

ABSTRACT
Interferon-gamma (IFN-γ) is a major effector molecule of immunity and a common feature of tumors responding to immunotherapy. Active IFN-γ signaling can directly trigger apoptosis and cell cycle arrest in human cancer cells. However, the mechanisms underlying these actions remain unclear. Here, we report that IFN-γ rapidly increases protein synthesis and causes the unfolded protein response (UPR), as evidenced by the increased expression of glucose-regulated protein 78, activating transcription factor-4, and c/EBP homologous protein (CHOP) in cells treated with IFN-γ. The JAK1/2-STAT1 and AKT-mTOR signaling pathways are required for IFN-γ-induced UPR. Endoplasmic reticulum (ER) stress promotes autophagy and restores homeostasis. Surprisingly, in IFN-γ-treated cells, autophagy was impaired at the step of autophagosome-lysosomal fusion and caused by a significant decline in the expression of lysosomal membrane protein-1 and −2 (LAMP-1/LAMP-2). The ER stress inhibitor 4-PBA restored LAMP expression in IFN-γ-treated cells. IFN-γ stimulation activated the protein kinase-like ER kinase (PERK)-eukaryotic initiation factor 2a subunit (eIF2α) axis and caused a reduction in global protein synthesis. The PERK inhibitor, GSK2606414, partially restored global protein synthesis and LAMP expression in cells treated with IFN-γ. We further investigated the functional consequences of IFN-γ-induced ER stress. We show that inhibition of ER stress significantly prevents IFN-γ-triggered apoptosis. CHOP knockdown abrogated IFN-γ-mediated apoptosis. Inhibition of ER stress also restored cyclin D1 expression in IFN-γ-treated cells. Thus, ER stress and the UPR caused by IFN-γ represent novel mechanisms underlying IFN-γ-mediated anticancer effects. This study expands our understanding of IFN-γ-mediated signaling and its cellular actions in tumor cells.
KEYWORDS: IFN-γ, lung adenocarcinoma, ER stress, LAMP, autophagy, apoptosis
Introduction
Interferon-gamma (IFN-γ) is a key moderator of cellular immunity and secreted by activated T lymphocytes, γδT cells, and natural killer cells. IFN-γ has attracted much attention because of its significant role in promoting antitumor immune responses. Numerous recent studies have indicated that active IFN-γ signaling is a common feature in tumors that respond to immune checkpoint blockades through antibodies targeting CTLA-4 and PD-1/PD-L1.1,2 Loss of IFN-γ pathway genes in tumor cells reduces the chance of response to the immunotherapy with anti-CTLA-4.3 New studies using state-of-the-art technology have shown that while IFN-γ secretion requires local antigen recognition, IFN-γ diffuses extensively to alter the tumor microenvironment in a distant area. These findings imply the feasibility of activated tumor-infiltrating CD8+ T cells, through IFN-γ to modulate the behavior of remote tumor cells.4,5 Substantial evidence shows that besides its role in activating cellular immunity and enhancing antitumor immune response, active IFN-γ signaling triggers apoptosis and cell cycle arrest in human cancer cells, and acts as a direct anticancer agent.6,7 The mechanism underlying the IFN-γ-mediated anticancer effects remains unclear.
IFN-γ engaging with its receptor leads to the recruitment and activation of JAK1 and JAK2, which results in the activation of STAT1, a pivotal transcription factor. STAT1 regulates the expression of a wide range of immune-related genes.8 IFN-γ signaling can also activate the MAPK, PI3K-AKT-mTOR, and NF-κB signaling pathways to regulate gene transcription and promote mRNA translation.9–11 Recently, Yu et al. have shown that IFN-γ promotes yes-associated protein (YAP) condensation in tumor cells after anti-PD-1 therapy. The YAP condensation creates a transcription hub that increases the expression of immunosuppressive target genes, independent of the canonical STAT1-IRF1 transcription program.12 Increased protein synthesis demands may disrupt endoplasmic reticulum (ER) homeostasis, leading to a condition referred to as ER stress.13 Whether IFN-γ induces ER stress in lung adenocarcinoma cells is yet to be determined.
ER plays a crucial role in protein folding, assembly, and secretion. Disruption of ER homeostasis may lead to the activation of a specific cellular process called the unfolded protein response (UPR) to restore and maintain ER homeostasis.14 The UPR in mammals is controlled by three ER-resident transmembrane proteins: inositol-requiring enzyme-1 (IRE1), protein kinase-like ER kinase (PERK), and activating transcription factor-6 (AFT6).15 Glucose-regulated protein 78 (GRP78) is a major ER chaperone and master regulator of ER homeostasis.16 Under homeostatic conditions, GRP78 binds to three UPR sensors IRE1, PERK, and ATF6 to silence these proteins to prevent UPR signaling.17 However, GRP78 combines with the misfolded proteins accumulated in the ER under stress conditions to keep the proteins in foldable states and release three UPR sensors.18 Each of the three released UPR sensors induces a different signaling pathway that mediates the UPR.
Activation of PERK leads to phosphorylation of eukaryotic initiation factor 2α (elF2α), which inhibits general protein translation and reduces the amount of nascent proteins directed into the ER. PERK also induces the expression of activating transcription factor 4 (ATF-4), which upregulates ER chaperones, including GRP78. ATF-4 plays a dominant role in the induction of C/EBP homologous protein (CHOP) expression in response to ER stress.19 CHOP is known to promote apoptotic cell death.20 IRE1 activation is responsible for the splicing of X-box binding protein 1 (XBP1) mRNA. This alternative spliced XBP1 encodes a transcription factor that targets chaperones.21 After disassociation from GRP78, ATF6 is cleaved to release its N-terminal fragment and is transported into the nucleus to turn on the expression of ER stress-targeted gene.22,23 The consequences of the UPR are two-fold: (1) the transient suppression of global protein synthesis rates and (2) the expression of new genes aimed at increasing protein folding and ER capacity.24 Loss of ER homeostasis also leads to the activation of two protein degradation pathways: the ubiquitin-proteasome system and the autophagy pathway.25 Prolonged ER stress or unrestored ER homeostasis triggers apoptotic cell death.26
The role of autophagy in removing misfolded proteins and protein aggregates is an essential protective mechanism during ER stress. Autophagy consists of two consecutive stages: (1) an early stage characterized by the engulfment of cytoplasmic constituents and formation of autophagosomes and (2) a later stage characterized by the formation of autolysosomes through fusion between autophagosomes and lysosomes and subsequent degradation of the engulfed contents by lysosomal hydrolases.27,28
In this study, we investigated intracellular events caused by IFN-γ in lung adenocarcinoma cells. Our study demonstrated that IFN-γ triggers the UPR through the activation of the JAK1/2-STAT1 and AKT-mTOR pathways. IFN-γ-induced ER stress impaired autophagic flux by downregulating LAMP-1 and LAMP-2. ER stress-induced by IFN-γ contributed to apoptotic cell death and cell cycle arrest in lung adenocarcinoma cells.
Materials and methods
Cell lines and cell culture
A549, H1975, HCC827, H2030, and UMC-11 cell lines were obtained from Cobioer Biosciences (Nanjing, China). A549 and H2030 harbor mutations in KRAS; H1975 and HCC827 harbor EGFR mutations. Authentication of A549, HCC827, and H1975 cell lines was performed using short tandem repeat (STR) DNA profiling at Cobioer Biosciences in 2017. Authentication of H2030 and UMC-11 cell lines was performed using STR in 2019. All cell lines were maintained in RPMI-1640 (Hyclone, Omaha, NE, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) and 100 U/mL penicillin/streptomycin (Hyclone, Logan, UT, USA). All cell lines were passaged for less than 2 months after thawing. For the experiments that required IFN-γ treatment, the cell cultures were supplemented with the indicated amount of IFN-γ (100–2000 IU/mL).
Antibodies and reagents
All antibodies and reagents are listed in Table S1.
siRNA transfections
siRNAs targeting JAK1, JAK2, STAT1, and DDIT3 were synthesized by RiboBio (Guangzhou, China). Transient transfection was used to deliver siRNA. Briefly, siRNA (50 nM) and Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) were gently premixed in a medium without serum according to the manufacturer’s guidelines. The transfection mixture was added to the culture plate, and subsequently, cell suspensions were seeded into culture plates and maintained in culture for 24–48 h.
Immunofluorescence
The cells were seeded on polylysine-coated coverslips and treated with the indicated reagents (IFN-γ, Baf A1, and rapamycin) for various time intervals. Subsequently, the cells were fixed with 4% paraformaldehyde (Servicebio, Wuhan, China) for 15 min at room temperature and then permeabilized with 0.3% Triton X-100 (Standard Reagent, Hyderabad, India) in phosphate-buffered saline (PBS) for 10 min. After fixation and permeabilization, the cells were blocked with 5% goat serum (Invitrogen, Carlsbad, CA, USA) in PBS for 30 min at room temperature. The cells were then incubated with primary antibodies at 4°C overnight, followed by incubation with fluorescent conjugated secondary antibodies. Finally, all cells were stained with DAPI (Servicebio, Wuhan, China). The cells were visualized using a wide-field fluorescence microscope (Carl Zeiss, Baden-Württemberg, Germany). The number of LAMP-1-positive puncta was quantified using the Analyze Particle plugin for FIJI (NIH, MD, USA) after creating binary images. We analyzed three randomly selected images from each culture condition.
Lysosomal tracking
The cells were seeded on coverslips and incubated with LysoTracker Red DND-99 (50 nM) (Yeason, Shanghai, China) for 30 min at 37°C. Hoechst 33,342 (Beyotime, Shanghai, China) staining was performed to counterstain the nucleus. We quantified the number of lysosomes using the same approach as mentioned for the quantification of LAMP1-positive puncta.
Autophagosome staining
The cells were seeded on coverslips and incubated with 2 µL/mL CYTO-ID® Green detection reagent (Enzo Life Sciences, Farmingdale, NY, USA) for 30 min at 37°C. Hoechst 33,342 staining was performed to counterstain the nucleus. Images were captured immediately after the staining. The area of the CYTO-ID® green puncta was quantified using the Analyze Particle plugin for FIJI. We analyzed three randomly selected images from each group. We quantified the area of autophagosomes using the same approach as mentioned previously for the quantification of LAMP1-positive puncta.
Premo™ autophagy tandem sensor RFP-GFP-LC3B kit
The Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B Kit was obtained from Invitrogen. The cells were transfected with BacMam reagent according to the manufacturer’s instructions. Subsequently, the cells were treated with Hank’s balanced salt solution (HBSS) and chloroquine (Enzo Life Sciences, Farmingdale, NY, USA). For the IFN-γ treatment group, the cells were pretreated with IFN-γ for 48 h and then transfected with BacMam reagents. After treatment, the cells were fixed with 4% paraformaldehyde and stained with DAPI. We quantified the numbers of red and yellow puncta using the Analyze Particle plugin for FIJI. A hue of 0–20 was counted as red puncta. A hue of 20–45 was counted as yellow puncta. We analyzed at least 20 randomly selected cells from each culture condition.
RNA isolation and quantitative real-time PCR analysis
Total RNA was extracted using TRIzol (Takara Bio Inc., Shiga, Japan). mRNA was reverse transcribed into cDNA using an RT reagent kit (Vazyme, Nanjing, China). mRNA expression was detected using quantitative real-time polymerase chain reaction (PCR) with a SYBR Green Master Mix Kit (Vazyme) via Applied Biosystems (Vilnius, Lithuania). The primers were obtained from TsingKe Biology Technology (Wuhan, China). The primer sequences used are listed in Table S2. Negative controls without a template were included, and all reactions were assayed in triplicate. We used ACTB as an internal control and determined the relative expression of the target genes according to the 2−ΔΔCt method.
Western blot analysis
The cells were lysed using RIPA lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS). The proteins were resolved using SDS-PAGE and transferred to Immobilon®-P membranes (Merck Millipore, Darmstadt, Germany). The blots were developed using an enhanced chemiluminescent detection system (Tanon 5200, Shanghai, China). To ensure that equal amounts of sample proteins were applied to each lane, we used β-actin as a loading control.
Histone extraction
The cells were harvested and washed twice with ice-cold PBS. Subsequently, the cells were resuspended in Triton Extraction Buffer (TEB: PBS containing 0.5% Triton X-100 and 2 mM PMSF) at a cell density of 107 cells/ml. Then the cells were lysed on ice for 10 min with gentle stirring and centrifuged at 400 g for 10 min at 4°C. The cell pellet was resuspended in 0.2 N HCl at a cell density of 4 × 107 cells/ml at 4°C overnight. The samples were centrifuged at 6,500 g for 10 min at 4°C. The supernatant was used for immunoblot analysis after neutralized with 2 M NaOH.
Immunoprecipitation assay
The cells were lysed using GST lysis buffer (10% glycerol, 1% NP40, 50 mM Tris-HCl pH 7.8, 150 mM NaCl, NP40 15, and 2 mM MgCl2) supplemented with a complete protease inhibitor cocktail. The samples were incubated with 5 µg of primary antibody overnight at 4°C, followed by the addition of Pierce TM protein A/G magnetic beads (Thermo Fisher Scientific, Rockford, IL, USA) for 2 h. Immunoprecipitates were washed three times with GST buffer. The magnetic beads were collected using a magnetic stand. Bound proteins were eluted by boiling in SDS-sample buffer and analyzed using western blotting.
Cell viability
Cell viability was measured using a CCK-8 kit (Dojindo Molecular Laboratories, Kumamoto, Japan). The cells were seeded (4000 cells in 100 µL/well) in 96-well plates and cultured overnight before IFN-γ stimulation. The absorbance was measured at 450 nm using a microplate reader (TECAN, Baldwin Park, CA, USA).
Apoptosis assays
We collected cells treated with or without IFN-γ and washed them twice with PBS. Subsequently, the cells were stained with an Annexin V-7-AAD Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA). The samples were analyzed within 1 h of processing using an Attune NxT Acoustic cytometer (Invitrogen). The percentage of cells in each quadrant of the dot plot was determined using FlowJo v10 (BD Biosciences, San Jose, CA, USA).
Cell cycle analysis
The cells were treated with or without IFN-γ for 24 h. The cells were then harvested and washed in PBS. Finally, 2 × 105 cells were suspended in 0.5 mL of cold PBS and 1.5 mL of 100% ethanol was added dropwise to the cell suspension with gentle vortexing. The cells were stored at −20°C for at least 2 h. Subsequently, the cells were centrifuged at 400 g for 10 min at 4°C and washed twice with cold PBS. The cells were resuspended in 500 µL PI/RNase Staining Buffer (BD Biosciences) and incubated at 37°C for 15 min. The samples were analyzed using an Attune NxT Acoustic cytometer and ModFit LT software (Verity software house, Topsham, ME, USA).
SUnSET (surface sensing of translation)
SUnSET is a method used to monitor the rate of protein synthesis.29 After the cells were cultured with IFN-γ, 10 mg/mL of puromycin was added to the culture medium for an additional 10 min. Subsequently, the cells were lysed using RIPA buffer. We detected puromycin incorporation using western blotting and an antibody against puromycin.
Ex vivo culture of patient-derived lung cancer explants
Fresh tissues were obtained from patients undergoing pulmonary resection prior to radiation or chemotherapy at the Department of Thoracic Surgery, Tongji Hospital. Ex vivo culture was performed as previously described.30 Briefly, we dissected the tumor tissues into 1 mm3 cubes and placed them on a gelatin sponge (Hushida, Jiangxi, China) bathed in RPMI-1640 media supplemented with 10% heat-inactivated FBS and 100 U/mL penicillin-streptomycin. The indicated amounts of IFN-γ were then added to the cultures. The tissues were cultured at 37°C for 48 h and collected for RNA and protein extraction. This study was performed in accordance with the Declaration of Helsinki. The use of human tissue samples was approved by the Institutional Ethics Committee of the Huazhong University of Science and Technology.
Activation of T lymphocytes in vitro
Peripheral blood mononuclear cells (PBMCs) were isolated from EDTA-K2 anticoagulated blood using Ficoll-Paque Medium (GE Healthcare, Uppsala, Sweden) and density gradient centrifugation. The PBMCs (2 × 106 cells/well, 6-well plate) were cultured alone or treated with 1 µg/mL CD3 monoclonal antibody (mAb) (BD Bioscience, San Jose, CA, USA) in RPMI 1640 medium supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. Two days later, the supernatants were collected and used for co-culture with lung cancer cells.
Detection of IFN-γ expression by enzyme-linked immunosorbent assay (ELISA)
The PBMCs were stimulated with or without anti-CD3 mAb for 48 h. The supernatants were collected using centrifugation. Production of IFN-γ in the supernatants was quantified using a commercial ELISA kit (Invitrogen), according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using Prism V6 software (GraphPad, La Jolla, CA, USA). A two-sided Student’s t-test was used. The data in the bar graphs are presented as the mean±SEM. Differences were considered statistically significant at p < .05.
Results
IFN-γ induces the UPR in lung adenocarcinoma
IFN-γ binding to the IFN-γ-receptor leads to activation of gene expression proceeds via the JAK-STAT pathway.31 Thus, we hypothesized whether IFN-γ could accelerate global protein synthesis in lung cancer cells. We performed the SUnSET assay, a robust and accurate method for measuring the relative rates of protein synthesis in cell culture,29 to evaluate the protein synthesis rates in A549 cells in response to IFN-γ stimulation. As shown in Figure S1a, IFN-γ treatment led to an increased protein synthesis rate, as quickly as 3 h after treatment in A549 cells.
Next, we examined whether IFN-γ triggers UPR. As shown in Figure 1a, IFN-γ significantly increased the transcription of UPR-related molecules, including HSPA5 (encoding GRP78 protein), sXBP1, and DDIT3 (encoding CHOP protein) genes in lung cancer cells. IFN-γ-induced transcription of HSPA5 in A549 and H1975 was dose related (Figure S1b). Additionally, the expression of ATF-4 and CHOP were increased dramatically as early as 24 h after exposure to IFN-γ (Figure 1b). Notably, IFN-γ did not affect UPR-related molecule expression in UMC-11 lung carcinoid cells. GRP78 is a chaperone and master regulator of the UPR. We found that GRP78 expression was upregulated by IFN-γ (Figure 1c). To confirm the results obtained using western blotting, we performed immunofluorescence staining for GRP78. Higher immunoreactivity of GRP78 was observed in IFN-γ-treated cells than in untreated cells (Figure 1d). ATF-6 is also involved in the UPR. Under ER stress conditions, ATF-6 is released from GRP78 and cleaved by site 1 and site 2 proteases in the Golgi body. The resultant N-terminal ATF-6 fragment is transported into the nucleus to turn on ER stress-targeted gene expression. As shown in Figure 1e, IFN-γ stimulation increased the N-terminal fragment of ATF-6. Thus, we showed that IFN-γ stimulation leads to a rapid increase in the protein synthesis rate, which may cause ER stress and trigger the UPR.
Figure 1.
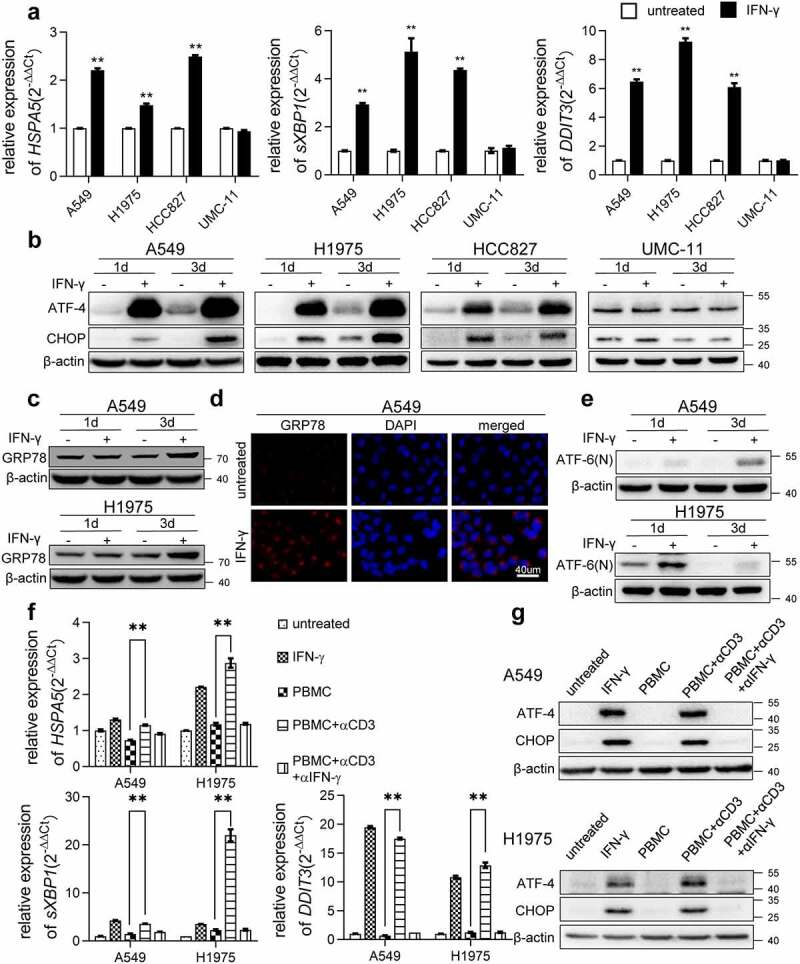
IFN-γ induces UPR in lung adenocarcinoma. (a) The indicated cells were treated with IFN-γ (1000 IU/mL) or untreated for 2 days. qRT-PCR was used to detect the expression of selected genes involved in ER stress. The expression of each gene of interest was corrected for ACTB expression. **, p < .01. (b) Immunoblots showing ATF-4 and CHOP protein expression in IFN-γ treated and untreated cells. β-actin was used as a loading control. (c) Western blot analysis showing increased expression of GRP78 in IFN-γ treated cells vs. untreated cells. (d) Immunofluorescence images of GRP78 in A549 cells treated with IFN-γ or untreated for 3 days (scale bar = 40 µm). (e) Western blot analysis for ATF-6(n) in IFN-γ treated A549 and H1975 cells vs. untreated cells. (f and g) Peripheral blood mononuclear cells (PBMCs) were stimulated with anti-CD3 monoclonal antibody (mAb) in the presence or absence of anti-IFN-γ antibody for 48 h. Subsequently, the supernatants were collected and cultured with A549 and H1975 cells for 2 days. qRT-PCR was used to assess the transcription of HSPA5, sXBP1, and DDIT3 (f). Western blot analysis was used to detect the expression of ATF-4 and CHOP (g)
IFN-γ is a key effector molecule of immunity and is secreted by activated T lymphocytes. Next, we examined whether the activation of T cells can trigger UPR in lung cancer cells. PBMCs were stimulated with anti-CD3 mAb for 2 days. A significant amount of IFN-γ was produced by activated T cells (Figure S1c). The supernatants collected from cultures of PBMCs stimulated with anti-CD3 mAb upregulated the transcription of the UPR-associated molecules, including HSPA5, DDIT3, and sXBP1 (Figure 1f), and increased the expression of ATF-4 and CHOP in lung cancer cells (Figure 1g). These effects were abolished in the presence of an IFN-γ neutralizing antibody. Our results provide direct evidence of how T cell activation affect the biological processes of tumor cells. In this case, we show that IFN-γ produced by activated T cells triggers UPR in cancer cells.
To further confirm the effect of IFN-γ on ER conditions, we cultured human-derived lung adenocarcinoma cells from seven patients ex vivo. We found that IFN-γ induced DDIT3 transcription in four out of seven tumor specimens (Figure S1d). Upregulation of ATF-4 expression was also observed (Figure S1e). The data reflect heterogeneity in tumors in response to IFN-γ treatment. Our data show that IFN-γ can induce UPR in some of the human adenocarcinomas.
IFN-γ-mediated JAK1/2-STAT1 and PI3K-AKT-mTOR signaling is required for the induction of the UPR
We examined whether IFN-γ-triggered UPR is JAK1/2-STAT1-dependent. We used siRNA-JAK1, siRNA-JAK2, and siRNA-STAT1 to knockdown JAK1, JAK2, and STAT1, respectively. We found that IFN-γ-induced CHOP expression was abrogated in cells transfected with JAK1, JAK2, and STAT1 siRNA (Figure S2a and Figure 2a). Our data indicate that the IFN-γ-mediated JAK1/2-STAT1 pathway is required for the induction of UPR. IFN-γ did not affect the expression of UPR-related molecules in UMC-11 cells (Figure 1a and b). We examined whether IFN-γ activates the STAT1 pathway in UMC-11 cells. As shown in Figure S2b, IFN-γ failed to induce the activation of STAT1 in UMC-11 cells, which further indicates the requirement of STAT1 activation for the induction of UPR by IFN-γ. IFN-γ also activates AKT-mTOR signaling in lung cancer cells.10 To determine whether the AKT-mTOR pathway is involved in IFN-γ-triggered UPR, we first used LY294002 to inhibit AKT activation. We found that LY294002 abrogated IFN-γ-induced CHOP expression in A549 and H1975 cells (Figure 2b). Next, we added the mTOR inhibitor rapamycin to the cells cultured with IFN-γ and we found that addition of rapamycin to the cell culture resulted in significant suppression of AKT-mTOR activity and CHOP expression (Figure 2c). Our data demonstrate that the AKT-mTOR pathway is also involved in IFN-γ-triggered UPR.
Figure 2.
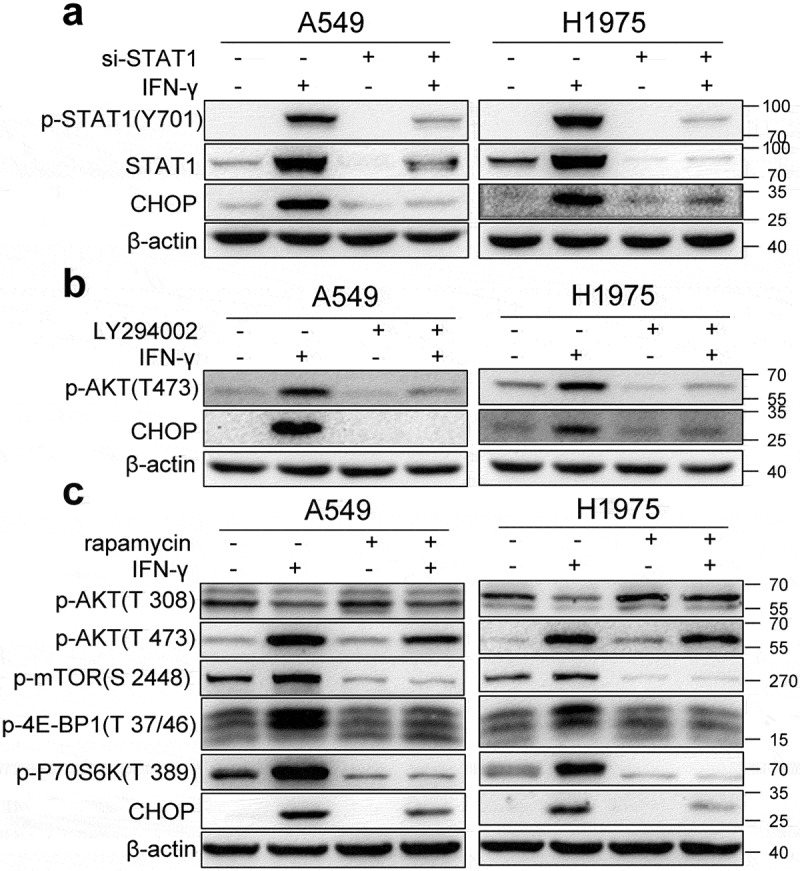
IFN-γ-mediated JAK1/2-STAT1 and PI3K-AKT-mTOR signaling pathways are required for the induction of the UPR. (a) A549 and H1975 cells were transfected with control siRNA or si-STAT1 for 24 h and then treated with IFN-γ (1000 IU/mL) for 3 days. The expression of STAT1 and CHOP was determined using western blotting. (b) A549 and H1975 cells were pretreated with 10 µM of LY294002 for 1 h and then treated with IFN-γ (1000 IU/mL) for 36 h. The expression levels of phosphorylated AKT and CHOP was determined using western blotting. (c) A549 and H1975 cells were pretreated with 500 nM of rapamycin for 1 h and then treated with IFN-γ (1000 IU/mL) for 36 h. Western blot analyses were used to determine the expression of p-AKT, p-mTOR, p-4E-BP1, p-70S6K, and CHOP
Accumulation of autophagosomes in IFN-γ-treated cells
ER stress is a potent trigger for autophagy, a self-degradative process that has an adaptive function.32 Therefore, we examined whether IFN-γ promotes autophagy in lung cancer cells. LC3 is commonly used to monitor autophagy. One approach is to detect LC3 conversion (LC3-I to LC3-II) by immunoblotting analysis because the amount of LC3-II is strongly associated with the number of autophagosomes.33 Treatment of lung adenocarcinoma cells (A549, HCC827, H1975, and H2030) with IFN-γ led to increased levels of LC3-II (Figure 3a). IFN-γ did not induce autophagosome accumulation in UMC-11 cells. To confirm the results obtained from western blotting, we used the CYTO-ID® green detection reagent, which selectively stains autophagic vesicles. Autophagosome accumulation was observed in IFN-γ-treated cells, similar to that observed in cells cultured in HBSS and chloroquine (CQ) (Figure 3b).
Figure 3.
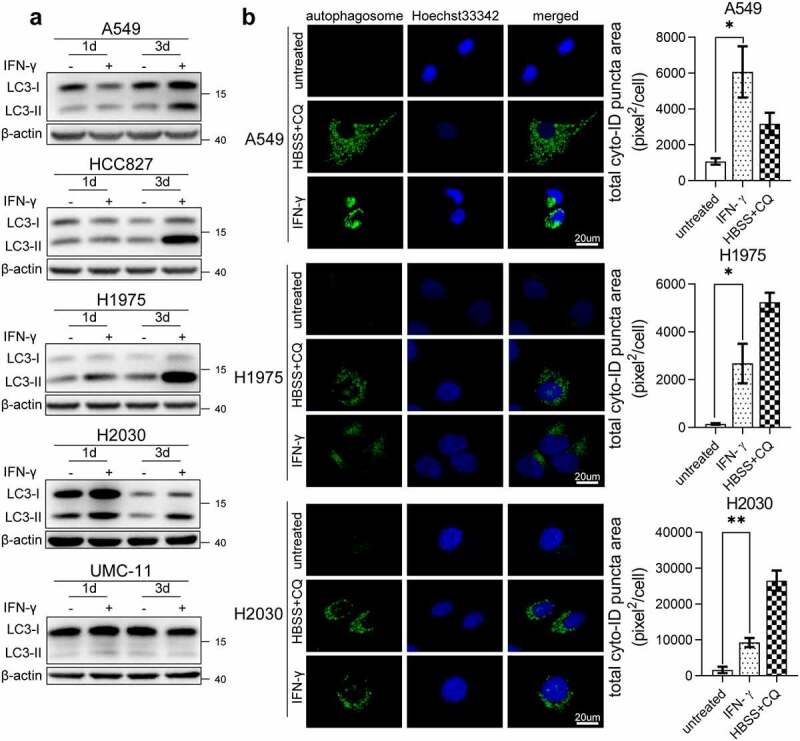
Accumulation of autophagosomes in IFN-γ-treated cells. (a) Immunoblots showing LC3 protein expression in IFN-γ treated and untreated cells. (b) The indicated cells were treated with IFN-γ (1000 IU/mL) or untreated for 3 days. At the end of the treatment, the cells were labeled with CYTO-ID® green detection reagent to detect autophagosomes. Cells treated with HBSS in the presence of CQ (60 µM) for 3 h were used as a positive control for the accumulation of autophagosomes. Puncta areas per cell from three random images for each culture condition are shown. *, p < .05; **, p < .01
IFN-γ impairs autophagic flux in lung cancer cells
LC3-II accumulation reflects either enhanced autophagosome formation or decreased fusion of autophagosomes with lysosomes. First, we evaluated whether IFN-γ treatment promoted autophagosome formation. Beclin-1 is involved in ER stress-promoted autophagy.34 As shown in Figure 4a and b, IFN-γ did not enhance Beclin-1 expression on day 3 of treatment. Several studies have shown that Bcl-2 can bind to the BH3 domain-containing Beclin-1 to regulate autophagy activation.35,36 Wei et al. reported that starvation induces the phosphorylation of Bcl-2. Subsequently, Beclin-1 dissociates from Bcl-2, and leading to autophagy activation.37 As shown in Figure S3a, IFN-γ did not significantly affect Beclin-1 and total Bcl-2 expression on day 2 of treatment. The lysates from IFN-γ-treated and untreated cells were immunoprecipitated with an anti-Bcl-2 antibody. Beclin-1 was detected at a comparable level in immunoprecipitates from IFN-γ-treated and untreated cells. Death-associated kinase 1 (DAK1) has also been reported to be involved in ER stress-promoted autophagy.38 DAK1 expression in cancer cells was not affected by IFN-γ (Figure S3b).
Figure 4.
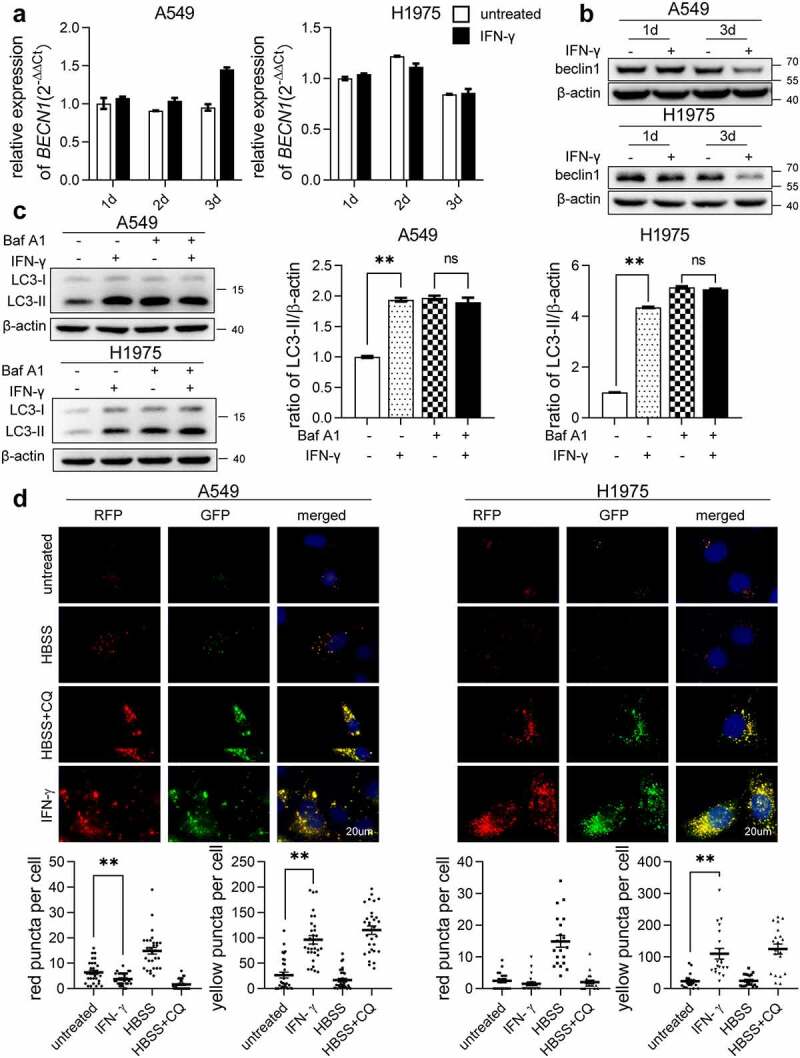
IFN-γ impairs autophagic flux in lung cancer cells. (a and b) The expression of Beclin 1 was assessed at the transcriptional level by qRT-PCR (a) and at the protein level by western blot analysis (b) in A549 and H1975 cells after treatment with IFN-γ (1000 IU/mL) or mock treated for the indicated duration. (c) LC3 expression was determined using western blotting. The bar graphs show a densitometric analysis of changes in the abundance of LC3-II normalized to β-actin level for loading variability. **, p <.01; ns, not significant. (d) The indicated cells were transfected with the Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B and cultured under the indicated conditions. Red puncta (RFP puncta only) and yellow puncta (RFP puncta colocalized with GFP puncta) were detected using fluorescence microscopy (scale bar = 20 µm). The number of LC3 yellow puncta and red puncta per cell was calculated from at least 20 cells from each culture condition. The bar graphs represent the mean ± SEM. **, p < .01
Next, we investigated autophagic flux status. We compared the ratio of LC3-II to β-actin in cells treated with IFN-γ to untreated cells in the presence or absence of bafilomycin A1 (Baf A1), an inhibitor of autophagosome-lysosome fusion. As shown in Figure 4c, the LC3II/β-actin ratio increased considerably in IFN-γ untreated cells in the presence of Baf A1 compared with IFN-γ untreated cells without Baf A1, indicating that there was a basal level of activated autophagic flux in IFN-γ-untreated cells. However, in IFN-γ-treated cells, the LC3-II/β-actin ratios between the presence and absence of Baf A1 were comparable, suggesting that IFN-γ treatment impairs autophagic flux.
We then investigated whether IFN-γ affects autophagosome-lysosomal fusion. The cells were transfected with the Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B. The GFP signal was quenched in the acidic autolysosome, whereas RFP was more stable. Colocalization of GFP and RFP indicates that an autophagosome is not fused with a lysosome. In contrast, an RFP signal without GFP corresponds to an autolysosome.39 As shown in Figure 4d, the culture of cells in HBSS led to an increased quenching of GFP signaling, which was caused by increased autophagosome-lysosome fusion, and yellow puncta were almost undetectable. CQ suppresses lysosomal degradation by increasing lysosomal pΗ. In cells cultured with HBSS in the presence of CQ, the quenched GFP signal was recovered, suggesting that CQ blocked the autophagosome-lysosomal fusion. There was a significantly increased colocalization of GFP and RFP fluorescence (yellow puncta) in cells treated with IFN-γ, compared to untreated cells, similar to the cells cultured in HBSS in the presence of CQ. Our data showed that IFN-γ impairs autophagic flux rather than promoting autophagy.
LAMP-1 and LAMP-2 expression is reduced in lung cancer cells in response to IFN-γ
LysoTracker is used to detect lysosomes in living cells.40 LysoTracker Red DND-99 was used to evaluate lysosomes in cells treated with IFN-γ. The immunoreactivity of LysoTracker was significantly reduced in cells treated with IFN-γ compared to untreated cells, similar to the cells cultured with Baf A1 (Figure 5a). Lysosomal membranes contain several highly N-glycosylated proteins, among which LAMP-1 and LAMP-2 are the best known.41 LAMP-1 and LAMP-2 play essential roles in autophagosome maturation. As shown in Figure 5b, LAMP-1 and LAMP-2 expression decreased in cells cultured with IFN-γ on day 3. IFN-γ did not affect LAMP expression in UMC-11 cells. To confirm the western blot analysis results, we used cell immunofluorescence to detect LAMP-1. The immunoreactivity of LAMP-1 reduced significantly in cells treated with IFN-γ compared to untreated cells (Figure 5c). Notably, IFN-γ did not reduce the transcription of LAMP1 and LAMP2 (Figure S4).
Figure 5.
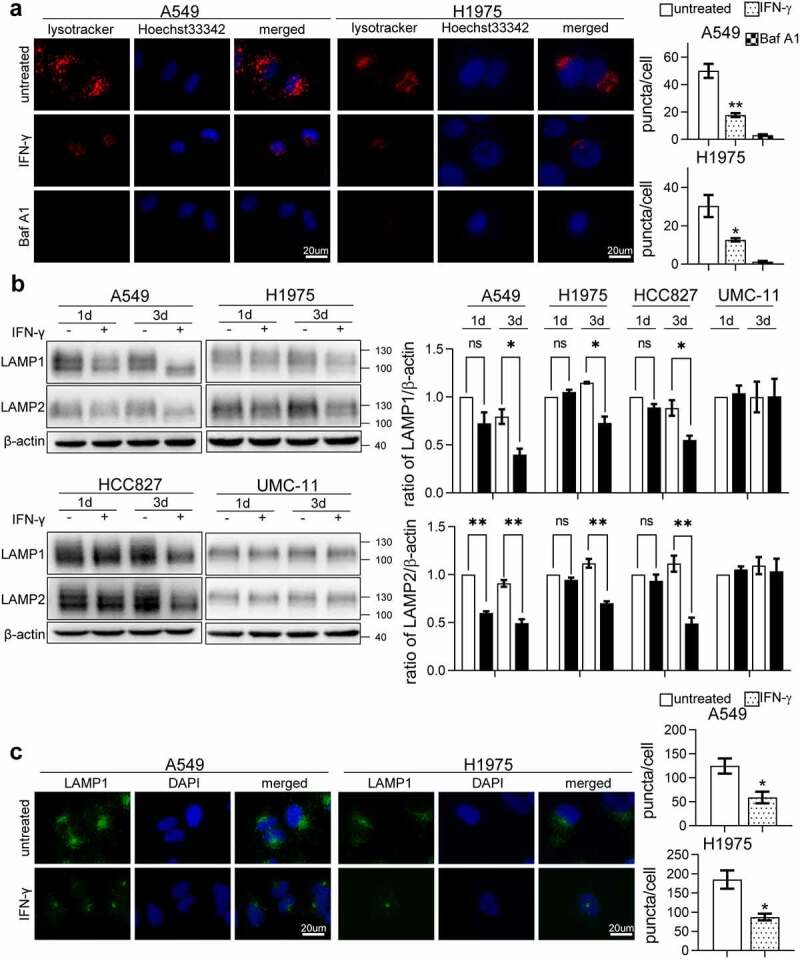
LAMP-1 and LAMP-2 expression is reduced in cells treated with IFN-γ. (a) The indicated cells treated or untreated with IFN-γ were loaded with LysoTracker Red DND-99 for 30 min and analyzed using fluorescence microscope. Cells treated with Baf A1 alone were used as an experimental control. (b) Representative immunoblots showing LAMP-1 and LAMP-2 expression in IFN-γ treated and untreated cells. The bar graphs show a densitometric analysis of 3–4 individual experiments in the abundance of LAMP after normalization to β-actin for loading variability. *, p < .05; **, p < .01; ns, not significant. (c) The expression of LAMP-1 was assessed using fluorescence microscopy (scale bar = 20 µm). The number of puncta per cell was counted from three random images from each culture condition. The bar graphs represent the mean ± SEM. *, p < .05; **, p <.01
Inhibition of ER stress restores IFN-γ-mediated downregulation of LAMP
To determine whether IFN-γ-triggered UPR contributes to the reduction in LAMP, the ER stress inhibitor 4-PBA was used. In addition to inhibiting ER stress, 4-PBA is also used as an inhibitor of histone deacetylase.42 As shown in Figure S5, -PBA at 2 mM and above significantly altered the histone H3 acetylation status. To minimize the influence of 4-PBA on histone acetylation, we used 1 mM 4-PBA in our experimental setting. Additional 4-PBA abrogated IFN-γ-induced ATF-4 expression and restored LAMP expression in cells treated with IFN-γ (Figure 6a). Furthermore, rapamycin significantly restored LAMP1 immunoreactivity in IFN-γ-treated A549 cells (Figure 6b and c). These results indicate that the IFN-γ-triggered UPR is responsible for the decline in LAMP expression.
Figure 6.
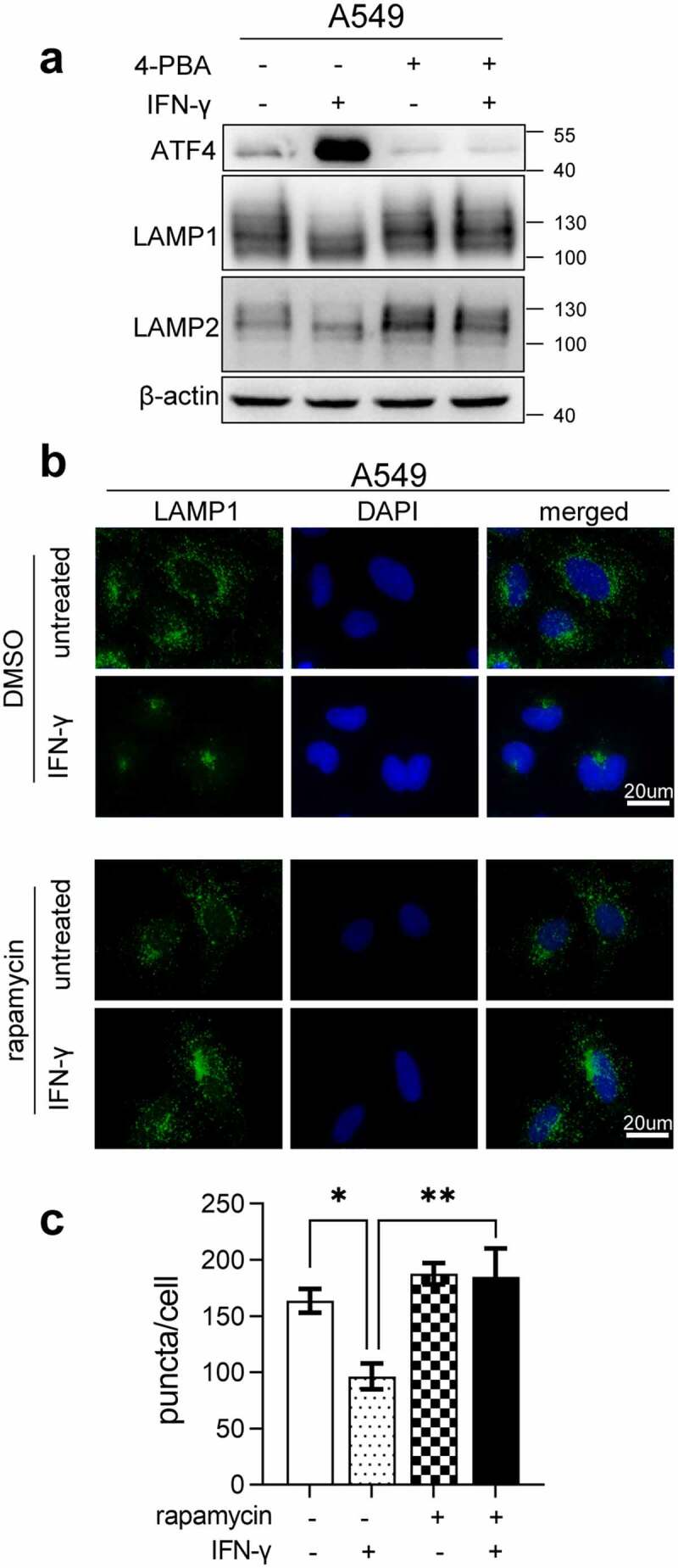
Inhibition of ER stress restores IFN-γ-mediated downregulation of LAMP. (a) A549 cells treated with IFN-γ or mock treated in the presence or absence of 1 mM 4-PBA for 3 days were subjected to western blot analyses of the expression of ATF-4, LAMP-1, and LAMP-2. (b and c) Immunofluorescence images of LAMP-1 in A549 cells treated with IFN-γ or mock treated in the presence or absence of rapamycin (b). Quantification of LAMP-1 puncta per cell is shown (c). *, p < .05; **, p < .01
IFN-γ-induced activation of PERK- eIF2α is involved in downregulation of LAMP expressions
As we demonstrated that IFN-γ-triggered UPR is responsible for regulating LAMP expression, we sought to determine how UPR regulates LAMP expression in IFN-γ-treated cells. First, we examined whether ER stress-mediated activation of the ubiquitin-proteasome system (UPS) is involved in the downregulation of LAMP. As shown in Figure S6a, ubiquitin activity increased in cells treated with IFN-γ. Furthermore, additional 4-PBA reversed the action of IFN-γ on the UPS, indicating that IFN-γ-induced activation of UPS is associated with UPR (Figure S6b). However, additional MG132 did not restore LAMP-1 and LAMP-2 expression in cells treated with IFN-γ (Figure 7a). The UPS may not be involved in the IFN-γ-induced downregulation of LAMP.
Figure 7.
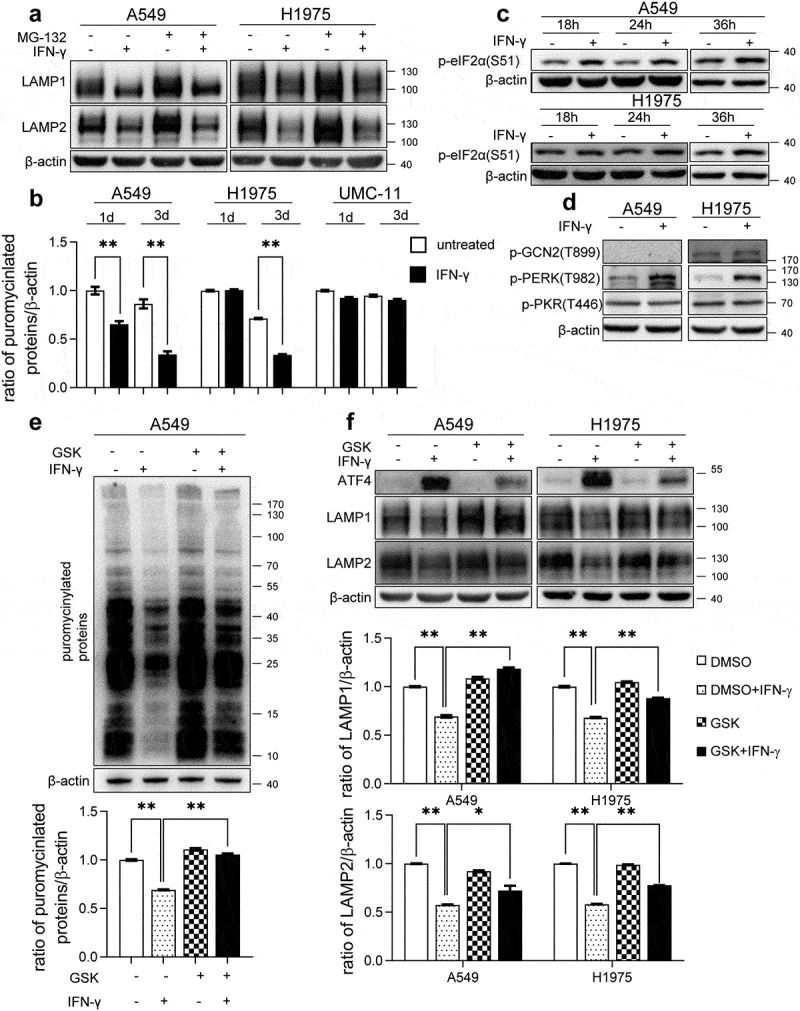
IFN-γ-induced activation of PERK-eIF2α is responsible for the reduction in the expression of LAMP. (a) The indicated cells were treated with IFN-γ or mock treated for 3 days followed by culturing with 10 μM MG132 for an additional 4 h. Immunoblots showing LAMP-1 and LAMP-2 expression. (b) The indicated cells were treated with IFN-γ or mock treated. Puromycin (10 µg/mL) was added during the last 10 min of incubation. Puromycinylated proteins were detected using western blotting and the bar graphs show densitometric analysis of the changes in the abundance of puromycinylated proteins normalized to β-actin for loading variability. **, p < .01. (c) Phosphorylated eIF2α was determined using western blot analyses of A549 and H1975 cells treated with or without IFN-γ (1000 IU/mL) for the indicated time intervals. (d) A549 and H1975 cells were treated with or without IFN-γ (1000 IU/mL) for 12 h. The expression levels of phosphorylated PERK, GCN2, and PKR were determined using western blot analysis. (e) A549 cells were treated with 1000 IU/mL of IFN-γ in the presence of 500 nM GSK2606414 for 24 h. Puromycin (10 µg/mL) was added during the last 10 min of incubation. Puromycinylated proteins were detected using western blotting and the bar graphs show densitometric analysis of the changes in the abundance of puromycinylated proteins normalized to β-actin for loading variability. **, p < .01. (f) The indicated cells were treated with 1000 IU/mL of IFN-γ in the presence of 500 nM GSK2606414 for 2 days. Immunoblots showing ATF-4, LAMP-1, and LAMP-2 expression. LAMP-1 and LAMP-2 expression was normalized to β-actin level for densitometric analyses and is presented as the mean ± SEM. *, p < .05; **, p < .01
Global inhibition of protein synthesis is one of the initial and key responses to cope with ER stress.43 Phosphorylation of elF2α is associated with the inhibition of protein synthesis.44 We first examined whether IFN-γ causes global inhibition of protein synthesis. As shown in Figures S7 and 7b, global protein synthesis rates were reduced significantly in A549 cells treated with IFN-γ for 24 h as determined by the SUnSET assay, and suppression of protein synthesis was even more remarkable following prolonged treatment with IFN-γ. In H1975 cells, a reduction in protein synthesis was observed on day 3 of IFN-γ treatment. IFN-γ did not affect protein synthesis in UMC-11 cells. Next, we examined whether IFN-γactivated eIF2α. IFN-γ markedly stimulated the phosphorylation of eIF2α at serine-51, as early as 18 h in A549 cells. In H1975 cells, we observed the phosphorylation of eIF2α 36 h after exposure to IFN-γ (Figure 7c). eIF2α can be activated by several stress-associated kinases, including PERK, GCN2, and PKR.45 IFN-γ stimulation induced the activation of PERK but not that of GCN and PKR (Figure 7d). Interestingly, the selective PERK inhibitor GSK2606414 significantly restored the global protein synthesis rates in cells treated with IFN-γ (Figure 7e).
We then investigated whether IFN-γ-induced activation of PERK- eIF2α signaling is involved in the decline of LAMP-1 and LAMP-2 expression. As shown in Figure 7f, GSK2606414 inhibited IFN-γ-induced ATF-4 expression and partially restored LAMP-1 and LAMP-2 expression levels in IFN-γ-treated cells. Our results suggest that IFN-γ-mediated activation of PERK-eIF2α signaling is involved in the reduction of expression of LAMP.
ER stress contributes to IFN-γ-induced apoptosis
Our data showed that IFN-γ-induced ER stress impairs autophagy. As unsolved ER stress is often associated with cell death and cell cycle arrest, we first examined whether IFN-γ triggers apoptotic cell death in lung cancer cells. IFN-γ treatment reduced cell viability in a dose-dependent manner (Figure 8a). We performed western blot analyses to examine the activated apoptotic marker cleaved caspase-3 in cells treated with IFN-γ. As shown in Figure 8b, cleaved caspase-3 level increased significantly in cells exposed to IFN-γ. Moreover, A549 and H1975 cells cultured with the supernatants collected from cultures of PBMCs stimulated with anti-CD3 mAb displayed an increased level of cleaved caspase-3 compared to the cells cultured with the supernatants collected from unstimulated PBMCs. Neutralization of IFN-γ in the supernatant led to a reduction in cleaved caspase-3 (Figure 8c).
Figure 8.
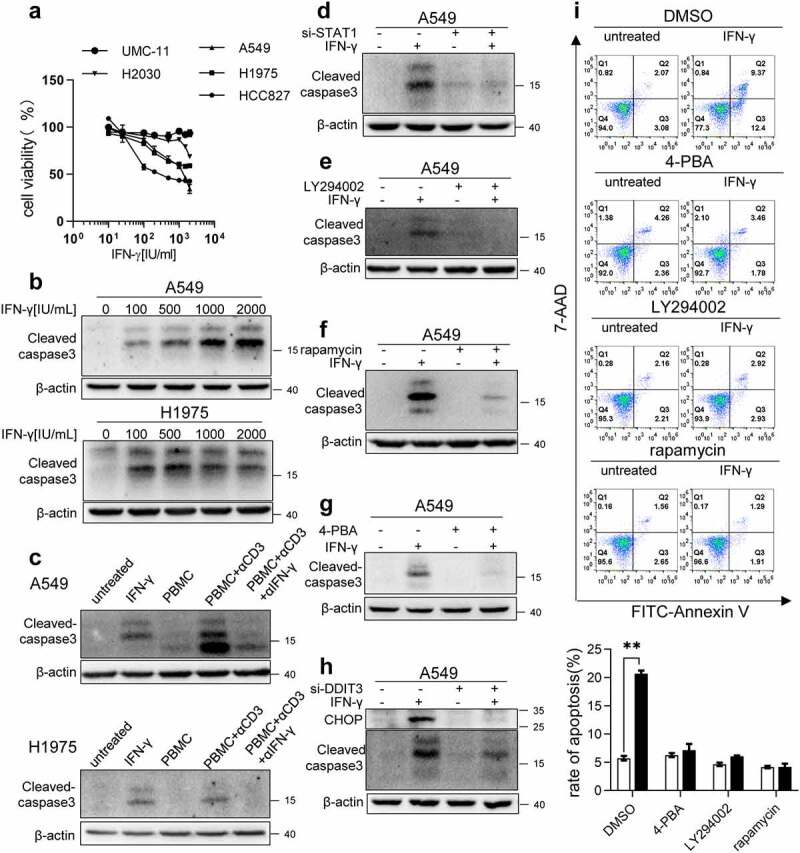
ER stress contributes to IFN-γ-induced apoptosis. (a) The indicated cells were treated with IFN-γ for 3 days followed by a CCK-8 assay. The untreated cells were used as controls (100%). (b) The indicated cells were treated with doses of IFN-γ (100–2000 IU/mL) for 3 days. The expression of cleaved caspase-3 was determined using western blotting. (c) PBMCs were stimulated with anti-CD3 mAb in the presence or absence of anti-IFN-γ antibody for 48 h. Subsequently, the supernatants were collected and cultured with A549 and H1975 cells for 2 days. Western blot analysis was performed to detect cleaved caspase-3. (d) A549 cells transfected with control siRNA or si-STAT1 were treated with IFN-γ (1000 IU/mL) for 3 days. Immunoblots showing cleaved caspase-3 expression. (e and f) A549 cells treated with IFN-γ in the presence or absence of LY294002 (e) or rapamycin (f) were subjected to western blotting to detect cleaved caspase-3 expression. (g) A549 cells treated with IFN-γ in the presence or absence of 4-PBA was subjected to western blot analysis to assess cleaved caspase-3 expression. (h) A549 cells transfected with control siRNA or si-DDIT3 were treated with IFN-γ for 3 days. Immunoblots showing CHOP and cleaved caspase-3 expression. (i) A549 cells were treated with IFN-γ (1000 IU/mL) for 3 days. Apoptotic cell death was evaluated using annexin V and 7-AAD staining assay followed by flow cytometry analysis. The data are presented as the means ± SEM from 3–4 individual experiments
IFN-γ-mediated cell death required the activation of JAK1/2-STAT1 (Figure S8a and 8d). Inhibition of AKT and mTOR signaling using LY294002 and rapamycin, respectively, also reduced the expression levels of cleaved caspase-3 in A549 cells (Figure 8e and f) and H1975 cells (Figure S8b and c). Next, we determined whether ER stress was responsible for IFN-γ-induced apoptosis. As shown in Figure 8g, the addition of 4-PBA abrogated IFN-γ-induced cleaved caspase-3 expression in A549 cells. Furthermore, CHOP knockdown by siRNA in A549 cells and H1975 cells led to a reduction in cleaved caspase-3 expression (Figure 8h and Figure S8d, respectively). The status of cell apoptosis induced by IFN-γ and the role of ER stress in the induction of cell apoptosis was confirmed using flow cytometry analyses (Figure 8i). Our data indicate that IFN-γ-triggered ER stress is responsible for the induction of apoptotic cell death.
ER stress is involved in IFN-γ-mediated suppression of cell cycle progression
We investigated whether ER stress is also involved in the IFN-γ-mediated inhibition of cell proliferation. As shown in Figure 9a, IFN-γ suppressed the expression of key molecules, including cyclin D1, cyclin E2, and cyclin B1 that were involved in the control of cell cycle progression. The suppressive action of IFN-γ required STAT1 signaling (Figure 9b). To further analyze the effect of IFN-γ on cell cycle progression, flow cytometry analysis using PI staining was performed to determine the changes in nuclear DNA content. The G1-phase cell numbers in cells treated with IFN-γ for 24 h were higher than those in cells not exposed to IFN-γ, whereas S- and G2/M-phase cell numbers decreased in cells treated with IFN-γ (Figure 9c and d). These results indicate that IFN-γ causes cell cycle arrest at the G1/S phase.
Figure 9.
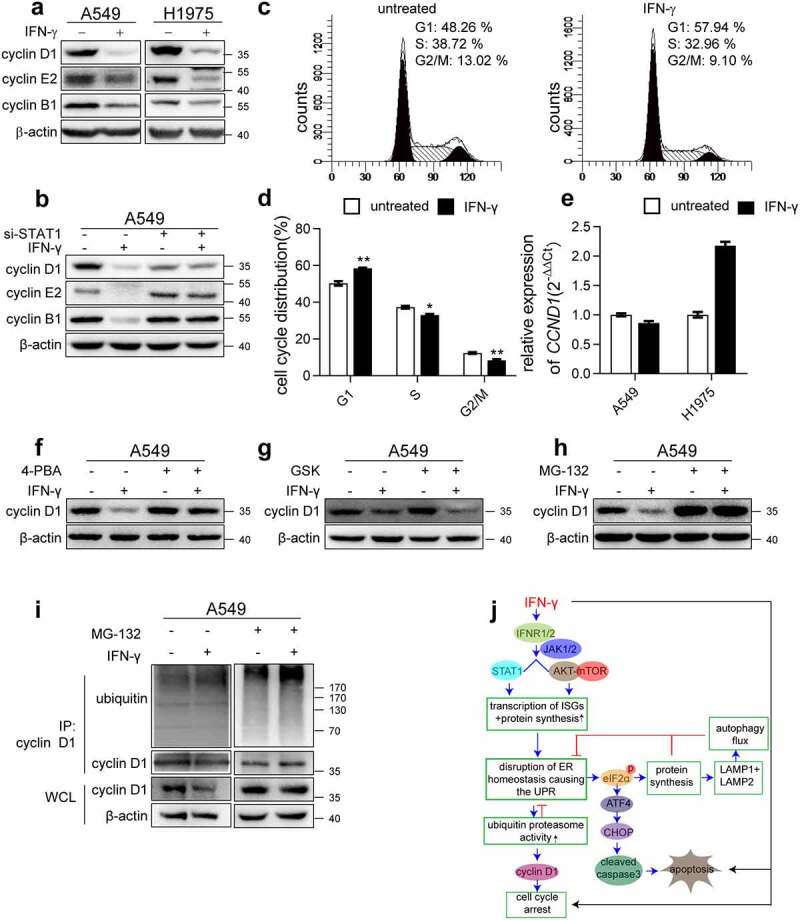
ER stress is involved in IFN-γ-mediated suppression of cell cycle progression. (a) Immunoblots showing cyclin D1, cyclin E2, and cyclin B1 expression in the indicated cells treated with IFN-γ vs. mock treated for 3 days. (b) The indicated cells transfected with control siRNA or si-STAT1 were treated with IFN-γ (1000 IU/mL) for 3 days. Immunoblots showing cyclin D1, cyclin E2, and cyclin B1 expression. (c) A549 cells were treated with IFN-γ (1000 IU/mL) for 24 h, and PI staining was performed to determine cell cycle progression, and (d) the data are presented as the means ± SEM from 3–4 individual experiments. (e) The expression of CCND1 was assessed using qRT-PCR in the indicated cells after treatment with IFN-γ (1000 IU/mL) or mock-treated for 24 h. (f) Cyclin D1 expression was assessed in A549 cells treated with IFN-γ in the presence or absence of 4-PBA. (g) A549 cells were treated with IFN-γ (1000 IU/mL) for 3 days in the presence of GSK2606414. Cyclin D1 expression was determined using western blotting. (h) A549 cells were treated with IFN-γ (1000 IU/mL) for 3 days followed by culturing with MG132 (10 μM) for an additional 4 h. Cyclin D1 expression was determined using western blotting. (i) Cell lysates were immunoprecipitated with anti-cyclin D1 antibody, and ubiquitination was detected using western blotting. (j) Schematic depicting the effect of IFN-γ signaling on lung adenocarcinoma cells. IFN-γ induces ER stress and UPR in lung adenocarcinoma cells through the activation of JAK1/2-STAT1 and AKT-mTOR signaling. This UPR consequently reduced LAMP-1 and LAMP-2 expression and led to impairment of autophagic flux. We also demonstrated that IFN-γ-induced ER stress contributes to IFN-γ-triggered apoptotic cell death and cell cycle arrest
Cyclin D1 is a crucial regulator of the cell cycle. IFN-γ significantly reduced cyclin D1 expression, as determined by western blotting (Figure 9a). Interestingly, IFN-γ did not decrease the transcription of CCND1 (Figure 9e). As a similar pattern was observed in LAMP, we speculated that cyclin D1 is also regulated by IFN-γ-induced ER stress. The ER stress inhibitor 4-PBA significantly restored the protein level of cyclin D1 (Figure 9f), suggesting that ER stress is involved in regulating the expression of cyclin D1. Next, we hypothesized that the reduction in cyclin D1 expression was due to ER stress-mediated suppression of protein synthesis. As shown in Figure 9g, GSK2606414 did not restore cyclin D1 expression in IFN-γ-treated cells. Interestingly, MG132 significantly restored cyclin D1 expression in IFN-γ treated cells (Figure 9h). Furthermore, we assessed the degree of cyclin D1 ubiquitination using immunoprecipitation with an anti-cyclin D1 antibody, followed by western blotting with an anti-ubiquitin antibody. As shown in Figure 9i, ubiquitination was detected and significantly increased in IFN-γ treated cells in the presence of MG132 compared to untreated cells. Our data indicate that ER stress-enhanced proteasome activity, but not the PERK-eIF2α-axis, may cause the downregulation of cyclin D1, thereby inhibiting cell cycle progression.
Discussion
Although the role of IFN-γ in the modulation of tumor immunity and its underlying mechanisms have been widely documented, the cellular and molecular actions of IFN-γ in tumor cells have not been fully investigated. Our study showed for the first time that IFN-γ induced ER stress and UPR in lung adenocarcinoma cells through the activation of JAK1/2-STAT1 and AKT-mTOR signaling. The UPR triggered by IFN-γ consequently reduced LAMP-1 and LAMP-2 expression and led to impairment of autophagic flux. We also demonstrated that IFN-γ-induced ER stress contributed to IFN-γ-triggered apoptotic cell death and cell cycle arrest (Figure 9j).
To date, very few studies have documented the role of IFN-γ in ER homeostasis.46,47 IFN-γ has been reported to increase UPR-associated molecule expression in conjunctival goblet cells, causing a failure in the expression of Muc2 and Muc5AC glycoproteins, which leads to dye eye in patients with Sjogren syndrome.47 It has long been recognized that the biological functions of IFN-γ rely on newly synthesized proteins, for example, increases in CD274 expression on tumor cells through the activation of STAT1 and mTOR signaling, to promote immune escape.10 Rapidly increased protein synthesis disrupts ER homeostasis. Indeed, our studies demonstrated that IFN-γ induces ER stress and UPR in lung cancer cell lines. Furthermore, induction of UPR by IFN-γ was also observed in human tumor specimens. Notably, we evaluated a total of seven human tumor specimens to determine the effect of IFN-γ on the induction of DDIT4 transcription. IFN-γ increased transcription of the DDIT3 gene in four tumors but failed to induce DDIT3 transcription in the other tumors. Our data reflect heterogeneity in tumors in response to IFN-γ treatment. One explanation could be that approximately one-third of melanoma and lung adenocarcinoma cell lines had inactivating mutations in the IFN-γ pathway components.48,49 In addition, studies have shown that in some tumor tissues, the elF2α-ATF-4-CHOP signaling pathway is in a highly activated state,50 which could undermine the effect of IFN-γ on ER stress. The precise underlying molecular mechanisms regarding the diverse response to IFN-γ in tumors could be more complex and require further investigation. Inhibition of JAK1/2-STAT1 and AKT-mTOR effectively diminished IFN-γ-induced UPR. It is worth noting that one of the consequences of UPR is the inhibition of global protein synthesis. Therefore, the overall biological functions of IFN-γ on tumor cells appear to be determined by both cellular processes.
Autophagy is an essential mechanism of ER stress. ER stress can either stimulate or inhibit autophagy.32 Miyagawa et al. showed that saturated fatty acids impaired autophagosome-lysosome fusion in the livers of mice, and this impairment was ameliorated by the reduction of ER stress.51 LAMP-1 and LAMP-2 are essential components of the lysosome and are involved in autophagy.52 Cui et al. have shown a decrease in LAMP-2 accompanied with the impaired autophagic flux in response to oxygen, glucose, or serum deprivation treatments. Overexpression of LAMP-2 significantly alleviated autophagy flux.53 Very recently, Nakashima et al. reported that in trophoblast cells, ER stress could reduce autophagic flux by suppressing the expression of LAMP, although the underlying mechanism remains elusive.54
Phosphorylation of eIF2α impairs cap-dependent translation and is responsible for inhibiting global protein synthesis in the ER and other stresses.55 Gennaro et al. found that sustained phosphorylation of eIF2α decreases the synthesis of proteins belonging to the endolysosomal system.56 We showed that suppression of IFN-γ-induced UPR by inhibiting AKT-mTOR or PERK-elF2α signaling restored the protein content of LAMP. The precise underlying molecular mechanism by which the PERK- eIF2α axis regulates LAMP-1 and LAMP2 warrants further investigation.
The role of IFN-γ in autophagy has been thoroughly investigated. IFN-γ induces autophagy in gastric epithelial cells through increased expression of Beclin-1.57 IFN-γ also induces autophagosome formation and promotes autophagic flux in hepatocellular carcinoma cells (HCC).58 Interestingly, IFN-γ treatment of HCCs activates the STAT1-IRF1 pathway but inhibits AKT-mTOR signaling, which is in contrast to the signaling transduced by IFN-γ in lung adenocarcinoma cells, indicating that the effects of IFN-γ on autophagy are cell type-specific. Autophagy can be a survival mechanism that allows cells to develop adaptive changes under various conditions.59 However, the relationship between autophagy and cell death is complex. For instance, the discovery of the interaction between Beclin-1 and Bcl-2/Bcl-xL implicates the crosstalk between autophagy and apoptosis.60 Bcl-2/Bcl-xL targets Bax and Bak to inhibit apoptosis.61,62 Bcl-2 can also bind to Beclin-1 and inhibit the activation of autophagy.63 Strappazzon et al. reported that serum starvation increases JNK1-mediated phosphorylation of Bcl-2 and leads to the release of Beclin-1 from Bcl-2 and induction of autophagy. Interestingly, under the conditions of extreme serum starvation, JNK1 induces hyperphosphorylation of Bcl-2, causing its dissociation from Bax to promote caspase 3-dependent apoptotic cell death.64,65 In addition, Beclin-1 can also be phosphorylated by DAPK, promoting its dissociation from Bcl-2, which leads to enhanced autophagosome formation.66 In this study, IFN-γ treatment impaired autophagy by blocking autophagosome-lysosomal fusion. The precise relationship between impaired autophagy and cell death in cancer cells under the influence of IFN-γ remains undetermined and warrants further investigation.
Prolonged ER stress or unrestored ER homeostasis leads to cell death. We showed that CHOP is responsible for IFN-γ-triggered apoptosis in our experimental setting. In our study, we showed that inhibition of the PI3K-AKT-mTOR signaling pathway effectively reduced IFN-γ-induced CHOP expression and apoptosis. Although the primary role of the PI3K-AKT-mTOR axis in tumor cells is to promote proliferation, cell growth, and resistance to apoptosis induction, PI3K-AKT-mTOR signaling is also involved in several biological processes that could ultimately determine the fate of tumor cells. For example, there is a complex relationship between the PI3K-AKT-mTOR and ER stress. Studies by Kitamura and coworkers have shown that mTORC1 causes ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway.67 In cases where ER stress cannot be reversed and cellular functions deteriorate, often resulting in cell death. Thus, there is an indirect relationship between cell death and AKT-mTOR activation. AKT-mTOR is also closely related to autophagy and serves as a negative modulator of autophagy.68 Although several studies have suggested that the autophagy participates in a caspase-independent cell death,69 autophagy is well recognized as a cell survival process that promotes tumor development. Taken together, whether activation of AKT-mTOR signaling is pro-tumor or antitumor may be context-dependent.
Our study provides evidence of an unappreciated effect of IFN-γ on ER homeostasis in lung cancer cells. Moreover, identifying the biological consequences of IFN-γ-triggered ER stress beyond cell death and cell cycle arrest may shed light on the use of IFN-γ to treat lung cancer.
Supplementary Material
Acknowledgments
Not applicable.
Funding Statement
This work was supported by the National Natural Science Foundation of China (Grant numbers 81874168, 81672808, and 81472652 for L.L.).
Abbreviations
IFN-γ: Interferon gamma
ER: Endoplasmic reticulum
UPR: unfolded protein response
ATF4: Activating transcription factor-4
CHOP: C/EBP homologous protein
GRP78: Glucose-regulated protein 78
sXBP-1: spliced X-box binding protein 1
LC3: Microtubule-associated protein light chain 3
LAMP: Lysosomal membrane protein
PERK: Protein kinase-like ER kinase
elF-2α: eukaryotic initiation factor 2 α subunit
ISGs: Interferon stimulated genes
Authors of contributions
C.F. planned and performed experiments. T.W., S.H., Z.Y., B.H., H.X., and Y.C. provided technical and material support. C.F. provided acquisition, analysis, and statistical analysis. C.F. and L.L. provided interpretation of data. All authors provided critical feedback and helped shape the research and analysis. L.L. and X.F. designed and directed the project. L.L. and C.F. took the lead in writing the manuscript with input from all authors.
Availability of data and materials
The analyzed datasets generated during the current study are available from the corresponding author on reasonable request.
Disclosure Statement
The authors declare no competing interests.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of Tongji Hospital of Huazhong University of Science and Technology.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Sharma P, Pachynski R, Narayan V.. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the checkMate 650 trial. Cancer Cell. 2020;38(4):489–499.e483. doi: 10.1016/j.ccell.2020.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Ayers M, Lunceford J, Nebozhyn M. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–18. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao J, Shi L, Zhao H. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404.e399. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thibaut R, Bost P, Milo I. Bystander IFN-γ activity promotes widespread and sustained cytokine signaling altering the tumor microenvironment. Nature Cancer. 2020;1(3):302–314. doi: 10.1038/s43018-020-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoekstra M, Bornes L, Dijkgraaf F. Long-distance modulation of bystander tumor cells by CD8 T cell-secreted IFNγ. Nature Cancer. 2020;1(3):291–301. doi: 10.1038/s43018-020-0036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wall L, Burke F, Barton C, Smyth J, Balkwill F. IFN-gamma induces apoptosis in ovarian cancer cells in vivo and in vitro. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2003;9:2487–2496. [PubMed] [Google Scholar]
- 7.Yin H, Jiang Z, Wang S, Zhang P. IFN-γ restores the impaired function of RNase L and induces mitochondria-mediated apoptosis in lung cancer. Cell Death Dis. 2019;10(9):642. doi: 10.1038/s41419-019-1902-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroder K, Hertzog P, Ravasi T, Hume D. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. [DOI] [PubMed] [Google Scholar]
- 9.Ramsauer K, Sadzak I, Porras A. p38 MAPK enhances STAT1-dependent transcription independently of ser-727 phosphorylation. Proc Natl Acad Sci U S A. 2002;99(20):12859–12864. doi: 10.1073/pnas.192264999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Y, Yang J, Cai Y. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. International Journal of Cancer. 2018;143(4):931–943. doi: 10.1002/ijc.31357. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Li Z, Qiu S. IFN-γ induces gastric cancer cell proliferation and metastasis through upregulation of integrin β3-mediated NF-κB signaling. Transl Oncol. 2018;11(1):182–192. doi: 10.1016/j.tranon.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu M, Peng Z, Qin M. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol Cell. 2021;81(6):1216–1230.e1219. doi: 10.1016/j.molcel.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 13.Arrieta A, Blackwood E, Glembotski C. ER protein quality control and the unfolded protein response in the heart. Curr Top Microbiol Immunol. 2018;414:193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science (New York, NY). 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 15.Schröder M, Kaufman R. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. [DOI] [PubMed] [Google Scholar]
- 16.Ni M, Lee A. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581(19):3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaufman R. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110(10):1389–1398. doi: 10.1172/JCI0216886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertolotti A, Zhang Y, Hendershot L, Harding H, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 19.Harding H, Novoa I, Zhang Y. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 20.Marciniak S, Yun C, Oyadomari S. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee A, Iwakoshi N, Glimcher L. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology. 2003;23(21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen J, Prywes R. Dependence of site-2 protease cleavage of ATF6 on prior site-1 protease digestion is determined by the size of the luminal domain of ATF6. J Biol Chem. 2004;279(41):43046–43051. doi: 10.1074/jbc.M408466200. [DOI] [PubMed] [Google Scholar]
- 23.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev Cell. 2002;3(1):99–111. doi: 10.1016/S1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 24.Smith H, Mallucci G. The unfolded protein response: mechanisms and therapy of neurodegeneration. Brain: A Journal of Neurology. 2016;139(8):2113–2121. doi: 10.1093/brain/aww101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senft D, Ronai Z. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–148. doi: 10.1016/j.tibs.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sano R, Reed J. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klionsky D, Emr S. Autophagy as a regulated pathway of cellular degradation. Science (New York, NY). 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt E, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6(4):275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 30.Ravindranathan P, Lee T, Yang L. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. 2013;4(1):1923. doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- 31.Schneider W, Chevillotte M, Rice C. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32(1):513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rashid H, Yadav R, Kim H, Chae H. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klionsky D, Abeliovich H, Agostinis P. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margariti A, Li H, Chen T. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288(2):859–872. doi: 10.1074/jbc.M112.412783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ciechomska I, Goemans G, Skepper J, Tolkovsky A. Bcl-2 complexed with beclin-1 maintains full anti-apoptotic function. Oncogene. 2009;28(21):2128–2141. doi: 10.1038/onc.2009.60. [DOI] [PubMed] [Google Scholar]
- 36.Pihán P, Carreras-Sureda A, Hetz C. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017;24(9):1478–1487. doi: 10.1038/cdd.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gade P, Manjegowda S, Nallar S, Maachani U, Cross A, Kalvakolanu D. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Molecular and Cellular Biology. 2014;34(21):4033–4048. doi: 10.1128/MCB.00397-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 40.Bampton E, Goemans C, Niranjan D, Mizushima N, Tolkovsky A. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1(1):23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- 41.Granger B, Green S, Gabel C, Howe C, Mellman I, Helenius A. Characterization and cloning of lgp110, a lysosomal membrane glycoprotein from mouse and rat cells. J Biol Chem. 1990;265(20):12036–12043. doi: 10.1016/S0021-9258(19)38504-7. [DOI] [PubMed] [Google Scholar]
- 42.Wright J, Zeitlin P, Cebotaru L, Guggino S, Guggino W. Gene expression profile analysis of 4-phenylbutyrate treatment of IB3-1 bronchial epithelial cell line demonstrates a major influence on heat-shock proteins. Physiol Genomics. 2004;16(2):204–211. doi: 10.1152/physiolgenomics.00160.2003. [DOI] [PubMed] [Google Scholar]
- 43.Han J, Back S, Hur J. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harding H, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 45.Donnelly N, Gorman A, Gupta S, Samali A. The eIF2α kinases: their structures and functions. Cellular and Molecular Life Sciences: CMLS. 2013;70(19):3493–3511. doi: 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Jamal S, Taylor E, Abd Elmageed Z. Interferon gamma-induced apoptosis of head and neck squamous cell carcinoma is connected to indoleamine-2,3-dioxygenase via mitochondrial and ER stress-associated pathways. Cell Div. 2016;11(1):11. doi: 10.1186/s13008-016-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coursey T, Tukler Henriksson J, Barbosa F, de Paiva C, Pflugfelder S. Interferon-γ-induced unfolded protein response in conjunctival goblet cells as a cause of mucin deficiency in sjögren syndrome. Am J Pathol. 2016;186(6):1547–1558. doi: 10.1016/j.ajpath.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplan D, Shankaran V, Dighe A. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95(13):7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zaidi M, Merlino G. The two faces of interferon-γ in cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2011;17(19):6118–6124. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang L, Chen T, Kuo W, Hua C. αThe protein expression of PDL1 is highly correlated with those of eIF2 and ATF4 in lung cancer. Dis Markers. 2018;2018:5068701. doi: 10.1155/2018/5068701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyagawa K, Oe S, Honma Y, Izumi H, Baba R, Harada M. Lipid-induced endoplasmic reticulum stress impairs selective autophagy at the step of autophagosome-lysosome fusion in hepatocytes. Am J Pathol. 2016;186(7):1861–1873. doi: 10.1016/j.ajpath.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Eskelinen E. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27(5–6):495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Cui L, Zhao L, Ye J. The lysosomal membrane protein lamp2 alleviates lysosomal cell death by promoting autophagic flux in ischemic cardiomyocytes. Frontiers in Cell and Developmental Biology. 2020;8:31. doi: 10.3389/fcell.2020.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakashima A, Cheng S, Kusabiraki T. Endoplasmic reticulum stress disrupts lysosomal homeostasis and induces blockade of autophagic flux in human trophoblasts. Sci Rep. 2019;9(1):11466. doi: 10.1038/s41598-019-47607-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wek R. Role of eIF2α kinases in translational control and adaptation to cellular stress. Cold Spring Harb Perspect Biol. 2018;10(7). doi: 10.1101/cshperspect.a032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gambardella G, Staiano L, Moretti M. GADD34 is a modulator of autophagy during starvation. Science Advances. 2020;6(39). doi: 10.1126/sciadv.abb0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tu S, Quante M, Bhagat G. IFN-γ inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011;71(12):4247–4259. doi: 10.1158/0008-5472.CAN-10-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li P, Du Q, Cao Z. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012;314:213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta. 2003;1603:113–128. [DOI] [PubMed] [Google Scholar]
- 60.Oberstein A, Jeffrey P, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282(17):13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 61.Finucane D, Bossy-Wetzel E, Waterhouse N, Cotter T, Green D. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274(4):2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 62.Willis S, Chen L, Dewson G. Proapoptotic bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang N, Nguyen M, Germain M, Shore G. Antagonism of beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010;29(3):606–618. doi: 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strappazzon F, Vietri-Rudan M, Campello S. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011;30(7):1195–1208. doi: 10.1038/emboj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4(7):949–951. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zalckvar E, Berissi H, Mizrachy L. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10(3):285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012;19(2):310–320. doi: 10.1038/cdd.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janku F, McConkey D, Hong D, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8(9):528–539. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 69.Lin L, Baehrecke E. Autophagy, cell death, and cancer. Molecular & Cellular Oncology. 2015;2(3):e985913. doi: 10.4161/23723556.2014.985913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The analyzed datasets generated during the current study are available from the corresponding author on reasonable request.