug testing methods details including formulas for generation of area under the curve (AUC) data and low-throughput testing at second lab site are provided in Supplementary Methods. We use the AUC for drug response as this metric combines information about the efficacy (how much cell viability is decreased by each drug) and potency (the amount of drug needed to reduce viability; EC50, IC50) of each drug.
The SEngine Precision Medicine internal pan-cancer database contained 57 samples from 16 tumor types, with n=8 (14%) derived from CRC. All tumour organoids were cultured using media containing standard core organoid components, with slight tissue-of-origin variations, and screened in an identical manner using the SEngine CLIA approved standard protocol, as described for peritonoids above. The drug library used was validated for activity and consistent for lot, delivery, and storage. This unique method of analysis allows for the detection of exceptional responses as well as sample specific sensitivities and is extensively validated semi-annually for both technically consistency and biological concordance with genomic biomarkers, as well as prospective and retrospective in vivo drug responses as part of the Clinical Laboratory Improvement Amendments (CLIA) process. Most recently, spearman correlations between technical replicates and biological replicates were 0.97 and 0.87, respectively. The primary method of quality control for each individual screen is determined by the ratio of signal range to noise in both the positive (bortezomib) and negative (vehicle) controls. Z’ or Z′ factor is a measure of statistical effect size, all tests presented here successfully passed quality control and achieved Z’>0.3.
Heat map visualization of peritonoid sensitivity to drug library
To assess shared and individual peritonoid drug sensitivities, AUC data from drug response curves was subjected to hierarchical clustering using the WGCNA R package version 1.68, R version 3.6.0 on Windows. The topological overlap matrix was calculated (Pearson’s correlation) with pairwise complete observations. The resulting dendrogram was superimposed on to the heatmap for visualisation (pheatmap version 1.0.12). Chi-square, one-way ANOVA and TukeyHSD tests were conducted to examine relationships between drug sensitivity and clinical or genomic variants. Z-score transformation of AUC data for each drug was performed using R version 3.6.0 on Windows and displayed using violin plots (FigS3A).
Results
Characterisation of patients with colorectal cancer peritoneal metastases
Twenty-eight patients (aged 43–81 years) with microsatellite stable (MSS) CRC and CRPM were recruited over 18 months and prospectively studied. Table S2 provides a clinical summary. With the exception of five patients who had unresectable CRPM, all underwent CRS and HIPEC. Five patients had synchronous primary CRC and CRPM at diagnosis and the remaining patients had metachronous peritoneal metastases. At the time tissue samples were obtained two patients were treatment naïve, however the majority had received multiple cycles of standard chemotherapeutic treatment including FOLFOX, FOLFIRI and/or CAPOX, the biologics cetuximab or bevacizumab, and/or chemoradiotherapy for those with rectal cancer (Table S2).
The majority of CRPMs are stroma-rich, poor prognosis, consensus molecular subtype four
CRC can be stratified by RNA expression into four consensus molecular subtypes (CMS) (32). While this has yet to result in subtype -specific interventions, the characterisation provides information about the underlying molecular pathways that are dysregulated in each tumor type and may provide rationale for drug sensitivities above existing biomarkers, such as RAS, BRAF mutation and microsatellite instability (MSI) status. We undertook mRNAseq of 14 of the CRPM tissue samples from this cohort. CMS analysis revealed the majority (71%, 10/14) to be CMS4 (FigS1A), a subtype known to confer a worse overall and relapse-free survival (32). Three matched synchronous primary and CRPM samples were examined. Of these, the primary tumors studied were CMS2, CMS3 and CMS4; however all three matching CRPM samples were classified as stromal-rich CMS4, suggesting evolution of the transcriptional signatures from the primary to the metastatic site (FigS1A). As would be expected for MSS disease, no samples were assigned to CMS1, which is associated with MSI, immune infiltration and activation (32). Gene-set enrichment analysis confirmed activation of transforming growth factor-β (TGFβ) pathway and epithelial to mesenchymal transition (EMT) genes in the CMS4 samples, an epithelial differentiation signature in CMS3 and WNT signature in CMS2. These findings are consistent with previously identified expression signatures for these CMS (32) (Fig S1B).
Characterisation of organoids derived from CRPM (peritonoids)
Using an optimised culture technique, peritonoids were successfully generated and validated from 19 of 28 patients (68%) (Fig S1C). This success rate is consistent with prior studies (26). Additionally, we isolated matched organoids (here tumoroids) from synchronously resected primary CRC for 2 patients. Peritonoids were validated with a combination of short tandem repeat (STR) analysis and immunohistochemistry (Cytokeratin-20, CK20 and Caudal Type homeobox2, CDX2) (Fig 1, Fig S1D–G). Tumorigenicity was confirmed by in vivo tumor growth following subcutaneous administration of peritonoids into immunocompromised mice (Fig S1D–G). Whole exome sequencing (WES) was undertaken on peritonoids (from 13 patients) and germline DNA from blood was used to evaluate DNA alterations that may predict sensitivity or resistance to targeted therapies. High-confidence calls of genomic variants and copy number alterations in 551 cancer associated or actionable genes are summarised in Table S1C and Fig S2A and were 100% concordant with KRAS, NRAS and BRAF testing undertaken in independent pathology laboratories using FFPE tumor tissue (n=9 CRPM tissue samples, Table S2). The peritonoids recapitulated genetic alterations commonly associated with CRC (33,34) (e.g. APC, TP53, KRAS, Table S1C–D). Furthermore, the samples had a mean somatic mutation rate of 1.8/MB (Table S1E), in line with their pathological characterisation as MSS disease (19). The COSMIC DNA mutation signature, that can suggest the molecular aetiology of DNA alterations in each sample, was also evaluated in the context of therapeutics that can target specific mechanisms of DNA alterations (e.g. PARP inhibitors for BRCA mutant cancers, Fig S2B). We concurrently undertook these genomic analyses with drug sensitivity testing.
Figure 1. Peritonoids are representative of the human tumors from which they are derived.
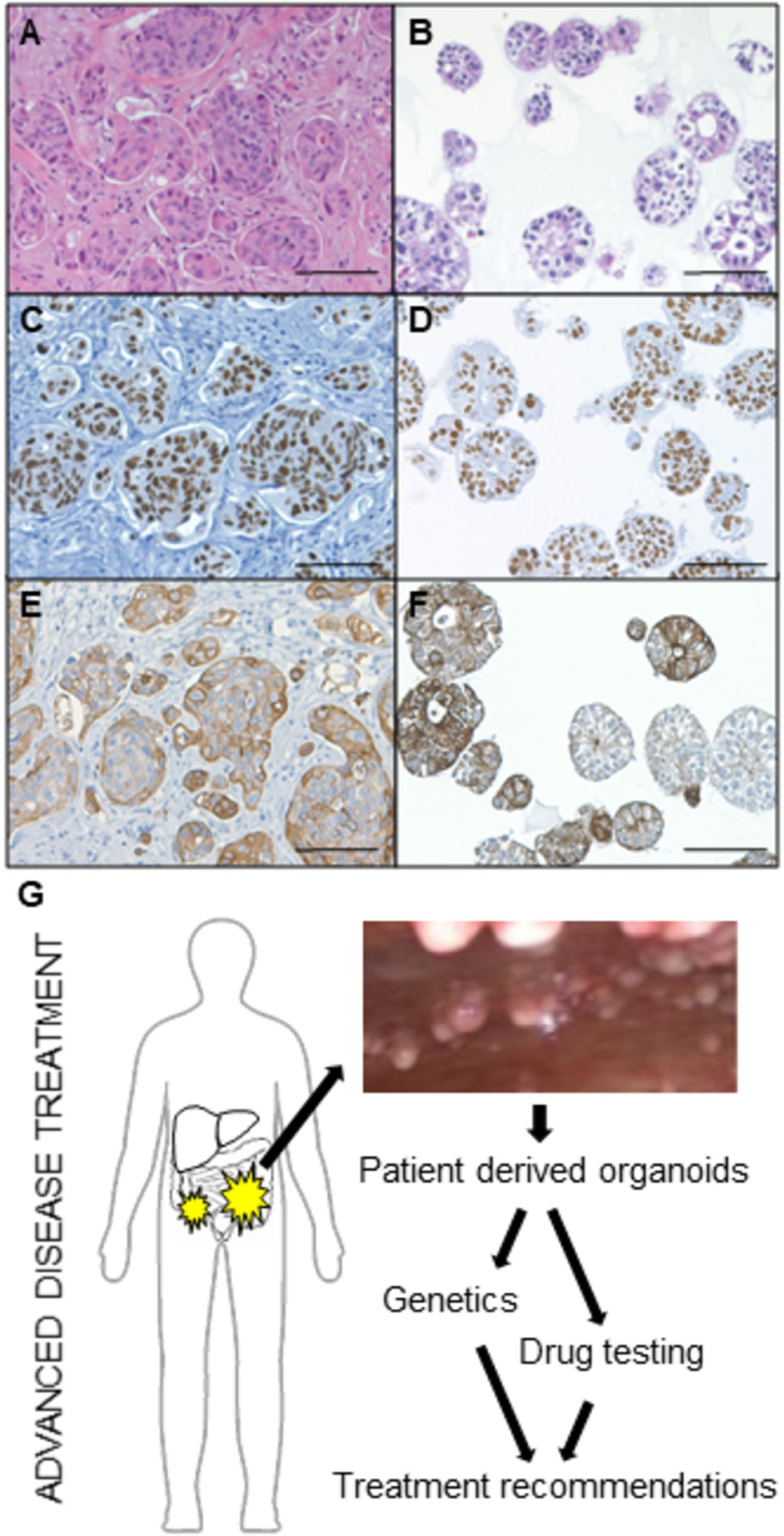
Peritonoids (B, D, F) resemble the tumor cell morphology of the native tumor (A, C, E) and express intestinal epithelial markers (A-B H&E, C-D CDX2, E-F CK20). Scale bar 100μm. (G) Schema depicting our precision medicine screening platform to guide patient-specific treatment for patients with worst prognosis CRC. Peritonoids and peripheral blood mononuclear cells undergo next-generation whole exome sequencing (WES) to identify genetic alterations found in the tumor and germline of each patient. This is combined with medium-throughput drug panel testing to identify specific drug sensitivities for each patient. Results are presented to the medical oncology team to provide treatment change options should patients exhaust standard care chemotherapy.
Medium throughput peritonoid drug sensitivity testing
To evaluate novel therapeutic options, peritonoids from 15 patients were subjected to medium throughput drug dose response screening using the CLIA-certified PARIS platform. Peritonoids from the first five patients, were challenged with an 87 pan-cancer drug panel consisting of both chemotherapies and targeted agents. A curated panel of 35 targeted agents was selected for subsequent patients based on availability in Australia as well as predicted efficacy in CRPM (Table S1A–B). Compounds that target the most common alterations found in mCRC, i.e. the ‘druggable’ landscape of mCRC (33), were well-represented in this smaller drug library (Table S1D).
A heat-map depicting unsupervised clustering of patient-derived organoid drug sensitivity for the 35 drugs, along with limited clinical and genomic characteristics for the 15 patients, is shown in Fig 2. Drugs with similar mechanisms of action generate similar responses across the peritonoids tested (for example, the two first generation EGFR inhibitors Gefitinib and Erlotinib, MEK inhibitors Cobimetinib and Trametinib, ALK inhibitors Ceritinib and Crizotinib and PARP inhibitors Olaparib and Rucaparib). In contrast, the majority of peritonoids also had unique and specific responses (Fig 2, FigS3A). Unique patient -specific findings along with common drug sensitivities across the cohort underpin the value of functional drug testing. Of note, replicate plating of peritonoids from patient 5 on two separate test dates 2-weeks apart generated very similar results against the drug panel (Pearson r=0.89, Fig S3B, Fig 2). Testing with selected drugs from the library at a second lab site, using the same peritonoid lines, also significantly correlated with results from the medium-throughput platform (Pearson r=0.86, P<0.05, Fig S3C). These results underscore the accuracy of the CLIA-accredited testing platform. Paired primary CRC tumoroids and peritonoids clustered near each other for patient 6 but were very distantly related for patient 7, suggesting that there may be shared drug sensitivity between the primary and peritoneal disease but this may also be patient and sample dependent. Peritonoids from patient 15 were broadly insensitive to all panel drugs ex vivo. This patient had aggressive disease, as assessed by early peritoneal recurrence despite initial low volume disease and successful CRS/HIPEC. We were unable to determine the accuracy of the peritonoid testing platform for this patient, as no promising drug candidates were uncovered through the screen. As such the patient was offered standard care FOLFOX treatment rather than organoid directed care.
Figure 2. Medium-throughput drug screening of peritonoids reveals shared and specific drug sensitivities to targeted agents.
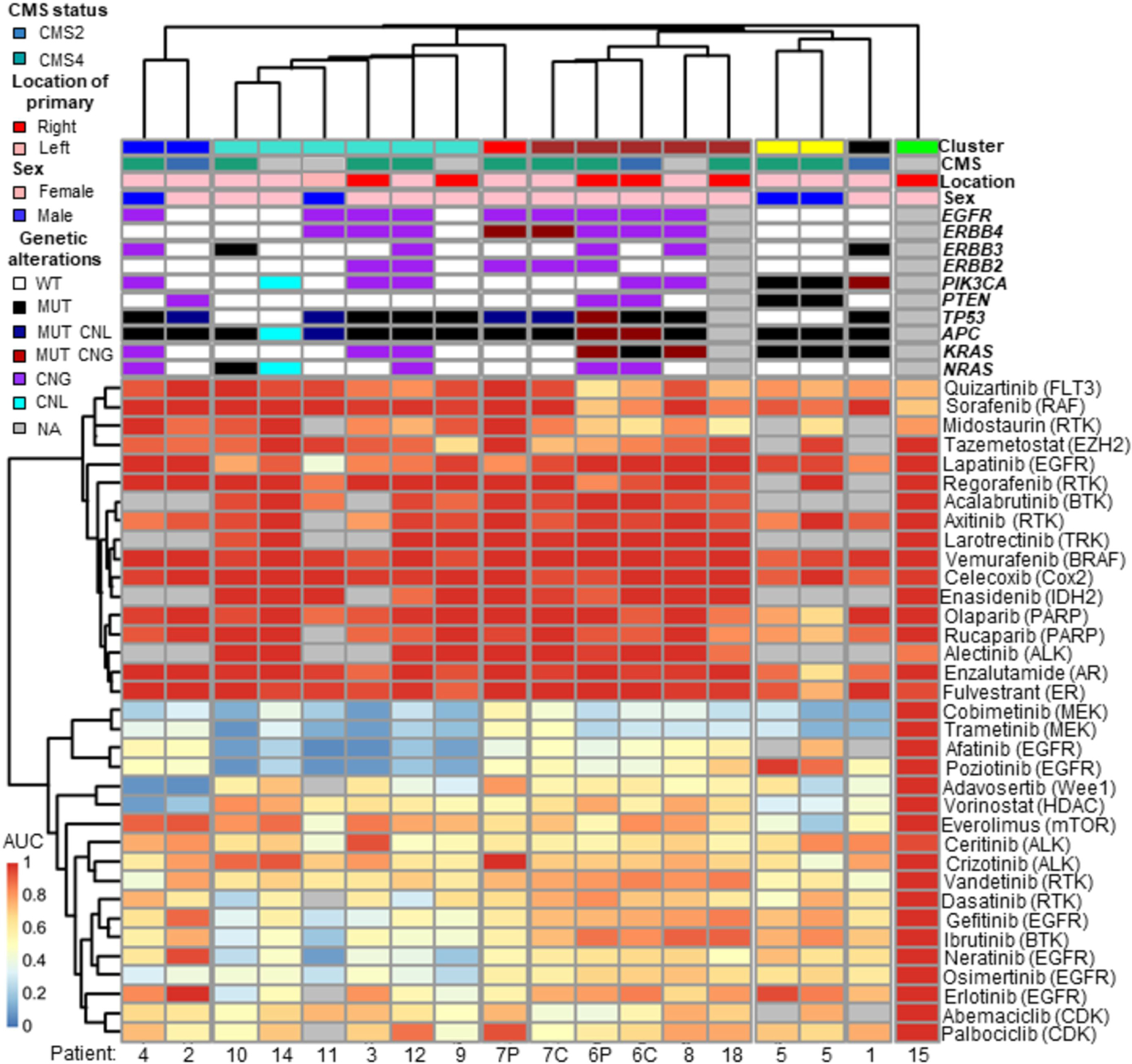
Clinical and molecular features summarised (top), non-supervised clustering of normalised dose response AUC data from ex vivo medium-throughput drug testing of peritonoids depicted below (red 100% viable cells to blue 0% viable cells, normalised to vehicle alone). Matched samples: C, tumoroid derived from primary colorectal cancer; P, peritonoid derived from CRPM. CMS, consensus molecular subtype, WT, wild type; MUT, mutant; CNG, copy number gain; CNL, copy number loss; NA, not available
Ex vivo, functional drug sensitivities are consistent with genomically predicted targets
Medium-throughput screening of targeted small molecule inhibitors identified several drug sensitivities that were unique to individual patients and consistent with their genetic biomarkers. For example, the presence of a PTEN mutation in Patient 5 was significantly associated with sensitivity to PARP inhibition (p< 0.0005, Fig 3A) in line with preclinical data (35,36). Alongside the truncating mutation in PTEN, peritonoids from patient 5 also harboured a PIK3CA frameshift mutation (N1068fs), and responded exceptionally well to both PI3K p110a catalytic subunit inhibitors present in the 87-drug panel (Alpelisib and Taselisib) and inhibitors of downstream targets AKT and mTOR (Fig S3D). Peritonoids from Patient 1 contained a pathogenic PIK3CA N1044K mutation and were also exceptionally sensitive to both p110a inhibitors as well as the AKT inhibitor MK2206 (Fig 3B, Fig S3E). Patient 11’s peritonoids harboured EGFR, ERBB2 and ERBB4 copy gain and demonstrated sensitivities to all 7 EGFR inhibitors tested with exceptional responses (>95% growth inhibition at lowest concentrations tested, 33nM) seen for the two inhibitors with ERBB4 activity, Afatinib and Poziotinib (Fig 3C). Many peritonoids demonstrated partial responses to one or more EGFR family inhibitors, consistent with the established dependency on EGFR signalling in CRC (37). However, activating mutations in KRAS were significantly associated with decreased sensitivity to 5 of 7 EGFR inhibitors, as would be expected from clinical trial data (37,38) (Fig 3D). While these specific genetic alterations have been previously shown to influence response to targeted agents, genomic alterations could explain some, but not all, observed drug sensitivities (Fig 2, Table S1C), a finding that underscores the need for functional testing in precision medicine.
Figure 3. Concordant patient specific genomic alterations and peritonoid drug responses.
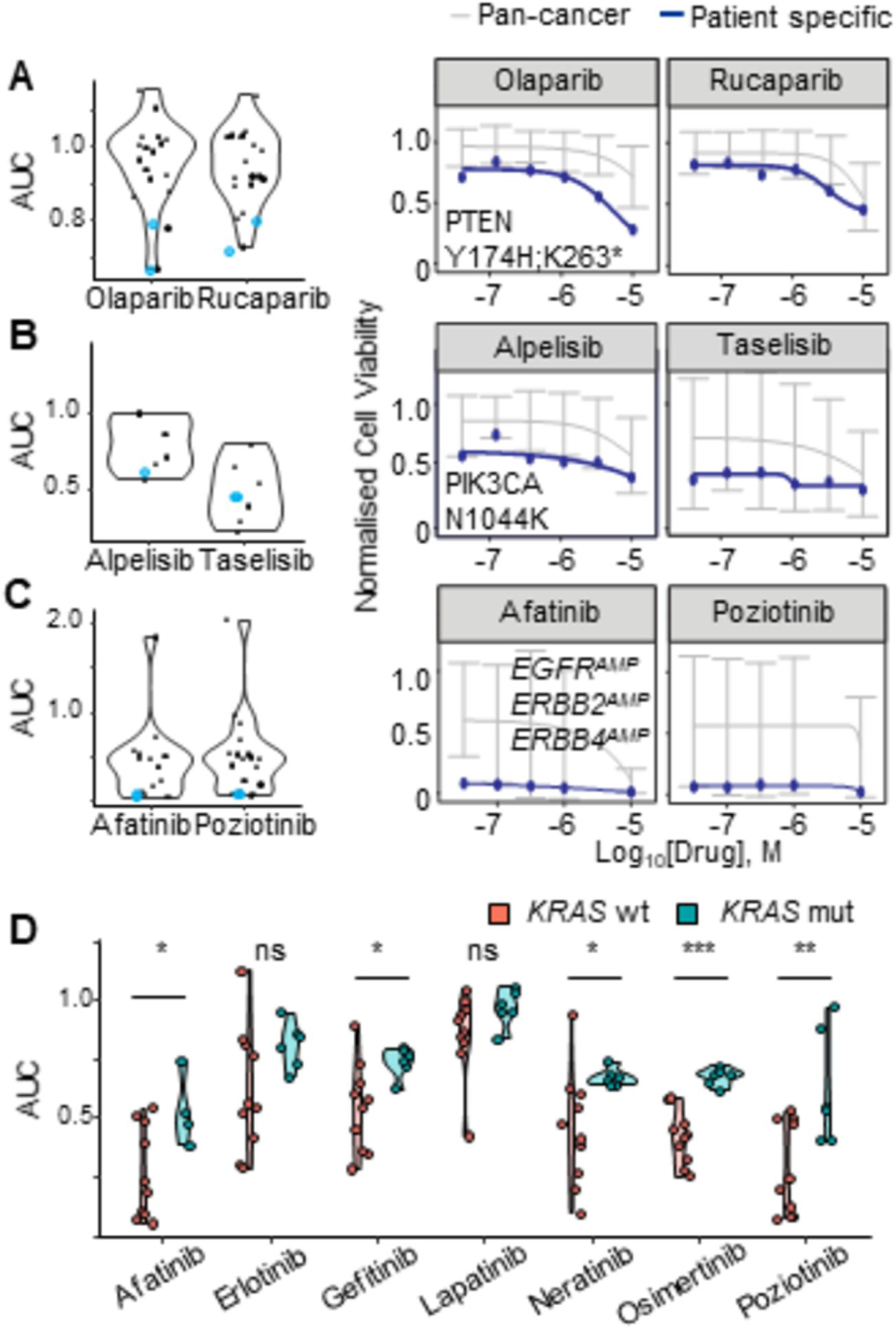
Peritonoid dose response AUC data displayed as a violin plot for inhibitors of: (A) PARP; (B) PI3K; (C-D) EGFR. (A-C) Blue data points indicate the peritonoid with dose response curves displayed on right: (A) Patient 5 PTEN Y174H,K263; (B) Patient 1 PIK3CA N1044K; (C) Patient 3 EGFRAMP, ERBB2AMP, ERBB4AMP. Blue line on dose response curves is patient specific response, grey line indicates average response for previously screened cancer organoid and cell lines. AUC, area under the curve, AMP, copy number gain.
Patient-derived peritonoid sensitivity to standard care chemotherapeutic regimes
Most patients were treated with standard care chemo(radio)therapy prior to tissue sampling, with many undergoing further chemotherapy as per standard practice following sample collection (Table S2). An assessment of peritonoid sensitivity to standard FOLFOX and FOLFIRI regimes ex vivo was undertaken for nine patients (Fig S4). Consistent with a recent study using organoids cultured from metastatic CRC (primarily liver) (26), ex vivo peritonoid FOLFOX sensitivity failed to clearly separate samples from patients who had in vivo partial responses or stable disease (PR/SD) versus progressive disease (PD) following FOLFOX treatment(Fig S4A–E). However, the two patients with clinically responsive disease had not received oxaliplatin therapies previously in the clinic and gave rise to two of the most sensitive peritonoid lines to FOLFOX ex vivo. Our patient cohort did not contain any patients with PR/SD following FOLFIRI treatment, hence we were unable to assess the predictive value of this testing platform for the FOLFIRI regimen (Fig S4F–M). In two patients with synchronously resected primary colorectal and metastatic tumors, both patients’ tumoroids derived from the primary tumor were more sensitive to FOLFIRI than their metastatic counterpart. For one patient, the tumoroid culture was also more sensitive to the FOLFOX regimen than the related peritonoid culture (Fig S4N–O).
Specific patient results and outcomes: using peritonoids to inform novel therapies for treatment-refractory CRPM
During the study period, thirteen of the 19 CRPM patients for whom peritonoids were successfully cultured had disease progression despite standard care chemotherapeutic treatment. Outside of our study, patient 3 was started on Regorafenib, a multikinase inhibitor, recently approved in Australia for treatment refractory mCRC. Peritonoid testing demonstrated insensitivity to Regorafenib with viability measurements similar to vehicle alone (AUC 1.07, Fig 2). Clinically, this patient failed to respond to Regorafenib, consistent with ex vivo findings. However, all peritonoid lines in our study were also insensitive to Regorafenib, possibly due to an inherent resistance of CRPM to this drug or because this drug targets stromal and angiogenic tumor properties that are not well modelled by the epithelial organoid cultures. Two patients exhibited specific sensitivities to agents in the functional screen (Fig S5A) and started off-label drug treatment based on peritonoid test results. Peritonoids from Patient 1 were KRASG12D mutant (Table S1C) and broadly insensitive to most monotherapy chemotherapeutics in the library (Fig S5A). Nevertheless, treatment with particular targeted agents, the MEK inhibitors (MEKi) Trametinib and Cobimetinib, and multi-tyrosine kinase inhibitor, Vandetanib, dramatically reduced organoid viability (Fig S5B–D). However, the inability to obtain compassionate access to MEKi, coupled with drug funding limitations prohibited us from offering a MEKi to Patient 1. Vandetanib is approved for use as a single agent in the treatment of advanced medullary thyroid cancer and was obtained under compassionate access for this patient. Treatment was changed to Vandetanib when the patient already had widespread, extensive disease progression on standard care chemotherapy. Unfortunately no disease response with four weeks of Vandetanib therapy was noted before the patient passed away.
Peritonoids from patient 2 were sensitive to multiple therapeutics, including chemotherapies that the patient had not been exposed to, despite this patient undergoing multiple rounds of standard care chemo(radio)therapy prior to CRPM sampling for organoid culture (Table S2, Fig S5A). Of note, these peritonoids displayed a striking sensitivity to an inhibitor of the WEE1 G2–M cell-cycle checkpoint kinase, Adavosertib, especially when compared to the average dose response across a reference set of all samples within the SEngine Precision Medicine internal database (Fig 4A). The database is used to highlight outstanding responses to a specific drug for a given sample to refine drugs of interest. By comparing results for a specific patient to this broader set of cancer organoid and cell viability data, we are able to identify drugs that are particularly effective for a specific sample, thereby allowing for a precision medicine approach. Adavosertib is currently in Phase II clinical trial for advanced CRC in the UK-based FOCUS4-C trial for patients with RAS & TP53 mutation or loss of histone marks (39). We were unable to obtain access to Adavosertib for our patient. Further therapeutic options based on peritonoid drug sensitivity for this patient, such as the EGFR inhibitor Osimertinib (Fig 4B) and the HDAC inhibitor Vorinostat (Fig 4C) were explored, but access to off-label use was restricted by funding. Finally the anti-metabolite, Gemcitabine, was offered to the patient based on peritonoid testing (Fig 4D) combined with drug access, cost, and toxicity considerations. Peritonoid sensitivity to Gemcitabine was also validated at a second lab site (Fig S5E–G) and the patient was treated with a Gemcitabine-Capecitabine combination. After 3 months of therapy there was a partial response demonstrated by FDG Positron Emission Tomography and Computed Tomography (PET-CT) scans, followed by disease progression following a further 2 months of treatment (Fig 4E–H). This was despite this patient showing prior, continual disease progression on standard care chemotherapy. These results illustrate the power of functional testing to identify effective chemotherapies outside of standard of care. We continue to monitor patients in this cohort and will provide peritonoid-directed therapy options should standard care be exhausted for the remainder of surviving patients.
Figure 4. Peritonoid-guided drug choice for chemo-refractory disease.
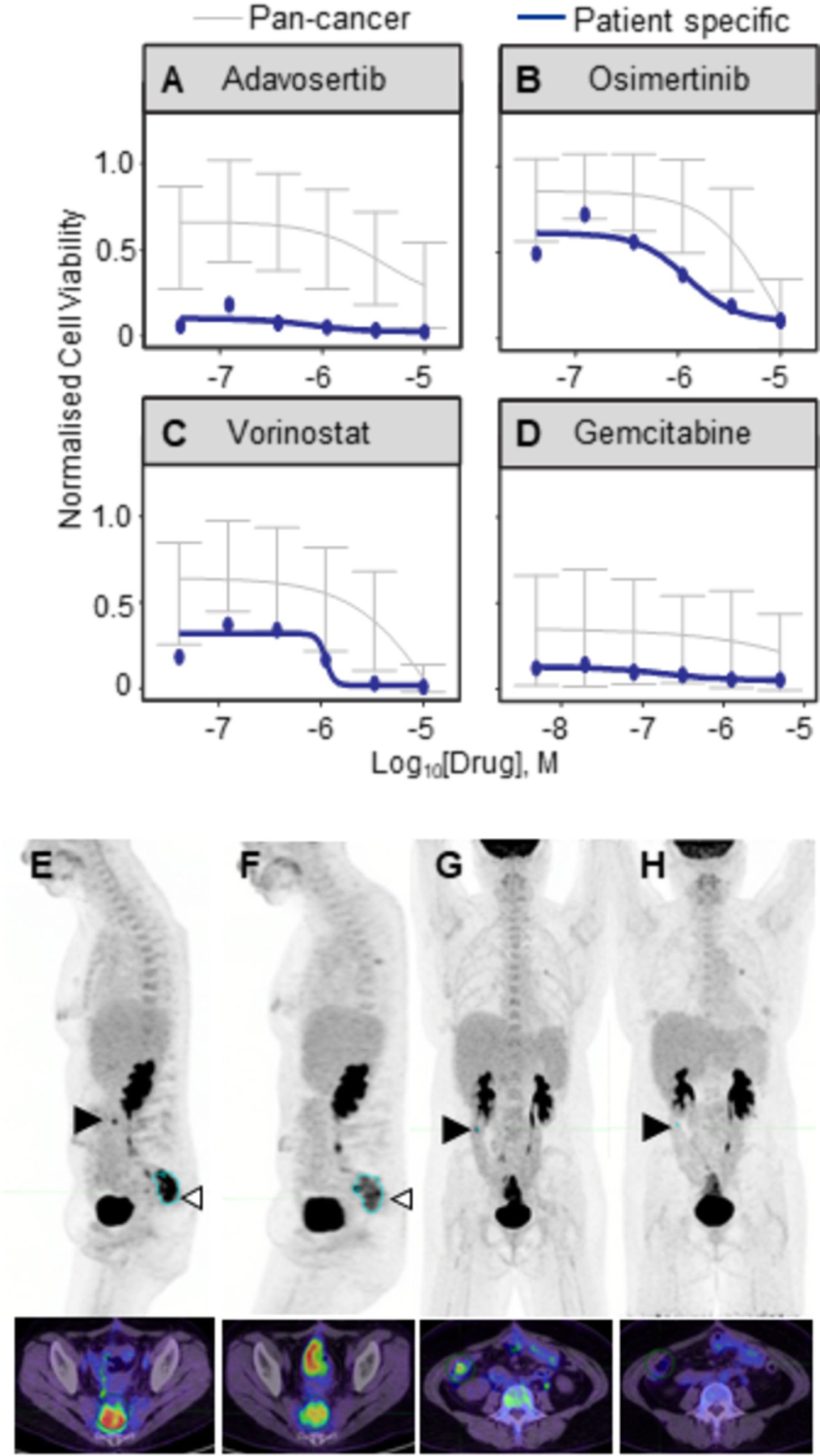
Disease in 43 year old patient 2 worsened despite standard care surgery and 5 rounds of chemo(radio)therapy, including EGFRi treatment. The medical oncology team considered candidate alternate therapies based on peritonoid testing results. Peritonoid dose response curves (in blue) for (A) Wee1 inhibitor Adavosertib, (B) Osimertinib, (C) Vorinostat and (D) Gemcitabine. In grey is average cell viability from prior SEngine testing of cancer cell and organoid lines. Error bars denote st dev. FDG-PET-CT scan of patient immediately prior to treatment change (E, G) and 3 months after change to Gemcitabine (F, H). (E, F) Sagittal and (G, H) frontal images. Position of the transverse axial slice (bottom) is indicated by open arrowhead in top image. Pelvic hotspot marked in blue (E, F) has reduced from 15 to 9.7 and right abdomen lesion (closed arrow, serosal deposit on bowel) is no longer visible.
Discussion
Historically, the inability to replicate tumor heterogeneity is postulated to be one of the key reasons that has limited the use of conventional cell lines to guide precision medicine. Here we generated ex vivo tumor models that more faithfully recapitulate the cellular and genomic heterogeneity present in the original tumor, by generating peritonoids from at least two different CRPM sites in operative specimens. Multiple retrospective studies have now linked ex vivo drug responses of patient-derived organoids to clinical outcomes across multiple solid cancers (24–26,28,29), albeit with fairly small patient cohorts to date. A major aim of the current body of work was to determine whether patient-derived peritonoids could be cultured and genomic and drug sensitivity analyses reported, within a time-frame that enabled alternate therapy options to be acted on by the treating physician for patients. To this end, peritonoid cultures were successfully grown in 68% (19/28) of cases, taking between 3–6 weeks to generate. Within 2 months of tissue sampling, a combined genomics and drug sensitivity report was ready for consideration by the medical oncologist.
The majority of patients in this cohort were recruited whilst still having standard care options available, allowing time for organoid testing well within the clinically actionable window for these patients. A shortfall is the timelag between tissue sampling and actioning the results. For patient 1, peritonoids were derived from CRPM tissue sampled 16 months prior to treatment change to the peritonoid-guided therapy. For patient 2, the time between tissue sampling and treatment change to peritonoid -guided therapy was reduced to 6 months and resulted in a partial response in the patient. The decision to change therapy is not only made on the availability of the data, but also the clinical circumstances of the patient. However, with this platform it is entirely feasible to re-biopsy accessible tumor deposit(s) in the event of disease progression to re-evaluate genomic changes and drug sensitivities. Further, as we attempted here, multiple sampling of disease should be included when possible to better guide selection of therapies that are efficacious across potentially heterogeneous disease sites.
Highlighting the utility of functional testing, the response of peritonoids to specific drugs with known genomic biomarkers was predictable in some, but not all samples. The MSK-Impact study of 1134 matched primary and metastatic CRC (including ~50 CRPM) revealed that CRPM were enriched for alterations to the phosphoinositide 3-kinase (PI3K) and mitogen-activated kinase (MAPK) pathways compared to other metastatic sites (33). These common genomic alterations in MAPK and PI3K pathway genes are also reflected in our CRPM cohort (Table S4). Concordantly, peritonoids with PIK3CA mutations (n=2) exhibited exceptional responses to PI3K inhibitors, while the cohort was broadly sensitive to MEK inhibitors (62% have mutation and/or copy gain to KRAS, NRAS or BRAF) and EGFR inhibitors (54% had EGFR copy gain), highlighting again the critical importance of EGFR/MAPK signalling for CRC. Mimicking the clinical situation, KRAS mutant peritonoids were, however, less sensitive to EGFR inhibition. Acting on these peritonoid drug sensitivity data for MAPK targets will require currently non-standard, combination treatments to constrain feedback loops that reactivate MAPK signalling in CRC, such as that trialled for BRAF mutant CRC (40) or in combination with immune checkpoint modulation in the future. We also uncovered unpredicted sensitivity to agents without validated biomarkers (such as Gemcitabine) that are not normally used for the treatment of CRC.
In summary, this study addresses a clinically unmet need to explore and evaluate novel treatment options for patients with CRPM. We have successfully established a patient-derived, peritonoid -based platform to direct personalised therapy in this poor prognosis cohort of patients. Our platform delivers functional testing and genomic data in a form and time frame that is clinically relevant for our current care pathways. We were limited in our impact by the anticipated difficulties in drug funding and access for off-label indications. Due to this limitation we were not able to change patient treatment to the most efficacious drugs identified from ex vivo peritonoid screening. As approaches such as ours mature, and are backed by larger randomised-controlled clinical trials, there will be the need to modernise drug approvals to include more tumor agnostic indications and one-off, personalised approvals. The tools and techniques exist, as we have shown here, to grow, propagate, transport and analyse living tumor samples. We now have the very exciting opportunity to search for new ways to better define individual drug sensitivities, linked to understanding tumor genomics and biology, to inform practise and help our patients.
Supplementary Material
Translational Relevance:
Recent studies have indicated that patient-derived organoid cultures can retrospectively predict treatment responses to standard care chemotherapies for various solid tumors such as gastric, colorectal, bladder, ovarian and pancreatic cancers. Here we show that genomics and medium-throughput drug screening of patient-derived organoids from colorectal peritoneal metastases can be used to prospectively guide innovative (off-label) therapy choices for this poor prognosis cohort. This study paves the way for a future phase II clinical trial to evaluate the utility of this organoid based platform to deliver personalized therapy in patients with colorectal peritoneal metastases, particularly in situations where standard care has been exhausted.
Acknowledgements:
We thank the patients and their families for taking part in this study; Prof. Cheri Ostroff for critical reading of the manuscript; Dr Andreas Shreiber and Paul Wang from the Australian Cancer Research Foundation Cancer Genomics Facility in the Centre for Cancer Biology, Adelaide for NGS analysis; Dr. Thio Niko and Magnus Zethoven from the Bioinformatics Consulting Core in the Cancer Research Division at Peter MacCallum Cancer Centre, Melbourne for NGS analysis.
Financial support:
Funding This study was supported by grants from the National Health and Medical Research Council (APP1156391 to RR, DLW, SLW), Cancer Council SA Beat Cancer Project on behalf of its donors and the State Government of South Australia through the Department of Health (MCF0418 to SLW, DLW), the Faculty of Health Science at the University of Adelaide (SLW), the Peter MacCallum Cancer Centre Foundation (RR) and NCI (R01CA144288 to CG). SEngine Precision Medicine provided organoid drug testing at no cost.
Footnotes
Potential conflicts of interest Outside of the research in this manuscript, TP reports grants from Amgen; MM from Ipsen Australia Pty Ltd, Merck Serono Australia and Amgen Australia Pty Ltd. RGR receives research support from Merck Serono Australia, Invion, Australia and Fisher and Paykel Healthcare, New Zealand. CG has patents pending regarding systems and methods for personalised cancer treatment and drug development. All other authors declare no competing interests.
Data and materials availability: NGS data are available in dbGaP (phs002023.v1.p1) and GEO (GSE147971). All data needed to evaluate the conclusions are present in the paper and/or the Supplementary Materials. Distribution of peritonoid lines is limited to existing approved sites through the signed informed consent of study participants. Approval for distribution may be granted by application to the HREC.
References
- 1.Siegel R, Miller K, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69(1):7–34 doi 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Simkens GA, Rovers KP, Nienhuijs SW, de Hingh IH. Patient selection for cytoreductive surgery and HIPEC for the treatment of peritoneal metastases from colorectal cancer. Cancer Manag Res 2017;9:259–66 doi 10.2147/CMAR.S119569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franko J, Shi Q, Goldman C, Pockaj B, Nelson G, Goldberg R, et al. Treatment of colorectal peritoneal carcinomatosis with systemic chemotherapy: a pooled analysis of north central cancer treatment group phase III trials N9741 and N9841. J Clin Oncol 2012;30(3):263–7 doi 10.1200/JCO.2011.37.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franko J, Shi Q, Meyers JP, Maughan TS, Adams RA, Seymour MT, et al. Prognosis of patients with peritoneal metastatic colorectal cancer given systemic therapy: an analysis of individual patient data from prospective randomised trials from the Analysis and Research in Cancers of the Digestive System (ARCAD) database. Lancet Oncol 2016;17(12):1709–19 doi 10.1016/S1470-2045(16)30500-9. [DOI] [PubMed] [Google Scholar]
- 5.Sugarbaker PH. Colorectal cancer: prevention and management of metastatic disease. Biomed Res Int 2014;2014:782890 doi 10.1155/2014/782890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugarbaker PH, Bijelic L, Chang D, Yoo D. Neoadjuvant FOLFOX chemotherapy in 34 consecutive patients with mucinous peritoneal carcinomatosis of appendiceal origin. J Surg Oncol 2010;102(6):576–81 doi 10.1002/jso.21679. [DOI] [PubMed] [Google Scholar]
- 7.Adam R, Delvart V, Pascal G, Valeanu A, Castaing D, Azoulay D, et al. Rescue surgery for unresectable colorectal liver metastases downstaged by chemotherapy: a model to predict long-term survival. Ann Surg 2004;240(4):644–57; discussion 57–8 doi 10.1097/01.sla.0000141198.92114.f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceelen W, Nieuwenhove Y, Putte D, Pattyn P. Neoadjuvant Chemotherapy with Bevacizumab May Improve Outcome after Cytoreduction and Hyperthermic Intraperitoneal Chemoperfusion (HIPEC) for Colorectal Carcinomatosis. Ann Surg Oncol 2014;21:3023–8. [DOI] [PubMed] [Google Scholar]
- 9.Leung V, Huo YR, Liauw W, Morris DL. Oxaliplatin versus Mitomycin C for HIPEC in colorectal cancer peritoneal carcinomatosis. Eur J Surg Oncol 2017;43(1):144–9 doi 10.1016/j.ejso.2016.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Alzahrani N, Valle S, Fisher O, Sugarbaker P, Yonemura Y, Glehen O, et al. Iterative cytoreductive surgery with or without hyperthermic intraperitoneal chemotherapy for colorectal peritoneal metastases: A multi-institutional experience. Journal of surgical oncology 2019;119(3):336–46. [DOI] [PubMed] [Google Scholar]
- 11.Passot G, Vaudoyer D, Villeneuve L, Kepenekian V, Beaujard A, Bakrin N, et al. What Made Hyperthermic Intraperitoneal Chemotherapy an Effective Curative Treatment for Peritoneal Surface Malignancy: A 25-Year Experience With 1,125 Procedures. Journal of surgical oncology 2016;113:796–803. [DOI] [PubMed] [Google Scholar]
- 12.Ubink I, Bolhaqueiro ACF, Elias SG, Raats DAE, Constantinides A, Peters NA, et al. Organoids from colorectal peritoneal metastases as a platform for improving hyperthermic intraperitoneal chemotherapy. Br J Surg 2019;106(10):1404–14 doi 10.1002/bjs.11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nowacki M, Alyami M, Villeneuve L, Mercier F, Hubner M, Willaert W, et al. Multicenter comprehensive methodological and technical analysis of 832 pressurized intraperitoneal aerosol chemotherapy (PIPAC) interventions performed in 349 patients for peritoneal carcinomatosis treatment: An international survey study. Eur J Surg Oncol 2018;44(7):991–6 doi 10.1016/j.ejso.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Alyami M, Gagniere J, Sgarbura O, Cabelguenne D, Villeneuve L, Pezet D, et al. Multicentric initial experience with the use of the pressurized intraperitoneal aerosol chemotherapy (PIPAC) in the management of unresectable peritoneal carcinomatosis. Eur J Surg Oncol 2017;43(11):2178–83 doi 10.1016/j.ejso.2017.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344(11):783–92 doi 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 16.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364(26):2507–16 doi 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003;348(11):994–1004 doi 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 18.Letai A Functional precision cancer medicine-moving beyond pure genomics. Nat Med 2017;23(9):1028–35 doi 10.1038/nm.4389. [DOI] [PubMed] [Google Scholar]
- 19.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011;141(5):1762–72 doi 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 21.Weeber F, van de Wetering M, Hoogstraat M, Dijkstra KK, Krijgsman O, Kuilman T, et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc Natl Acad Sci U S A 2015;112(43):13308–11 doi 10.1073/pnas.1516689112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science 2019;364(6444):952–5 doi 10.1126/science.aaw6985. [DOI] [PubMed] [Google Scholar]
- 23.Li M, Izpisua Belmonte JC. Organoids - Preclinical Models of Human Disease. N Engl J Med 2019;380(6):569–79 doi 10.1056/NEJMra1806175. [DOI] [PubMed] [Google Scholar]
- 24.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018;359(6378):920–6 doi 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganesh K, Wu C, O’Rourke KP, Szeglin BC, Zheng Y, Sauvé C-EG, et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nature Medicine 2019;25(10):1607–14 doi 10.1038/s41591-019-0584-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Science Translational Medicine 2019;11(513):eaay2574 doi 10.1126/scitranslmed.aay2574. [DOI] [PubMed] [Google Scholar]
- 27.Yao Y, Xu X, Yang L, Zhu J, Wan J, Shen L, et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2019. doi 10.1016/j.stem.2019.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov 2018;8(9):1112–29 doi 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Driehuis E, Kolders S, Spelier S, Lohmussaar K, Willems SM, Devriese LA, et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov 2019;9(7):852–71 doi 10.1158/2159-8290.CD-18-1522. [DOI] [PubMed] [Google Scholar]
- 30.Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018;172(1–2):373–86 e10 doi 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 2017;7(5):462–77 doi 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21(11):1350–6 doi 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018;33(1):125–36 e3 doi 10.1016/j.ccell.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009;1(6–7):315–22 doi 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med 2010;2(53):53ra75 doi 10.1126/scitranslmed.3001538. [DOI] [PubMed] [Google Scholar]
- 37.Messersmith WA, Ahnen DJ. Targeting EGFR in colorectal cancer. N Engl J Med 2008;359(17):1834–6 doi 10.1056/NEJMe0806778. [DOI] [PubMed] [Google Scholar]
- 38.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359(17):1757–65 doi 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 39. www.focus4trial.org.
- 40.Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. 2019;381(17):1632–43 doi 10.1056/NEJMoa1908075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.