

ABSTRACT
Next-generation multi-specific antibody therapeutics (MSATs) are engineered to combine several functional activities into one molecule to provide higher efficacy compared to conventional, mono-specific antibody therapeutics. However, highly engineered MSATs frequently display poor yields and less favorable drug-like properties (DLPs), which can adversely affect their development. Systematic screening of a large panel of MSAT variants in very high throughput (HT) is thus critical to identify potent molecule candidates with good yield and DLPs early in the discovery process. Here we report on the establishment of a novel, format-agnostic platform process for the fast generation and multiparametric screening of tens of thousands of MSAT variants. To this end, we have introduced full automation across the entire value chain for MSAT engineering. Specifically, we have automated the in-silico design of very large MSAT panels such that it reflects precisely the wet-lab processes for MSAT DNA library generation. This includes mass saturation mutagenesis or bulk modular cloning technologies while, concomitantly, enabling library deconvolution approaches using HT Sanger DNA sequencing. These DNA workflows are tightly linked to fully automated downstream processes for compartmentalized mammalian cell transfection expression, and screening of multiple parameters. All sub-processes are seamlessly integrated with tailored workflow supporting bioinformatics. As described here, we used this platform to perform multifactor optimization of a next-generation bispecific, cross-over dual variable domain-Ig (CODV-Ig). Screening of more than 25,000 individual protein variants in mono- and bispecific format led to the identification of CODV-Ig variants with over 1,000-fold increased potency and significantly optimized production titers, demonstrating the power and versatility of the platform.
KEYWORDS: Bispecific antibodies, protein engineering, developability, high throughput screening, lab automation, data analytics
Introduction
Protein engineering has paved the way for the development of new classes of biotherapeutics. In particular, multi-specific antibody therapeutics (MSAT) have emerged as successful treatment options for complex, multifactorial illnesses including cancer and chronic inflammatory diseases.1–5 Since MSATs are engineered to combine the functional activities of two or more monoclonal antibodies (mAbs) into one molecule, they provide therapeutic advantages over conventional, mono-specific antibody therapeutics. However, unlike mAbs for which industrial production platforms have been established over the past decades, technical development for highly engineered MSAT formats is challenging. Due to their inherent complexity in design, MSATs are often associated with poor yields, increased aggregation potential and chemical instability.6–8 Likewise, the design of the MSAT format can have substantial effects on function.9,10 For optimal physiological efficacy, testing multiple formats by varying design parameters, such as valency, geometry and flexibility, while probing key variables for each design is often necessary.9,11,12 As consequence, huge protein engineering efforts may be needed to generate potent MSAT molecules with drug-like properties (DLP). This is best exemplified by the case-study of emicizumab, a marketed bispecific antibody for the treatment of hemophilia A.13,14 Emicizumab triggers coagulation in FVIII-deficient patients by binding simultaneously to both activated factor IX and factor X, thereby mediating activation of the latter. Substantial engineering efforts were expended over a period of ~10 years to arrive at emicizumab. About 40,000 bispecific variants had to be screened to identify a bispecific starting molecule that could mimic the activity of FVIII in triggering coagulation.15 Subsequently, the starting variant underwent seven rounds of protein optimization, including testing of ~2,400 bispecific derivatives to meet critical developability criteria such as productivity and stability while keeping its FVIII-like activity.15
From this example, it is evident that there is a great need for novel, industry-standard platform processes that enable format-agnostic MSAT engineering in the highest possible throughput to facilitate the development of next-generation biotherapeutics. To address this need, we developed an automated platform process for the fast generation and characterization of very large MSAT panels. In contrast to other recently published platforms, where format diversity is generated on the protein level through mix and match of semi-stable building blocks,16 controlled Fab arm exchange17 or bioconjugation technologies,18 the platform introduced here applies compartmentalized expression of predefined plasmid DNA combinations. Here, we highlight functionalities for automated in-silico design and physical generation of the in-silico designed variant libraries on plasmid DNA level. We describe how DNA library generation technologies were combined with automated procedures for transfection and expression in HEK293 cells followed by compartmentalized multiparametric screening in the highest throughput to achieve very challenging target protein profiles.
Results
Conceptual workflow design
Design features of next-generation MSAT molecules can vary greatly depending on the desired biological functionalities.9,12 An industry-standard platform for HT-engineering of MSATs must therefore be able to accommodate any current or future format.11 The workflow we describe is purely based on the combination of pre-defined DNA expression cassettes and, consequently, fully agnostic to any format or process limitation. Furthermore, it enables the generation of very large sets of compounds (>10,000), which is not practical for workflows that include cumbersome protein purification steps16 and may require further biochemical manipulations.18
In-silico design and DNA library generation workflow
Figure 1 schematically shows the flow of the individual work steps along the MSAT engineering value chain, which are all supported by a customized version of Genedata Biologics™ (GDB)23 software. In-silico MSAT design, as well as all data management activities and associated automated lab unit operations (LUO), are illustrated by the example of a bispecific tetravalent CODV-Ig molecule.19 The first step comprises the in-silico creation of MSAT reference libraries. In our example, the configuration of the intended CODV-Ig format (Figure 1a) is first registered within GDB (Supplementary Figure 1). The registered format comes with an in-silico design template that allows selection of pre-registered sequence modules of variable regions (VRs), constant regions (CRs) or pre-defined generic regions (GRs; here of the peptide linker-type GR-L) and assembly of them combinatorially on the amino acid level into full CODV-Ig designed molecules (DMOL) (Figure 1b and Supplementary Figures 1 and 2). Template-generated DMOL design spaces can be further filtered for preferred DMOL subsets (i.e., variant libraries). Individual light chain (LC) and heavy chain (HC) designs are deduced from selected cognate DMOL subsets and created in the data base as new designed chain (DCHAIN) entities. For this unique set of DCHAINs, the expected DNA sequences are generated via inhouse bioinformatic tools that can now act as references for DNA library deconvolution (see below).
Figure 1.
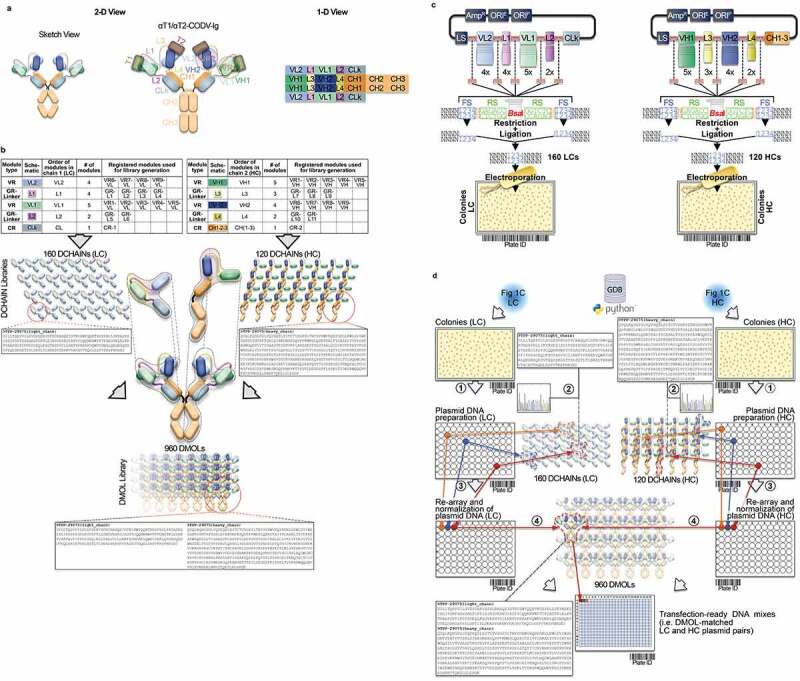
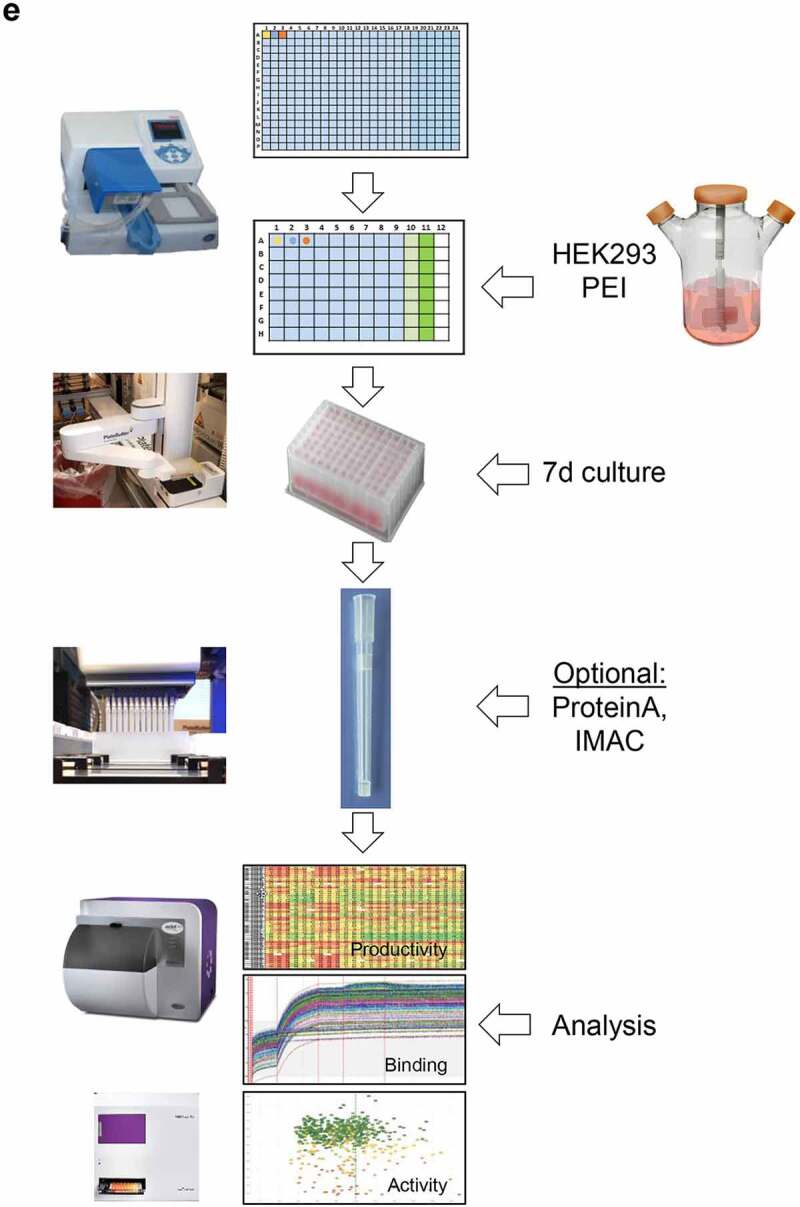
Conceptional process design for format-agnostic HT-engineering of complex MSATs. a) One-dimensional and two-dimensional views of a tetravalent, bispecific CODV molecule.19 In contrast to conventional, mono-specific immunoglobulins (IGs), in bispecific CODV-Igs one parental monoclonal antibody (mAb) here directed against target 1 (αT1; green) is extended N-terminally on both, heavy and light chains, by an additional fragment variable (Fv) domain derived from a second parental antibody directed against target 2 (α-T2; blue). However, when compared to the CODV heavy chains (HC), where the variable heavy chain domains of αT1 (VH1) are located N-terminally followed by VH2s and the heavy chain constant domains (CH; brown), the light chains (LC) carry the αT1 variable light chain domains (VL1) inserted between the VL2 domains and the constant domains of the light chains (CL; gray) resulting in a crossed-over arrangement of the variable regions (VRs) in the CODV format. Per definition, VLs and CLs are connected by peptide linkers L1 and L2, VHs and the first constant domains (CH1) of the HCs are connected by linkers L3 and L4, respectively. Full αT1- αT2-CODV-Igs are oligomerization products of two LCs and two HCs. b) In-silico MSAT reference library design by modular assembly templates. The pre-configured template for the CODV-Ig protein format includes information on the arrangement and pairing of chains, the sequential order of functional modules on chain level (i.e., variable regions (VRs), constant regions (CRs) and linker generic regions (GRs)) as well as the arrangement of functional modules between chains (e.g., cross-over pairing of VL1 with VH1 and VL2 with VH2 as depicted in a)). Pre-registered sequence building blocks can be chosen from the database (Supplementary Figure 2). As schematically shown in the idealized example five binding domains for the first CODV-Ig position (VR1 – VR5; green), four binding domains for the second CODV-Ig position (VR6-VR9; blue), one constant LC domain (CR-1), four linker variants for L1 (GR-L1 – GR-L4), two linker variants for L2 (GR-L5 – GR-L6), three linker variants for L3 (GR-L7 – GR-L9), two linker variants for L4 (GR-L10 – GR-L11), and one constant HC domain (CR-2) have been selected by the operator. The original view of the in-silico CODV-Ig design template as it appears within GDB is shown in Supplementary Figure 1b. After selection of building blocks, the system assembles the building blocks in a combinatorial fashion generating in-silico designs of the complete CODV-Ig molecules (Designed Molecules; DMOLs) according to the sequential order in the design template. 960 DMOLs covering the full combinatorial design space (here combinatorial assembly of 5 VRs x 4 VRs x 4 L1 linkers x 2 L2 linkers x 3 L3 linkers x 2 L4 linkers) are registered in the data base including their annotated sequences and information on the individual building blocks used to create respective DMOLs. In a further step the unique chain designs (DCHAINs) are derived from the DMOLs and registered in a chain specific manner (i.e., separate entries for light and heavy chains are created) holding similar descriptive information (e.g., annotated sequences and composition of building blocks) as their parental DMOLs. In our example 160 HC designs (4 x 4 × 5 x 2 × 1) and 120 LC designs (5 x 3 × 4 x 2 × 1) derived from the DMOLs are registered within GDB. By using pre-configured templates and pre-registered sequence modules, this workflow can be generically applied to any type of multi-chain MSAT format (see Supplementary Figure 1c). c) Chain library generation by modular cloning. The chain designs are realized on the DNA level using modular cloning technologies as described recently.20 Importantly, using this technology it is possible to assemble up to 10 DNA fragments in sequential, pre-defined order reflecting the in-silico design template.21 In brief, DNA building blocks that represent the pre-registered functional modules shown in b) are equipped with inversely oriented BsaI TypeIIS restriction sites. BsaI recognition sites (RS) are in green, one nucleotide spacers are indicted in orange, while the 4 base-pair cleavage/fusion sites (FS) within the DNA modules are indicated by “1234” in blue that allow the seamless and directed assembly of the 16 LC and 15 HC building blocks (i.e., the VLs, VHs, L1-4s and CRs) in one tube restriction/ligation reactions to result in pools of fully assembled 160 LCs and 120 HCs subcloned in an expression vector. The subcloned plasmid pools are transformed into E. coli and plated on omni-tray agar plates. d) Chain library deconvolution and molecule assembly on plasmid level. DNA sequences are compared against the in-silico generated reference libraries of DNA sequences for the theoretical LC and HC chain variants (2). Our Python-based algorithm identifies individual DNA constructs that match HC or LC reference designs and establishes a direct connection between the reference sequence and its physical DNA clone representative using bar-coded plate IDs and well positions of the latter. In a last step the identified clone sequences for all well positions are translated into amino acid sequences and uploaded to GDB where a connection of the physical clone variant to the corresponding DCHAIN entity is established. Identified HC and LC clone representatives (colored dots in plates) are then re-arrayed into new plates and their DNA concentrations are automatically normalized (3). In the next step our algorithm identifies corresponding HC and LC chains that match a MSAT DMOL reference and generates worklists for the liquid handling system to combine matched plasmid pairs in one cavity of a 384-well plate that will result in transfection ready DNA mixes (4). Matching HC/LC plasmid pairs are indicated by same color. e) Schematic representation of the automated LUOs covering transfection, expression, purification and multi parametrical analysis. For transient transfection plasmid pre-mixes are first transferred to 96 DWP and then combined with 1 ml of HEK293 suspension cells and PEI using a Multidrop dispensing unit. The DWPs are then closed with Duetz sandwich covers which ensure sufficient gas-exchange and prevent evaporation to result in mini-bioreactor like compartments22 (not shown). After 5–7 days incubation at 37°C the expression plates are harvested by short centrifugation. The supernatants are then transferred to new 96 DWPs and are either directly re-arrayed for various analytical and functional assays or, alternatively, first subjected to automated protein purification on the HamiltonStar LHD using PhyTips® with matched matrices (e.g., IMAC, Protein A) before further analysis. Quantification and binding data are usually obtained via label-free BLI technology using an OctetHTX device. HTRF-based data, such as from cell-based activity assays can be generated on a BMG Clariostar reader
Figure 1.
(Continued)
In the next step HC and LC CODV-Ig chain libraries are generated at the DNA level. We use one-step modular cloning technologies to assemble DNA building blocks that represent the pre-registered VRs, CRs or GRs provided as single plasmids in one-pot bulk reactions (Figure 1c).20 HC and LC chain libraries are deconvoluted into their individual DCHAIN representatives using high-throughput Sanger sequencing in combination with an inhouse bioinformatic tool for automated data analysis (Figure 1d and Supplementary Figure 3). Matched HC and LC plasmids are then transfected into HEK293 cells. After 7 days of cultivation, expression supernatants are harvested and analyzed for multiple parameters or subjected to one-step purification (Figure 1e and Supplementary Figure 6c) for further biochemical analyses.
Automation
Our robotic platform was designed to automate the entire value chain for MSAT engineering and accordingly accepts diverse types of 96- or 384-microwell-plates used for E. coli colony picking, preparation of DNA and its concentration determination, normalization, and re-arraying in 96- or 384-well plates, as well as transfection of mammalian cells and protein expression with optional purification in 96-deep well plates and subsequent multi-parameter analysis of the produced molecules. To this end, three individual robotic systems have been established that handle LUOs associated with the different entities that appear along the value chain. End-to-end integration of all LUOs with automation is realized through seamless 1- or 2-D sample barcodes that are stored in GDB. The design of the platform, including a comprehensive description of the integrated components and how it supports underlying LUOs, is detailed in Figure 2a-b.
Figure 2.
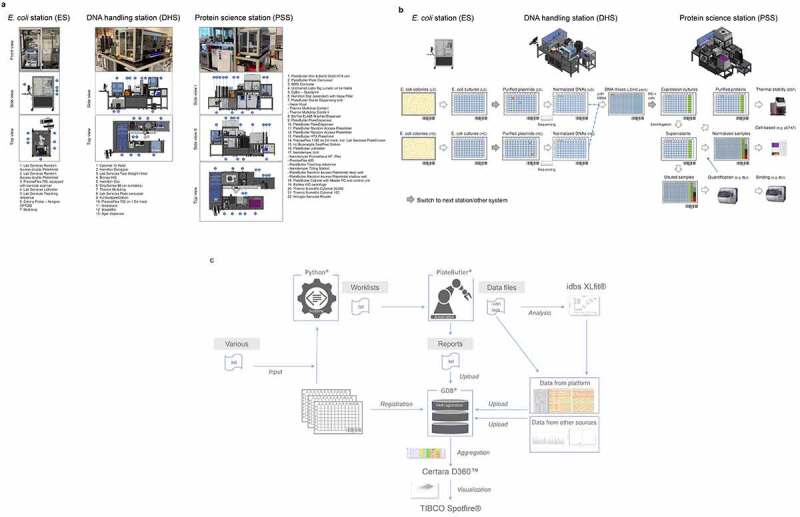
Designs of robotic platforms and automated lab unit operations. a) Robotic platforms designs. CAD drawings of the three robotic stations are shown providing side and top views including sequential numbering of integrated components. The E. coli station (ES) is designed as standalone unit to handle all E. coli-related lab unit operations (LUOs). The station is used to pick individual bacterial colonies from SBS (Society for Biomolecular Screening) compatible agar plates into 384- or 96-well plates and to inoculate 96-well-based plate formats from glycerol stocks by a cherry picking-like process using a Norgren CP7200 colony picker. Plate handling is done by a PlateButler® PreciseFlex robotic arm. A maximum of approximately 10,000 picking events can be realized per day. The fully automated DNA handling station (DHS) is designed to realize all plasmid DNA-related workflows. The station is particularly used to carry out plasmid DNA preparations using Phynexus PhyTip® chromatography technology, determine concentrations of plasmid DNAs by UV/Vis (DropSense96), re-array plasmid DNA samples and normalize the DNA concentrations of the samples in a single step, create pre-defined plasmid DNA mixtures, and generate plasmid DNA stocks for long term storage (cherry picking). To this end we integrated an automated Hamilton™ Star liquid handling device with other commercially available components for DNA concentration measurement and sample management and a PlateButler® PreciseFlex robotic arm for plate handling. The DHS accepts diverse plate and tube types which can be used with lids or seals. A maximum of 2,000 plasmid DNA preparations and 10,000 plasmid DNA normalizations can be performed per regular five-day work week. The protein science station (PSS) is a flexible robotic platform designed to conduct all cell and protein related LUOs in a fully automated fashion. The station is used to transfect mammalian HEK293 cells in 96 deep well plates in a sterile environment, to harvest protein containing cell supernatants by centrifugation and to transfer or dilute samples into other 96- or 384-well plates. Expression supernatants can be quantified by BLI- (ForteBio Octet HTX) or ELISA- (BMG CLARIOStar) based technologies. Furthermore, protein expression titers in cell supernatants can be normalized or diluted and successively applied to BLI- or ELISA-based binding assays. If required, proteins can be purified from expression supernatants using Phynexus Phytip® column technology. The purified proteins can then be quantified by UV-Vis (Unchained Labs Big Lunatic), normalized reformatted and analyzed by nanoDSF (Nanotemper Prometheus). As for the DHS, we integrated an automated Hamilton™ Star liquid handling device with other commercially available components for protein concentration measurement, analytics, and sample management and a PlateButler® PreciseFlex robotic arm for plate handling. In addition, the workstation is used to label labware with specific barcodes and to seal the samples for long term storage. In general, plates are handled with lids or seals for sterility reasons. For facility maintenance, instrument drawers and turntables are included where appropriate. To increase flexibility of the PSS, the ForteBio Octet HTX analyzer is located on a connector trolley that can be docked to the robotic station via the Lab Services PlateButler® Pier & BarQ system. This trolley can be easily disconnected and replaced by alternative instruments on further connector trolleys. Capacities of the PSS comprise over 12,000 HEK293 transfections and expression supernatant normalizations per regular five-day work week. All ES, DHS or PSS components, as listed and described in more detail in the materials and methods section and in Supplementary Table 1, are integrated and controlled by the PlateButler® software. By using a two-level principle, our ES, DHS, or PSS multi-instrument systems are set up in a distributed environment where the main application acts as the supervisor that integrates all devices. The instruments themselves are controlled by their own dedicated software which can, however, also be accessed directly by the operating scientist. All plate flows are tracked by PlateButler® software which also creates customized reports of automated experimental runs to allow for simple upload to the GDB-based laboratory information management system. b) Plateflows and automated lab unit operations. ES-operated processes: E. coli colony picking and inoculation: First, HC and LC DNA libraries are transformed into E. coli and plated onto SBS compatible omni-tray agar plates. A customized colony picking unit (Figure 2a; E. coli station (ES)) then mediates the transfer of individual colonies to 96-deep well plates (DWP) which have been supplied with 2YT growth media through an integrated multi-drop liquid dispenser. Inoculated bacterial cultures are then grown at 37°C overnight. Alternatively, glycerol stock plates are generated first from the bacterial cultures for long term storage and new 96 DWPs are inoculated from the glycerol stock plates (not shown; see materials and methods section). DHS-operated processes: DNA preparation and handling: In the next step, plasmid DNAs are extracted and purified automatically from sedimented E. coli cultures using Phynexus PhyTip® chromatography columns and subjected to DNA sequencing. Plasmid DNAs of sequence-verified clones (colored dots) are measured, normalized, and transferred to new 96-well plates. Following normalization of the plasmid DNA concentration, DNA LC/HC pairs which correspond to an in-silico MSAT DMOL reference are combined in individual cavities of 384-well plates together with oriP/EBNA vectors for stable plasmid propagation. PSS-operated processes: HEK293 cell transfection, protein expression and analysis: 384-well plates are re-arrayed to 96 DWPs that already include appropriate process and assay controls in pre-defined wells (columns 10–12). Subsequently, DNAs are mixed with HEK293F cells and PEI transfection reagent and cultivated for seven days at 37°C. After cultivation, cells are sedimented by centrifugation and cell expression supernatants are transferred to new 96 DWPs. From the new DWPs, aliquots of the samples are directly diluted into 384-well plates and first quantified by biolayer-interferometry (BLI) using an Fortebio Octet HTX instrument. Following quantification, samples are frequently normalized to appropriate concentrations before being applied to cell-based (e.g., protein phosphorylation) assays or qualitative and quantitative binding experiments using BLI- or ELISA-based technologies. For other biophysical characterization methodologies such as Nanotemper-based thermal stability assessment, samples are first immunoaffinity purified using Phynexus PhyTip® chromatography columns. c) Data processing workflow. Figure 2c describes the integrated data flow between the robotic systems (PlateButler® software), the data base (GDB®), internally developed bioinformatic tools (Python®-based) and commercial software for data analysis and visualization (idbs XLfit®, Certara D360™ and TIBCO Spotfire®). All plate and well information (including barcodes and plate hierarchies) along the screening process are consecutively fed into the data base using an a-posteriori workflow. After completion of all plate operations, the PlateButler® software generates reports as .txt files with relevant process information (e.g., barcodes of source and target plates, well to well mapping, plate types and transferred volumes) which can be uploaded into the data base (directly or after undergoing a quality control step of the pipetting operation via custom Python-based scripts). Pipetting and plate operations on the robotic systems can be initiated in different ways: 1) via pre-configured methods within the PlateButler® software (e.g., colony picking, plate copying and 96- to 384-well plate re-array operations); or 2) via a combination of pre-configured methods and worklists (e.g., hit picking, normalization, and transfection operations). Worklists are generated in .txt file format using Python-based tools and include information on source and target well positions and barcodes, as well as on plate types and volumes to be transferred. The required information for the worklist generation is automatically extracted from reports of previous plate operations or the central data repository GDB. Assay data files (including raw data) generated on the robotic system are pre-processed and analyzed via idbsXLfit® before being uploaded into the data base. Certara D360™ (Certara, Princeton, NJ) and TIBCO Spotfire® (Tibco, Palo Alto, CA) solutions are used to aggregate visualize and analyze all data generated along the value chain for the candidate molecules
Workflow support and data processing pipeline
In order to ensure a maximum of format and workflow flexibility, we developed a toolbox of Python-based bioinformatic applications for automation support and customized GDB to capture all data generated along the MSAT value chain by applying FAIR data principles.24 This refers to all data concerning molecules, materials, conditions, as well as robotics or liquid handling device (LHD) operations. We established a formal registry for MSAT molecule designs and associated materials, which we combined with tools for sequence annotation and analysis, as well as service programs for automated LHD worklist generation and for management of complex plasmid logistics. Together with pipelines for LHD quality control, all applications are either integrated within the GDB registration engine or implemented as external custom tools using Python as programming language. Additional interfaces allow processed data to be imported from other sources or the export of data for aggregation visualization, or modeling purposes. Figure 2c describes the current data processing pipeline.
Example of HT engineering – multi-parametricnext-generation bispecific CODV-Ig
Primary optimization goals were to increase potency and productivity of a prototypic CODV-Ig construct directed against two targets, T1 and T2 (αT1-αT2-CODV-Ig). In particular, the neutralization activity against T2 should be improved by at least a factor of 30 to be comparable to αT1 while expression titers should be increased 2–3 fold concomitantly (Figure 3). To this end, we applied a two-step engineering strategy. In a first step, we aimed to improve the functional activity of the mono-specific αT2 binder on the antigen-binding fragment (Fab) level to allow re-use of the optimized αT2 VR as a building block in further multi-specific combinations also including non-CODV formats (Figure 3a). In a second step, we intended to accommodate optimized αT2 VRs with αT1 in the bispecific CODV-Ig format by modulating composition and length of peptide linkers (Figure 3b).
Figure 3.
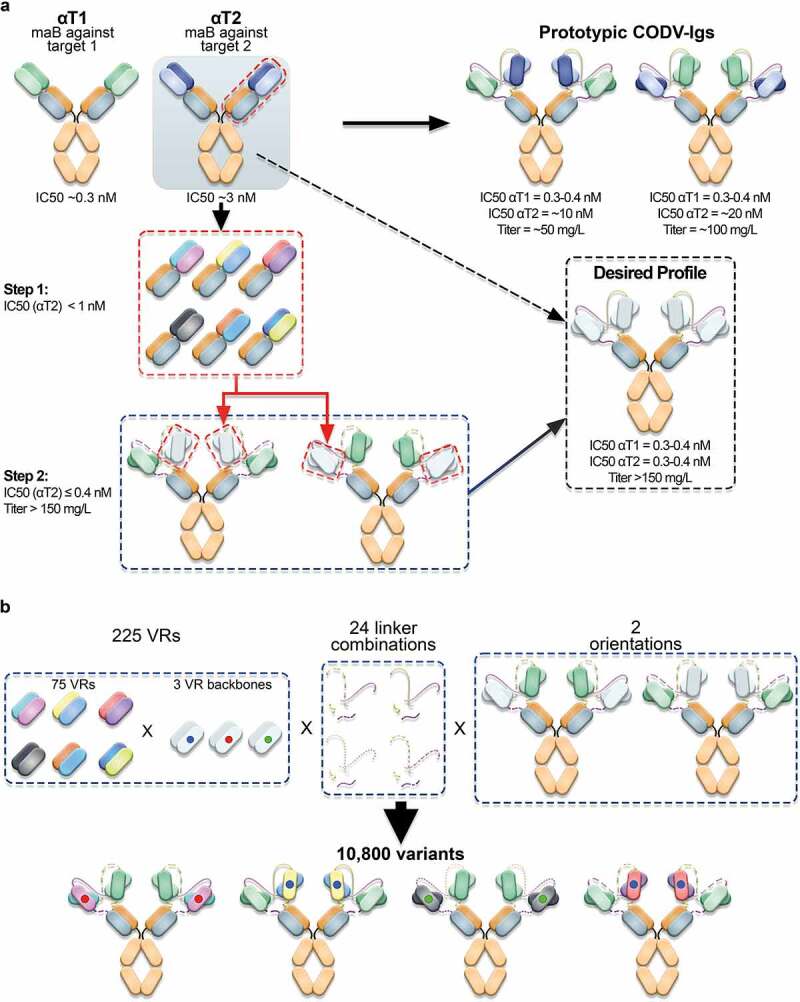
Optimization of prototypic CODV-Ig variants. a) Engineering strategy. At start, two antagonistic monoclonal antibodies (αT1 and αT2) directed against two unrelated antigens (target 1 (T1) and target 2 (T2)) were combined in a cross-over fashion in two orientations to allow for simultaneous neutralization of both targets (Prototypic CODV-Igs). The antagonistic drug potencies of the monoclonal antibodies were in the picomolar (αT1) to low nanomolar (αT2) range as determined in cell-based functional assays reflecting the inhibition of T1- and T2-mediated reporter gene activation respectively. However, when combining both antibodies in the CODV-Ig format, the inhibitory potency against T2 dropped by more than 3 fold while the IC50 against T1 remained largely unaffected. Interestingly, the bispecific αT1-αT2 CODV antibody could be produced at significantly higher titers (100 mg/L vs. 50 mg/L) when presenting the binding domain against T2 on the outer position of the CODV, which indicated additional structural constraints. Primary optimization goals were to increase potency and productivity of the prototypic CODV-Ig constructs to meet the desired profile by applying a two-step engineering approach. First the neutralization activity of the αT2 antibody should be increased to sub-nanomolar levels on the Fab level followed by accommodation of optimized αT2-VRs in the CODV-Ig format by modifying the composition of the VR connecting peptide linkers on the HCs and LCs while providing both possible VR orientations. b) Structural diversification. 75 mutational VRs designs from step1 displaying varying combinations of beneficial CDR mutations were selected to be combined with three different VR backbones in the CODV format. The VR backbones included in addition to the wildtype αT2 backbone two alternative, sequence-optimized αT2 VR backbones where potential post-translational modifications had been addressed by neutral substitutions (75 VR designs x 3 backbones = 225 VRs). The resulting 225 αT2 VR alterations were assembled with the αT1 VR in the CODV-Ig format in two orientations using 24 different peptide linker combinations, giving rise to 10,800 possible variants
Optimization step 1: isolation of αT2-Fab variants with increased potency
To increase the neutralization activity of the αT2-Fab to sub-nanomolar levels, we applied a two-staged mutagenesis approach that includes a first round to gather position-specific complementarity-determining region (CDR) mutations followed by a second round in which beneficial mutations are recombined. For positional library generation, we diversified the CDRs of αT2 as described by Votsmeier et al.25 with the difference that we used single-NNK saturation mutagenesis for each individual amino acid position. Deconvolution against all potential DCHAIN variants yielded 1,054 individual mutants representing 84% of the theoretical design space (represented by 1,254 registered DMOLs), which were quantified and then tested for T2 binding and in cell-based assays to inhibit T2-dependent signal transducer and activator of transcription (STAT) protein phosphorylation. Applying single-dose measurements, 56 improved αT2 variants were identified that displayed greater than 130% inhibitory activity when compared to the originating αT2WT-Fab (Figure 4a). Increased activities also translated well into reduced IC50 values in full-dose response measurements (Figure 4b). In contrast, binding and potency improvements were often not correlated (Figure 4c), further indicating a complex interplay of structural and functional features regarding the interaction with the cognate target T2. Accordingly, many potency improving substitutions did not map to the T2 antigen-αT2 antibody interface (Figure 4d). In conclusion, exhaustive genotype/phenotype landscaping of the αT2 CDRs led to the identification of single amino acid substitutions that significantly improved potencies.
Figure 4.
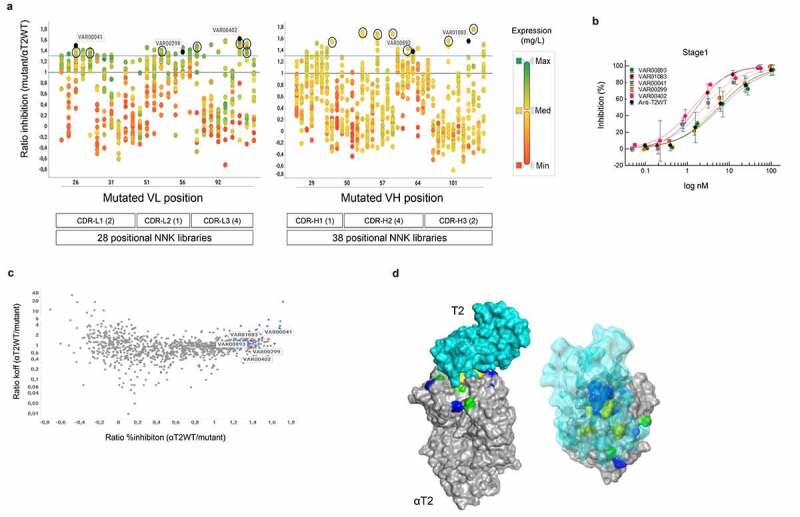
Compartmentalized positional Fab library screening. a) Expression activity correlation. 66 positional-NNK libraries (representing 28 variable light chain (VL) domain and 38 variable heavy chain (VH) domain libraries) addressing all complementarity-determining region (CDR) positions of the His-tagged anti-T2WT Fab were deconvoluted into their single amino acid substitutions using the same workflow as described for CODV-Igs in Figure 1 and Supplementary Figure 2. 1,054 of 1,254 potential variants (pre-registered as DMOLS) arising out of 19 alternative amino acid substitutions for all 66 CDR positions could be recovered. Individual substitutions that correspond to distinct CDRL1-L3 or CDRH1-H3 positions are represented by vertically aligned dots. CDR definition according to Kabat scheme slightly modified as described by Votsmeier et al.25 HEK293 expression supernatants of the single NNK Fab variants were quantified by anti-His detection using BLI technology and then analyzed in cell-based HTRF reporter gene assays to inhibit T2-dependent STAT protein phosphorylation. The expression quantification results are represented by different colors. Expression ratios relative to the quantified His-tagged anti-T2WT Fab construct are shown by a color gradient from low (red; VL & VH: Min (Minimum) = 0.04), medium (yellow; VL Med (Median) = 1.38, VH Med = 1.14) to high (green; VL Max (Maximum) = 2.61, VH Max = 3.74). Activity ratios of the corresponding mutants relative to the wildtype calculated from STAT protein phosphorylation inhibition in THP-1 cells after T2 stimulation in HTRF assay (single dose measurements) are displayed on the y-axis. The solid blue line represents the ratio of 1.3. Distinct CDR variants (VAR) marked in dark blue are representatives of full dose response measurements (Figure 4b). Black circles indicate highly active variants that were selected for recombination in the αT2-Fab scaffold (see below Figure 5). Ratios ≤0 depict variants with no measurable STAT protein phosphorylation inhibition. b) Dose response curves of STAT protein phosphorylation inhibition in THP-1 cells after T2 stimulation by selected Fab mutants. Supernatants containing the expressed Fab mutants were serially diluted and tested as described in materials and methods. Low control, containing only THP-1 cells without T2 stimulation is defined as 100% inhibition and high control with T2 stimulation as 0% inhibition. Averages of duplicate measurements are shown in the plot. Standard deviations are represented as error bars. Note that increased activity levels seen in a) correlated well with decreased 50% inhibitory concentrations (IC50). For example, variants exhibiting over 150% of the wild activity (VAR00402 or VAR01083) also showed a more than 5-fold decrease of the corresponding wild type IC50 values. c) Affinity potency correlation. Activity ratios of 1,054 individual Fab variants were plotted against the dissociation rate constants (koff) quotient from the anti-T2WT Fab and the individual variants as measured by BLI (antigen capture mode) with increased ratios indicating reduced off-rates. The 56 variants which displayed greater 130% activity are marked in blue. Five variants for which full dose responses have been determined are indicated in red. Note that slower off-rates (indicative of improved affinities) often did not correlate with increased activities. d) Structural analysis. Positional library screening yielded a comprehensive and multi-dimensional data set that constitutes a basis to deeper explore structure-function relationships of the αT2 binder. As an application example, a solid surface representation of a crystallographic structure of the anti-T2WT Fab fragment (gray) in complex with T2 (a. turquoise – solid surface; b. turquoise – transparent surface) was generated. CDR positions for which substitutions were found that: 1) significantly increase potency and affinity are shown in green; 2) increase affinity only are colored yellow; 3) increase potency only are depicted in blue. Note that many potency-improving residues are located outside the paratope-epitope interaction interface
To screen for combinations of synergistic or additive mutations, we next selected 14 dispersed potency-improving mutations (7 VL, 7 VH) addressing 13 CDR positions for recombination in the αT2WT-Fab scaffold (Figure 4a). The design of the recombination library included the selected beneficial substitutions, as well as the parental amino acids, for each of the 13 CDR positions, resulting in 96 VL and 128 VH recombination (rec) mutants. VH and VL rec mutants were individually cloned as full HC and LC constructs and subsequently combined on the plasmid DNA level, giving rise to 12,288 (96 x 128) variants that had been pre-registered as DMOL representatives in GDB. Matched HC and LC plasmid pairs were transfected and expressed in HEK293 cells and quantified as before. A total number of 4,294 variants were then selected based on expression level, potency, and species cross-reactivity in bio-layer interferometry (BLI) binding experiments (Figure 5a and Supplementary Figure 4), and subsequently screened to inhibit STAT protein phosphorylation applying single-dose measurements (Supplementary Figure 4b). From this primary screen, 443 variants with significantly increased potencies were subjected to full dose response measurements. Here, the best variants displayed up to 50-fold decreased IC50 values (Figure 5c). We found that the number of mutations that variants carry was not correlated with their potency. Variants with as few as two mutations could have a 20-fold increased potency, while top variants typically carried between 4 and 11 mutations (Figure 5c). Yet, from first analyses of the sequences belonging to significantly improved rec mutants, no apparent potency-increasing mutational patterns were detectable (not shown). This is consistent with the above observation that potency improvements can result from both direct and allosteric effects that might not act synergistically. Taken together, our two-stage directed-evolution approach yielded a large and diverse panel of significantly improved αT2 binders, many of which clearly exceeded the targeted potency level already as monovalent Fab.
Figure 5.
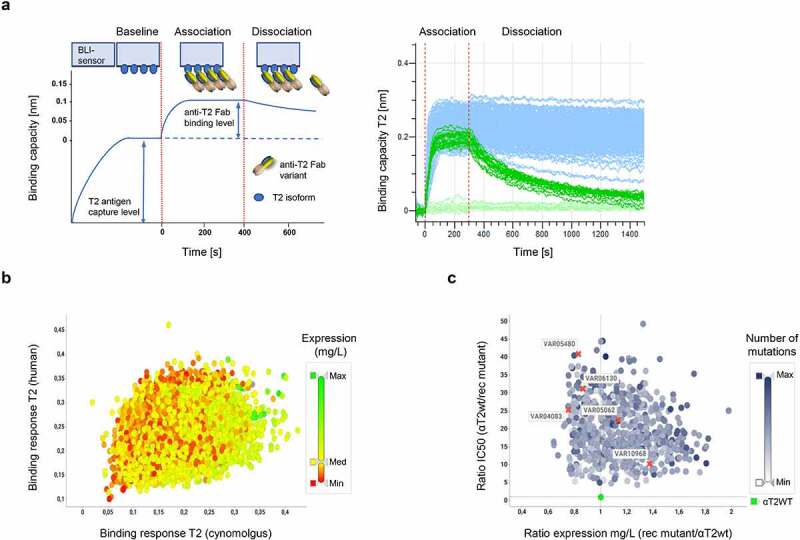
Compartmentalized recombination Fab library screening. a, b) Species cross-reactivity. a) To safeguard that recombination mutants which carry potency-improving substitutions against the human isoform of T2 are still cross-reactive against the cynomolgus T2 isoform, all 12,288 expression supernatants were quantified and then analyzed for their binding capacities against human and cynomolgus T2 isoforms using BLI technology in an antigen capture format.26 Here, the signal increase of the binding curve over baseline after addition of normalized samples to BLI sensors decorated with captured T2 antigen was measured as wavelength shift [nm]. The spectral shift is indicative for the binding capacity of the analyzed variant as schematically represented on the left. An aggregated view of 384 exemplary binding curves is depicted on the right. Note the increased binding capacities and reduced dissociation rates of many anti-T2WT Fab recombination variants (light blue curves) when compared to anti-T2WT Fab curves (light green; 16 replicates). b) Representation of 4,294 recombination mutants selected from 12,288 combinatorial variants. Only potency-improved variants were selected that displayed equal or better signal increases to human T2 (>0.1 nm) and cynomolgus T2 (>0.05 nm) when compared to the anti-T2WT Fab while expressing at titers >100 mg/L. Expression as determined by anti-His detection using BLI technology is shown by a color gradient from low (red; Min ≤ 0.1 mg/L), medium (yellow; Med = 150 mg/L) to high (green; Max = 750 mg/L). c) Expression activity correlation. Of the 4,294 variants which had been verified to be cross-reactive, 443 rec mutants with significantly increased activities as determined in single dose experiments (SSupplementary Figure 5b) were subjected to full dose response measurements. Activity ratios of the 443 variants were plotted against the expression rate quotient from the anti-T2WT Fab and the individual variants. The number of mutations that variants carry is shown by a color gradient from low (white; Min = 1 mutation) to high (blue; Max = 11 mutations). The anti-T2WT Fab is marked in green. Variants, for which full dose response curves are depicted in d) are indicated by a red cross and respective VAR identifiers. No correlations were found between expression levels, IC50 values and number of mutations. d) Dose response curves of STAT protein phosphorylation inhibition in THP-1 cells after T2 stimulation by selected Fab recombination mutants. Assay description as detailed in Figure 4b. Recombination of potency improving single substitutions leads to further activity improvements
Figure 5.
(Continued)
Optimization step 2: accommodation of optimized VRs in CODV-Ig format
Previous experiments using the prototypic αT1-αT2-CODV-Ig antibodies suggested that the spatial arrangement of αT1 and αT2 VRs can have an impact on potency and productivity (Figure 3a). In order to maintain potencies of the optimized αT2 VRs and at the same time ensure high expression titers in the bispecific CODV-Ig format, we wanted to explore a large CODV design space where optimized αT2 VRs of different mutational designs are combined with the fixed αT1 VR by varying their relative spatial arrangement. To this end, 75 mutational VRs designs derived from the Fab screen were transferred to three alternative backbones of αT2 and assembled with the αT1 VR in two orientations using 24 different peptide linker combinations (Figure 3b). After deconvolution, 10,538 of 10,800 theoretical CODV DMOLs (75 x 3 × 2 x 24) could be realized by the controlled pairing of 1,237 HCs and 182 LCs (Supplementary Figure 5a, note that some HCs and LCs are re-used in different designs), which represents 98% of the theoretical CODV-library design space. After transfection, 10,538 cell culture supernatants were quantified for CODV-Ig variant expression (Supplementary Figure 5b). The observed expression patterns clearly indicate a strong influence of the peptide linker configuration on CODV-Ig expression (Figure 6a). Combination of certain peptide linkers with distinct, optimized αT2 VRs led to a 2- to 5-fold increase in expression. It is also noteworthy that many variants that present optimized αT2 VRs at the inner CODV position (Orientation 2) expressed at much higher titers compared to the corresponding initial prototypic CODV-Ig. This further indicates that, by optimizing linker compositions, spatial VR arrangements can be tweaked to resolve potential structural constraints that hamper efficient expression.
Figure 6.
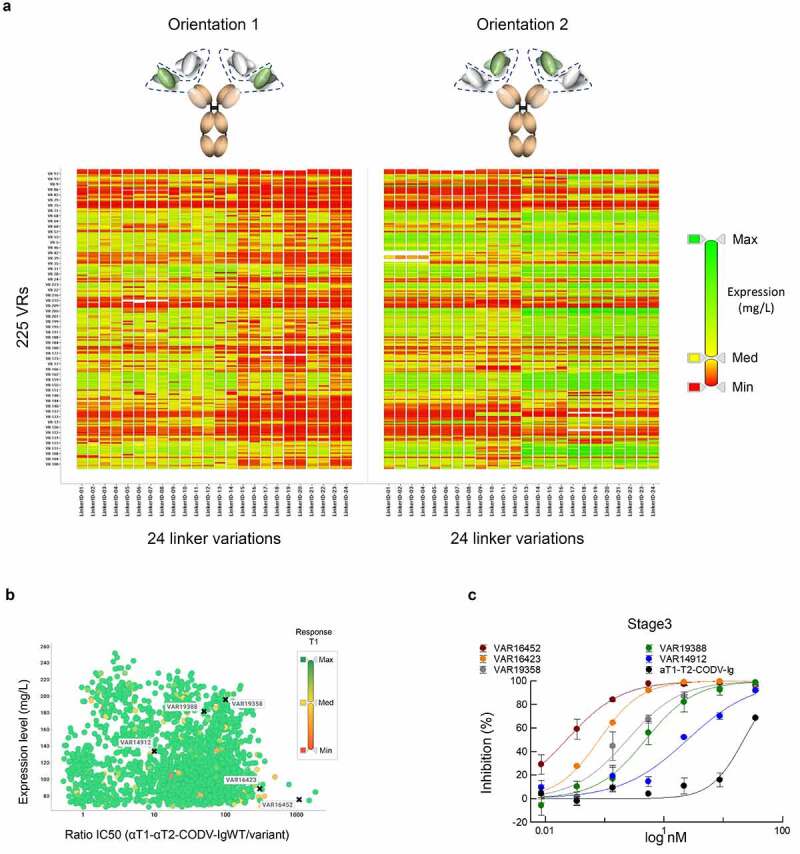
Compartmentalized CODV-Ig library screening. a) Impact of CODV-Ig design on expression. Graphical representation of expression values across 10,538 CODV-Ig variants. Individual tiles represent the different CODV-Ig designs with 225 optimized VRs corresponding to the rows of the matrix and 24 linker configurations corresponding to the columns. The two CODV-Ig orientations are shown separately. Individual expression values contained in tiles of the grid are scaled by a color gradient from low (red; Min ≤ 0.1 mg/L), medium (yellow; Med = 29 mg/L) to high (green; Max = 233 mg/L). Note that linker variegations can lead to significantly improved expression titers for same VR combinations in different orientations indicating that expression titers can be optimized by structural adaptations. b) Expression activity correlation. Full dose response measurements of STAT protein phosphorylation inhibition in THP-1 cells after T2 stimulation by 1,621 CODV-Ig variants selected based on expression (>75 mg/L). Activity ratios of the corresponding 1,621 variants relative to the prototypic αT1-αT2-CODV-Ig (Orientation 2) were plotted against the expression rate (mg/L). Binding capacities against T1 were confirmed by BLI in an antigen capture format. Binding response ratios of the variants relative to the prototypic αT1-αT2-CODV-Ig (Orientation 2) are shown by a color gradient from low (red; Min = 0.04), medium (yellow; Med = 0.52) to high (green; Max ≥ 1). Note that the vast majority of CODV-Ig variants displayed binding capacities against T1 similar to the αT1-αT2-CODV-Ig starting variant. Variants, for which full dose response curves are depicted in c) are indicated by a black cross and respective VAR identifiers. Note that accommodation of optimized VRs in CODV-Ig format led to the identification of variants showing up to 1,000-fold improved activities. c) Dose response curves of STAT protein phosphorylation inhibition in THP-1 cells after T2 stimulation by selected CODV-Ig variants. Assay description as detailed in Figure 4b
Supernatants of 1,621 variant samples were then selected based on their expression level and screened to inhibit STAT protein phosphorylation in full-dose response measurements. Several potency-matured CODV-Ig variants were determined to neutralize T2-induced STAT protein phosphorylation up to 1,000 times more efficiently than the prototypes (Figure 6b and c). All variants were further confirmed to maintain T1 binding (Figure 6b) and desired species cross reactivity (not shown). Importantly, observed high neutralization potencies were not caused by multimerization of CODV-Ig variants, as evidenced by size-exclusion chromatography (SEC) of one-step protein A affinity-purified samples (Supplementary Figure 6). In addition, improvements in functional activities and production titers seen at the level of micro-scale expression supernatants were confirmed at protein level for selected variants purified from large-scale expression cultures as previously described27 (data not shown).
In summary, our two-step engineering approach yielded a large and diverse panel of significantly improved αT1-αT2-CODV-Igs, many of which exceeded the targeted potency level by far while expressing at increased titers. This demonstrates the power and versatility of compartmentalized screening in very high throughput for MSAT optimization. This panel will now serve as a starting point to screen sequence and structurally differentiated, optimized variants for additional developability criteria based on desired candidate profiles.
Discussion
We report the development of a novel, industry-standard platform for the automated generation and screening of very large panels of complex antibody-based therapeutics. In contrast to existing platforms used elsewhere,16,28,29 our platform process has several properties that makes it stand out. In particular, it enables us to 1) more tightly integrate with downstream development processes; 2) de-risk the progression of engineered candidates from research to development while simultaneously allowing maximum flexibility as to the type of examined modalities; 3) use compartmentalized screening of deconvoluted variant libraries as a foundation for multi-parametric analysis of compounds; and 4) generate large consistent data sets that allow fast and sophisticated data analysis.
The implemented process is predictive for downstream biotherapeutic development platforms since HT screening is obligatorily performed in the final product format and all molecules are expressed as soluble proteins in mammalian production hosts. In this way, anticipated post-translational modifications that are potentially important for function of the final product are preserved. On the other hand, unwanted modification patterns or altered physicochemical properties are avoided ab-initio. Such alterations can result from cell surface display-based selection procedures30 or at later stages when displayed constructs, typically fragments of the envisioned product, are reformatted into the desired soluble product format.31,32 In addition, mini-bioreactor-like compartments22 are used for cell cultivation to approximate as best as possible large-scale fermentation processes. Using this approach, compatibility of the engineered molecules with development processes is safeguarded from the beginning.
Multi-specific antibody therapeutics are at high risk for chemical or physical stability issues,6,8 which are difficult to predict from the primary amino acid sequence alone.7 Usually, such issues become apparent at late discovery stages during comprehensive developability assessments of the drug pre-candidates. To mitigate the risk of late-stage failure, it is therefore desirable to select variants for developability assessments from large variant panels that comprise significant diversity on the sequence or structural level. Moreover, it could be important to be able to select from pools of engineered MSAT variants that cover a wide range of affinities or potencies for their individual targets. Thus, the target-specific activities can be balanced or fine-tuned to achieve optimal biological efficacy15 or to enable novel therapeutic mechanisms.33 In our example, we could single out variants with different potencies, affinities, production titers, configurations, mutational designs, or combinations thereof, from a panel of more than 1,600 variants (Figure 6b). Having access to this wide selection of variants will substantially increase the probability of identifying viable MSAT drug candidates that can be progressed to development.34
Our example suggests that independent diversification of the αT2 binding domain in Fab format followed by an optimization step within the final multi-specific CODV-Ig format is required to identify variants combining excellent biological activity with good developability profiles. All 75 re-combinatorial variants of the αT2 binding domain that were selected for the CODV accommodation step showed good to excellent expression in Fab format. As shown in Figure 6a, the expression titers of the optimized Fab molecules did not transfer into the CODV-Ig format for all variants, and even minor differences in few point mutations between individual αT2 variants could significantly affect CODV expression titers. A similar observation was made when comparing the potency improvement of αT2 variants between formats. In some cases, the potency of a binding domain is reduced in CODV format as compared to the Fab molecule, which can be explained by geometry, spatial constraints, or accessibility of the binding domain within the multi-specific format. However, even for binding domains for which properties like expression, stability or biological activity did not fully correlate between Fab and CODV-Ig format, we could restore or even improve the specific properties by optimizing linker setups or orientations within the final multi-specific format (as shown for expression in Figure 6a). Specifically, the co-optimization of molecular interactions via mutational variation and geometrical features through linker diversification and changes in orientation resulted in stronger improvements of properties like binding and potency within the multi-specific format as compared to the Fab variants. Accordingly, reduction of the design space via pre-selection of a preferred orientation and linker setup prior to the building block optimization would have led to a loss in hit variants with desired profiles. Furthermore, generating diversity and optimizing the αT2 binding domain at the level of the Fab format came with the additional advantage that a toolbox of αT2 variants covering a wide range of different biophysical properties was generated for use within other multi-specific protein formats or for the exploration of further target combinations.
The number of multi-targeting modalities in clinical development is increasing.1,3,12 At the same time, the number of experimental MSAT formats for the treatment of complex diseases is constantly growing.11 Several technologies have been described for the generation of IgG-like bispecific antibodies, including knob-into-hole-mediated combination of half-antibodies,35,36 Fab Arm exchange28,29 and bioconjugation-mediated assembly.18 Recently, Dengl et al. introduced a novel approach where various bispecific antibody formats can be generated in parallel through the exchange of semi-stable placeholder chains.16 Clearly, this approach represents considerable technical progress to address the need for exploring diversified MSAT formats for optimal biological activity.9,37 Along these lines, our process design takes account of the demand for format-agnostic platform technologies for HT-engineering purposes. We designed the platform in such a way that different therapeutic formats, i.e., multi-chain proteins, can be flexibly realized by automated pairing of individual protein chains on the DNA level (Figure 1d). We showcased the applicability of our system using two examples of distinct two-chain formats, the potency maturation of monovalent Fabs and the accommodation of optimized VRs in complex, bispecific CODV-Igs. Accordingly, in these examples, the system was configured to support two-chain formats (Supplementary Figure 1a and b). The settings, however, can be easily reconfigured to support any multi-chain protein of higher complexity, such as four-chain Tri-specific CODV-Igs38 (Supplementary Figure 1b). Hence, through its inbuilt chain-paring logic, the process is readily adaptable to accept any currently existing or future MSAT format and can support both format accommodation applications and exploration of novel format designs.
Compartmentalization is central for successful multi-parametric screening because it allows unbiased determination of functional activities and biophysical properties of individual variants. Different strategies for compartmentalization have been described, including agar plate-, microtiter plate-, water-in oil droplet- or whole cell-based technologies.30,39,40 Also, automated microtiter plate-based HT screens of non-deconvoluted variant libraries have proven to be successful in the maturation of antibodies to ultra-high, femtomolar-binding affinities.25 In our microtiter plate-based approach we explicitly use deconvoluted plasmid libraries instead, as it is a pre-requisite for the systematic pairing of plasmids that code for the individual chains of in-silico designed molecules. An additional advantage is that, in deconvoluted libraries, oversampling during screening is not required since all variants that comprise the library are already known ab-initio. This approach thus saves considerable time and resources. Of utmost importance, however, is the fact that it enables informed screening, i.e., real-time genotype/phenotype landscaping: the genotype of a distinct variant can be instantly correlated with its respective phenotypes through the registered plate-well position. By this, live observations such as production titers or kinetic measurements can be used immediately to feed computational methods for rational design41 (Figure 4d) or machine learning approaches,42 all of which can significantly accelerate subsequent protein engineering cycles.
One major aim of this project was to advance protein engineering strategies by providing data sets of large volume with great predictive capability.43,44 Through rigorous automation of lab unit operations, we were able to generate and characterize over 25,000 variants in two formats in a period as short as 12 months. We performed exhaustive genotype/phenotype landscaping over three engineering cycles to generate a large and diverse panel of viable MSAT variants to select from for downstream developability assessments. Concomitantly, HT-screening resulted in a very large set of multi-dimensional curated and contextualized data in machine-readable form. Attention should be paid to the fact that our compartmentalized process preserves data of weakly performing variants, which is important to efficiently train machine-learning models. In conclusion, having this process in place confers considerable advantages, as it allows us not only to screen large sets of MSATs in a very short period of time but at the same time set the foundation for data-driven rational design, more sophisticated virtual screening approaches and various strategies for deep data mining based on machine learning models, amongst others.30,42,44
Materials and methods
Molecular biology reagents and E. coli strains
T4 DNA ligase, Deoxynucleotide (dNTP) Solution Mix and 10x NEBuffer 2.1 were purchased from NEB (Ipswich, MA, Cat No. M0202M/N0447S/B7202S). Restriction enzymes Eco31I (BsaI) and DpnI were purchased from Thermo Fisher Scientific (Waltham, MA, Cat. No. FD0294/ER1701). PfuUltra II Fusion HS DNA Polymerase was purchased from Agilent Technologies (Santa Clara, CA, Cat. No. 600,670). Ampligase DNA Ligase was purchased from Illumina (Epicenter, San Diego, CA, Cat. No. 111,025 (A32250)).
Gene modules were designed as human codon optimized DNA constructs lacking BsaI restriction sites and ordered with flanking BsaI recognition sites and specific fusion sites in plasmids backbones carrying kanamycin resistance at Thermo Fisher Scientific (Waltham, MA).
For cloning in E. coli and gene expression in HEK293F cells, a pTT5 vector45 obtained from the National Research Council Canada (Ottawa, Ontario, Canada) was used. Manual plasmid DNA preparation were processed using the QIAGEN Plasmid Plus Midi Kit (Qiagen, Hilden, Germany, Cat. No. 12,943) following the manufacturer´s protocol. Manual measurements of plasmid DNA concentrations were done using a Little Lunatic spectrophotometer (Unchained Labs, Pleasanton, CA). Synthetic oligonucleotides were purchased from Merck KGaA (Sigma Aldrich, St. Louis, MO). NEB® 10-beta Competent E. coli (High Efficiency) cells were purchased from NEB (Ipswich, MA, Cat. No. C3019) and used for standard cloning. One Shot™ TOP10 Electrocomp™ E. coli cells were purchased from Thermo Fisher Scientific (Waltham, MA, CAt. No. C404052) and used for library cloning. One Shot ccdB Survival 2T1 cells (Thermo Fisher Scientific, Waltham, MA, Cat. No. A10460) were used for cloning and replication of ccdB containing library template plasmids.
Standard modular cloning
All modular cloning reactions were performed as described21 in one-pot restriction/ligation reactions containing 100 ng destination plasmid (~6 kb), 50 ng of each insert module (~2.6 kb), 10x T4 ligase buffer, 700 U T4 DNA ligase, 3.5 U of Eco31I (BsaI), in a total volume of 10 µl. The reaction was conducted in a thermo cycler according to the following cycling steps: 15 min/37°C, 7 min/37°C followed by 7 min/16°C (40 repeats), 45 min/37°C, 10 min/65°C. 0.5 µl of the modular cloning reaction were used for chemical transformation of 50 µl of NEB 10-beta Competent E. coli cells. Unique clones were analyzed by restriction analysis, colony PCR, and Sanger sequencing.
Saturation mutagenesis
Saturation mutagenesis was done by ligation during amplification (LDA) PCR using synthetic NNK-oligonucleotides (NNK-primers; N = A/G/T/C, K = G/T) each representing a sub-library for a single codon position. NNK primers were designed with 19–24 DNA template complementary nucleotides 5ʹ and 3ʹ of the NNK codon and a 5ʹ-phosphorylation. For a 50 µl LDA reaction, 100 ng of the template DNA plasmid was mixed with 0.5 µl PfuUltra II Fusion HS DNA Polymerase, 10 pmol NNK-Primer, and 2.5 U Ampligase DNA Ligase in PfuUltra II Fusion HS DNA Buffer, 200 µM dNTPs, and 1 mM NAD+ (Nicotinamide adenine dinucleotide). The reaction was conducted in a thermo cycler according to the following cycling steps: 3 min/95°C followed by 18 repeats of 1 min/95°C, 1 min/55°C, and 12 min/65°C. Subsequently, 25 U DpnI were added to the reaction mix and incubated at 37°C for 2 h. Then, DNA was purified by ethanol precipitation and the resulting DNA pellet was resuspended in 40 µl of water. For electroporation, 10 µl of the resuspended DNA and 45 µl of water were added to a 15 µl aliquot of TOP10 E. coli cells. After transformation, glycerol stocks were generated (stored at −80°C) and colony forming units (cfus) determined using titration on selective agar. A quality control for all sub-libraries was performed via Sanger sequencing using an overnight culture started from glycerol stocks. For colony picking, calculated volumes of the glycerol stocks were spread on agar OmniTrays (Thermo Fisher Scientific, Waltham, MA) to receive ~200 cfus per plate.
Modular library cloning
All modular libraries were generated using modular cloning technologies as previously described.20 In brief, a reaction with a total volume of 20 µl contained 20 fmol pTT5 destination plasmid and 20 fmol of each required module or module pre-mix, T4 ligase buffer, 1,400 U T4 DNA ligase, and 7 U of Eco31I (BsaI). The reaction was conducted in a thermo cycler according to the following cycling steps: 5 min/37°C, followed by 40 repeats of 7 min/37°C and 7 min/16°C, 30 min/37°C, and 10 min/65°C. For the electroporation of TOP10 E. coli cells, 1 µl of the modular cloning reaction was mixed with 54 µl water for 30 sec and chilled on ice. 15 µl TOP 10 E. coli cells were added and resuspended thoroughly before electroporation. 300 µl SOC medium was added to cells immediately after the electroporation, followed by careful resuspension and incubation for 1 h at 37°C and 800 rpm in a thermomixer. After transformation, glycerol stocks were generated by adding 300 µl of medium containing 34% (v/v) glycerol to the cell solution, mixing thoroughly, and aliquoting in new reaction tubes, before storage at −80°C. Cfus in glycerol stocks were determined using titration on selective agar plates. A quality control for all sub-libraries was performed via Sanger sequencing using an overnight culture started from the glycerol stocks. For colony picking, calculated volumes of the glycerol stocks were spread on agar OmniTrays (Thermo Fisher Scientific, Waltham, MA) to receive ~200 cfus per plate.
Automated colony picking and plasmid DNA preparation on the E. coli station and DNA handling station
The colonies were automatically picked by the E. coli station (ES; Figure 2a) built by Lab Services (Breda, The Netherlands). Plate handling was done by PlateButler® PreciseFlex750 robotic arm (Precise Automation, Fremont, CA; integration by Lab Services, Breda, The Netherlands); the integrated barcode reader was used for inventory scan. Source and destination plates were stored in random access plate hotels. A vacuum-based lid holder was used for lid handling. Maximum of 96 colonies per agar OmniTray (Thermo Fisher Scientific, Waltham, MA) were transferred into 96-deep well plates (DWPs; Thermo Fisher Scientific, Waltham, MA) that had been prefilled with 1.5 mL 2YT growth media (Becton Dickinson, Franklin Lakes, NJ) supplemented with ampicillin (Sigma-Aldrich, St. Louis, MO) using the integrated multi drop dispenser (Thermo Fisher Scientific, Waltham, MA). Norgren CP7200 (Wagner Life Science, Middleton, MA) represents the integrated picking unit. When the picking process was finalized, all inoculated plates were sealed with a semi-permeable membrane (Thermo Fisher Scientific, Waltham, MA) and incubated at 37°C and 800 rpm in a Microtron Shaker (Infors HT, Basel, Switzerland) overnight. The E. coli cells were harvested by centrifugation at 4,600 rpm for 5 min. Cell pellets were used for plasmid DNA isolation.
A set of four plates could be processed in parallel on the DNA handling station (DHS; Figure 2a) built by Lab Services (Netherlands). Plate handling was done by a PlateButler® PreciseFlex750 robotic arm (Lab Services, Breda, The Netherlands). Source and destination plates were stored in a Cytomat10 hotel (Thermo Fisher Scientific Waltham, MA) that performed the inventory scan. All pipetting steps were carried out by an integrated Hamilton STAR liquid handling device (Hamilton, Bonaduz, Switzerland) using a 96-channel pipetting head. PhyNexus (San Jose, CA) technology was used for plasmid DNA isolation. In a first step, cells were resuspended in 150 µL resuspension buffer, then lysed in 180 µL lysis buffer and precipitated by 210 µL of neutralization buffer. PhyNexus tips were equilibrated in PhyNexus Wash Buffer. The plasmid DNA was isolated directly from precipitated cell suspension by pipetting up and down slowly. PhyNexus tips were washed three times with 700 µL PhyNexus Wash Buffer and bound plasmid DNA was eluted with 150 µL elution buffer (10 mM Tris-Cl, pH 8.5; Qiagen, Hilden, Germany) into 96-well plates (Greiner Bio-One, Frickenhausen, Germany). Elution plates were sealed using a HJ SealPeel Station and Polyolefin foil (HJ-BIOANALYTIK GmbH, Erkelenz, Germany; Cat. No. 900,360) to avoid evaporation.
Measurement of plasmid DNA concentration on the DHS
A DropSense96 device (Trinean, Gentbrugge, Belgium) integrated in the DHS (Figure 2a) was used to determine plasmid DNA concentrations. Plate handling was done by a PlateButler® PreciseFlex750 robotic arm. Plates were stored in a Cytomat10 hotel that also performed the inventory scan. Due to the risk of condensation and cross contamination plates containing DNA were spun shortly for 1 min at 1,000xg (BioNex HIG™; BioNex Solutions, San Jose) and subsequently cover foils were peeled at the HJ SealPeel Station. All pipetting steps were carried out by an integrated Hamilton STAR liquid handling device using a 96-channel pipetting head. First 5 µL of elution buffer (10 mM Tris-Cl, pH 8.5; Qiagen, Hilden, Germany) that was provided in a reagent reservoir on the LHD deck were transferred into the cavities of a 96 High Lunatic plate (Unchained Labs, Gent, Belgium) and UV/Vis absorption was measured at the DropSense96 giving blank values. DNA solution from each plate was transferred and measured subsequently. DNA plates were sealed again using Polyolefin foil (HJ-BIOANALYTIK GmbH, Erkelenz, Germany; Cat. No. 900,360).
Normalization of DNA concentration on the DHS
Plate handling was done by a PlateButler® PreciseFlex750 robotic arm. Plates were stored in a Cytomat10 hotel that performs the inventory scan. Due to the risk of condensation and cross contamination plates containing DNA were spun shortly (BioNex HIG™ centrifuge, 1,000xg, 1 min) and subsequently cover foils were peeled at the HJ SealPeel Station. All pipetting steps were carried out by an integrated Hamilton STAR liquid handling device using a 96-channel pipetting head. DNA samples of known concentration were typically normalized to 36 ng/µL. A pipetting worklist was used to provide information of source positions, sample volume, and diluent volume. EB buffer (Qiagen, Hilden, Germany) was utilized to dilute the samples. EB buffer was transferred first from a reagent reservoir on the Hamilton deck, followed by sample transfer. Source and target plates were immediately sealed using Polyolefin foil (HJ-BIOANALYTIK GmbH, Erkelenz, Germany; Cat. No. 900,360) after processing.
Generation of DNA mixtures on the DHS
Plate handling was done by a PlateButler® PreciseFlex750 robotic arm. Plates were stored in a Cytomat10 hotel that performs the inventory scan. Due to risk of condensation and cross-contamination, plates containing DNA were spun shortly (BioNex HIG™ centrifuge, 1,000 x g, 1 min), and then cover foils were peeled at the HJ SealPeel Station. All pipetting steps were carried out by an integrated Hamilton STAR liquid handling device using a 96-channel pipetting head. Up to five destination plates were loaded on Hamilton STAR deck. Source plates were loaded one after the other. Combinations of plasmid DNAs coding for HC and LC were created according to a pipetting list. The pipetting list provided information on source positions, sample volumes, and destination positions. In total, 150 µL of transfection DNA mix was prepared, resulting in two possible transfection reactions each containing 1.2 µg plasmid DNA in 50 µL. First, 50 µL of oriP/EBNA vector46 (4 ng/µL) was transferred into 384-well destination plates (Corning, NY; Cat. No. 3342) using a 96 pipetting head. Second, 50 µL of each DNA sample was pipetted into single cavities of the destination plate using the 8-channel head. Each source plate was immediately sealed using Polyolefin foil (HJ-BIOANALYTIK GmbH, Erkelenz, Germany; Cat. No. 900,360) at HJ PeelSeal station. All sample plates (hereinafter named the transfection ready DNA (TD) plates) were sealed at the end of the run. All plate transfer operations were tracked in GDB by uploading automatically generated DHS report files.
Sequence deconvolution workflow
The whole sequence data analysis procedure was automated using an inhouse implemented bioinformatic tool (Python-based), which allowed the analysis and identification of thousands of clones in less than a day (Supplementary Figure 3). The sequence analysis workflow started with the assembly of all possible DNA reference sequences by combining the DNA fragments of building blocks in a combinatorial fashion for modular cloning approaches (as shown above). In the case of mutational libraries, all theoretical DNA sequences were generated on a DNA level by introducing all NNK codons in specified positions of the wild-type sequence string. The generated reference sequences were stored as multi-sequence FASTA file, which served as input for the following steps. For each well (every well should only contain one specific variant), all available Sanger reads (provided by the DNA sequencing CRO Qiagen, Hilden, Germany) were organized as separate FASTA files. All individual well positions of the 96-well sequencing plates were analyzed consecutively, and the results of individual reads were combined in a last step to identify the clone variant. Before starting the analysis of the individual reads, reversed reads were transformed into the reversed complement sequence strings. In a first step, a predefined number (usually 25–50 depending on the read quality and position of the sequencing primers) of nucleotides from the beginning of the read were removed to get rid of positions with potentially high uncertainty. The resulting trimmed string was compared against the whole library of DNA reference sequences. If no 100% substring match could be identified, 10 nucleotides from the end of the read were removed and the resulting trimmed sequence string was again compared against the full library of DNA reference sequences. This procedure of removing the last 10 nucleotides and comparing the resulting trimmed string against the reference library was repeated until one or more 100% substring matches within the reference library were identified or until the length of the trimmed read fell below a predefined threshold. This trimming and mapping procedure has been conducted for all available reads for a specific variant (well), resulting in a hitlist of potential reference sequence matches for each read. The hitlists for the individual reads contain all 100% identity substring matches that could be identified. In a last step, the intersection of all hitlists was calculated, i.e., if all available hitlists contained exactly one shared reference sequence match, this match represented the identity of the examined clone variant. Custom Python code used in this study is available from the corresponding author upon reasonable request.
Cell culture and transfection on the protein science station
Suspension-adapted HEK293-F cells (Thermo Fisher Scientific Waltham, MA; Cat. No. R79007) were cultivated in Freestyle F17 Expression medium (Thermo Fisher Scientific, Waltham, MA; Cat. No. A1383502) containing 6 mM glutamine (Thermo Fischer Scientific, Waltham, MA; Cat. No. 25,030,024). The day prior to transfection 800 ml of cells were seeded at a density of 1.3E6 cells/ml in 3 L Fernbach Erlenmeyer flasks with vent cap (Corning, NY; Cat. No. 431,252) and incubated overnight at 37°C with agitation at 110 rpm (50 mm orbit), 8% CO2 and 75% humidity in a Multitron Pro Shaker (Infors HT, Basel, Switzerland). In a next step, the transfection-ready DNA plates were thawed, and the protein science station (PSS) (Figure 2a) was used to transfer 50 µl DNA of each sector of a TD plate to four 96-deep well expression plates (EX) (Thermo Fisher Scientific, Waltham, MA; Cat. No. 10,447,181); Lid (Greiner Bio-One, Frickenhausen, Germany; Cat. No. 691,161). This process was automatically repeated for up to 24 cycles per day. The handling of 24 TD plates resulted in a maximum number of 96 EX plates. The DNA-containing EX plates were stored in the Cytomat 10 C incubator (Thermo Fisher Scientific, Waltham, MA) overnight at 8°C. The TD plates were sealed, manually removed from the PSS for long-term storage at −20°C. The DNA transfer of 24 TD plates to 96 EX plates took about 2 hours.
At the day of transfection, a 1 mg/ml polyethylenimine (PEI) 25 K stock solution pH 6.6 (Polysciences Europe GmbH, Hirschberg, Germany; Cat. No. 23,966–2) was diluted to 17 µg/ml with Freestyle F17 Expression medium (Thermo Fisher Scientific, Waltham, MA; Cat. No. A1383502) containing 6 mM glutamine (Thermo Fisher Scientific, Waltham, MA: Cat. No. 25,030,024) and installed on a Midi MR1 magnetic stirrer (IKA®-Werke GmbH & Co. KG, Staufen, Germany) of the PSS sterile dispensing unit in an opaque 2 L glass bottle (Schott AG, Mainz, Germany; Schott Duran 2l80663). The PEI was kept in solution at room temperature by rigorous stirring, while avoiding foaming. In a next step, the HEK293F cells were adjusted to 2.1E+06 cells/ml with F17 expression medium containing 6 mM glutamine. Three liters of the cells were transferred into a disposable 3 L Spinner Flask (Thermo Fisher Scientific, Waltham, MA; Cat. No. 10,337,203) and installed on the Midi MR1 magnetic stirrer 2 of the PSS sterile dispensing unit. The cells were kept in suspension by rigorous stirring, while avoiding foaming. To avoid cooling of the cells, the spinner flask was wrapped with a custom heating jacket (Horst GmbH, Lorsch, Germany), where the temperature control was set to 37°C. In a next step, the 2 m long tubing cassettes (Thermo Fisher Scientific, Waltham, MA; Cat. No. 24,072,677) of the Multidrop devices of the sterile dispensing unit were installed and the ends of the tubing were inserted into the HEK293F cell and PEI solution. The PSS was then used to dispense 200 µl (2.8 µg) of PEI solution with Multidrop device 1 to column 1–11 of EX plates. After 12 minutes incubation, 950 µl µl cells were dispensed with Multidrop device 2 to the DNA-PEI mix in column 1–11 of the same plate.
This process was automatically repeated for up to 96 cycles per day, while the PlateButler® Software managed the scheduling of the process. To handle 96 EX plates, the 3 L Spinner flask must be exchanged 3 times with a new one. This was done on the fly while the PSS system was running. The transfected cells in the EX plates were successively removed manually from the PlateButler® PlateDispenser (Lab Services, Breda, The Netherlands) and the plastic lids were exchanged by metallic low-evaporation DUETZ system lids (Enzyscreen BV, Heemstede, The Netherlands; Cat. No. CR1296a). The plates were mounted on an Enzyscreen Cover clamp system (Enzyscreen BV, Heemstede, The Netherlands; Cat. No. CR1700) installed on a Multitron Pro shaker and incubated for 7 days at 37°C, 8% CO2, 75% humidity and 1,000 rpm shaking with 3 mm orbit. Each shaker provided space for 16 DWPs. The automated transfection of 96 EX plates took about 6.5 hours. All plate handling operations were done by a PlateButler® PreciseFlex1160 robotic arm (Precise Automation, Fremont, CA; integration by Lab Services, Breda, The Netherlands) and plate transfer operations were tracked in GDB by uploading automatically generated PSS report files.
Harvesting and preparation of quantification assay plates on the PSS
After 7 days of expression, the EX plates were manually removed from the Multitron shakers and the DUETZ lids were exchanged by regular plastic lids. Cell suspension containing EX plates, 96-deep well plates for the supernatant (SN) (Thermo Fisher Scientific, Waltham, MA; Cat. No. 10,462,214) and 384-well assay plates for the BLI quantification (BQ) (Thermo Fisher Scientific, Waltham, MA; Cat. No. 10,090,212) were loaded onto the PSS station. The PSS station was then used to spin the EX plates (5 minutes at 1,000 g) and to transfer 1,000 µl supernatant of the sedimented cell suspension to the SN plates using the 96-channel head of the Hamilton LHD. In the same run, a 10 µl sample aliquot was transferred to one sector of the BQ plate. One BQ plate finally contained the supernatant of four SN plates. SN and BQ plates were sealed and the EX plates were discarded. The process described was automatically repeated for up to 24 cycles to manage 96 EX plates per day, whereas the PlateButler® Software managed the scheduling of the process. Finally, all plates were removed from the PSS and are stored at 4°C (BQ plates) or −20°C (SN plates). The automated harvesting and BQ plate preparation of 96 EX plates took about 6 hours. All plate handling operations were done by a PlateButler® PreciseFlex1160 robotic arm and plate transfer operations were tracked in GDB by uploading automatically generated PSS report files.
Quantification of supernatants on the PSS
The protein-containing supernatants were quantified by BLI using the Octet HTX system (Sartorius FortéBio, Göttingen, Germany). The quantification of Fab molecules by using HIS2 biosensors (Sartorius FortéBio, Göttingen, Germany; Cat. No. 18–5116) was performed as follows: 10 µl supernatant on BQ plates was diluted 1:10 with 90 µl Dulbecco’s phosphate-buffered saline (D-PBS) (Thermo Fisher Scientific, Waltham, MA; Cat. No. 14,190–094) + 0.1% bovine serum albumin (BSA) (Miltenyi Biotec 130–091-376). Quantification time was set to 80 sec, regeneration/neutralization of biosensors was done with 10 mM glycine/HCl, pH 1.7 and D-PBS for 5 sec, using three cycles before the first measurement and between all following measurements. The quantification assay was performed at 25°C while shaking at 1,000 rpm. The sensor offset was set to 5 mm. The experiment started after a delay of 600 sec to equilibrate the plate for 10 min (25°C and shaking). Two BQ plates were analyzed with one biosensor and 96 channels in high-throughput acquisition mode.
The quantification of CODV molecules by using ProA biosensors (Sartorius FortéBio, Göttingen, Germany; Cat. No. 18–5013) was performed as follows: 10 µl supernatant on BQ plates were diluted 1:10 with 90 µl D-PBS (Thermo Fisher Scientific, Waltham, MA; Cat. No. 14,190–094) + 0.1% BSA (Miltenyi Biotec B.V. & Co. KG, Bergisch Gladbach, Germany; Cat. No. 130–091-376). Quantification time was set to 120 s, regeneration/neutralization of biosensors was done with 10 mM glycine/HCl, pH 1.7 and D-PBS for 5 s, using three cycles before the first measurement and between all following measurements. The assay was performed at 30°C while shaking at 1,000 rpm. The sensor offset was set to 5 mm. The experiment started after a delay of 600 s to equilibrate the plate for 10 min (30°C and shaking). Five BQ plates were analyzed with one biosensor and 96 channels in high-throughput acquisition mode.
The PSS station was used for sample dilution and Octet HTX instrument loading. The automated assay preparation and analysis of 24 BQ plates took about 8 hours.
A format-specific external standard curve was generated for each campaign. Binding rates were calculated via initial slope and standard curve fitting was done with 5PL unweighted fit. The low concentration threshold binding rate was set at 0.003. The binding rates of the samples were measured and interpolated from the standard curve to determine the concentration. Data analysis was done using FortéBio Data Analysis HT 11.0.0.50 software. Assay data were uploaded to GDB. All plate handling operations were done by a PlateButler® PreciseFlex1160 robotic arm and plate transfer operations were tracked in GDB by uploading automatically generated PSS report files.
Hitpicking of supernatants on the PSS
The PSS station was used for cherry picking of selected hit supernatants from SN plates into 96-well supernatant hit selection plates (Thermo Fisher Scientific, Waltham, MA; Cat. No. 10,462,214). Volumes, and source and destination plate well locations were predefined in a worklist generated by GDB. SN and SH plates were sealed after supernatant transfer using the HJ SealPeel Station. The process described was automatically repeated until the worklist had been completed, while the PlateButler® Software managed the scheduling of the process. All plate handling operations were done by a PlateButler® PreciseFlex1160 robotic arm and plate transfer operations were tracked in GDB by uploading automatically generated PSS report files.
Normalization of supernatants on the PSS
After quantification, the concentrations of the hit supernatants were normalized to 60 µg/ml in a final volume of 150 µl (stage 2, CODV library). The 8-channel head of the PSS LHD was used for the pipetting of Mock diluent (provided in a separate reservoir on the LHD deck) and supernatant from the SH plate to an empty normalization supernatant plate (NS) (Thermo Fisher Scientific, Waltham, MA; Cat. No. 0030504119). The described process was automatically repeated until the worklist had been completed, while the PlateButler® Software managed the scheduling of the process. Finally, all plates were removed from the PSS and were stored at 4°C (NS) or −20°C (SH plates). All plate handling operations were done by a PlateButler® PreciseFlex1160 robotic arm and plate transfer operations were tracked in GDB by uploading automatically generated PSS report files.
Preparation of binding and cell-based assay plates on the PSS
All binding and cell-based assays were performed in 384-well format. The PSS was used to prepare the BLI binding (BB) and cell-based assay (CA) plates by transferring the supernatants from the 96-well sample plates to 384-well assay plates with the 96-channel head of the Hamilton LHD. In stage 1 (Fab level), 10 µl supernatant of 4 individual SN or 4 SH 96-well plates were transferred into 1 BB 384-well plate and 50 µl supernatant of 4 individual SN or 4 SH 96-well plates into 1 CA plate. In stage 2 (CODV-Ig level), 10 µl 1:2 in Mock diluted supernatant of individual 2 SN or 2 SH plates were reformatted into sector 1 and 2 of 1 BB plate and 50 µl SN of 4 individual SN or 4 SH plates into 1 CA plate. All plates were sealed after transfer. The process described was automatically repeated up to 48 cycles, while the PlateButler® Software managed the scheduling of the process. All plate handling operations were done by a PlateButler® PreciseFlex1160 robotic arm and plate transfer operations were tracked in GDB by uploading automatically generated PSS report files.
BLI Binding Assay
The Octet® HTX system (Sartorius FortéBio, Göttingen, Germany) was used for T1 and T2 off-rate screenings. In stage 1 (Fab level), the supernatants were diluted 1:10 (round 1) or 1:50 (round 2) in D-PBS (Thermo Fisher Scientific, Waltham, MA; Cat. No. 14,190–094) + 0.1% BSA (Miltenyi Biotec B.V. & Co. KG, Bergisch Gladbach, Germany; Cat. No. 130–091-376). 0.2 µg/ml recombinant human T2 was loaded on SA biosensors (Sartorius FortéBio, Göttingen, Germany; Cat. No. 18–5021) for 300 sec. The SA Biosensors were then regenerated and neutralized with 10 mM glycine/HCl, pH 1.7 and D-PBS for 5 s and 3 times each, prior to the first measurement and between all following measurements. After a 60 sec baseline step in D-PBS + 0.1% BSA, the biosensors were dipped into the diluted Fab supernatants on the BB plates for 300 s to record the association kinetics. The dissociation kinetics were then reported by dipping the biosensors in D-PBS + 0.1% BSA for 600 sec (round 1) or 1,200 s (round 2) followed by a regeneration step to wash off the remaining Fabs. Then, the next sector was analyzed accordingly (Figure 5, baseline – association – dissociation – regeneration). All samples were measured in a single-referenced manner by using D-PBS + 10% mock SN + 0.1% BSA as analyte. One biosensor was used to analyze 1 BB plate. Binding curves were analyzed with a local full fit using a 1:1 model.
In stage 2 (CODV-Ig level), 10 µl of the 30 µg/ml normalized and diluted supernatants on BB plates were further diluted 1:10 with 90 µl D-PBS (Thermo Fisher Scientific, Waltham, MA; Cat. No. 14,190,094) + 0.1% BSA (Miltenyi Biotec B.V. & Co. KG, Bergisch Gladbach, Germany; Cat. No. 130–091-376) to a final concentration of 3 µg/ml. The expressed CODVs in the diluted samples were loaded on an AHC biosensor (Sartorius FortéBio, Göttingen, Germany; Cat. No. 18–5064) for 300 sec (Loading step). After a 60 sec baseline step in D-PBS + 0.1% BSA, the biosensors were dipped into 15 nM recombinant T2 or 5 nM recombinant human T1 for 300 s to record the association kinetics. The dissociation kinetics were then reported by dipping the biosensors in D-PBS + 0.1% BSA for 1,800 sec. The AHC Biosensors were regenerated and neutralized with 10 mM glycine/HCl, pH 1.7 and D-PBS for 5 s and 3 times each, before the first measurement and between all following measurements to wash away both analyte and ligand. The second plate sector was then analyzed accordingly (baseline – loading – baseline – association – dissociation – regeneration). All samples of round 2 were measured in a single-referenced manner by using D-PBS + 10% mock SN + 0.1% BSA as ligand. Binding curves were analyzed with a local full fit using a 1:1 model. One biosensor was used to analyze 2 BB plates. All data were uploaded to GDB.
Inhibition of T2-mediated STAT protein phosphorylation in vitro from THP-1 Cells
THP-1 is a human monocytic cell line derived from an acute monocytic leukemia patient and naturally expresses the T2-receptor (T2R). Incubation of THP-1 cells with human T2 results in the increase of phosphorylation of a STAT signaling protein, which can be measured using commercially available HTRF phospho STAT kits (CisBio, Codolet, France). THP-1 cells are routinely cultured in assay medium (RPMI 1640 containing 10% fetal bovine serum). On the day the assay was performed, cells were harvested after centrifugation for 6 min at 1,000 rpm, and then resuspended in serum-free RPMI 1640 (without phenol red) at 6E+06 cells per ml. 5 µl of 30,000 cells was added to a well of a small volume 384-well plate (Greiner bio-one, Frickenhausen, Germany; Cat. No. 781,080) and followed by the addition of 5 µl of test sample. After preincubation of the plate at 37°C (with 5% CO2, 95% humidity) for 30 minutes, 5 µl per well of T2 at a final concentration of 3.3 ng/ml was added and the plate was incubated for another 30 minutes at 37°C (with 5% CO2, 95% humidity). Measurement of STAT protein phosphorylation by HTRF was performed according to manufacturer instructions with a standalone PheraStar FXS device or a PSS-integrated Clariostar reader (both BMG LABTECH, Ortenberg, Germany) at 620 nm/665 nm. Results were expressed as the concentration where 50% of the T2-induced STAT protein phosphorylation was inhibited (IC50s) and the dose response curves were fitted using 4 parameters sigmoidal fit of the data using XLfit® (idbs, Guildford, Surrey, UK) software. All data were uploaded to GDB.
Supplementary Material
Acknowledgments
The authors are grateful to the following individuals for their contributions to this work: Anja Wille (Sanofi), Katrin Reichel (Sanofi) and Robbert van de Worp (LS Labservices).
Abbreviations
| BB plates | BLI binding plates |
| BLI | Biolayer interferometry |
| CA plates | Cell-based assay plates |
| CDR | Complementarity-determining region |
| Cfu | Colony forming unit |
| CODV-Ig | Cross-over dual variable domain-Immunoglobulin |
| CR | Constant region |
| CV | Coefficient of variation |
| DCHAIN | Design chain |
| DHS | DNA handling station |
| DLP | Drug-like property |
| DMOL | Design molecule |
| DWP | Deep well plate |
| ES | E. coli station |
| EX plate | Expression plate |
| Fab | Fragment antigen-binding |
| Fc | Fragment crystallizable |
| FS | Cleavage/fusion site |
| GDB | GeneData Biologics |
| GR | Generic region |
| GR-L | Generic region of linker peptide type |
| HC | Heavy chain |
| HEK cells | Human Embryonic Kidney Cells |
| HT | High throughput |
| IC50 | 50% inhibitory concentration |
| koff | dissociation rate constants |
| Ig | Immunoglobulin |
| LC | Light chain |
| LHD | Liquid handling device |
| LUO | Lab unit operation |
| M | Molar |
| mAb | Monoclonal antibody |
| Max | Maximum |
| Med | Median |
| Min | Minimum |
| MSAT | Multi-specific antibody therapeutic |
| NS plate | Normalization supernatant plate |
| PEI | Polyethylenimine |
| PSS | Protein Science Station |
| Rec | Recombination |
| RS | Recognition site |
| SBS | Society for Biomolecular Screening |
| SD | Standard deviation |
| SH plate | Supernatant hit selection plate |
| STAT | Signal transducer and activator of transcription |
| T1 | Target number one |
| T2 | Target number two |
| TD plate | Transfection ready DNA plate |
| TF | Transfection |
| VH | Variable domain of heavy chain |
| VL | Variable domain of light chain |
| VR | Variable region |
Disclosure statement
All authors are employees of Sanofi and may hold shares and/or stock options in the company.
Author contributions
N.F., M.S., N.S., W.D.L., I.F., A.D., W.D., J.K., T.S., and J.B. designed built and implemented the automated platform processes. N.F., W.D.L, J.M., D.D., K.K. and J.B. designed and implemented the data management and analytics pipeline. N.F., M.S., N.S., B.S., Z.L., D.D., K.K., A.D., C.L. and J.B. designed performed and analysed experiments. M.M. solved the crystallographic structure. M.S., N.F., Z.L. and J.B. designed the experimental strategy and supervised analysis. N.F., M.S., B.S., N.S., Z.L. and J.B. wrote the manuscript. All authors participated in manuscript review and revisions.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Labrijn AF, Janmaat ML, Reichert JM, Parren P.. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov PMID: 31175342. 2019;18:585–19. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 2.Suurs FV, Lub-de Hooge MN, De Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther PMID: 31028837. 2019;201:103–19. doi: 10.1016/j.pharmthera.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs PMID: 31847708. 2020;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Runcie K, Budman DR, John V, Seetharamu N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol Med PMID: 30249178. 2018;24(1):50. doi: 10.1186/s10020-018-0051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheridan C. Bispecific antibodies poised to deliver wave of cancer therapies. Nat Biotechnol PMID: 33692520. 2021;39(3):251–54. doi: 10.1186/s10020-018-0051-4. [DOI] [PubMed] [Google Scholar]
- 6.Wang Q, Chen Y, Park J, Liu X, Hu Y, Wang T, McFarland K, Betenbaugh MJ. Design and production of bispecific antibodies. Antibodies (Basel). 2019;8. PMID: 31544849. doi: 10.3390/antib8030043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawant MS, Streu CN, Wu L, Tessier PM. Toward drug-like multispecific antibodies by design. Int J Mol Sci. 2020;21. PMID: 33053650. doi: 10.3390/ijms21207496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y. A brief introduction of IgG-like bispecific antibody purification: methods for removing product-related impurities. Protein Expr Purif PMID: 30513344. 2019;155:112–19. doi: 10.1016/j.pep.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 9.Dickopf S, Georges GJ, Brinkmann U. Format and geometries matter: structure-based design defines the functionality of bispecific antibodies. Comput Struct Biotechnol J PMID: 32542108. 2020;18:1221–27. doi: 10.1016/j.csbj.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol PMID: 25637431. 2015;67(2):95–106. doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs PMID: 28071970. 2017;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brinkmann U, Kontermann RE. Bispecific antibodies. Science PMID: 34045345. 2021;372(6545):916–17. doi: 10.1126/science.abg1209. [DOI] [PubMed] [Google Scholar]
- 13.Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, Yoshihashi K, Okuyama-Nishida Y, Saito H, Tsunoda H, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570–74. PMID: 23023498. doi: 10.1038/nm.2942. [DOI] [PubMed] [Google Scholar]
- 14.Kitazawa T, Shima M. Emicizumab, a humanized bispecific antibody to coagulation factors IXa and X with a factor VIIIa-cofactor activity. Int J Hematol PMID: 30350119. 2020;111(1):20–30. doi: 10.1007/s12185-018-2545-9. [DOI] [PubMed] [Google Scholar]
- 15.Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8(2):e57479. PMID: 23468998. doi: 10.1371/journal.pone.0057479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dengl S, Mayer K, Bormann F, Duerr H, Hoffmann E, Nussbaum B, Tischler M, Wagner M, Kuglstatter A, Leibrock L, et al. Format chain exchange (FORCE) for high-throughput generation of bispecific antibodies in combinatorial binder-format matrices. Nat Commun. 2020;11(1):4974. PMID: 33009381. doi: 10.1038/s41467-020-18477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geuijen CAW, De Nardis C, Maussang D, Rovers E, Gallenne T, Hendriks LJA, Visser T, Nijhuis R, Logtenberg T, de Kruif J, et al. Unbiased combinatorial screening identifies a bispecific igg1 that potently inhibits her3 signaling via her2-guided ligand blockade. Cancer Cell. 2018;33(5):922–36e10. PMID: 29763625. doi: 10.1016/j.ccell.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann T, Krah S, Sellmann C, Zielonka S, Doerner A. Greatest hits-innovative technologies for high throughput identification of bispecific antibodies. Int J Mol Sci. 2020;21. PMID: 32911608. doi: 10.3390/ijms21186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinmetz A, Vallee F, Beil C, Lange C, Baurin N, Beninga J, Capdevila C, Corvey C, Dupuy A, Ferrari P, et al. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. MAbs. 2016;8(5):867–78. PMID: 26984268. doi: 10.1080/19420862.2016.1162932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber E, Birkenfeld J, Franz J, Gritzan U, Linden L, Trautwein M. Modular Protein Expression Toolbox (MoPET), a standardized assembly system for defined expression constructs and expression optimization libraries. PLoS One PMID: 28520717. 2017;12(5):e0176314. doi: 10.1371/journal.pone.0176314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S. A modular cloning system for standardized assembly of multigene constructs. PLoS One PMID: 21364738. 2011;6(2):e16765. doi: 10.1371/journal.pone.0016765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duetz WA. Microtiter plates as mini-bioreactors: miniaturization of fermentation methods. Trends Microbiol PMID: 17920276. 2007;15(10):469–75. doi: 10.1016/j.tim.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Wendt M, Cappuccilli G. Data-Driven Antibody Engineering Using Genedata Biologics. Methods Mol Biol PMID: 28255885.. 2017;1575:237–50. doi: 10.1007/978-1-4939-6857-2_15. [DOI] [PubMed] [Google Scholar]
- 24.Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten JW, Da Silva Santos LB, Bourne PE, et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data. 2016;3(1):160018. PMID: 26978244. doi: 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Votsmeier C, Plittersdorf H, Hesse O, Scheidig A, Strerath M, Gritzan U, Pellengahr K, Scholz P, Eicker A, Myszka D, et al. Femtomolar Fab binding affinities to a protein target by alternative CDR residue co-optimization strategies without phage or cell surface display. MAbs. 2012;4(3):341–48. PMID: 22531438. doi: 10.4161/mabs.19981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci PMID: 25821140. 2015;104(6):1885–98. doi: 10.1002/jps.24430. [DOI] [PubMed] [Google Scholar]
- 27.Becker W, Scherer A, Faust C, Bauer DK, Scholtes S, Rao E, Hofmann J, Schauder R, Langer T. A fully automated three-step protein purification procedure for up to five samples using the NGC chromatography system. Protein Expr Purif PMID: 30102973. 2019;153:1–6. doi: 10.1016/j.pep.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. 2013;110(13):5145–50. doi: 10.1073/pnas.1220145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labrijn AF, Meesters JI, Priem P, de Jong RN, van den Bremer ET, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat Protoc PMID: 23479652. 2014;9:2450–63. doi: 10.1073/pnas.1220145110. [DOI] [PubMed] [Google Scholar]
- 30.Dyson MR, Masters E, Pazeraitis D, Perera RL, Syrjanen JL, Surade S, Thorsteinson N, Parthiban K, Jones PC, Sattar M, et al. Beyond affinity: selection of antibody variants with optimal biophysical properties and reduced immunogenicity from mammalian display libraries. MAbs. 2020;12(1):1829335. PMID: 33103593. doi: 10.1080/19420862.2020.1829335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beerli RR, Bauer M, Buser RB, Gwerder M, Muntwiler S, Maurer P, Saudan P, Bachmann MF. Isolation of human monoclonal antibodies by mammalian cell display. Proc Natl Acad Sci U S A PMID: 18812621. 2008;105(38):14336–41. doi: 10.1073/pnas.0805942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowers PM, Horlick RA, Neben TY, Toobian RM, Tomlinson GL, Dalton JL, Jones HA, Chen A, Altobell L 3rd, Zhang X, et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc Natl Acad Sci U S A. 2011;108(51):20455–60. PMID: 22158898. doi: 10.1073/pnas.1114010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuch de Zafra CL, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL, Stevens J, Case R, Pearson JT, Liu S, et al. Targeting multiple myeloma with AMG 424, a novel anti-CD38/CD3 bispecific T-cell-recruiting antibody optimized for cytotoxicity and cytokine release. Clin Cancer Res. 2019;25(13):3921–33. PMID: 30918018. doi: 10.1158/1078-0432.CCR-18-2752. [DOI] [PubMed] [Google Scholar]
- 34.Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. MAbs. 2020;12(1):1743053. PMID: 32249670. doi: 10.1080/19420862.2020.1743053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng PMID: 8844834. 1996;9(7):617–21. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 36.Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol. 2013;31(8):753–58. PMID: 23831709. doi: 10.1038/nbt.2621. [DOI] [PubMed] [Google Scholar]
- 37.Deshaies RJ. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature PMID: 32296187. 2020;580(7803):329–38. doi: 10.1038/s41586-020-2168-1. [DOI] [PubMed] [Google Scholar]
- 38.Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, Lord DM, Wei RR, Deng G, Louder M, et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358(6359):85–90. PMID: 28931639. doi: 10.1126/science.aan8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clackson T, Wells JA. In vitro selection from protein and peptide libraries. Trends Biotechnol PMID: 7764900. 1994;12(5):173–84. doi: 10.1016/0167-7799(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 40.Krebber A, Burmester J, Pluckthun A. Inclusion of an upstream transcriptional terminator in phage display vectors abolishes background expression of toxic fusions with coat protein g3p. Gene PMID: 8921894. 1996;178(1–2):71–74. doi: 10.1016/0378-1119(96)00337-x. [DOI] [PubMed] [Google Scholar]
- 41.Norman RA, Ambrosetti F, Bonvin A, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform PMID: 31626279. 2020;21(5):1549–67. doi: 10.1093/bib/bbz095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurumida Y, Saito Y, Kameda T. Predicting antibody affinity changes upon mutations by combining multiple predictors. Sci Rep PMID: 33177627. 2020;10(1):19533. doi: 10.1038/s41598-020-76369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obrezanova O, Arnell A, de la Cuesta RG, Berthelot ME, Gallagher TR, Zurdo J, Stallwood Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. MAbs PMID: 25760769.1. 2015;7(2):352–63. doi: 10.1080/19420862.2015.1007828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narayanan H, Dingfelder F, Butte A, Lorenzen N, Sokolov M, Arosio P. Machine learning for biologics: opportunities for protein engineering, developability, and formulation. Trends Pharmacol Sci PMID: 33500170. 2021;42(3):151–65. doi: 10.1016/j.tips.2020.12.004. [DOI] [PubMed] [Google Scholar]
- 45.Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res PMID: 11788735. 2002;30(2):E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Young JM, Cheadle C, Foulke JS Jr., Drohan WN, Sarver N. Utilization of an Epstein-Barr virus replicon as a eukaryotic expression vector. Gene PMID: 2835291. 1988;62(2):171–85. doi: 10.1016/0378-1119(88)90556-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.