

Abstract
Several Candida species can undergo a heritable and reversible transition from a ‘white’ state to a mating proficient ‘opaque’ state. This ability relies on highly interconnected transcriptional networks that control cell-type-specific gene expression programs over multiple generations. Candida albicans, the most prominent pathogenic Candida species, provides a well-studied paradigm for the white-opaque transition. In this species, a network of at least eight transcriptional regulators controls the balance between white and opaque states that have distinct morphologies, transcriptional profiles, and physiological properties. Given the reversible nature and the high frequency of white-opaque transitions, it is widely assumed that this switch is governed by epigenetic mechanisms that occur independently of any changes in DNA sequence. However, a direct genomic comparison between white and opaque cells has yet to be performed. Here, we present a whole-genome comparative analysis of C. albicans white and opaque cells. This analysis revealed rare genetic changes between cell states, none of which are linked to white-opaque switching. This result is consistent with epigenetic mechanisms controlling cell state differentiation in C. albicans and provides direct evidence against a role for genetic variation in mediating the switch.
Keywords: Candida albicans, epigenetic switch, comparative genomics
Introduction
The ability to generate and maintain diverse cell types is a central theme in biology. Eukaryotic cell fate is often regulated by epigenetic mechanisms that do not involve a change in the primary DNA sequence. Instead, heritable changes are defined by gene expression programs driven by transcription factor networks that act in combination with alterations in histone and DNA modifications (Christophersen and Helin 2010; Rhee and Ho 2017). Autoregulation of the transcription factors within these networks is a key feature of cell fate determination that enables stable propagation of cell states (Crews and Pearson 2009).
Although epigenetic mechanisms have been extensively studied in multicellular eukaryotes, single-celled eukaryotes can also undergo epigenetic transitions between heritable cell states. This is thought to occur in several Candida species that have the ability to reversibly transition between alternative ‘white’ and ‘opaque’ states (Slutsky et al. 1987; Pujol et al. 2004; Porman et al. 2011; Xie et al. 2012). The white-opaque switch has been most extensively studied in Candida albicans, a commensal fungus of the human gastrointestinal tract (GI) but also a prevalent opportunistic pathogen (Pappas et al. 2018). Here, formation of the opaque state is governed by a complex network of at least eight transcriptional regulators (Srikantha et al. 2000; Zordan et al. 2006, 2007; Wang et al. 2011; Hernday et al. 2013; Lohse et al. 2013; Hernday et al. 2016; Lohse et al. 2016). Switching between white and opaque states occurs stochastically and is also driven by multiple environmental cues including temperature, CO2 and N-acetyl glucosamine (Rikkerink et al. 1988; Ramírez-Zavala et al. 2008; Huang et al. 2009; Alby and Bennett 2009a, 2009b; Huang et al. 2010).
Candida albicans white and opaque states display distinct cellular morphologies (opaque cells are elongated relative to the more spherical white cells), and are named for their colony appearance as opaque colonies are darker and flatter than brighter, dome-shaped white colonies (Slutsky et al. 1987). In addition to morphological differences, white and opaque cells show stark differences in their mating ability, with opaque cells being able to undergo mating a million times more efficiently than white cells (Miller and Johnson 2002; Scaduto et al. 2017). White and opaque cells also differ in their metabolic capacity (Lan et al. 2002; Ene et al. 2016), drug susceptibility (Ene et al. 2016), interaction with immune cells (Sasse et al. 2013; Mallick et al. 2016), and adherence to host tissue (Kennedy et al. 1988). Accordingly, these cell types display differing host niche preferences, with white cells displaying increased relative fitness across multiple niches, whereas opaque cells appear better adapted for colonization of the skin (Kvaal et al. 1999; Pande et al. 2013; Noble et al. 2017; Takagi et al. 2019).
Given the importance of the white-opaque switch to C. albicans biology, its regulation has been a major research focus. Candida albicans is typically a heterozygous diploid species and initial studies indicated that a homozygous mating locus (MTLa/a or alpha/alpha) is a prerequisite for white-to-opaque switching (Miller and Johnson 2002). In MTL homozygous strains, white-to-opaque switching occurs every 104–105 generations under standard laboratory conditions (Rikkerink et al. 1988). More recently it was shown that MTL heterozygous (MTLa/alpha) strains can also undergo the white-opaque switch under select conditions (Xie et al. 2013; Sun et al. 2016). Switching is highly regulated at the transcriptional level via a network of interconnected transcriptional regulators including Wor1, Wor2, Wor3, Wor4, Ahr1, Ssn6, Efg1, Czf1 (Srikantha et al. 2000; Huang et al. 2006; Srikantha et al. 2006; Zordan et al. 2006; Vinces and Kumamoto 2007; Zordan et al. 2007; Lohse et al. 2013; Hernday et al. 2016; Lohse et al. 2016). Multiple positive feedback loops between these transcription factors contribute to the stable inheritance of cell states (Zordan et al. 2007; Hernday et al. 2013).
Currently, the white-opaque switch is presumed to be epigenetic, yet a genomic comparison of white and opaque cells has yet to be conducted, despite examples of genetically regulated cell state transitions in other microbial species (van der Woude 2011; Darmon and Leach 2014; Norman et al. 2015). This question is also relevant given the high levels of genomic plasticity observed in C. albicans strains (Forche et al. 2009, 2011; Bennett et al. 2014; Hirakawa et al. 2015; Todd et al. 2019). The recent discovery of a mutational transition in C. albicans driven by loss-of-function mutations in a known white-opaque regulator (Liang et al. 2019) also illustrates how genetic changes can generate distinct phenotypic states that could be mistaken for epigenetic switching.
Here we present the first comparative genomic analysis of white and opaque C. albicans isolates using deep Illumina whole-genome sequencing. We conducted a comprehensive analysis of copy number variants, structural variants (SVs), loss of heterozygosity events and point mutations. Limited genetic differences were identified between cell states from matched white/opaque cells, and these differences were not shared across lineages nor were they located in genes with known roles in white-opaque switching. These findings are consistent with the current paradigm that white-opaque switching is epigenetic and occurs without genomic alterations.
Materials and methods
Strain construction and selection of white and opaque colonies
Candida albicans strains were derived from isolate SC5314 either by targeted deletion of the MTLalpha locus generating strain A (MTLa/-), or via sorbose-mediated selection to generate strain B [RBY1118 (a/a) (Schaefer et al. 2007)]. SC5314 was transformed with plasmid pRB102 (Sherwood and Bennett 2008) to delete MTLalpha and resulting in integration of the SAT1 gene (SAT1R). Correct integration was checked by PCR using oligos 51–54 as previously described (Alby and Bennett 2009a). Colonies were grown in YPD (2% peptone, 1% yeast extract, 2% glucose, 25 μg/ml uridine) to allow excision of the SAT1 cassette and plated on YPD plates supplemented with nourseothricin (25 μg/ml) to identify SAT1S colonies. Excision of the SAT1 cassette was verified by PCR using oligos 51–54 (Alby and Bennett 2009a), giving rise to strain A.
Opaque cells of A and B isolates were obtained from glycerol stocks of pure opaque populations by streaking out for single colonies on SCD solid medium [0.7% yeast nitrogen base without amino acids, 2% glucose, amino acid mix (uracil, uridine, histidine, leucine, arginine)] and growing for six days at room temperature. For each lineage, a full opaque colony was re-streaked onto a fresh SCD plate and allowed to grow for six days, at which point colonies were inspected for switching to the white state. A full opaque and a full white colony (indicating that cells formed from a recent opaque-to-white switch) were harvested from this plate and grown overnight (∼18 h) in liquid SCD for genomic DNA extraction and microscopy. Only cultures displaying a minimum of >95% purity in cell identity were used for subsequent analysis.
Microscopy
Selected white and opaque colonies were removed with a toothpick from solid SCD media, mixed with sterile H2O and wet-mounted on glass slides. Cells were imaged using differential interference contrast (DIC) on a Zeiss Axio Observer Z1 inverted microscope.
Whole-genome sequencing
To extract genomic DNA, white and opaque cells were grown overnight in SCD liquid media at 25° and DNA was isolated from ∼109 cells using a Qiagen Genomic Buffer Set and a Qiagen Genomic-tip 100/G kit according to manufacturer’s instructions. Libraries were made using the Nextera XT DNA Library preparation kit protocol (Illumina) with an input of 2 ng/μL in 10 μL. Each isolate was sequenced on an Illumina HiSeq X generating 101 bp paired reads. The nuclear genome sequences and General Feature Files (GFF) for C. albicans SC5314 reference genome (version A22-s07-m01-r69) were obtained from the Candida Genome Database (www.candidagenome.org). Reads were aligned to the SC5314 reference genome (haplotype A chromosomes) using Burrows-Wheeler Aligner (BWA) v0.7.4-r385 mem (Li and Durbin 2009), and converted to sorted BAM format using Samtools v0.1.9 (r783) (Li et al. 2009). Briefly, reads were trimmed using trimmomatic 0.36 (with default parameters except for slidingwindow: 10:25 and minlen: 75) (Bolger et al. 2014) and Picard Tools (http://broadinstitute.github.io/picard/). AddOrReplaceReadGroups, MarkDuplicates, CreateSequenceDictionary and ReorderSam were used to preprocess the alignments. We used GATK4 (Brouard et al. 2019) RealignerTargetCreator and IndelRealigner for resolving misaligned reads close to indels on parental-progeny pairs of isolates to avoid discrepancies between isolates. This resulted in an average of 16.76 (SD ± 5.79) million paired reads and 166.7X (SD ± 57.5X) coverage per sample.
Variant calling and validation
The Genome Analysis Toolkit (GATK4) (McKenna et al. 2010; Brouard et al. 2019) was used to call both variant and reference bases from the alignments. GATK Haplotype Caller and Pilon (Walker et al. 2014) (with diploid genotyper ploidy setting) were run with both SNP and INDEL genotype likelihood models (GLM). We then merged and sorted all the calls from Haplotype Caller and ran VariantFiltration with the following filters QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < –12.5, ReadPosRankSum < −8. Next, we removed any base that had less than a minimum genotype quality (QUAL) of 50, or a minimum read depth (DP) of 20. Finally, we removed any positions that were called by both GLMs (i.e., incompatible indels and SNPs), any marked as “LowQual” by GATK, nested indels, or sites that did not include a PASS flag. Similar filtering was performed for Pilon calls, removing low-quality sites and setting a minimum read depth of 20. A minimum allelic frequency difference of 25% between compared genome pairs was used to establish mutations. All mutations identified between white and opaque strains were visually inspected using IGV (Robinson et al. 2011).
GATK4 outputs were used to calculate allele frequencies for each heterozygous position across all eight C. albicans chromosomes. Thus, all heterozygous positions identified in the reference strain (SC5314 A22) were examined across isolates and the percent of reads mapping to each chromosome homolog was determined for each heterozygous position. Allele frequencies for each chromosome homolog were plotted using GraphPad Prism 8. To identify large LOH events, we calculated the number of heterozygous positions per 1 kbp region and averaged across 10 kbp windows (Figure 2A, red lines). Average heterozygosity levels for each isolate were plotted using GraphPad Prism 8.
Figure 2.

Chromosome copy numbers and heterozygosity levels for white and opaque isolates. (A) Coverage was normalized to disomic levels and relative read depth was plotted across all eight Candida albicans chromosomes to determine their relative copy numbers, plotted in blue. The number of heterozygous positions was examined for each chromosome and average heterozygosity per 10 kbp window is plotted in red. Lines and tick marks denote chromosomes and centromere positions, respectively, on each chromosome. Shaded boxes indicate LOH regions unique to the B-1 and B-2 lineages. The parental strain SC5314 is included for reference. WH, white state; OP, opaque state. (B) Allele frequencies were determined for white/opaque cells and parental strain SC5314. Frequencies were compared across chromosomes as indicated for homolog A (blue) and homolog B (green) for each isolate. Note that allele frequencies have a median value of ∼50% for heterozygous disomic chromosomes, whereas median values are skewed toward 0% or 100% for chromosomes that are homozygous. Center lines represent median frequencies, upper and lower lines indicate 25th and 75th percentiles, shaded boxes indicate LOH regions unique to the B-1 and B-2 lineages.
Ploidy and copy number variation analysis
To examine copy number variation across the genome, the Illumina read alignment depth was calculated for 1 kbp windows across the genome using BEDTools 2.28 (Quinlan and Hall 2010) and SAMtools 1.3.1 (Li et al. 2009). Read depths were calculated as the number of bases aligned per window divided by the length of the window (1000 bp or size of coding regions) and normalized to the average read depth for each genome and by GC content using pybedtools 0.8.1 (Quinlan and Hall 2010; Dale et al. 2011). The normalized alignment depth for each 1 kbp was averaged across 10 kbp for plotting (GraphPad Prism 8) and analysis of large-scale CNV. Both 1 kbp windows and chromosomal elements were examined for small CNV. Chromosomal elements examined included 6213 coding regions (ORFs), 156 tRNAs, 75 snoRNAs, six rRNAs, five ncRNAs and five snRNAs, as defined by the Candida Genome Database for SC5314 A22. Regions were considered to be a site of CNV if coverage for a region on the opaque state genome was two standard deviations above or below the coverage determined for the corresponding region of the white state genome.
SV calling
SVs were called using Pindel 0.2.0 (Ye et al. 2009) with potential variants including inversions, deletions, insertions, duplications and replacements. Pindel was ran with default options setting and -M 10 as previously described (Ghoneim et al. 2014). All variants called were filtered based on allele frequency differences between the two cell types and those showing differences higher than 25% were visually inspected using IGV.
Data availability
Strains are available upon request. Data generated under this study can be found under NCBI BioProject PRJNA686122, including whole-genome assemblies and respective annotations for white and opaque isolates. Raw reads for the parental isolate SC5314 have been previously published under NCBI BioProject PRJNA193498 (Hirakawa et al. 2015). Supplementary material includes Supplementary Table S1, which contains additional details on CNV analysis (uploaded to figshare: https://doi.org/10.25387/g3.13501908).
Results and discussion
Sequencing of Candida albicans white and opaque isolates
We generated MTL hemizygous or MTL homozygous derivatives of C. albicans strain SC5314 (MTLa/alpha) by deletion of the MTLalpha locus (isolate A; MTLa/-) or via sorbose selection and outgrowth on YPD medium [isolate B; MTLa/a (Schaefer et al. 2007)]. The derived strains undergo stochastic switching between the white and opaque cell states. Opaque cells of each strain were inoculated on SCD medium and grown for six days at 22°. From this plate, a single opaque colony was restruck on SCD and again grown for six days at 22°. A subset of colonies presented in the white state (i.e., had switched to the white state from the opaque state) based on colony appearance and cellular morphology. Cells from opaque colonies were larger and more elongated whereas cells from white colonies were smaller and more spherical, as expected (Figure 1). Verified white and opaque colonies from each strain were cultured overnight (∼18 h) and genomic DNA isolated. Using this strategy, any mutations identified between white and opaque cells would have occurred either during the 12 days of growth on SCD or during liquid culturing prior to DNA isolation. In addition, we performed these experiments in duplicate, generating independent, paired white/opaque versions for isolate A (lineages A-1 and A-2) and isolate B (lineages B-1, B-2).
Figure 1.
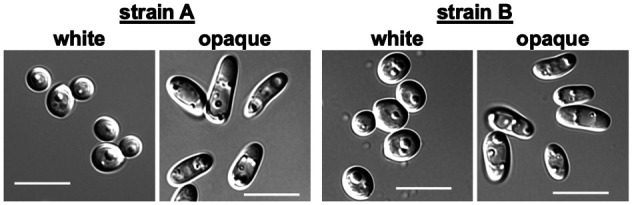
Cell morphologies of Candida albicans white and opaque cells. Cells were obtained from SC5314 derivatives that are MTLa/- (lineage A1) or MTLa/a (lineage B1). Scale bars, 10 μm.
DNA from white and opaque cells was sequenced on the Illumina HiSeq X platform using 101 bp paired end reads. Reads from each genome (white and opaque forms for A-1, A-2, B-1, and B-2) were aligned to the SC5314 reference genome (assembly 22) with an average of 98.29% (SD ± 0.52%) of paired reads successfully mapped across the genome reference, and an average of 99.87% (SD ± 0.017%) of the reference genome covered by reads. This resulted in average coverage levels of 166.7X (SD ± 57.5X) across the sequenced white and opaque cell genomes.
Karyotype and heterozygosity levels are similar between white and opaque cell genomes
As aneuploidy and large losses of heterozygosity (LOH) events can frequently arise in diploid C. albicans isolates following transformation (Bouchonville et al. 2009) or sorbose selection (Hickman et al. 2015), we first examined the sequenced genomes for evidence of such large-scale events. Mean coverage levels were calculated per 10 kbp regions across the eight chromosomes and normalized by the mean read depth for each genome (Figure 2a, blue lines). This analysis indicated that the evaluated genomes showed disomic levels across all eight chromosomes in each case.
LOH events involve the loss of genetic information from one homolog across a segmental region or even a whole chromosome. This can occur either by deletion/loss of a chromosomal region or via recombination between homologs (e.g., gene conversion or break-induced replication) and is detected by the loss of heterozygous positions across the impacted region. To identify LOH events, heterozygous positions were mapped using GATK and the number of heterozygous positions calculated for each genome using 1 kbp regions and averaged across 10 kbp windows (Figure 2A). This revealed homozygous regions across the right half of chromosomes (Chr) 3 and R in all eight analyzed genomes, regions that are also homozygous in the parental SC5314 lineage (Figure 2A) (Hirakawa et al. 2015). Additional LOH regions in the B1 and B2 lineages included the whole of Chr 5 as well as the left arm of Chr 2 (Figure 2A). LOH of Chr 5 is likely a result of using sorbose selection to generate the MTL homozygous isolate B, as growth on this medium frequently results in the loss of one homolog of Chr 5 (Kabir et al. 2005; Andaluz et al. 2007; Hickman et al. 2015). Importantly, however, genomes from white/opaque pairs displayed matching patterns of heterozygosity indicating that large LOH events did not accompany the white-opaque switch.
Mean allele frequencies for each chromosome were compared to those in the parental SC5314 genome, as these can reveal both LOH and changes in chromosome copy number. The mean frequencies were close to 50% for most chromosomes, consistent with these chromosomes being disomic and heterozygous in the evaluated genomes (Figure 2B). B-1 and B-2 lineages (both white and opaque cells) displayed mean allele frequencies close to 0% and 100% for Chr 2 and 5, respectively, consistent with large LOH events having occurred in the sorbose-selected parent of these lineages. Overall, however, no notable differences in allele frequencies were observed between genomes from matched white/opaque pairs, establishing similar heterozygosity levels between the two cell states.
Rare single nucleotide variants exist between matched white and opaque cells
To assess small-scale variants such as single nucleotide polymorphisms (SNPs) and indels (insertions/deletions) that may have occurred during the opaque-to-white transition, variants distinguishing white/opaque states were identified using GATK (McKenna et al. 2010) and Pilon (Walker et al. 2014), and visually confirmed using IGV. This revealed one mutation (1 SNP) between A-1 white/opaque cells and no mutations between A-2 white/opaque cells. For the B lineages, six mutations (one insertion and five SNPs) were identified between B-1 white/opaque cells and four mutations (one SNP and three insertions) were identified between B-2 white/opaque cells (Figure 3). None of these mutations were shared across multiple lineages. For B-1 and B-2 lineages, these mutations involved an equal number of LOH events and de novo base substitutions, the latter resulting in a gain of heterozygosity, or GOH, event. This is consistent with previous reports that LOH and GOH events often occur at similar frequencies during C. albicans microevolution (Ene et al. 2018). All mutations with the exception of two SNPs in the B-1 white/opaque comparison were located in intergenic regions. None of the identified indels therefore resulted in frameshifts. The two B-1 mutations located in coding regions were the result of short-tract LOH events that produced nonsynonymous mutations, thereby leading to changes in the encoded amino acid in only one chromosome homolog. One of the genes affected is an uncharacterized open reading frame (ORF, orf19.928), whereas the other is a putative NADH dehydrogenase based on sequence homology (orf19.5713, YMX6). None of the genes impacted by nonsynonymous mutations or located proximal to intergenic mutations (orf19.873 and orf19.2664, Figure 3) have any known connections to white-opaque switching and are not differentially expressed between the two cell states (Tuch et al. 2010), yet further investigation may be warranted to fully rule out a role for these genetic changes in switching. Similarly, a search for structural variants including indels, inversions, duplications and replacements using Pindel (Ye et al. 2009) did not identify any variants between matched white/opaque pairs, indicating that this type of structural events are not associated with white-opaque switching.
Figure 3.

Differences in DNA sequence identified between paired white and opaque cells of each lineage. Mutations were identified with either GATK or Pilon and were inspected in IGV. For mutations identified in close proximity to ORFs, the distance relative to the respective gene is specified. LOH, loss of heterozygosity; GOH, gain of heterozygosity; SNP, single nucleotide polymorphism; nonsyn, nonsynonymous mutation. Shaded rows indicate nonsynonymous mutations.
Copy number variation between white/opaque states
Copy number variation (CNV) has emerged as a powerful strategy for genetic variation and recent yeast studies have shown that amplification of particular gene regions can drive rapid adaptation to stress (Todd and Selmecki 2020) or nutrient limitation (Lauer et al. 2018). These CNVs are transient so that cells can revert after removal of the selective pressure (Todd and Selmecki 2020). This raises the possibility that a similar mechanism could enable the white-opaque switch. To look for CNV between genomes from white/opaque cells, we examined differences in coverage across both 1 kbp windows and chromosomal elements including gene coding regions and small RNAs (tRNAs, rRNAs, ncRNAs, snoRNA and snRNAs). We identified a small number of regions showing CNV between white and opaque cells from the same lineage, as well as CNV regions when comparing cells of each type from different lineages (Figure 4, A and C). Regions displaying elevated or reduced copy number were compared between the four lineages using two standard deviations as a cutoff. This comparison identified one 1 kbp region and two small RNAs (one tRNA and one snoRNA) as having differential coverage in white/opaque pairs in all four lineages (Figure 4, B and D). However, none of these CNV regions showed consistent directionality (elevated or decreased) with respect to coverage levels in white v. opaque cells (Supplementary Table S1). Additional CNV comparisons were carried out between genomes of the same cell type but different lineages from the same sequencing run (e.g., A-1 white vs B-1 white). This analysis identified 12 1 kbp regions and 46 chromosomal elements that were common across the four comparisons (Supplementary Table S1). In contrast to comparisons between cell states, this analysis revealed consistent CNV directionality and multiple lineage-specific differences, thus validating these CNV regions. Lineage-specific CNVs included those associated with sorbose selection, differences in auxotrophic makers (HIS1, URA3), differences in MTLa copy number (reflecting MTLa/- in the A-1 and A-2 lineages versus MTLa/a in the B-1 and B-2 lineages), as well as CNVs associated with repeat regions (Supplementary Table S1). Although further investigation of these CNV regions may be warranted, the limited number of regions identified as well as the lack of consistent directionality in coverage levels between white and opaque cell genomes do not point to a strong link between CNV and white-opaque switching.
Figure 4.

Copy number variation (CNV) between white and opaque cells. CNVs were identified using read coverage depth across 1 kbp windows (A) and different chromosomal elements (C) for the four A and B lineages (using two standard deviations above or below coverage levels as cutoffs for CNV). Comparisons were made between white/opaque states within a lineage and between A and B lineages for cells of the same type within the same sequencing run. Numbers indicate the total number of CNV regions identified in each comparison (above or below two standard deviations), lines indicate means and standard deviations. OP, opaque; WH, white. Venn diagrams indicate the number of shared CNVs identified between cell states for 1 kbp windows (B) and for chromosomal elements (D). Control comparisons between lineages as well as a full list of the common regions along with corresponding Candida albicans genome annotations are included in Supplementary Table S1.
Here, we provide a comparative genomic analysis of C. albicans white and opaque cells. The lack of consistent genetic changes between the two cell states (including changes in karyotype, copy number, structural variants or point mutations) is in line with the current epigenetic model for regulation of white-opaque switching. Although we cannot rule out a role for the genetic variants identified here in contributing to switching or to phenotypic differences between states, we consider this unlikely given that genetic changes were not shared by the independent lineages examined here, as well as the lack of a clear pattern for CNVs that present in all lineages.
Supplementary Material
Acknowledgments
The authors thank the Broad Institute Microbial 'Omics core for library construction and the Broad Genomics Platform for Illumina sequencing. We also thank the Computational Biology Core at Brown University for bioinformatic support.
Funding
This work was supported by NIH NIAID R21AI139592 and NIH NIGMS IDeA award (P20GM109035) to I.V.E., and by NIH NIAID R01 AI081704 and R01 AI141893 to R.J.B. C.A.C. was supported by NIH NIAID award U19AI110818 to the Broad Institute.
Conflicts of interest: None declared.
Literature cited
- Alby K, Bennett RJ.. 2009a. Stress-induced phenotypic switching in Candida albicans. Mol Biol Cell. 20:3178–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alby K, Bennett RJ.. 2009b. To switch or not to switch?: Phenotypic switching is sensitive to multiple inputs in a pathogenic fungus. Commun Integr Biol. 2:509–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andaluz E, Gomez-Raja J, Hermosa B, Ciudad T, Rustchenko E, et al. 2007. Loss and fragmentation of chromosome 5 are major events linked to the adaptation of RAD52-DeltaDelta strains of Candida albicans to sorbose. Fungal Genet Biol. 44:789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RJ, Forche A, Berman J.. 2014. Rapid mechanisms for generating genome diversity: whole ploidy shifts, aneuploidy, and loss of heterozygosity. Cold Spring Harb Perspect Med. 4:a019604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchonville K, Forche A, Tang KE, Selmecki A, Berman J.. 2009. Aneuploid chromosomes are highly unstable during DNA transformation of Candida albicans. Eukaryot Cell. 8:1554–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouard JS, Schenkel F, Marete A, Bissonnette N.. 2019. The GATK joint genotyping workflow is appropriate for calling variants in RNA-seq experiments. J Anim Sci Biotechnol. 10:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophersen NS, Helin K.. 2010. Epigenetic control of embryonic stem cell fate. J Exp Med. 207:2287–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews ST, Pearson JC.. 2009. Transcriptional autoregulation in development. Curr Biol. 19:R241–R246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale RK, Pedersen BS, Quinlan AR.. 2011. Pybedtools: a flexible python library for manipulating genomic datasets and annotations. Bioinformatics. 27:3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon E, Leach DR.. 2014. Bacterial genome instability. Microbiol Mol Biol Rev. 78:1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Farrer RA, Hirakawa MP, Agwamba K, Cuomo CA, et al. 2018. Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc Natl Acad Sci USA. 115:E8688-E8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Lohse MB, Vladu AV, Morschhauser J, Johnson AD, et al. 2016. Phenotypic profiling reveals that Candida albicans opaque cells represent a metabolically specialized cell state compared to default white cells. MBio. 7:e01269–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Abbey D, Pisithkul T, Weinzierl MA, Ringstrom T, et al. 2011. Stress alters rates and types of loss of heterozygosity in Candida albicans. mBio. 2: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Magee PT, Selmecki A, Berman J, May G.. 2009. Evolution in Candida albicans populations during a single passage through a mouse host. Genetics. 182:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneim DH, Myers JR, Tuttle E, Paciorkowski AR.. 2014. Comparison of insertion/deletion calling algorithms on human next-generation sequencing data. BMC Res Notes. 7:864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Lohse MB, Fordyce PM, Nobile CJ, DeRisi JL, et al. 2013. Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol Microbiol. 90:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Lohse MB, Nobile CJ, Noiman L, Laksana CN, et al. 2016. Ssn6 defines a new level of regulation of white-opaque switching in Candida albicans and is required for the stochasticity of the switch. MBio. 7:e01565–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MA, Paulson C, Dudley A, Berman J.. 2015. Parasexual ploidy reduction drives population heterogeneity through random and transient aneuploidy in Candida albicans. Genetics. 200:781–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa MP, Martinez DA, Sakthikumar S, Anderson MZ, Berlin A, et al. 2015. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 25:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Srikantha T, Sahni N, Yi S, Soll DR.. 2009. CO(2) regulates white-to-opaque switching in Candida albicans. Curr Biol. 19:330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Wang H, Chou S, Nie X, Chen J, et al. 2006. Bistable expression of WOR1, a master regulator of white-opaque switching in Candida albicans. Proc Natl Acad Sci USA. 103:12813–12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Yi S, Sahni N, Daniels KJ, Srikantha T, et al. 2010. N-acetylglucosamine induces white to opaque switching, a mating prerequisite in Candida albicans. PLoS Pathog. 6:e1000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir MA, Ahmad A, Greenberg JR, Wang YK, Rustchenko E.. 2005. Loss and gain of chromosome 5 controls growth of Candida albicans on sorbose due to dispersed redundant negative regulators. Proc Natl Acad Sci USA. 102:12147–12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Rogers AL, Hanselmen LR, Soll DR, Yancey RJ Jr.. 1988. Variation in adhesion and cell surface hydrophobicity in Candida albicans white and opaque phenotypes. Mycopathologia. 102:149–156. [DOI] [PubMed] [Google Scholar]
- Kvaal C, Lachke SA, Srikantha T, Daniels K, McCoy J, et al. 1999. Misexpression of the opaque-phase-specific gene PEP1 (SAP1) in the white phase of Candida albicans confers increased virulence in a mouse model of cutaneous infection. Infect Immun. 67:6652–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan CY, Newport G, Murillo LA, Jones T, Scherer S, et al. 2002. Metabolic specialization associated with phenotypic switching in Candida albicans. Proc Natl Acad Sci USA. 99:14907–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer S, Avecilla G, Spealman P, Sethia G, Brandt N, et al. 2018. Single-cell copy number variant detection reveals the dynamics and diversity of adaptation. PLoS Biol. 16:e3000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, 1000 Genome Project Data Processing Subgroup, et al. 2009. The sequence alignment/map format and samtools. Bioinformatics. 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SH, Anderson MZ, Hirakawa MP, Wang JM, Frazer C, et al. 2019. Hemizygosity enables a mutational transition governing fungal virulence and commensalism. Cell Host Microbe. 25:418–431.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MB, Ene IV, Craik VB, Hernday AD, Mancera E, et al. 2016. Systematic genetic screen for transcriptional regulators of the Candida albicans white-opaque switch. Genetics. 203:1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MB, Hernday AD, Fordyce PM, Noiman L, Sorrells TR, et al. 2013. Identification and characterization of a previously undescribed family of sequence-specific DNA-binding domains. Proc Natl Acad Sci USA. 110:7660–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick EM, Bergeron AC, Jones SK Jr., Newman ZR, Brothers KM, et al. 2016. Phenotypic plasticity regulates Candida albicans interactions and virulence in the vertebrate host. Front Microbiol. 7: 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. 2010. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MG, Johnson AD.. 2002. White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell. 110:293–302. [DOI] [PubMed] [Google Scholar]
- Noble SM, Gianetti BA, Witchley JN.. 2017. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol. 15:96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman TM, Lord ND, Paulsson J, Losick R.. 2015. Stochastic switching of cell fate in microbes. Annu Rev Microbiol. 69:381–403. [DOI] [PubMed] [Google Scholar]
- Pande K, Chen C, Noble SM.. 2013. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet. 45:1088–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, Kullberg BJ.. 2018. Invasive candidiasis. Nat Rev Dis Primers. 4:18026. [DOI] [PubMed] [Google Scholar]
- Porman AM, Alby K, Hirakawa MP, Bennett RJ.. 2011. Discovery of a phenotypic switch regulating sexual mating in the opportunistic fungal pathogen Candida tropicalis. Proc Natl Acad Sci USA. 108:21158–21163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol C, Daniels KJ, Lockhart SR, Srikantha T, Radke JB, et al. 2004. The closely related species Candida albicans and Candida dubliniensis can mate. Eukaryot Cell. 3:1015–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Zavala B, Reuß O, Park Y-N, Ohlsen K, Morschhäuser J.. 2008. Environmental induction of white-opaque switching in Candida albicans. PLoS Pathog. 4:e1000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee CK, Ho T.. 2017. Transcriptional regulation of the first cell fate decision. J Dev Biol Regen Med. 1:102. [PMC free article] [PubMed] [Google Scholar]
- Rikkerink EH, Magee BB, Magee PT.. 1988. Opaque-white phenotype transition: a programmed morphological transition in Candida albicans. J Bacteriol. 170:895–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, et al. 2011. Integrative genomics viewer. Nat Biotechnol. 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse C, Hasenberg M, Weyler M, Gunzer M, Morschhauser J.. 2013. White-opaque switching of Candida albicans allows immune evasion in an environment-dependent fashion. Eukaryot Cell. 12:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaduto CM, Kabrawala S, Thomson GJ, Scheving W, Ly A, et al. 2017. Epigenetic control of pheromone MAPK signaling determines sexual fecundity in Candida albicans. Proc Natl Acad Sci USA. 114:13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer D, Cote P, Whiteway M, Bennett RJ.. 2007. Barrier activity in Candida albicans mediates pheromone degradation and promotes mating. Eukaryot Cell. 6:907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RK, Bennett RJ.. 2008. Microtubule motor protein Kar3 is required for normal mitotic division and morphogenesis in Candida albicans. Eukaryot Cell. 7:1460–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky B, Staebell M, Anderson J, Risen L, Pfaller M, et al. 1987. "White-opaque transition": a second high-frequency switching system in Candida albicans. J Bacteriol. 169:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikantha T, Borneman AR, Daniels KJ, Pujol C, Wu W, et al. 2006. Tos9 regulates white-opaque switching in Candida albicans. Eukaryot Cell. 5:1674–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikantha T, Tsai LK, Daniels K, Soll DR.. 2000. Efg1 null mutants of Candida albicans switch but cannot express the complete phenotype of white-phase budding cells. J Bacteriol. 182:1580–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Gadoury C, Hirakawa MP, Bennett RJ, Harcus D, et al. 2016. Deletion of a YCI1 domain protein of Candida albicans allows homothallic mating in mtl heterozygous cells. mBio. 7:e00465–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi J, Singh-Babak SD, Lohse MB, Dalal CK, Johnson AD.. 2019. Candida albicans white and opaque cells exhibit distinct spectra of organ colonization in mouse models of infection. PLoS One. 14:e0218037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd RT, Selmecki A.. 2020. Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs. Elife. 9:e58349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd RT, Wikoff TD, Forche A, Selmecki A.. 2019. Genome plasticity in Candida albicans is driven by long repeat sequences. Elife. 8:e45954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, Mitrovich QM, Homann OR, Hernday AD, Monighetti CK, et al. 2010. The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PLoS Genet. 6:e1001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Woude MW.2011. Phase variation: How to create and coordinate population diversity. Curr Opin Microbiol. 14:205–211. [DOI] [PubMed] [Google Scholar]
- Vinces MD, Kumamoto CA.. 2007. The morphogenetic regulator Czf1p is a DNA-binding protein that regulates white opaque switching in Candida albicans. Microbiology (Reading). 153:2877–2884. [DOI] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song W, Huang G, Zhou Z, Ding Y, et al. 2011. Candida albicans Zcf37, a zinc finger protein, is required for stabilization of the white state. FEBS Lett. 585:797–802. [DOI] [PubMed] [Google Scholar]
- Xie J, Du H, Guan G, Tong Y, Kourkoumpetis TK, et al. 2012. N-acetylglucosamine induces white-to-opaque switching and mating in Candida tropicalis, providing new insights into adaptation and fungal sexual evolution. Eukaryot Cell. 11:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Tao L, Nobile CJ, Tong Y, Guan G, et al. 2013. White-opaque switching in natural MTLa/alpha isolates of Candida albicans: evolutionary implications for roles in host adaptation, pathogenesis, and sex. PLoS Biol. 11:e1001525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z.. 2009. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 25:2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Galgoczy DJ, Johnson AD.. 2006. Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc Natl Acad Sci USA. 103:12807–12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, Johnson AD.. 2007. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol. 5:e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Strains are available upon request. Data generated under this study can be found under NCBI BioProject PRJNA686122, including whole-genome assemblies and respective annotations for white and opaque isolates. Raw reads for the parental isolate SC5314 have been previously published under NCBI BioProject PRJNA193498 (Hirakawa et al. 2015). Supplementary material includes Supplementary Table S1, which contains additional details on CNV analysis (uploaded to figshare: https://doi.org/10.25387/g3.13501908).