

ABSTRACT
Adoptive transfer of tumor-infiltrating lymphocytes (TIL) elicits the regression of metastatic malignancies, yet a low proportion of patients achieve complete durable responses. The high incidence of relapse in these patients highlights the need to better understand mechanisms of tumor escape from T cell control. While melanoma has provided the foundation for developing TIL therapy, much less is known about TIL efficacy and relapse in other malignancies. We sought to investigate TIL characteristics in mouse tumors which have not been studied in this setting. Here, we expanded murine TIL ex vivo in IL-2 from fragments of multiple tumor models, including oral cavity cancer models of varying immunogenicity. Additionally, TIL was expanded from pmel-1 mice bearing B16F10 melanoma, yielding an enriched population of tumor-infiltrating TCR transgenic T cells. Murine TIL are similar to human TIL in that they express high levels of inhibitory receptors (PD-1, Tim-3, etc.) and can be expanded ex vivo in IL-2 extensively. Of clinical relevance, we draw parallels between murine and human oral cavity cancer TIL, evaluating relationships between inhibitory receptor expression and function. This platform can be used by labs even in the absence of clinical specimens or clean cell facilities and will be important to more broadly understand TIL phenotypes across many different malignancies.
KEYWORDS: Adoptive T cell therapy, TIL therapy, tumor-infiltrating lymphocyte, solid tumors, T cell, ACT
Introduction
The adoptive transfer of large numbers of tumor-infiltrating lymphocytes (TIL) is a promising clinical immunotherapy strategy as objective responses of up to 50% have been reported in patients with metastatic disease.1–3 Modern TIL expansion protocols were derived from early discoveries that large numbers of T cells could mediate long-lived immunity to tumors, especially when supported by exogenous IL-2.4,5 Multiple methods of TIL expansion have been described, each seeking to improve cell yield and reactivity to tumors. Two distinct methods include either culturing single-cellular digests of TIL and tumor cells or generating “tumor microcultures”; the latter allows T cells to egress from tumor fragments, and is more efficient for identifying tumor-reactive clones.6 While the cell-suspension method has been routinely tested in murine sarcoma, leukemia, and colorectal cancer models since the 1980s,4,5 the feasibility of establishing microcultures from tumor fragments in other murine cancers as preclinical models of TIL therapy is relatively undescribed.
Beyond direct isolation of T cells from mouse tumors, TIL models have expanded to include adoptive transfer of transgenic T cells, obtained from mice modified to express T cell receptors (TCRs) specific to antigens expressed in tumor lines.7–10 Two transgenic models for melanoma, the pmel-1 and the TRP-1 models, provide a source of T cells which target naturally expressed melanocyte differentiation antigens from gp100 or tyrosinase.9,10 Other T cell-transgenic models are created via nuclear transfer, which allows versatility in selecting a range of TCRs with varied affinities against antigens of interest.11,12 These models have been a critical proxy for TIL therapy as they allow study of tumor-reactive T cells and their function after adoptive transfer into recipients with established malignancies.
However, there remain disadvantages for using TCR transgenic mice to model TIL therapy. Transgenic T cells are often obtained from the animal’s spleen or lymph nodes prior to adoptive transfer, which provides a source of antigen-specific T cells, yet the cells have not encountered tumor unlike a patient’s TIL. As T cells obtained peripherally are likely to be inherently distinct from those that reside in the tumor, the starting T cell product may be less clinically relevant compared to patient TIL. Additionally, TIL models for cancers beyond melanoma are underdeveloped; therefore, there remains poor understanding of the mechanisms underlying regression and relapse after ACT therapy in other solid tumors.
Herein, we reveal the feasibility of expanding TIL from murine tumor fragments, including a clinically relevant approach to expand transgenic T cells from tumors on transgenic mice. By presenting both the feasibility of expanding TIL ex vivo, and the characteristics of expanded TIL from both murine and human tumors, we describe a platform which can be used to evaluate outcomes to TIL therapy in combination with checkpoint blockade or other immune modulating strategies in weakly immunogenic tumors.
Results
Modeling clinical TIL expansion with murine solid tumors
We posited that murine TIL could be expanded from tumor fragments similar to human TIL expansion.6 We implanted three different tumor cell lines on syngeneic immunocompetent mice, including two oral cavity squamous cell carcinoma (hereafter referred to as OCSCC or OCC) lines (Moc2 and Moc22) and one Lewis Lung carcinoma cell line (LLC-A9F1). After growing for at least 1 week on mice (Figure 1a), tumors were harvested and diced into fragments approximately 3–4mm2. Each tumor fragment was rinsed in fresh media and then plated in individual wells of a 24-well plate in 2 mL of media with 6000IU IL-2/mL (Figure 1b).
Figure 1.
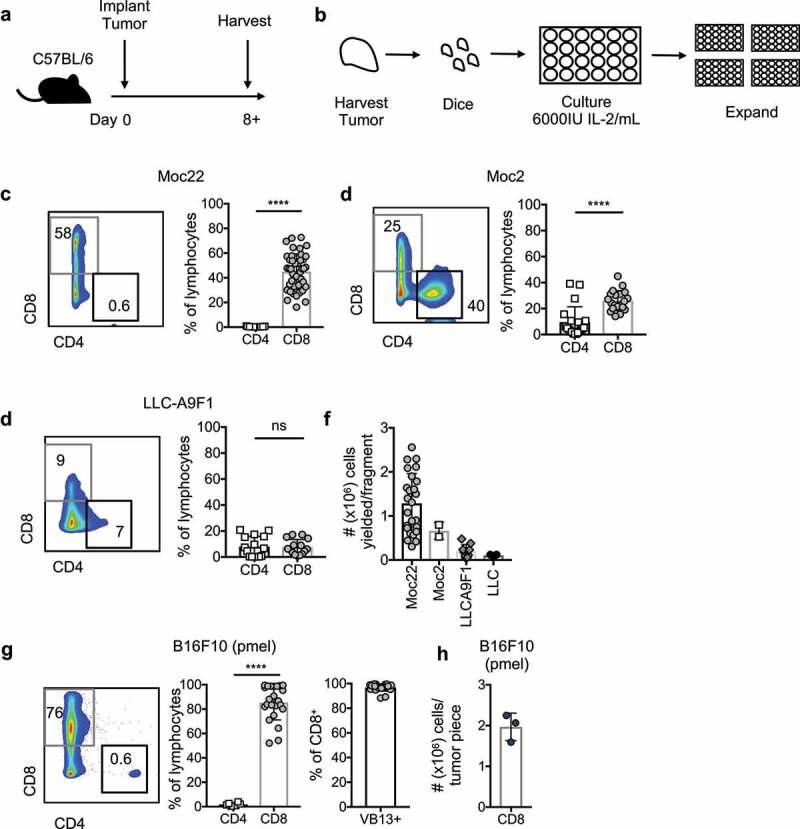
Characteristics of TIL expanded ex vivo from murine solid tumors
a) Model schematic. Tumors are established in mice for one week or more prior to harvest for TIL expansion. b) Schematic of ex vivo TIL expansion. Tumors are diced into ~3–4 mm2 fragments. Each fragment is plated in a separate well of a 24-well plate with media containing 6000IU IL-2/mL. These cells are expanded 3–4 weeks prior to analysis. c) T cell populations expanded from Moc22 tumors. n = 43 TIL cultures established from tumors across 26 mice. D) TIL populations expanded from Moc2 tumors. n = 20 TIL cultures from 10 mice. e) T cell populations expanded from LLC-A9F1 tumors; n = 15 cultures from five mice. f) Yield of cells after 3 weeks of expansion based on tumor cell line. g and h) TIL expanded from B16F10 tumors established on pmel-1 transgenic mice, where the TCR subunit Vβ13 is used by the TCR transgenic T cells specific for melanoma. n = 23 TIL cultures from three mice. Statistical tests: C-H, Mann Whitney U test, ****p < .0001; ns, not significant.
After 3 weeks of expansion, TIL were collected and assayed for T cell phenotypes. Overall, the frequencies of CD4+ and CD8+ T cells were variable and dependent upon tumor origin. Within the oral cavity cancer cell lines, TIL expanded from Moc22 tumors was predominantly CD8+ T cells, while Moc2 tumors expanded a mix of CD4+ and CD8+ T cells (Figure 1c-d). These findings are concordant with previously reported T cell populations infiltrating OCSCC tumors prior to harvest.13 TIL expanded less efficiently from LLC-A9F1 using this protocol (Figure 1e). Total yields of CD8+ T cells were higher for oral cavity cancers relative to lung cancers and ranged from <0.1–3×106 cells per tumor fragment (Figure 1f).
We next questioned whether transgenic, antigen-specific CD8+ T cells could be expanded from a tumor growing on a pmel-1 TCR transgenic mouse, where the CD8+ T cells within the animal express an H2-Db restricted TCR specific for gp100, expressed in melanoma and healthy melanocytes.9 Previous studies have demonstrated that a B16F10 melanoma tumor grows unabated on the pmel-1 mouse,9 yet the ability to expand TIL ex vivo from a tumor on this mouse remains unexplored. Indeed, expansion of pmel-1 T cells from B16F10 tumors was efficient, yielding predominantly CD8+ T cells, nearly all of which expressed Vβ13 of the gp100-specific pmel-1 TCR (Figure 1g). Total yield of pmel-1 T cells neared approximately 2 × 106 cells per tumor fragment (Figure 1h). Thus, TIL can be expanded ex vivo from murine tumor fragments using protocols mirroring clinical TIL expansion, and TIL composition varies across tumor cell lines.
Murine TIL are enriched in inhibitory receptors relative to peripheral T cells
We determined whether TIL had different inhibitory receptor expression profiles relative to peripheral T cells. To address this, we focused on Moc22, Moc2, and B16F10 cell lines as TIL were reproducibly expanded from these tumors. As shown in the schematic in Figure 2a, Moc2 or Moc22 tumors were established for 12 d, after which tumors and peripheral blood were collected. TIL were expanded ex vivo from tumors for 4 weeks and assayed for surface marker expression compared to peripheral blood at the time of harvest (Figure 2a). As expected, CD8+ TIL expressed significantly higher Tim-3 and PD-1 compared to peripheral blood from the host (Figure 2b-c). Interestingly, in Moc22 tumors, up to 40% of TIL co-expressed these inhibitory receptors while in Moc2 tumors, less than 20% co-expressed PD-1 and Tim-3 (Figure 2b-c). While expression of these markers on T cells in the tumor has been reported previously,14 it remained possible that the high doses of IL-2 could have driven some of the differences in phenotype we observed between TIL and peripheral blood.
Figure 2.
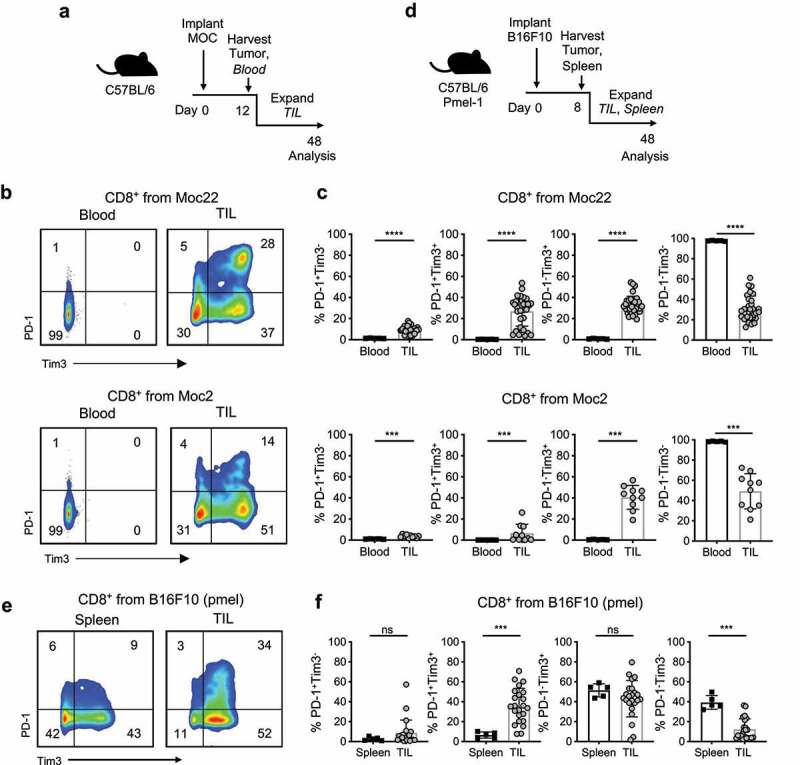
Inhibitory receptor expression in murine TIL exceeds peripheral T cells
a) Moc2 and Moc22 tumors were established to a palpable subcutaneous tumor in C57BL/6 animals for 12 d prior to tumor harvest and plating for TIL expansion. Blood was obtained from time of harvest for a peripheral T cell control. b and c) Inhibitory receptor expression in Moc22 and Moc2 CD8+ TIL relative to blood. Blood: n = 5, TIL: Moc22 n = 34 cultures from 21 mice, combined from two independent experiments, Moc2 n = 10 cultures from 5 mice. d) Pmel-1 transgenic mice were inoculated with B16F10 melanoma tumor. Transgenic T cells infiltrating tumors or spleens were expanded ex vivo and characterized. e and f) Surface expression of inhibitory receptors after 3–4 weeks of expansion in IL-2. Spleen n = 5 mice, TIL cultures n = 23 from three mice. Statistical tests: c,f) Mann Whitney U test; ns not significant, *p < .01, ***p < .001, ****p < .0001.
Therefore, we next questioned if expansion in IL-2 was responsible for the large differences in T cell phenotypes comparing expanded TIL to peripheral T cells. We addressed this question in two ways; first, we harvested Moc2 and Moc22 TIL directly from the tumor and peripheral tissues without ex vivo expansion (Supplementary Figure 1) and analyzed surface expression of PD-1 and Tim3. As expected, few CD8+ T cells from the spleens, lymph nodes, or peripheral blood expressed PD-1 or Tim3 (Supplementary Figure 1). In contrast, in the tumor, T cells expressing PD-1 or Tim3 were readily found (Supplementary Figure 1). In Moc22 TIL, a population of cells co-expressing PD-1 and Tim3 were detected (Supplementary Figure 1a-b) which were present at only low levels in Moc2 TIL (Supplementary Figure 1c-d). The PD-1+Tim3+ population was also lower in Moc2 expanded TIL versus Moc22 expanded TIL (Figure 2c). Indeed, few Moc2 TIL at harvest expressed PD-1, which may relate to the resistance of this cell line to PD-1 blockade13 (Supplementary Figure 1c-d).
Secondly, we tested if IL-2 was driving Tim3/PD-1 expression by expanding T cells from either the tumor (TIL) or the spleen of the pmel-1 transgenic animal in high doses of IL-2 (Figure 2d). Naïve pmel-1 transgenic cells, without any manipulation, do not express Tim3 or PD-1 (Supplementary Figure 2). However, after 4 weeks of expansion in IL-2, approximately 80% of pmel-1 TIL expressed Tim-3 while approximately 40% expressed PD-1 or co-expressed the two (Figure 2e-f). Spleen-derived T cells, expanded in high-dose IL-2, less frequently co-expressed Tim-3 and PD-1 than in expanded TIL populations, and a greater proportion of cells were double negative relative to TIL (Figure 2e-f). TIL harboring the Vβ13 chain of the transgenic TCR existed within all subsets of cells expressing PD-1, Tim3, or combinations of the two. There was a statistically significant but biologically minor enrichment of tumor-specific TIL within the PD-1+Tim3+ and PD1−Tim3+ populations (Supplementary Figure 3).
Thus, tumor exposure was associated with high PD-1 and Tim-3 on TIL as expansion in IL-2 was not sufficient to drive expression of these molecules on peripheral T cells to the level seen in expanded TIL. However, IL-2 expansion does impact the expression of PD-1 and Tim3 relative to T cells isolated directly from the tumors.
Ex vivo TIL expansion drives alterations in activation status relative to inhibitory receptor expression
To next understand the differences in activation for TIL expressing PD-1 and Tim3 and the impact of ex vivo expansion on these phenotypes, we characterized T cells obtained both prior to and after ex vivo expansion for expression of CD44 (Supplementary Figure4). In Moc22 TIL directly harvested from the tumor, the cells that expressed the highest CD44 were among the PD1+Tim3− and PD1+Tim3+ subsets, while cells that expressed only Tim3 or were double negative did not express CD44 (Supplementary Figure 4a-b). Similar trends were observed in freshly harvested Moc2 TIL, though the low frequency of PD1+Tim3− and PD1+Tim3+ cells made characterization more variable (Supplementary Figure 4b-c). With ex vivo expansion, however, Moc22 TIL were almost all CD44+ regardless of IR expression, while most Moc2 TIL were CD44lo or CD44− (Supplementary Figure 4b,d). Overall, the expression of the activation marker CD44 within inhibitory receptor (IR) subsets was higher in Moc22 TIL relative to Moc2 TIL, particularly among the PD1+Tim3− or PD1+Tim3+ cohorts, and appeared to be upregulated with ex vivo TIL expansion.
CD4+ and CD8+ T cells express distinct combinations of inhibitory receptors in human TIL
As heterogeneous populations existed in murine OCC TIL, we next investigated how the expression of IRs including PD-1, Tim3, and Lag3 compared among CD4+ and CD8+ TIL expanded ex vivo from OCC patients. After 4 weeks of successful TIL expansion from nine oral cavity tumors (Figure 3a), we profiled the frequency of CD4+ and CD8+ populations within TIL and determined the PD-1 expression within each population. We found that the frequency of CD4+ and CD8+ populations varied depending on the patient (Figure 3b). Interestingly, a greater frequency of CD4+ TIL expressed PD-1 relative to CD8+ TIL (Figure 3c). No difference in Tim-3 between T cell populations was observed in this patient cohort, while Lag-3 was more highly expressed on CD8+ T cells (Figure 3c). Frequencies of cells expressing inhibitory receptors in prostate TIL were similar to those seen in OCC patients, suggesting that these qualities were not specific to the malignancy (Supplementary Figure 5).
Figure 3.
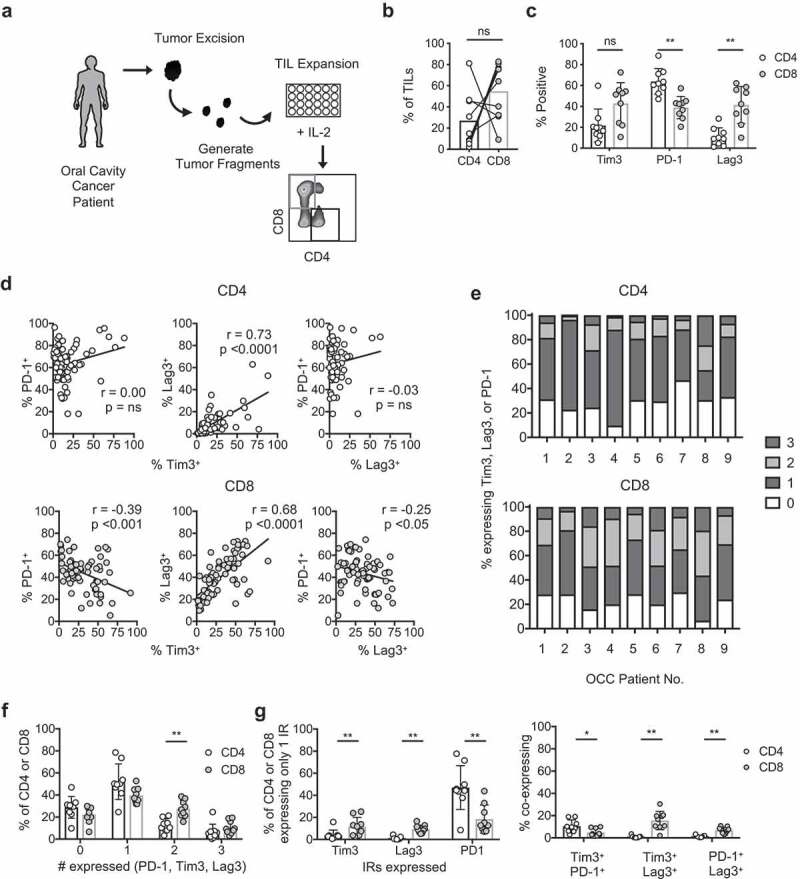
Inhibitory receptor expression dynamics in T cell subsets expanded from human OCSCC tumors
a) TIL expansion schematic. Tumor biopsies were obtained from patients with oral cavity squamous cell carcinoma (OCC). Tumors were diced and plated in 6000IU IL-2/mL and expanded for 4 weeks prior to analysis. Gating strategy for CD4+ and CD8+ T cell subsets shown. b) Frequency of CD4+ and CD8+ T cells in expanded TIL. n = 9 OCC. c) Frequency of cells expressing inhibitory receptors in OCC patients. d) Correlation of cells expressing pairs of inhibitory receptors from all individual TIL cultures of OCC patients. e–g) OCC TIL were assessed for co-expression of Tim-3, PD-1, and Lag-3. Cells can express from 0 to 3 of these receptors. e) Fraction of all TIL within CD4+ and CD8+ T cell populations expressing 0, 1, 2, or 3 IRs displayed per OCC patient. n = 9 OCC patients. f) Statistical comparison of data displayed in E. g) Frequency of T cells expressing only one or two inhibitory receptors, defined and compared between T cell subsets. n = 9 OCC patients. Statistical tests: B, C, F, G) Wilcoxon sign rank test, d) Spearman correlation. **p < .01; ns, not significant.
We next investigated PD-1, Tim-3, and Lag-3 expression dynamics within our OCC patient population. Our questions were threefold: 1) does expression of one IR directly correlate with the others, 2) do most of the TIL express more than one of these receptors, and 3) if TIL express 1 or more IRs, which combinations are the most frequent? Correlation patterns of PD-1, Tim-3, and Lag-3 pairs were distinct for CD4+ and CD8+ TIL expanded ex vivo (Figure 3d). In CD4+ TIL, Tim-3 and Lag-3 expression correlated but the frequency of cells involved was relatively low. Additionally, the co-expression of PD-1/Tim-3 or PD-1/Lag-3 did not show a visible trend (Figure 3d). In contrast, for CD8+ TIL, PD-1 expression negatively correlated with Tim-3 and Lag-3, while the Lag-3 and Tim-3 pair demonstrated a strong positive correlation (Figure 3d). Overall, these data suggested that expanded TIL expressing PD-1 did not frequently co-express Tim-3 or Lag-3, while TIL expressing Tim-3 were highly likely to co-express Lag-3.
For both CD4+ and CD8+ TIL, over 40% of the cells expressed only one IR, followed in frequency by populations expressing 0, 2, or 3 IRs, revealing that only a small population concurrently expressed PD-1, Tim-3, and Lag-3 (Figure 3e-f)). Within cells expressing only one IR (Figure 3g), more CD8+ T cells singly expressed Tim-3 and Lag-3 relative to CD4+ TIL, while the opposite relationship was observed for PD-1. Finally, of TIL expressing 2 IRs, more CD4+ TIL co-expressed Tim-3 and PD-1 relative to CD8+ TIL, while more CD8+ TIL co-expressed Tim-3/Lag-3 and PD-1/Lag-3 relative to CD4+ TIL (Figure 3g). Such expression patterns may have therapeutic implications for patients, especially in the design of combination therapies targeting specific T cell populations.
Cytokine production is highest in TIL co-expressing PD-1 and Tim-3
We next questioned whether cytokine production by patient TIL expanded ex vivo was related to their expression of negative regulatory molecules (Figure 4a). We hypothesized that T cells co-expressing these markers represented exhausted T cells, which would correspond with reduced effector function.15 To address this question in an unbiased way, we conducted scRNAseq on a cohort of seven different OCC patient TIL expanded ex vivo in IL-2. A total of 14,665 single cells were sequenced and analyzed into a total of 15 distinct clusters, which we then interrogated for inhibitory receptor or cytokine transcript expression (Figure 4a-b). We identified a particular cluster, cluster 7, which co-expressed heightened transcripts for multiple molecules of interest, including PDCD1, LAG3, HAVCR2, and CTLA4 (Figure 4c). This cluster contained both CD4 and CD8 transcripts (Figure 4b). Interestingly, cluster 7 also had a high relative expression of cytotoxic molecule transcripts like IFNG, TNF, and GZMB (Figure 4c), and was nearly the sole cluster to express IL10 (Figure 4c). These cells in cluster 7 also expressed ICOS, CD69, and TNFRSF4, and clustered separately from stem-like cells in cluster 4, which were notable for expressing TCF7, FAS, SELL, and CCR7 (Supplementary Figure 6). Thus, in addition to revealing the heterogeneity of T cells within OCC TIL, a subset of TIL were found to co-express transcripts for cytokines, cytotoxic molecules, and multiple inhibitory receptors.
Figure 4.
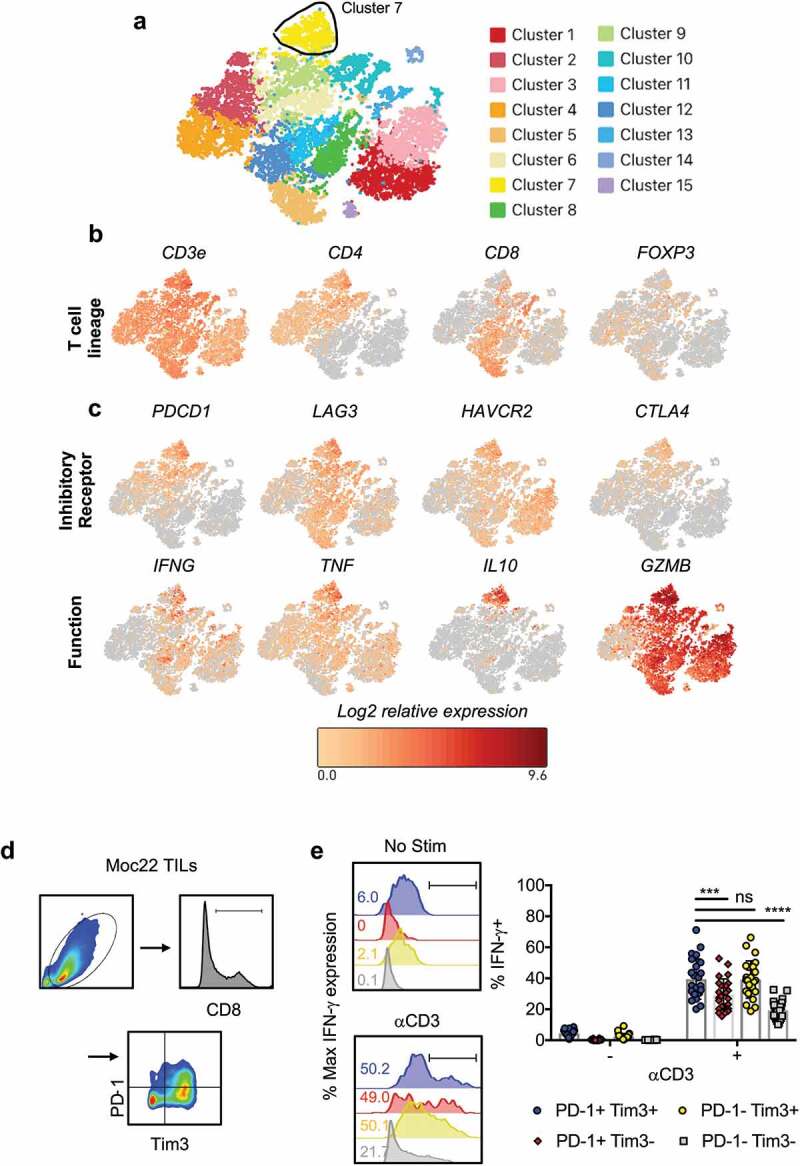
Cytokine production corresponds with inhibitory receptor expression in mouse TIL and OCC patient TIL
a) Seven separate OCC patient TIL were expanded ex vivo for 6 weeks and then analyzed by scRNA-seq. Approximately ~15,000 cells were sequenced in total. t-SNE clustering identified 15 clusters within the TIL. t-SNE plot overlaid with mRNA transcripts for b) T cell lineage, c) inhibitory receptors and cytokine/cytotoxic molecules (10X Loupe Cell Browser, 3.1.0). d) Gating strategy. Lymphocytes expanded from Moc22 tumors were gated on live cells, FSC/SSC and CD8 expression prior to gating for expression of Tim-3 or PD-1. Cells were activated with 1 mg/mL plate-bound aCD3 overnight with Golgi stop prior to analysis by flow cytometry. e) Histogram and biological replicates displaying IFN-γ production from cells activated overnight or at resting state by inhibitory receptor expression. Representative gating strategy depicted in histograms. n = 24 TIL cultures from 16 mice. Statistical tests: Wilcoxon matched-pairs sign rank test, ***p < .001, ****p < .0001.
Given the functional phenotype of patient TIL, we posited that we would observe similar functional patterns of cytokine production in murine TIL. To address this, Moc22 TIL products expanded ex vivo were activated with plate-bound aCD3 agonist overnight. PD-1+Tim-3+ double positive T cells had the largest cell size and highest granularity, while cells negative for both markers had the smallest size and granularity (Supplementary Figure 7a-b). Most cells expressed an effector memory phenotype (CD44+CD62L−) with activation (Supplementary Figure 7c-d). As suggested by transcript levels in patient TIL (Figure 4a-c), expression of one or more IRs was associated with cytokine production (Figure 4d-e). A similar fraction of cells co-expressing PD-1 and Tim-3 or expressing Tim3 only expressed IFN-γ, while fewer cells expressing only PD-1 produced IFN-γ (Figure 4e).
Collectively, this work highlights that TIL expanded ex vivo exist within various stages of development and activation in both murine and human patient cohorts. Expanded TIL from both mouse and human OCC tumors contain populations expressing high levels of inhibitory receptors and show similarities in functional dynamics. Using preclinical models, like expanding endogenous murine TIL ex vivo from tumor fragments followed by adoptive transfer, could be a useful tool to study methods to enhance the therapeutic efficacy of TIL products in future clinical trials.
Discussion
The first evidence that autologous TIL could elicit an antitumor response in patients dates back to 1988 in a cohort of patients with metastatic melanoma.16 Since then, TIL expansion and treatment protocols have incrementally improved to include the incorporation of lymphodepletion preparative regimens, enrichment of tumor-reactive T cells, and addition of costimulatory agents into the rapid T cell expansion process.17,18 Even so, only a small subset of patients experience durable responses to TIL therapy, which highlights the ongoing need to develop TIL products with improved efficacy.18
Improvements in TIL therapy could be licensed by either streamlining the expansion process or identifying agents which could combine with TIL to extend durability of response. For example, one important question is whether TIL therapy would benefit from combination with checkpoint inhibitors, or whether TIL therapy is even effective in patients after checkpoint blockade.3,19–21 As the process of generating TIL on a patient-specific basis is costly and time-consuming, use of preclinical protocols which 1) mirror the clinical process of ex vivo TIL expansion, 2) are controlled and reproducible, and 3) could apply across multiple tumor models could help address these questions prior to clinical trial execution.
Herein, we expanded TIL ex vivo from murine OCSCC and melanoma tumor fragments, while expansion from one lung tumor line yielded few cells. Of the two oral cavity cancer cell lines investigated, CD8+ TIL expanded in higher numbers from the Moc22 tumor, which is known to be immunogenic, infiltrated by CD8+ T cells, and is responsive to PD-1 blockade.13 In comparison, we obtained fewer TIL from the less immunogenic Moc2.13 Even so, we were surprised to expand any TIL ex vivo from the Moc2 tumor as it had been described to have a scant T cell infiltrate.13 Moc22 TIL were phenotypically activated – expressing higher PD-1, Tim-3, and CD44 relative to Moc2 TIL – which could indicate potential for tumor reactivity. In contrast to TIL from the OCC cell lines, TIL from the Lewis lung carcinoma cell line expanded poorly or in many cases not at all. Whether the failure of TIL expansion is related to T cell infiltration, or infiltration of suppressive immune cells remains to be explored. Future work could include manipulation of expansion conditions, including additional cytokine growth factors, or addition of costimulatory agonists or checkpoint inhibitors to improve TIL expansion yields. These experiments would be informative for patient tumors that fail to expand TIL.
Expanded murine TIL overall expressed higher PD-1 and Tim-3 relative to peripheral blood or secondary lymphoid organs. These differences were consistent for TIL either isolated directly from tumors or TIL expanded in IL-2, suggesting that IL-2 expansion is not solely responsible for driving expression of PD-1 or Tim3. While these characteristics describe bulk oral cavity TIL – unselected for tumor-reactivity – we found the same expression patterns in melanoma-specific TIL, where gp100-specific TIL maintained high PD-1 and Tim-3 expression relative to gp100-specific T cells obtained from the spleen. To improve the clinical relevance of transgenic TIL models, researchers could consider isolating transgenic T cells from tumors rather than lymphoid organs in order to recapitulate phenotypes resembling human TIL. This approach is likely to provide a preclinical tool to understand details of tumor-reactivity, memory lineage, and T cell exhaustion more deeply, as well as how these programs are related regarding experience of tumor antigen. Translationally, a deeper understanding of the relationships between inhibitory receptors, costimulatory molecules, and cytokine receptors and how these can be modulated would be helpful to design the optimal methods to harness T cell immunity against patient tumors.
In OCSCC patient TIL, only a small frequency of cells expressed all three PD-1, Tim-3, and Lag-3 receptors and most TIL expressed only one of the receptors. Based on the differences in surface expression profiles between CD4+ and CD8+ TIL, it remains likely that CD4+ TIL and CD8+ TIL could be suppressed by different inhibitory pathways after infusion into the patient. In both murine and human OCC expanded TIL, cells which expressed the highest IRs also simultaneously expressed the highest cytokine and cytotoxic molecules, including IFN-γ. Within tumors, TIL expressing both PD-1 and Tim-3 are highly dysfunctional, and combination PD-1/Tim-3 blockade promotes their functional activity.22,23 Therefore, when ex vivo expanded TIL products expressing PD-1 and Tim3 among other IRs are transferred into patients, it is possible that the T cells would benefit from co-administration of IR blockade to promote T cell activation and improved cytokine release in the tumor.
Our work challenges researchers to continue investigating how to make TIL therapy more feasible and effective, and highlights that these goals could be addressed using accessible preclinical models. We assume all cancer types will not have the same routes of immune evasion; therefore, multiple models of ACT beyond melanoma are important for clinical translation. Questions we seek to address in the future include whether tumors relapse after ACT because of 1) T cell exhaustion/dysfunction in the tumor (via Lag-3, Tim-3, PD-1, or others), 2) selective pressure on the tumor, or 3) loss/death of tumor-specific T cells. Overall, developing TIL protocols for all types of cancer and determining the settings in which combination therapies are most effective can converge to extend survival of patients worldwide.
Materials and methods
Animal studies
C57BL/6 mice were purchased from Jackson labs or Taconic labs, and pmel-1 TCR transgenic mice (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J) were purchased from Jackson labs. Male or female mice were bred and housed in the Hollings Cancer Center at the Medical University of South Carolina. Mice were euthanized using carbon dioxide followed by cervical dislocation as a secondary measure. Euthanasia occurred for experimental endpoints to collect tissues on either day 8 or 12 after tumor inoculation, which was determined based on tumor growth kinetics for each tumor model in order to harvest a palpable, three-dimensional tumor for ex vivo TIL expansion. No animals in this study died prior to meeting endpoints for euthanasia. Mice were between 6 and 8 weeks of age. All experiments were reviewed and approved via the Institutional Animal Care & Use Committee (IACUC) at the Medical University of South Carolina (Protocol #3039). Housing and experiments were conducted in accordance with MUSC’s IACUC procedures and with the supervision and support of the Division of Laboratory Animal Resources.
Tumor lines
Moc2 and Moc22 cell lines were obtained from Ravindra Uppaluri at the Dana-Farber Cancer Institute and were cultured in sterile media containing the following elements as described previously:24 2:1 mixture of IMDM: Hams nutrient mixture containing 1% Pen/Strep, 5% FBS, and 5ug/mL insulin, 5 ng/mL EGF, and 40 ng/mL hydrocortisone. LLCA9F1 was a gift from Mark Rubinstein and Eric Bartee at the Medical University of South Carolina and was cultured with DMEM containing 10% FBS and 1% Pen/Strep. B16F10 was a gift from Nicholas P. Restifo, NCI Surgery Branch, and were cultured in RPMI complete medium. Murine tumor cell lines were confirmed mycoplasma and pathogen free.
Tumor transplantation
All cell lines were injected subcutaneously in PBS in the abdomen of recipient animals. Moc22 and Moc2 were injected into Taconic mice and dosed at 1 × 106 and 0.1 × 106 cells per mouse respectively due to differences in growth kinetics, in order to analyze and compare tumors at similar sizes. LLC was given to C57BL/6 from Jackson labs in a dose of 0.4–0.5 × 106 cells/mouse. B16F10 was given in doses of 0.4–0.5 × 106 cells/pmel mouse which originated from Jackson labs and were bred in our facility (Medical University of South Carolina).
Ex vivo TIL expansion
Murine tumors were harvested prior to tumor size reaching a length of 10 mm by width of 10 mm (maximum tumor size was 100 mm2). Tumors were removed from mice under sterile conditions and incubated in sterile RPMI complete medium on ice. Mouse tumors were then diced into pieces with length and width measuring approximately 2 mm, washed, and plated in media containing recombinant human IL-2 (6000IU/mL) prior to incubation at 37°C (NCI preclinical repository). Cultures were left undisturbed for a minimum of 5 d, after which half of the media was changed at least 2–3 times per week. TIL were split upon reaching confluency and were maintained at approximately 1 × 106 cells/mL. The remaining tumor was removed at the time of first split or at three weeks into culture, depending on rate of cell egress from the tumor. All TIL cultures were maintained as individual cultures after plating from a distinct tumor fragment. Human TIL was generated in a similar manner, modeling protocols established previously.6
Tissue distribution
Peripheral blood was collected via small lancet puncture of the mandibular vein into a tube containing 0.125 M EDTA. Spleens and lymph nodes were collected into RPMI complete medium, processed to a single cell suspension over a 70 μM filter. Remaining RBCs were lysed. For analysis of T cells directly isolated from tumors, murine oral cavity tumors were harvested and digested in 1 mg/mL collagenase type IV (Worthington) at 37°C for 1 hour. The remaining sample was crushed over a 70uM filter (Corning) to a single-cell suspension. All samples were filtered (40 μM, Corning) prior to analysis.
Clinical samples
Samples were collected after informed consent and were deidentified prior to delivery to the research lab. All studies were approved by the Institutional Review Board at the Medical University of South Carolina (Approval #00082245).
Flow cytometry
A BDFACSVerse instrument was used for data collection, and analysis was conducted using FlowJo software (BD). Samples were stained for extracellular proteins by suspension in FACS buffer (PBS + 2% FBS) with Fc block (1:500 dilution), and incubation with antibodies at 4°C for 25 minutes. For cytokine production analysis, samples were activated with aCD3 agonist (clone 145–2 C11, Biolegend) overnight and incubated with Monensin (2 μM) and Brefeldin A (5 μg/mL) (Biolegend) for 4 hours, followed by fixation and permeabilization according to manufacturer’s protocol (Biolegend). A complete list of antibodies is found in the Supplementary Materials.
Single cell RNA-sequencing of TIL
Experimental procedures followed established techniques using the Chromium Single Cell 3ʹ Library v2 Kit (10x Genomics). Briefly, cells in single-cell suspension were loaded onto a 10X Genomics Chip A and emulsified with 3ʹ Single Cell GEM beads using a Chromium™ Controller (10x Genomics). Libraries were constructed from the barcoded cDNAs (Translational Science Laboratory at the Medical University of South Carolina). RNA sequencing was performed on each sample (approximately 50,000 reads/cell) using a NovaSeq S4 flow cell (Illumina) at the VANTAGE facility (Vanderbilt University Medical Center).
Statistical analysis
Statistical analysis was performed with Graphpad Prism (v7.0). Continuous measures compared between two groups were made using Mann Whitney U test or t tests depending on whether assumptions for the t-test were met. For functional comparisons of T cell subsets expressing inhibitory receptors, the Wilcoxin signed-rank test was used as a pairwise comparison within biological replicates. Comparisons across multiple tissues were made using a repeated-measures one way-ANOVA. Given the data are exploratory in nature and it was desired not to be overly restrictive, hypothesis tests were not adjusted for multiple comparisons except in situations where an ANOVA was performed, as indicated. P values less than 0.05 were considered significant. Plots display mean values and error bars represent standard deviations. Outliers were not omitted in the data presented.
Supplementary Material
Acknowledgments
We are thankful to Eric Bartee and Dimitri Arhontoulis for experimental support, as well as Kent Armeson for support on statistical design. We are also grateful for support from Corrine Levingston and Kirsten McDanel at the Flow Cytometry & Cell Sorting Core at the Medical University of South Carolina.
Funding Statement
This work was supported by NCI F30 CA243307, NIH T32 GM008716 and DE017551, and the American Head and Neck Society (HMK); NIH R50 CA233186 (MMW); NCI F31 CA232646-01A1 and Hollings Cancer Center Graduate Fellowship (ASS); NIH T32 AI132164-01 (CJD); NIH T32 GM008716 (GORR); NCI R01 CA222817 (MPR); NCI R01 CA175061, R01 CA208514 (CMP); NIDCR K08 DE26542 and SCTR UL1TR001450 (DMN); NIDCR R21 DE029592 (CMP and DMN); supported in part by the Cell Evaluation & Therapy and Translational Science Shared Resources, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313).
Disclosure Statement
Chrystal Paulos is a co-founder for Ares Immunotherapy. All other authors declare no conflicts of interest.
Author contributions
HK, DN and CP conceptualized and designed this work. HK, AMRR, MW, AS, RC, CD, MB, GORR, JH, MR, and CT performed experiments and collected data. ML provided clinical samples and oversight. MR provided experimental reagents, guidance and critical feedback. All authors critically read and approved the manuscript prior to submission.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–10. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of Tumor-Infiltrating lymphocytes for patients with metastatic melanoma. Journal of Clinical Oncology. 2016;34(20):2389–2397. doi: 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarnaik AA, Hamid O, Khushalani NI, Lewis KD, Medina T, Kluger HM, Thomas SS, Domingo-Musibay E, Pavlick AC, Whitman ED, et al. Lifileucel, a Tumor-Infiltrating lymphocyte therapy, in metastatic melanoma. JCO. Journal of Clinical Oncology. 2021;21:00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheever MA, Thompson DB, Klarnet JP, Greenberg PD.. Antigen-driven long term-cultured T cells proliferate in vivo, distribute widely, mediate specific tumor therapy, and persist long-term as functional memory T cells. J Exp Med. 1986;163(5):1100–1112. doi: 10.1084/jem.163.5.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233(4770):1318. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 6.Dudley M, Wunderlich J, Shelton T, Even J, Rosenberg S. Generation of Tumor-Infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 8.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 9.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muranski P, Boni A, Antony P, Cassard L, Irvine K, Kaiser AD, Paulos CM, Palmer DC, Touloukian CE, Ptak K, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112(2):362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sehrawat S, Kirak O, Koenig P-A, Isaacson Marisa K, Marques S, Bozkurt G, Simas J, Jaenisch R, Ploegh H. CD8+ T cells from mice transnuclear for a TCR that recognizes a single H-2Kb-Restricted MHV68 epitope derived from gB-ORF8 help control infection. Cell Rep. 2012;1(5):461–471. doi: 10.1016/j.celrep.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho H-I, Jaenisch R, Celis E, Ploegh HL. Transnuclear TRP1-Specific CD8 T cells with high or low affinity TCRs show equivalent antitumor activity. Cancer Immunology Research. 2013;1(2):99. doi: 10.1158/2326-6066.CIR-13-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zolkind P, Przybylski D, Marjanovic N, Nguyen L, Lin T, Johanns T, Alexandrov A, Zhou L, Allen CT, Miceli AP, et al. Cancer immunogenomic approach to neoantigen discovery in a checkpoint blockade responsive murine model of oral cavity squamous cell carcinoma. Oncotarget. 2018;9(3):4109–4119. doi: 10.18632/oncotarget.23751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of Tumor-Infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. New England Journal of Medicine. 1988;319(25):1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 17.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science (New York, NY). 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (New York, NY). 2015;348(6230):62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, et al. Adoptive transfer of Tumor-Infiltrating lymphocytes in patients with metastatic Melanoma: intent-to-Treat analysis and efficacy after failure to prior immunotherapies. Clinical Cancer Research. 2013;19(17):4792. doi: 10.1158/1078-0432.CCR-13-0380. [DOI] [PubMed] [Google Scholar]
- 20.Forget M-A, Haymaker C, Hess KR, Meng YJ, Creasy C, Karpinets T, Fulbright OJ, Roszik J, Woodman SE, Kim YU, et al. Prospective analysis of adoptive TIL therapy in patients with metastatic melanoma: response, impact of anti-CTLA4, and biomarkers to predict clinical outcome. Clinical Cancer Research. 2018;24(18):4416–4428. doi: 10.1158/1078-0432.CCR-17-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borch TH, Andersen R, Ellebaek E, Met O, Donia M, Marie Svane I. Future role for adoptive T-cell therapy in checkpoint inhibitor-resistant metastatic melanoma. J Immunother Cancer. 2020;8(2):e000668. doi: 10.1136/jitc-2020-000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–2186. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr., Dunn GP, Bui JD, Sunwoo JB, Uppaluri R, et al. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012;72(1):365–374. doi: 10.1158/0008-5472.CAN-11-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.