

Abstract
Background
We investigatedthe association between time-averaged area under the curve (AAUC) of cytomegalovirus (CMV) viral load (VL) by day 100 and overall survival (OS) at 1-year after hematopoietic cell transplantation (HCT).
Methods
In a retrospective cohort study, including patients receiving HCT between June 2010 and December 2017 from Memorial Sloan Kettering Cancer Center, AAUC was calculated for patients with detected VL. Patients were categorized into non-controllers (Q4) and controllers (Q1–Q3) using the highest AAUC quartile as cutoff. Cox models were used to estimate the association between AAUC and OS. Patients with non-detected CMV VL were categorized into elite-controllers (recipient+ [R+] or R−/donor+ [D+]) and R−/D−.
Results
The study (N = 952) included 282 controllers, 93 non-controllers, 275 elite-controllers, and 302 R−/D−. OS was 80.1% and 58.1% for controllers and non-controllers, respectively. In multivariable models, non-controllers had worse OS versus controllers (adjusted hazard ratio [HR] = 2.65; 95% confidence interval [CI], 1.71–4.12). In landmark analyses, controllers had similar OS as elite-controllers (HR = 1.26; 95% CI, .83–1.91) or R−/D− (HR = 0.98; 95% CI, .64–1.5).
Conclusions
Non-controllers had worse OS 1-year post-HCT. Controllers had similar OS as elite-controllers or R−/D−. Future studies are needed to validate our AAUC cutoff across different cohorts and CMV management strategies.
Keywords: cytomegalovirus (CMV), hematopoietic cell transplantation (HCT), averaged area under the curve (AAUC), viral load (VL), end-organ disease (EOD), overall survival (OS), non-relapse mortality (NRM)
The highest quartile of the averaged area under the curve (AAUC) of cytomegalovirus viral load by day 100 post-HCT was independently associated with worse overall survival (OS) at 1-year post-HCT. OS was similar across the 3 lower AAUC quartiles.
(See the Editorial Commentary by Prono and Avery, on pages 563–4.)
Cytomegalovirus (CMV) seropositivity of the recipient (R) or donor (D) adversely impacts overall survival (OS) after hematopoietic cell transplantation (HCT) [1–3]. CMV viremia post-HCT is associated with decreased OS and increased non-relapse mortality (NRM) [4, 5]. Parameters of viral load (VL) kinetics such as the magnitude, duration, or slope of viral decay correlate with CMV end organ disease (EOD) and mortality post-HCT [4, 6–8]. Conversely, prompt resolution of CMV viremia with antiviral preemptive therapy (PET) is associated with improved survival [9]. Collective evidence from these and other mainly retrospective studies, supported the use of clinically significant CMV viremia (cs-CMV) as a surrogate end-point in recent clinical trials of CMV prevention [10–12]. In the phase 3 study of letermovir for CMV prevention, letermovir recipients had better survival at 24 weeks compared to placebo, providing the first evidence from a randomized controlled study that prevention of CMV viremia is associated with improved survival [11]. Interestingly, a post hoc survival analysis of this study showed a survival advantage specifically among letermovir recipients who developed cs-CMV [13]. While these results are somewhat contradictory to the current notion that CMV viremia is a predictor of mortality, it is possible that differences in VL kinetics among letermovir recipients may at least partially account for this finding.
The time-averaged area under the curve (AAUC) of CMV VL incorporates both the amplitude and the duration of viremia. High AAUC for double-stranded viruses was previously shown to independently correlate with increased overall mortality post-HCT [6, 14]. We investigated the association between the AAUC of CMV VL by day 100 (D100) and OS at 1-year post-HCT. Our objectives were to (1) establish an AAUC cutoff that correlates with CMV EOD, CMV-related mortality, and OS; (2) evaluate the impact of the AAUC cutoff on OS and NRM in multivariable models; and (3) compare OS and NRM between groups of patients with CMV viremia based on the selected AAUC cutoff and patients without CMV viremia.
METHODS
Study Cohort
The cohort consisted of consecutive adult recipients of first marrow or peripheral blood HCT at Memorial Sloan Kettering Cancer Center (MSKCC) between June 2010 and December 2017. Patients with multiple myeloma as the underlying disease were excluded. Data were extracted from patients’ electronic medical records and MSKCC research databases. Patients were followed for 1 year after HCT or death, whichever occurred earlier. The study was reviewed and approved by the MSKCC Institutional Review Board (No. 16–920).
Institutional Standard of Care
Patients with acute leukemia in first complete remission and patients with myelodysplastic syndrome preferentially received CD34+ selected T-cell depleted (TCD) HCT if able to receive myeloablative cytoreductive conditioning and had a ≥8/10 HLA matched donor. Patients with leukemia or myelodysplastic syndrome not eligible for TCD and patients with lymphoma, chronic leukemia, myeloproliferative syndromes, or nonmalignant indications (eg, aplastic anemia), received unmodified HCT with reduced intensity conditioning [15, 16]. Recipients of conventional allografts received a calcineurin inhibitor (in combination with sirolimus in some cases) and methotrexate as graft versus host disease (GVHD) prophylaxis. Recipients of TCD allografts did not receive additional pharmacologic GVHD prophylaxis. Acute GVHD was scored by standard criteria [17].
All patients received acyclovir prophylaxis starting on admission for HCT and continued for at least 12 months. HCT recipients who were CMV seropositive (R+) or seronegative with a seropositive donor (R−/D+) were routinely monitored at least weekly by CMV quantitative polymerase chain reaction (qPCR) starting on D14 post-HCT. Per institutional guidelines the initiation of PET was indicated when ≥2 consecutive PCR showed >500 copies/mL in whole blood or >300 IU/mL in plasma for patients with low risk for CMV (unmodified HCT from HLA matched donor) and ≥2 consecutive PCR at any value for patients at high risk for CMV (TCD or HLA mismatched donor HCT) [18]. PET was initiated with induction valganciclovir or ganciclovir (vGCV) (oral valganciclovir 900mg every [q] 12 hours [h] or intravenous ganciclovir 5 mg/kg q12h) or intravenous foscarnet (90 mg/kg q12h) when vGCV was contraindicated (mainly due to cytopenia). Induction was continued for 10–14 days or until CMV VL became nonquantifiable for 2 consecutive measurements (whichever longer). Maintenance doses of the same agent (valganciclovir 900 mg q24h, ganciclovir 5 mg/kg q24h, or foscarnet 90 mg/kg q24h) were typically continued for 4–6 weeks based on patient’s CMV risk. Antibacterial and antifungal prophylaxis was administered as previously described [19].
Laboratory Methods
CMV IgG levels were determined using an automated semiquantitative enzyme-linked immunosorbent assay (ELISA; VIDAS; Biomerieux). qPCR for CMV was performed by the Clinical Microbiology Laboratory at MSKCC using Roche analyte specific reagent (Roche Diagnostics) in whole-blood samples before March 2013, and Cobas Ampliprep/Cobas Taqman CMV qPCR in plasma (Roche Molecular Diagnostics) from March 2013 onward. The lower limit of quantification and linear range were ≥500 to 1.0 × 106 copies/mL for whole blood and ≥137 to 9.1 × 106 IU/mL for plasma [20]. For detectable qPCR results in the unquantifiable range (<500 copies/mL for whole blood and <137 IU/mL for plasma), we assumed worst-case scenario and calculated as 500 copies/mL or 137 IU/mL, respectively.
Time Averaged Area Under the Curve Calculation
AAUC was calculated as the sum of the areas of trapezoids formed by plotting the log10 VL over time divided by the number of days from first test day to last test day until D100 or death, whichever occurred earlier [21]. We calculated AAUC for each patient using all available VL measurements by D100 post-HCT. For each viremia episode, AAUC was calculated starting from the last undetectable VL to the last detectable VL. The AAUC between 2 non-detected measurements was 0. In case of missing values, time-weighted average of the measurements before and after the missing value was used to calculate the trapezoid.
Definition of Study Groups
We defined 4 study groups. Patients with CMV viremia by D100 were categorized into 2 mutually exclusive groups using the highest AAUC quartile (Q4) as cutoff, to controllers (Q1–Q3) and non-controllers (Q4). Patients without CMV viremia by D100 were categorized into 2 mutually exclusive groups based on CMV risk, as elite-controllers (R+ or R−/D+) and R−/D−.
Study End Points
The primary end points were OS and NRM at 1-year post-HCT. Secondary end points included: the incidence of CMV EOD by 1-year post-HCT, late CMV, PET initiation by D100, and CD4+ T-cell counts at D100.
Late CMV was defined as a new cs-CMV episode, separated from the previous episode by ≥2 weeks of undetectable CMV PCR and occurring from D100 to D180. CMV EOD was defined according to previously describe standard criteria [22]. The cause of death was scored as previously described [23]. For patients with relapse, death was attributed to relapse. For patients with GVHD and no relapse, death was attributed to GVHD. In patients with GVHD, if a concomitant infection contributed to death, the death was attributed to GVHD plus infection. CMV was considered the cause of death when death was directly attributed to CMV.
Statistical Methods
Descriptive statistics were used to summarize demographics, clinical characteristics, and the cause of death. We used χ 2 tests and Mann-Whitney rank-sum tests to compare categorical and continuous variables, respectively.
Patients with AAUC > 0 were divided into 4 quartiles based on the CMV AAUC. We generated Kaplan-Meier curves of OS for each AAUC quartile and calculated pairwise P values using logrank tests with Benjamini-Hochberg adjustment for each pair of quartiles separately. Using the fourth AAUC quartile (Q4) as a cutoff to define controllers and non-controllers, we generated Kaplan-Meier curves and cumulative incidence curves for OS and NRM, respectively, and calculated P values using logrank tests and Gray's tests, respectively. For NRM, relapse was considered as competing risk. We performed univariable and multivariable logistic regression analyses to evaluate risk factors associated with non-controller status.
Next, the AAUC cutoff was evaluated in univariable and multivariable models as a predictor of OS using Cox proportional hazards regression models and of NRM using Fine and Gray proportional subdistribution hazards regression models. Variables with P < .3 in the univariable models entered the multivariable models. Forward selection was used to keep variables with P < .1 in the final models. As a variable of major interest, the AAUC cutoff was kept in the multivariable models regardless of significance. Factors entered to the model included: age, sex, stem cell source, HLA match, underlying disease, conditioning intensity, GVHD, recipient and donor CMV serostatus, study period, and AAUC cutoff (controllers as reference). We performed subgroup analysis for TCD and unmodified HCT to account for inherent differences in CMV VL kinetics. To compare OS between the 4 study groups, we used landmark analyses at D100 to account for immortal time bias [24]. Statistical analyses were performed with R version 4.0.3 (R foundation for Statistical Computing; https://www.rproject.org/).
RESULTS
Of 956 patients who met inclusion criteria, 3 R+/D+ patients and 1 R−/D+ patient did not have any CMV VL assessment (due to early demise or loss to follow-up) and were excluded from analyses.
The cohort consisted of 952 patients, including 282 controllers, 93 non-controllers, 275 elite-controllers and 302 R−/D−. Baseline characteristics are summarized in Table 1.
Table 1.
Baseline Characteristics for the Entire Cohort and for Each Study Group
| Characteristics | Overall (N = 952) | CMV Controllers (n = 282) | CMV Non-controllers (n = 93) | Elite-controllers (n = 275) | R−D− (n = 302) | P Value |
|---|---|---|---|---|---|---|
| Demographic characteristics | ||||||
| Age, y, median (IQR) | 57 (47–65) | 58 (48–66) | 58 (48–66) | 56 (45–64) | 57 (47–65) | .191 |
| Sex | .006 | |||||
| Female | 388 (40.8) | 118 (41.8) | 48 (51.6) | 121 (44.0) | 101 (33.4) | |
| Male | 564 (59.2) | 164 (58.2) | 45 (48.4) | 154 (56.0) | 201 (66.6) | |
| Transplant characteristics | ||||||
| CMV R serostatus | <.001 | |||||
| R− | 443 (46.5) | 13 (4.6) | 2 (2.2) | 126 (45.8) | 302 (100.0) | |
| R+ | 509 (53.5) | 269 (95.4) | 91 (97.8) | 149 (54.2) | 0 (0.0) | |
| CMV D serostatus | <.001 | |||||
| D− | 511 (53.7) | 92 (32.6) | 45 (48.4) | 72 (26.2) | 302 (100.0) | |
| D+ | 441 (46.3) | 190 (67.4) | 48 (51.6) | 203 (73.8) | 0 (0.0) | |
| Underlying disease | .003 | |||||
| Leukemia | 523 (54.9) | 161 (57.1) | 61 (65.6) | 157 (57.1) | 144 (47.7) | |
| Lymphoma | 197 (20.7) | 52 (18.4) | 7 (7.5) | 64 (23.3) | 74 (24.5) | |
| MDS | 172 (18.1) | 45 (16.0) | 19 (20.4) | 43 (15.6) | 65 (21.5) | |
| Othera | 60 (6.3) | 24 (8.5) | 6 (6.5) | 11 (4.0) | 19 (6.3) | |
| Stem cell source | .219 | |||||
| Bone marrow | 106 (11.1) | 39 (13.8) | 7 (7.5) | 32 (11.6) | 28 (9.3) | |
| Peripheral blood | 846 (88.9) | 243 (86.2) | 86 (92.5) | 243 (88.4) | 274 (90.7) | |
| Donor HLA matching | .393 | |||||
| Matched related | 318 (33.4) | 88 (31.2) | 32 (34.4) | 95 (34.5) | 103 (34.1) | |
| Matched unrelated | 476 (50.0) | 151 (53.5) | 39 (41.9) | 139 (50.5) | 147 (48.7) | |
| Mismatchedb | 158 (16.6) | 43 (15.2) | 22 (23.7) | 41 (14.9) | 52 (17.2) | |
| Graft manipulation | <.001 | |||||
| Unmodified | 557 (58.5) | 173 (61.3) | 29 (31.2) | 181 (65.8) | 174 (57.6) | |
| CD34+ selection | 395 (41.5) | 109 (38.7) | 64 (68.8) | 94 (34.2) | 128 (42.4) | |
| Conditioning intensity | .002 | |||||
| Myeloablative | 582 (61.1) | 169 (59.9) | 75 (80.6) | 151 (54.9) | 187 (61.9) | |
| Reduced | 264 (27.7) | 80 (28.4) | 16 (17.2) | 89 (32.4) | 79 (26.2) | |
| Nonmyeloablative | 106 (11.1) | 33 (11.7) | 2 (2.2) | 35 (12.7) | 36 (11.9) | |
| Acute ≥ grade 2 GVHD | .949 | |||||
| No | 552 (58.0) | 166 (58.9) | 53 (57.0) | 156 (56.7) | 177 (58.6) | |
| Yes | 400 (42.0) | 116 (41.1) | 40 (43.0) | 119 (43.3) | 125 (41.4) |
Data are No. (%) except where indicated. CMV control among patients with CMV viremia by day 100 was defined by AAUC quartiles. CMV controllers comprised patients with AAUC in the lower 3 quartiles. CMV non-controllers comprised patients with AAUC in the fourth quartile. Elite controllers comprised of R+, or R−/D+ without CMV viremia by day 100 post-HCT. P values were calculated using χ 2 tests.
Abbreviations: AAUC, averaged area under the curve of CMV viral load; CMV, cytomegalovirus; D, donor CMV serostatus; GVHD, graft versus host disease; HCT, hematopoietic cell transplant; HLA, human leucocyte antigens; IQR, interquartile range; MDS, myelodysplastic syndrome; R, recipient CMV serostatus.
aOther included myeloproliferative disorders in 41 patients, aplastic anemia in 16 patients, and nonmalignant hematologic disorders in 3 patients.
bMismatched included unrelated nonidentical donors in 110 patients, related nonidentical donors in 5 patients, and related haploidentical donors in 43 patients.
Overall, the median age was 57 years (interquartile range [IQR], 47–65). Eight hundred forty-nine (89%) patients received peripheral blood and 107 (11%) received bone marrow from matched related (n = 320, 33%), matched unrelated (n = 478, 50%), or mismatched (n = 158, 17%) donors, for acute leukemia (n = 525, 55%), myelodysplastic syndrome (n = 173, 18%), lymphoma (n = 198, 21%), or other hematologic diseases (n = 60, 6%). Three hundred ninety-eight patients (42%) received a CD34+ selected TCD allograft. Five hundred twelve (54%) patients were CMV recipient seropositive (R+) and 445 (47%) had a CMV seropositive donor (D+). The distribution of R/D pairs is provided in Supplementary Table 1.
Defining a Clinically Relevant AAUC Cutoff
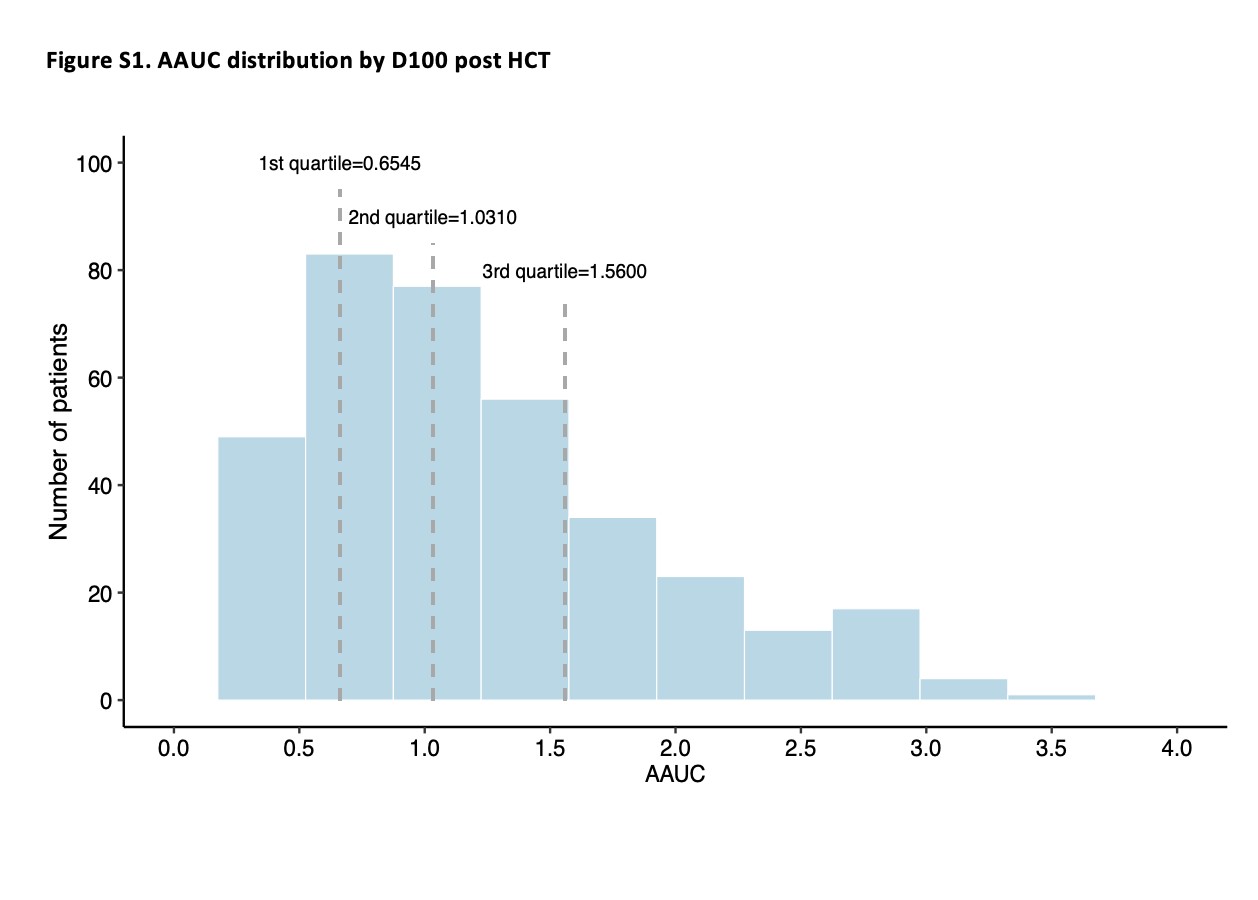
For the 375 patients who had CMV viremia by D100, the median AAUC was 1.03 (IQR, 0.65–1.56). Supplementary Figure 1 shows the distribution of AAUC in patients with CMV viremia.
We categorized patients with AAUC > 0 into 4 AAUC quartiles (Q1–Q4) and generated Kaplan-Meier survival curves for each quartile. The OS for Q4 (AAUC > 1.56) was 57.4% compared with 79.8% for Q1 (P = .003), 85.1% for Q2 (P = .0001), and 76.3% for Q3 (P = .03). In contrast, OS was similar across the 3 lowerquartiles (P = nonsignificant; Figure 1).
Figure 1.

Overall survival at 1 year by AAUC quartile. AAUC was calculated for all patients with CMV viremia by day 100 post-HCT (n = 375). Patients were categorized by quartiles based on AAUC values. Kaplan-Meier curves of overall 1-year survival were generated and P values calculated using log rank test with Benjamini-Hochberg adjustment for each pair of AAUC quartiles separately. Abbreviations: AAUC, averaged area under the curve of CMV viral load; CI, confidence interval; CMV, cytomegalovirus; HCT, hematopoietic cell transplant; HR, hazard ratio.
We defined patients in Q1–Q3 as controllers and patients in Q4 as non-controllers. The median AAUC for controllers and non-controllers were 0.84 (IQR, 0.55–1.15) and 2.12 (IQR, 1.78–2.6), respectively (P < .0001).
Next, we evaluated the AAUC cutoff as a predictor of mortality at 1 year post-HCT. Non-controllers had lower OS compared with controllers (58.1% and 80.1%, respectively, P < .0001; Figure 2A). Similarly, NRM was significantly higher for non-controllers compared with controllers (33.6% and 10.2%, respectively, P < .0001; Figure 2B).
Figure 2.

Comparison of overall survival and non-relapse mortality amongst all patients with CMV viremia by day 100 post-HCT (n = 375) between CMV controllers and non-controllers. A, Kaplan-Meier curves for overall survival at 1 year by CMV controller status. B, Cumulative incidence curves for non-relapse mortality at 1 year by CMV controller status. Abbreviations: CI, confidence interval; CMV, cytomegalovirus; HCT, hematopoietic cell transplant; HR, hazard ratio.
In multivariable analyses predictors for non-controller status were HLA mismatch (adjusted odds ratio [aOR], = 2.18; 95% confidence interval [CI], 1.05–4.53; P = .037) and TCD (aOR = 3.62; 95% CI, 1.74–7.52; P = .001), while CMV D+ was protective (aOR = 0.45; 95 % CI, .27–.75; P = .002; Supplementary Table 2).
Multivariable Analyses for OS and NRM among Patients with CMV Viremia
Using univariable and multivariable models, we evaluated the association of the AAUC cutoff with OS and NRM among the 375 patients with CMV viremia by D100 post-HCT (Supplementary Table 3 and Table 2). Non-controllers had worse OS compared to controllers after adjusting for covariates (adjusted hazard ratio [aHR] = 2.65; 95% CI, 1.71–4.12; P < .0001). Other factors associated with worse OS were acute GVHD (aHR = 1.96; 95% CI, 1.28–3; P = .002) and CMV D+ (aHR = 1.67; 95% CI, 1.07–2.62; P = .03). Underlying disease other than acute leukemia, myelodysplastic syndrome, or lymphoma was associated with better OS (aHR = 0.12; 95% CI, .02–.83; P = .03; Table 2).
Table 2.
Multivariable Analyses for Overall Survival and Non-relapse Mortality for Patients with CMV Viremia by Day 100 (N = 375)
| Overall survivala | Non-relapse mortalityb | |||||
|---|---|---|---|---|---|---|
| Characteristics | aHR | 95% CI | P value | aHR | 95% CI | P value |
| Age, y | ||||||
| 18–39 | ||||||
| 40–64 | 0.74 | (.37, 1.48) | .39 | .67 | (.26, 1.70) | .40 |
| 64+ | 1.93 | (.99, 3.75) | .05 | 2.24 | (.92, 5.45) | .07 |
| Underlying disease | ||||||
| Leukemia | ||||||
| Lymphoma | 0.76 | (.39, 1.47) | .41 | |||
| MDS | 0.87 | (.52, 1.46) | .60 | |||
| Other | 0.12 | (.02, .83) | .03 | |||
| Acute ≥ grade 2 GvHD | ||||||
| No | ||||||
| Yes | 1.96 | (1.28, 3) | .002 | 3.34 | (1.83, 6.08) | < .0001 |
| CMV D serostatus | ||||||
| D– | ||||||
| D+ | 1.67 | (1.07, 2.62) | .03 | 2.75 | (1.45, 5.22) | .002 |
| Study period | ||||||
| Before 2013 | ||||||
| After 2013 | 0.66 | (.42, 1.04) | .07 | 0.49 | (.27, .89) | .02 |
| CMV control status | ||||||
| Controllers | ||||||
| Non-controllers | 2.65 | (1.71, 4.12) | < .0001 | 5.02 | (2.82, 8.92) | < .0001 |
Abbreviations: CI, confidence interval; CMV, cytomegalovirus; GVHD, graft versus hostdisease; HR, hazard ratio; MDS, myelodysplastic syndrome.
aCox proportional hazards regression models were used for overall survival.
bFine and Gray proportional subdistribution hazards regression models were used for non-relapse mortality. Relapse was considered as competing risk event.
Similarly, non-controllers had higher NRM after adjusting for covariates (aHR = 5.02; 95% CI, 2.82–8.92; P < .0001). Other factors associated with higher NRM were acute GVHD (aHR = 3.34; 95% CI, 1.83–6.08; P < .0001) and CMV D+ (aHR = 2.75; 95% CI, 1.45–5.22; P = .002). HCT performed in 2013 or later was associated with less NRM compared with HCT performed prior to 2013 (HR = 0.49; 95% CI, .27–.89; P = .02; Table 2).
When stratifying patients with CMV viremia by graft manipulation, non-controllers had worse OS compared with controllers in unmodified and TCD HCT recipients (Supplementary Figures 2).
CMV Outcomes
When comparing VL kinetics between controllers and non-controllers, non-controllers had higher maximal VL (9056 vs 948.5 IU/mL; P < .0001) and longer duration of CMV viremia (42 vs 15 days; P < .0001; Figure 3A).
Figure 3.

Comparison of CMV kinetics and outcomes between CMV controllers (n = 282) and non-controllers (n = 93). CMV AAUC, maximum CMV VL, duration of CMV viremia, PET initiation by day 100, late CMV infection (occurring > day 100–180), CMV end-organ disease, and CMV-related mortality by 1-year post-HCT were compared between the 2 groups. All patients with CMV viremia by day 100 post-HCT were included (n = 375). A, Comparison of CMV viral kinetics by day 100. Horizontal lines represent median, boxes interquartile ranges, and whiskers range. P values were calculated with the Mann-Whitney U tests. B, Number (%) of CMV-related outcomes in CMV controllers and non-controllers. C, Unadjusted ORs (95% CI) for CMV outcomes in non-controllers (CMV controllers is the reference group). Abbreviations: AAUC, averaged area under the curve of CMV viral load; CI, confidence interval; CMV, cytomegalovirus; EOD, end-organ disease; HCT, hematopoietic cell transplant; OR, odds ratio; PET, preemptive therapy; VL, viral load.
Compared with controllers, more non-controllers received PET (98.9% vs 70.6%; P < .0001), had late CMV (60.2% vs 18.8%; P < .0001), had CMV EOD (25.8% vs 6.4%; P < .0001), and died of CMV (8.6% vs 0.7%; P < .0001; Figure 3B and 3C). By 1 year post-HCT, 10 patients died of CMV; 8/10 patients (80%) were non-controllers.
Comparison of Survival Between Controllers, Non-controllers, Elite-Controllers, and R−/D−
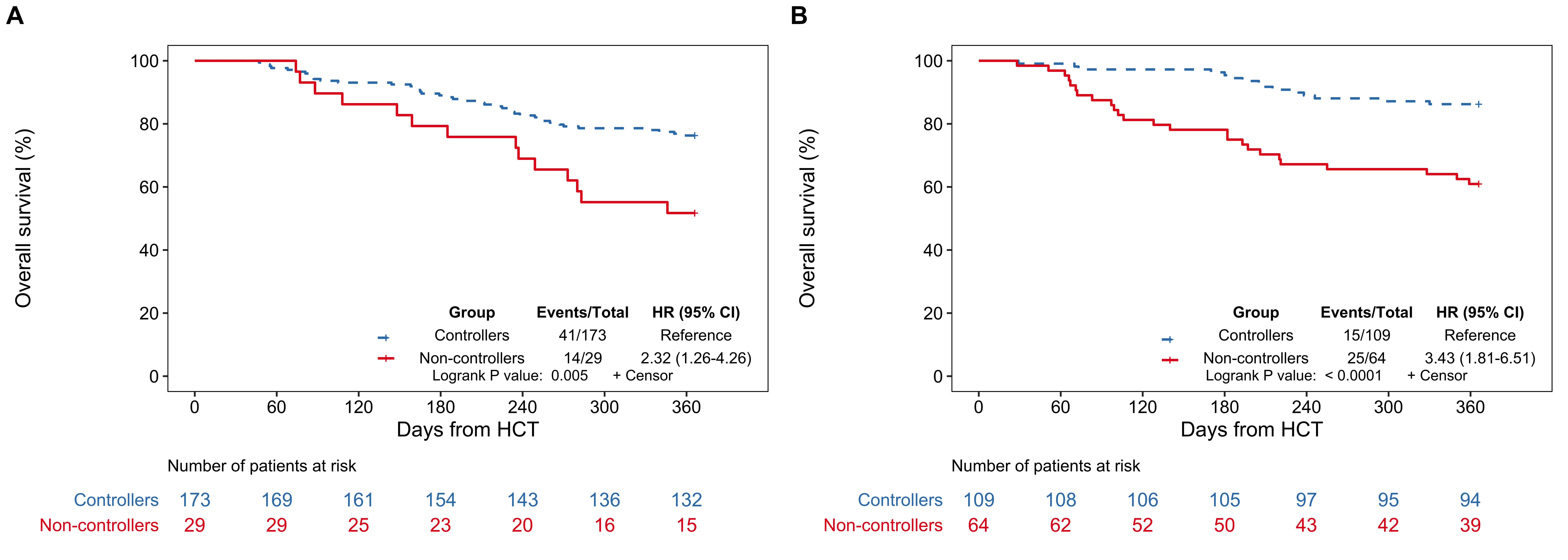
We compared the outcomes of controllers and non-controllers with patients who did not have CMV viremia, including elite-controllers and R−/D− patients. To account for immortal time bias (death is a competing risk for CMV infection) we performed landmark analyses including only patients alive at D100 for OS and NRM at 1 year. OS and NRM were similar for controllers, elite-controllers, and R−/D−, but significantly worse for non-controllers compared to each of the other groups (Figure 4).
Figure 4.

Landmark analyses for overall survival and non-relapse mortality across the 4 groups including patients alive by day 100 (n = 883). One-year overall survival and non-relapse mortality were compared between CMV non-controllers, controllers, elite-controllers, and R−/D−. P values for pairwise comparison between groups were calculated using logrank tests and Gray's tests with Benjamini-Hochberg adjustment for overall survival and non-relapse mortality, respectively. A, Kaplan-Meier curves for overall survival for the 4 groups. B, Cumulative incidence curves for non-relapse mortality for the 4 groups. Abbreviations: CI, confidence interval; CMV, cytomegalovirus; D, donor; HCT, hematopoietic cell transplant; HR, hazard ratio; R, recipient.
Causes of Death
By 1 year post-HCT, 233 patients died. The most common cause of death was relapse (n = 96, 41%), followed by infection (n = 44, 19%), GVHD (n = 45, 19%) including GVHD with infection contributing to death (n = 18, 8%), and other causes (n = 44, 19%). Table Supplementary 3 summarizes the cause of death across the 4 groups. Relapse was the most common cause of death in all study groups. Among non-controllers a higher proportion of patients died from infection other than CMV (13%) compared to controllers (3.5%, P = .0008), elite-controllers (6%, P = .026), and R−/D− (5%, P = .005).
CD4+ T-Cell Counts
We compared CD4+ counts at D100 across the study groups (Figure 5). To account for the known major differences in post-HCT T-cell recovery between unmodified and TCD, we evaluated CD4+ at D100 separately for these 2 groups. CD4+ values between D80 and D150 were available for 699 patients. Overall, the median CD4+ count was significantly lower for TCD compared with unmodified HCT (63cells/mcL vs 183 cells/mcL, respectively, P < .0001). In unmodified HCT recipients, the median CD4+ count was higher for controllers (202.5 cells/mcL) compared to non-controllers (67 cells/mcL, P = .0019) and similar to elite-controllers (172 cells/mcL, P = nonsignificant) and R−/D− (185 cells/mcL, P = nonsignificant; Figure 5A). In TCD recipients, the median CD4+ count was higher for CMV controllers (143 cells/mcL) compared to all other groups (CMV non-controllers 17 cells/mcL, P < .0001; elite-controllers 40 cells/mcL P < .0001; R−/D− 55 cells/mcL, P < .0001; Figure 5B).
Figure 5.

Comparison of CD4+ counts across the 4 groups. CD4+ counts at day 100 were compared between CMV controllers and each of the 3 remaining study groups for unmodified and TCD HCT recipients separately. CD4+ results between day 80 and day 150 were included in the analyses. If > 1 result was available for the same patient within this time frame, only the CD4 number obtained closest to day 100 was included. Horizontal lines represent medians, boxes interquartile ranges, and whiskers ranges. P values were calculated using Mann-Whitney test. A, Unmodified HCT (n = 342). B, TCD HCT (n = 357). Abbreviations: CMV, cytomegalovirus; D, donor; HCT, hematopoietic cell transplant; R, recipient; TCD, T-cell depleted.
DISCUSSION
We examined the utility of the AAUC of CMV VL by D100 as a predictor of mortality at 1 year after HCT. Based on AAUC quartiles we categorized patients into controllers (Q1–Q3) and non-controllers (Q4) and showed that non-controllers had worst survival compared with controllers. Classification as non-controller was associated with CMV EOD and CMV-related mortality further supporting our selection of the Q4 cutoff as clinically relevant. After adjusting for covariates, classification as non-controller was associated with 2.65 times higher risk of death and 5 times higher risk of NRM compared with controllers. These findings add to the increasing strength of evidence that VL could serve as a surrogate for clinical outcomes and that VL suppression is associated with improved outcomes [8].
Prior studies have evaluated the correlation of AAUC for CMV and other viruses with various outcomes after transplantation [6, 14, 25–28]. CMV AAUC correlated with CMV disease and mortality in solid organ transplant and HCT recipients [6, 28]. Hill et al reported that AAUC was a predictor of early and late mortality following HCT after adjusting for immune reconstitution and GVHD [27]. In contrast, Giménez et al found no association between CMV AAUC and post-HCT mortality [29]. Differences in AAUC calculation, sample size, and transplant setting may at least partially explain differences in results. Our data support the utility of AAUC as a predictor for post-HCT outcomes.
Survival disparity between R−/D− and R+ or D+ patients has been documented in cohort and registry studies [1–3]. After establishing that CMV controllers had better OS than non-controllers, we sought to compare the OS of controllers with elite-controllers (R+ or R−/D+ with no CMV viremia) and R−/D−. We found similar OS across the 3 groups. Hence, in CMV controllers, CMV viremia did not adversely impact survival.
Previous studies demonstrated a dose-response relationship between CMV viral burden measures such as maximal VL, mean VL, or duration of viremia and OS. Maximal CMV VL at any value was associated with worse survival compared with no viremia [4]. While some studies have shown that CMV viremia correlated with lower rates of relapse in HCT recipients with acute myeloid leukemia [30], these findings were not supported in registry studies [5]. In our cohort, controllers had similar NRM compared with patients without CMV viremia including R−/D−. While small numbers precluded any formal comparison of cause of death, it is notable that non-controllers had more deaths due to infection compared with the other groups; CMV and non-CMV infections accounted for almost one-fifth of deaths among non-controllers compared with 4%–6% in other groups. CMV is associated with bacterial and fungal infections in HCT recipients [31, 32]. Moreover, CMV non-controllers are likely to receive more myelosuppressive antiviral therapy, which could contribute to infection risk and associated mortality.
In prior studies, early T-cell function recovery predicted improved survival after TCD and the rate of T-cell recovery correlated with protection from opportunistic infections [33, 34]. We next compared immune reconstitution at D100 between the study groups. We show that CMV controllers had significantly higher CD4+ counts at D100 compared with non-controllers. Interestingly, in TCD recipients, CMV controllers had higher CD4+ counts compared with recipient with no CMV (elite controllers and R−/D−) suggesting better immune recovery possibly driven by CMV.
Our results are consistent with prior reports that CMV viremia enhances the tempo of T-cell recovery and the breadth of T-cell repertoire. In recipients of alemtuzumab-based, in vivo TCD HCT after reduced intensity conditioning, CMV viremia promoted T-cell immunity and influenced chimerism status [35]. In CMV seropositive patients with no GVHD, CMV-specific T cells triggered alloimmune responses promoting conversion to complete donor chimerism [36]. In unmodified HCT, CMV viremia by D60 promoted CD8+ T-cell subset recovery including higher expansion of CD8+ effector memory cells [37].
In our cohort, CMV non-controllers had lower D100 CD4+ counts, more CMV EOD, lower OS, and more deaths due to infections. Recently, immune monitoring assays have been evaluated as predictors of self-limiting viremia after HCT [38]. AAUC may be used as an additional tool to assess patients’ immune status and risk for complications late after HCT.
Our study has limitations inherent to the observational, retrospective design. Our cutoff for CMV control classification was chosen a priori based on quartile analysis and as such may inaccurately define the exact clinically relevant cutoff. Different cutoffs may apply in other centers based on patient and HCT types and laboratory methods. Multicenter studies with cross-validation are required to identify the optimal AAUC cutoff that could serve as a marker for CMV control and predictor of post-HCT outcomes.
In our cohort, 42% of patients received CD34+ selected TCD allografts. We have previously reported earlier CMV reactivation, higher VL, and longer viremia duration in TCD compared with unmodified HCT [39]. In addition, our large proportion of TCD probably affected the rates and severity of GVHD. Nevertheless, in multivariable analyses, non-controllers remained associated with increased mortality after adjusting for TCD and GVHD. When we looked separately at TCD and unmodified HCT, our selected AAUC cutoff was a predictor of mortality in both groups. During our study, the qPCR method for CMV monitoring changed from whole blood to plasma in 2013. When comparing individual patients tested with both methods, we have shown that the slopes of VL over time were similar, but the absolute values differed [20]. In our study, each individual patient was assessed by the same method for all VL measurements. Furthermore, all VL values were log-transformed to calculate the AAUC resulting in attenuation of potential differences between the 2 methods. Nonetheless, to account for potential differences in qPCR method, we entered HCT timing as a covariate in our multivariable models. Non-controller classification remained an independent predictor of mortality after adjusting for HCT timing.
While CMV controllers had favorable survival, PET administered for CMV control is associated with substantial toxicities and increased health care utilization, which need to be factored when assessing the net benefit of CMV prevention [18, 40]. PET was previously associated with increased risk for neutropenia and acute kidney injury [8]. The relative contribution of antiviral-related toxicities to survival was beyond the scope of this study. This is an important area of current research as safer antivirals for CMV prevention and treatment are being implemented. Maribavir appears to be safer than currently available antivirals and is in late development for PET [41]. Letermovir was recently implemented for CMV prophylaxis in many centers and is associated with lower rates of cs-CMV post-HCT [11]. Preliminary studies suggest that letermovir prophylaxis may impact timing of onset and VL kinetics of cs-CMV following HCT [42]. A post hoc survival analysis of the letermovir phase 3 study showed a survival advantage among letermovir recipients who developed cs-CMV, supporting the notion that CMV viremia in certain circustances may be beneficial [13]. Further studies are needed to assess the impact of these new CMV antivirals on survival.
In summary, being CMV non-controller was an independent predictor of mortality at 1-year post-HCT. CMV non-controllers had lower CD4+ counts and more deaths due to CMV and non-CMV infections. In contrast, CMV controllers did not have survival disparity compared with R−/D−. Our data support that controlled CMV viremia is not an adverse prognostic factor for survival at 1-year after HCT. Prospective cross-validating studies are needed to define clinically relevant AAUC cutoffs as predictors of immune reconstitution and HCT outcomes.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
Notes
Author contribution. A. S. and Y. S. designed the research, collected and analyzed data, and wrote the paper. H. D. and J. F. helped in data collection and analysis. R. T., A. A. J., C. C., S. G., and M. A. P provided critical review of the manuscript. G. A. P. contributed to and supervised all aspects of the study.
Financial support. This work was supported in part by the National Institutes of Health National Cancer Institute (grant number P30 CA008748).
Potential conflicts of interest. S. G. receives research funding from Miltenyi Biotec, Takeda Pharmaceutical, Co, Celgene Corp., Amgen Inc, Sanofi, Johnson and Johnson, Inc, Actinium Pharmaceuticals, Inc; and is on the Advisory Boards for Kite Pharmaceuticals, Inc, Celgene, Corp, Sanofi, Novartis, Johnson and Johnson, Inc, Amgen Inc, Takeda Pharmaceutical, Co, Jazz Pharmaceuticals, Inc, and Actinium Pharmaceuticals, Inc. M. A. P. reports receiving institutional research support for clinical trials from Incyte Corporation; honoraria from AbbVie, Bellicum, Bristol-Myers Squibb, Incyte Corporation, Merck, Novartis, Nektar Therapeutics, and Takeda; serving on data and safety monitoring boards for Servier and Medigene; and serving on scientific advisory boards for MolMed and NexImmune. G. A. P is an investigator for Merck and Shire and has received grant support, and consulting and other fees from Merck & Co, Astellas, Chimerix, Ampyx, AlloVir Octapharma, Partners Therapeutics, Cidara, ADMA Biologics, and Shionogi. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Boeckh M, Nichols WG. The impact of cytomegalovirus serostatus of donor and recipient before hematopoietic stem cell transplantation in the era of antiviral prophylaxis and preemptive therapy. Blood 2004; 103: 2003–8. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt-Hieber M, Labopin M, Beelen D, et al. . CMV serostatus still has an important prognostic impact in de novo acute leukemia patients after allogeneic stem cell transplantation: a report from the Acute Leukemia Working Party of EBMT. Blood 2013; 122:3359–64. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt-Hieber M, Tridello G, Ljungman P, et al. . The prognostic impact of the cytomegalovirus serostatus in patients with chronic hematological malignancies after allogeneic hematopoietic stem cell transplantation: a report from the Infectious Diseases Working Party of EBMT. Ann Hematol 2019; 98:1755–63. [DOI] [PubMed] [Google Scholar]
- 4.Green ML, Leisenring W, Xie H, et al. . Cytomegalovirus viral load and mortality after haemopoietic stem cell transplantation in the era of pre-emptive therapy: a retrospective cohort study. Lancet Haematol 2016; 3:e119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teira P, Battiwalla M, Ramanathan M, et al. . Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood 2016; 127:2427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill JA, Mayer BT, Xie H, et al. . Kinetics of double-stranded DNA viremia after allogeneic hematopoietic cell transplantation. Clin Infect Dis 2018; 66:368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emery VC, Sabin CA, Cope AV, Gor D, Hassan-Walker AF, Griffiths PD. Application of viral-load kinetics to identify patients who develop cytomegalovirus disease after transplantation. Lancet 2000; 355:2032–6. [DOI] [PubMed] [Google Scholar]
- 8.Duke ER, Williamson BD, Borate B, et al. . CMV viral load kinetics as surrogate endpoints after allogeneic transplantation. J Clin Invest 2021; 131:e133960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camargo JF, Kimble E, Rosa R, et al. . Impact of cytomegalovirus viral load on probability of spontaneous clearance and response to preemptive therapy in allogeneic stem cell transplantation recipients. Biol Blood Marrow Transplant 2018; 24:806–14. [DOI] [PubMed] [Google Scholar]
- 10.Duke ER, Williamson BD, Wychera C, et al. . CMV viral load kinetics as surrogate endpoints for antiviral prophylaxis trials. Biol Blood Marrow Transplant 2020; 26(3 Suppl):):S327–8. [Google Scholar]
- 11.Marty FM, Ljungman P, Chemaly RF, et al. . Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med 2017; 377:2433–44. [DOI] [PubMed] [Google Scholar]
- 12.Marty FM, Winston DJ, Chemaly RF, et al. ; SUPPRESS Trial Clinical Study Group . A randomized, double-blind, placebo-controlled phase 3 trial of oral brincidofovir for cytomegalovirus prophylaxis in allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2019; 25:369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ljungman P, Schmitt M, Marty FM, et al. . A mortality analysis of letermovir prophylaxis for cytomegalovirus (CMV) in CMV-seropositive recipients of allogeneic hematopoietic cell transplantation. Clin Infect Dis 2020; 70:1525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YJ, Fang J, Zavras PD, et al. . Adenovirus viral kinetics and mortality in ex-vivo T-cell depleted hematopoietic cell transplant recipients with adenovirus infection from a single center. J Infect Dis 2020; 222:1180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hobbs GS, Kaur N, Hilden P, et al. . A novel reduced intensity conditioning regimen for patients with high-risk hematological malignancies undergoing allogeneic stem cell transplantation. Bone Marrow Transplant 2016; 51:1010–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montoro J, Ceberio I, Hilden P, et al. . Ex vivo T cell-depleted hematopoietic stem cell transplantation for adult patients with acute myelogenous leukemia in first and second remission: long-term disease-free survival with a significantly reduced risk of graft-versus-host disease. Biol Blood Marrow Transplant 2020; 26:323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowlings PA, Przepiorka D, Klein JP, et al. . IBMTR severity index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol 1997; 97:855–64. [DOI] [PubMed] [Google Scholar]
- 18.Zavras P, Su Y, Fang J, et al. . Impact of preemptive therapy for cytomegalovirus on toxicities after allogeneic hematopoietic cell transplantation in clinical practice: a retrospective single-center cohort study. Biol Blood Marrow Transplant 2020; 26:1482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seo SK, Xiao K, Huang YT, et al. . Impact of peri-transplant vancomycin and fluoroquinolone administration on rates of bacteremia in allogeneic hematopoietic stem cell transplant (HSCT) recipients: a 12-year single institution study. J Infect 2014; 69:341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Babady NE, Cheng C, Cumberbatch E, Stiles J, Papanicolaou G, Tang YW. Monitoring of cytomegalovirus viral loads by two molecular assays in whole-blood and plasma samples from hematopoietic stem cell transplant recipients. J Clin Microbiol 2015; 53:1252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zecca M, Wynn R, Dalle JH, et al. . Association between adenovirus viral load and mortality in pediatric allo-HCT recipients: the multinational advance study. Bone Marrow Transplant 2019; 54:1632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ljungman P, Boeckh M, Hirsch HH, et al. ; Disease Definitions Working Group of the Cytomegalovirus Drug Development Forum . Definitions of cytomegalovirus infection and disease in transplant patients for use in clinical trials. Clin Infect Dis 2017; 64:87–91. [DOI] [PubMed] [Google Scholar]
- 23.Copelan E, Casper JT, Carter SL, et al. . A scheme for defining cause of death and its application in the T cell depletion trial. Biol Blood Marrow Transplant 2007; 13: 1469–76. [DOI] [PubMed] [Google Scholar]
- 24.Gleiss A, Oberbauer R, Heinze G. An unjustified benefit: immortal time bias in the analysis of time-dependent events. Transpl Int 2018; 31:125–30. [DOI] [PubMed] [Google Scholar]
- 25.Kimura SI, Takeshita J, Kawamura M, et al. . Association between the kinetics of cytomegalovirus reactivation evaluated in terms of the area under the curve of cytomegalovirus antigenemia and invasive mold infection during the post-engraftment phase after allogeneic hematopoietic stem cell transplantation. Transpl Infect Dis 2020; 22:e13387. [DOI] [PubMed] [Google Scholar]
- 26.Deambrosis D, Davies E, Turner A, et al. . Burden of adenoviraemia predicts survival in paediatric recipients of allogeneic haematopoietic stem cell transplant. J Clin Virol 2020; 127:104373. [DOI] [PubMed] [Google Scholar]
- 27.Hill JA, Mayer BT, Xie H, et al. . The cumulative burden of double-stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood 2017; 129:2316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McBride JM, Sheinson D, Jiang J, et al. . Correlation of cytomegalovirus (CMV) disease severity and mortality with CMV viral burden in CMV-seropositive donor and CMV-seronegative solid organ transplant recipients. Open Forum Infect Dis 2019; 6:ofz003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giménez E, Solano C, Vinuesa V, et al. . Cytomegalovirus DNAemia burden and mortality following allogeneic hematopoietic stem cell transplantation: an area under a curve-based investigational approach. Clin Infect Dis 2018; 67:805–7. [DOI] [PubMed] [Google Scholar]
- 30.Green ML, Leisenring WM, Xie H, et al. . CMV reactivation after allogeneic HCT and relapse risk: evidence for early protection in acute myeloid leukemia. Blood 2013; 122:1316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marr KA, Carter RA, Boeckh M, Martin P, Corey L. Invasive aspergillosis in allogeneic stem cell transplant recipients: changes in epidemiology and risk factors. Blood 2002; 100:4358–66. [DOI] [PubMed] [Google Scholar]
- 32.Nichols WG, Corey L, Gooley T, Davis C, Boeckh M. High risk of death due to bacterial and fungal infection among cytomegalovirus (CMV)-seronegative recipients of stem cell transplants from seropositive donors: evidence for indirect effects of primary CMV infection. J Infect Dis 2002; 185:273–82. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg JD, Zheng J, Ratan R, et al. . Early recovery of T-cell function predicts improved survival after T-cell depleted allogeneic transplant. Leuk Lymphoma 2017; 58:1859–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Small TN, Papadopoulos EB, Boulad F, et al. . Comparison of immune reconstitution after unrelated and related T-cell-depleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood 1999; 93:467–80. [PubMed] [Google Scholar]
- 35.Sellar RS, Vargas FA, Henry JY, et al. . CMV promotes recipient T-cell immunity following reduced-intensity T-cell-depleted HSCT, significantly modulating chimerism status. Blood 2015; 125:731–9. [DOI] [PubMed] [Google Scholar]
- 36.Ogonek J, Varanasi P, Luther S, et al. . Possible impact of cytomegalovirus-specific CD8+ T cells on immune reconstitution and conversion to complete donor chimerism after allogeneic stem cell transplantation. Biol Blood Marrow Transplant 2017; 23:1046–53. [DOI] [PubMed] [Google Scholar]
- 37.Jain T, Cho C, Hilden P, et al. . Cytomegalovirus reactivation promotes CD8+ T cell subset recovery after unmodified allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2019; 25(3 Suppl):):S326–7. [Google Scholar]
- 38.Chemaly RF, El Haddad L, Winston DJ, et al. . Cytomegalovirus (CMV) cell-mediated immunity and cmv infection after allogeneic hematopoietic cell transplantation: the REACT study. Clin Infect Dis 2020; 71:2365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang YT, Neofytos D, Foldi J, et al. . Cytomegalovirus infection after CD34+-selected hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016; 22:1480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fang J, Su Y, Zavras PD, et al. . Impact of preemptive therapy for cytomegalovirus on hospitalizations and cost after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2020; 26:1937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maertens J, Cordonnier C, Jaksch P, et al. . Maribavir for preemptive treatment of cytomegalovirus reactivation. N Engl J Med 2019; 381:1136–47. [DOI] [PubMed] [Google Scholar]
- 42.Zavras PD, Stern A, Su Y, et al. . 1742. kinetics of CMV viremia with letermovir prophylaxis in the first 100 days post hematopoietic cell transplantation (HCT): a single-center experience. Open Forum Infect Dis 2019; 6(Suppl 2):):S638. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.