

Abstract
Mutation of the human genome results in three classes of genomic variation: single nucleotide variants; short insertions or deletions; and large structural variants (SVs). Some mutations occur during normal processes, such as meiotic recombination or B cell development, and others result from DNA replication or aberrant repair of breaks in sequence-specific contexts. Regardless of mechanism, mutations are subject to selection, and some hotspots can manifest in disease. Here, we discuss genomic regions prone to mutation, mechanisms contributing to mutation susceptibility, and the processes leading to their accumulation in normal and somatic genomes. With further, more accurate human genome sequencing, additional mutation hotspots, mechanistic details of their formation, and the relevance of hotspots to evolution and disease are likely to be discovered.
Introduction
In 1983, the HTT gene was mapped to human chromosome 4 [1]. Single allele loss-of-function mutations (see Glossary) at this locus result in autosomal dominant Huntington’s disease. It was later found that a CAG tract within HTT can expand to more than 35 times the normal length, and that the expanded allele encodes a nonfunctional protein [2]. This trinucleotide expansion is seen in ~10% of Huntington’s disease cases and occurs due to slippage of the DNA polymerase during replication of the tandem repeat [3,4]. The HTT CAG tract expansion is one example of the many different mutation hotspots scattered throughout our genome. These hotspots can lead to a ~100-fold difference in germline mutation rates across human DNA, and are typically generated by distinct mutational processes [5]. Understanding these processes is not only important for basic biology and evolutionary dynamics, but can also inform preventative and therapeutic approaches to genetic disease. In this review, we discuss mutation hotspots in the human genome, with an emphasis on mechanistic inferences.
Classes and Mechanisms of Genetic Mutation and Variation
Mutational processes and consequences are commonly observed by examining variation present between two individuals in a population. Genomic variants are largely defined by their size and mechanism of formation (Figure 1). Single nucleotide variants (SNVs) are one base pair substitutions in DNA, which are further classified as transitions (i.e., purine to purine or a pyrimidine to pyrimidine change) or transversions (i.e., a purine to pyrimidine switch or vice versa). They are the most numerous variant in the human genome, comprising ~80% of the differences between two individuals, and occur at a rate of ~1 × 10–9–1× 10–8 mutations per base pair per generation [6,7]. SNV mutational hotspots include germline variants in CpG-rich loci [6,8] and somatic mutations caused by activation-induced cytidine deaminase (AID) or apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC)-mediated deamination [9,10].
Figure 1. Types of Genetic Variation.
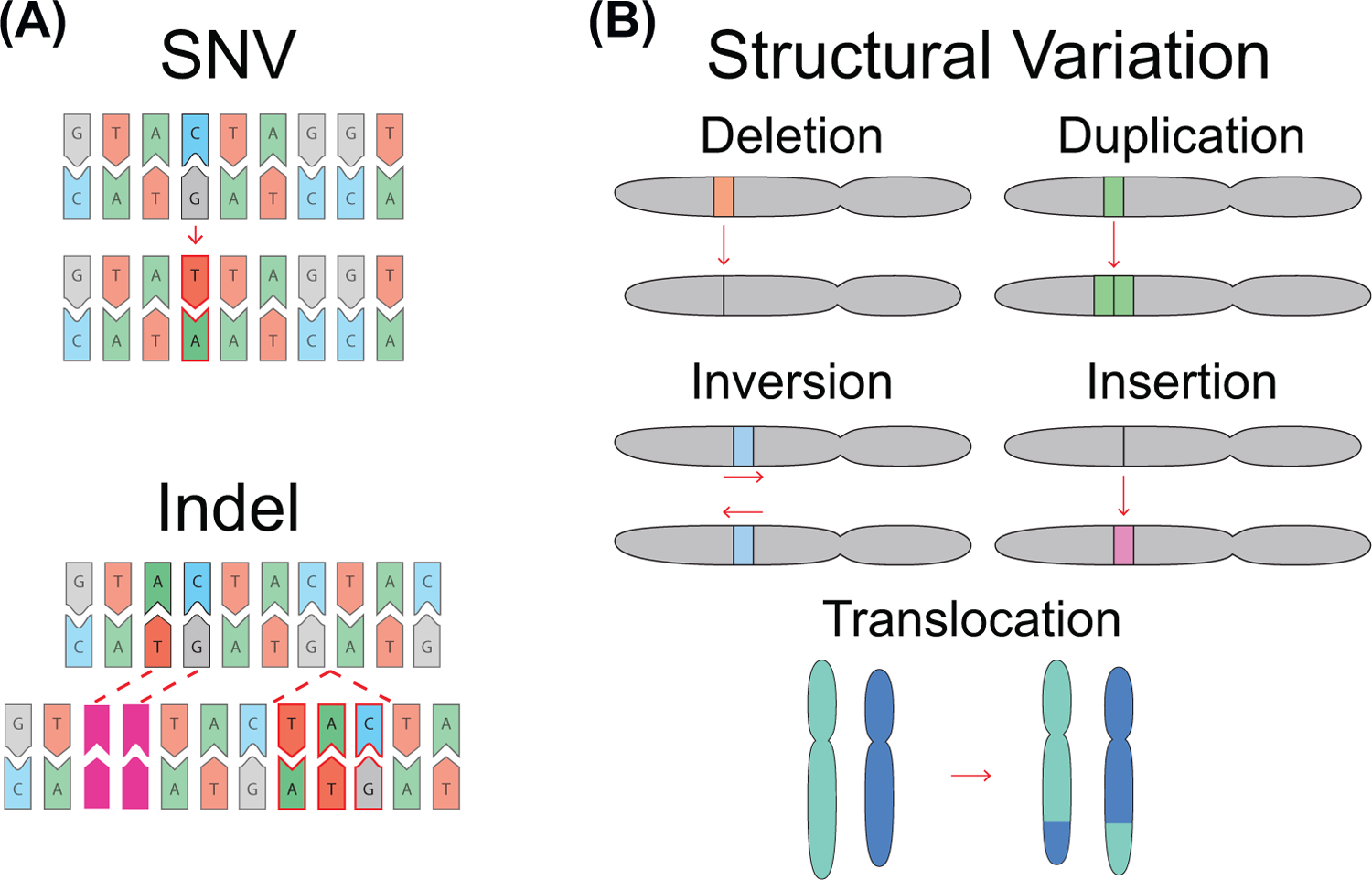
Single nucleotide variants (SNVs) and indels are changes that affect between one and 50 base pairs in a single event. (A) Example of a C:T SNV, and a two base pair deletion and a three base pair insertion indel. Examples of events over ≥50 base pairs that constitute SVs are shown in (B); these events include deletions, duplications, inversions, insertions, translocations, and complex combinations of these basic variant types. In each example, the top chromosome is the reference, and the variation is highlighted and displayed beneath.
An insertion or deletion (indel) is the gain or loss of one or more nucleotides spanning <50 base pairs. An analysis of 33 families found a de novo indel rate of ~9 × 10–10 mutations per nucleotide per generation [7]. Approximately 45% of indels are concentrated in ~4% of the genome and most can be explained by polymerase slippage [11] (Figure 2A, Key Figure). Sequences prone to polymerase slippage, such as homopolymeric runs and tandem repeat regions, harbor70% of indels [11]. Coding region indels are under stronger negative selection than are SNVs [12]. Based on the dbSNP database, SNVs in the coding regions outnumber short indels by sevenfold, while the density of SNVs and indels is almost the same in intronic regions [12].
Figure 2.
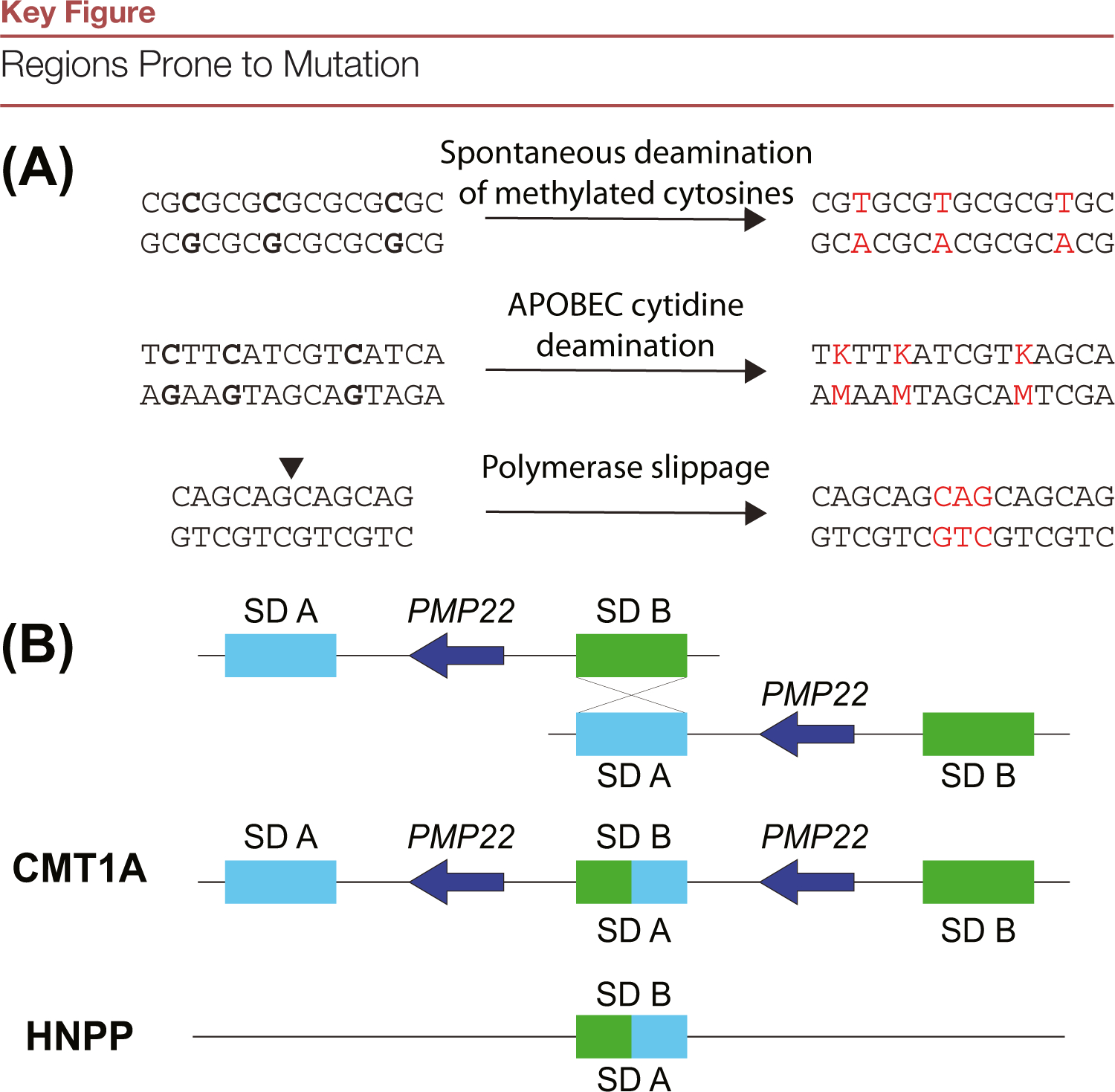
(A) The mechanism(s) of variant formation can be inferred through sequence context as well as the resultant variant. For example, spontaneous deamination of methylated cytosines result in C:T transitions at the GC-rich loci. Apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC) cytidine deaminases act upon tCw motifs changing them to tTw or tGw (IUPAC code K). This pattern is observed in kataegis. Finally, repetitive DNA regions are subject to indels when the polymerase dissociates from the template and pairs with the incorrect region upon reassociation, resulting in expansions or contractions. (B) Nonallelic homologous recombination can lead to rearrangements via incorrect repair between repeats. This example highlights a 1.5-Mb region in chromosome 17p11-p12. Two segmental duplications (SDs), denoted as SD A and SD B, represent the proximal and distal ~24-Kb CMT1A-REP repeats. These SDs ectopically pair during meiosis, and repair by nonallelic homologous recombination (NAHR), leading to duplication or deletion of the dosage-sensitive gene PMP22, resulting in Charcot-Marie-Tooth disease 1A (CMT1A) or hereditary neuropathy with liability to pressure palsies (HNPP) respectively.
SVs are large-scale changes of the genome (>50 base pairs), including deletions and duplications (copy number variations), inversions, insertions, and translocations. The estimated SV mutation rate is ~0.29 per generation [13]. Although SVs occur less frequently than SNVs and indels, they account for the variation in a larger number of nucleotides when comparing any two human genomes [14]. SVs are hallmarks of many cancers and can involve complex mutations, such as those observed in chromothripsis and whole-genome duplication events early on in cancer lineages [15,16].
Inherited variants of all of these types in the human population have been subject to selective pressures, leading to a change in frequency; this process can lead to variation hotspots (Box 1) that can be distinct from mutation hotspots. Specific mechanisms and environmental influences have been associated with distinct somatic mutational signatures identified in cancer, and are mentioned here in the sections corresponding to their mechanisms of formation [17]. In some instances, more than one mechanism can contribute to a mutational hotspot (Box 2).
Box 1. The Power of Positive Selection.
Mutations that result in altered cellular and organismal fitness are either preferentially lost or retained in a population. Selection has resulted in traits that differentiate humans from other species, and can lead to the clonal expansion of cells with advantageous mutations [106].
Selective forces acting on mutation can give rise to sites of recurrent variation in a population. Copy number amplification of the AMY1 locus in several species is caused by ectopic recombination between repeats and correlates with a starch-rich diet. AMY1 encodes the salivary amylase enzyme, and exists in variable copy numbers in the human population. Positive selection favors a higher copy number of AMY1 in Japanese and European individuals with starch-rich diets. Populations that consume less starch lack selective pressure for AMY1 amplification and, therefore, have a lower copy number [107].
Positive selection in tumorigenesis can result in a clonal population containing one or more driver mutations [108]. Some of these mutations are recurrent across many individuals and facilitate discovery of cancer-associated genes and therapeutic targets [108–110]. Recurrent mutations and amplifications of oncogenes are also seen in different tumor types [111]; in particular, gene amplification through double-minute or extrachromosomal circular DNA formation can result in higher order gene copy number amplification and increased oncogenic potential [112]. Positive selection through clonal hematopoiesis can result in distinct subpopulations of cells with driver mutations [113]. Tissue-specific indel mutations have also been observed in cancer, specifically occurring in noncoding regions of highly expressed genes [114]. Some driver mutations create cancer-specific drug targets. For example, >95% of patients with chronic myelogenous leukemia harbor a common reciprocal translocation between the BCR and the ABL1 genes on chromosomes 22 and 9, and the resultant fusion protein is a cancer-specific target [115]. Other recurrent driver mutations with clinically tested druggable targets include BRAF V600E, KRAS G12C, and METex14 skipping [116–120].
Box 2. A Hallmark Example of a Mutagenesis Hotspot.
Generating a diverse and highly specific set of antibodies in B cells involves all three classes of genetic variation. These mutations result from V(D)J recombination and somatic hypermutation at antibody-producing immunoglobulin loci. The gene segments V (variable), D (diversity), and J (joining) are responsible for antigen-binding regions and antibody diversity. Recombination signal sequences (RSS) that flank these segments are joined together to create a functional immunoglobulin gene, deleting the intervening sequence [121]. Key enzymes for V(D)J recombination are the recombination activating genes (RAG1 and RAG2), the high-mobility group box chromatin protein (HMGB), and the nonhomologous end-joining (NHEJ) DNA repair machinery [122,123].
The process of affinity maturation generates targeted SNVs that may allow production of more specific antibodies after V (D)J recombination [124]. At the transcriptionally active V(D)J locus, AID hydrolyzes cytosines on the nontemplate DNA strand into uracil [125]. The converted residue mimics thymidine during DNA replication and the U:G mismatch triggers the DNA repair process, creating random point mutations in the immunoglobin gene. AID increases the mutation rate specifically at the immunoglobin locus from one in 10–9 base pairs to one in 10–3 base pairs per cell division [126]. This creates approximately one mutation at the immunoglobin locus in each daughter cell [126]. This heightened mutation rate is compounded by the increased rate of active B cell division [126]. Somatic mutations of the genes encoding antibodies are under positive selection if they encode an antibody with higher affinity for the antigen compared with the antibody encoded by the non-mutated gene [9].
Figure I. V(D)J Recombination and Somatic Hypermutation.

Euchromatic H3K4me3 histone modifications at the immunoglobulin locus recruit the recombination complex during B cell development. RAG1 and RAG2 (pink and red ovals) bind two recombination signal sequences (RSS; dark-green and light-green arrows) that flank each V, D, or J coding segment dark-green, light-green, and yellow rectangles, respectively). These segments are processed and ligated together by the NHEJ machinery, usually deleting the intervening sequences. Occasionally, cuts in the RSS (indicated by red triangles in RSS) leave an uneven overhang. Translesion synthesis then fills in the missing information, potentially forming indels during the ligation process. The resulting process generates the antibody-defining coding joint and a signal joint that is circularized to prevent further recombination. After a B cell recognizes an antigen, the immunoglobulin locus may undergo somatic hypermutation to increase antibody specificity. During this process, AID (purple oval) hydrolyzes cytosines on the nontemplate DNA strand into uracil (orange triangles). The converted residue mimics thymidine during DNA replication and the U:G mismatch triggers the DNA repair process, creating point mutations in the immunoglobin gene. Positive selection acts on B cells that best bind a corresponding antigen.
DNA Sequences Prone to Mutation
GC-Rich Regions
Two mechanisms promote mutagenesis of GC-rich regions: spontaneous deamination (cancer mutational signature SBS1 [17]) of methylated cytosines and GC-biased gene conversion [8,18]. Cytosines have a germline mutation rate that is approximately ten times higher than other nucleotides and these changes are most prevalent at CpG dinucleotides [19]. CpG islands are a hotspot for mutation, are commonly located within and upstream of genes, and can regulate gene expression via their methylation status [20]. These loci comprise ~1.5% of our genome, and ~10.6% of all CpG dinucleotides are within islands [21]. Despite spontaneous 5-methylcytosine deamination resulting in C to U lesions, SNVs in CpG islands tend to be approximately seven times less frequent than in other CpGs [21]. Genome-wide methylation analyses have shown that the lower mutation rate of CpG islands is driven primarily by strong negative selection due to their role in gene expression regulation [22]. Although methylated CpGs are a mutation hotspot, their regulatory role means that C to U lesions are under negative selection within CpG islands.
GC-biased gene conversion is an example of a mutational mechanism occuring at recombination hotspots and is a major driver of base composition heterogeneity [23]. In this process, meiotic recombination favors GC-rich alleles over AT-rich alleles and facilitates local GC-content increases [24,25]. During the pairing step of gene conversion, there may be G:T or C:A mismatches resolved in favor of the G or C alleles [24]. After analysis of autosomes in >1000 sequenced genomes, AT to GC changes were enriched compared with GC to AT [24]. This simultaneously increases local GC-content as well as SNVs.
Microsatellites
Microsatellites comprise ~3% of the human genome [26]. These repetitive sequences are prone to polymerase slippage and indel formation, and therefore, are hotspots of mutation in both somatic and germline events. Indels in microsatellites can give rise to the inherited trinucleotide repeat expansions and contractions seen in Huntington’s disease, Fragile X syndrome, and X-linked spinal muscular atrophy [4,27,28]. There are 43 human genes that contain microsatellites in their coding sequence and may act as mutation hotspots for other diseases [11]. Some cancers display somatic microsatellite instability that results from functionally impaired mismatch repair enzymes (cancer mutational signatures SBS15, SBS21, SBS44, DBS7, DBS9, ID1, and ID2 [17]). This phenotype is present in ~15–30% of certain tumor types, resulting in an estimated 200–300-fold increase in mutation rate per cell division as measured in mismatch repair-deficient yeast and mice [29,30].
Meiotic Recombination Hotspots and Nonallelic Homologous Recombination
Meiotic recombination occurs at specific GC-rich loci that contain binding motifs for PR domain zinc finger protein 9 (PRDM9) [31,32]. PRDM9 binds its 12-base pair zinc finger motif, leaves euchromatic H3K4me3 and H3K36me3 marks, and recruits SPO11 to cut the DNA at one of two homologous chromosomes [33]. Holliday junctions form between the homologs, with DNA repair machinery facilitating crossover and repair with the intact homolog [31].
PRDM9 motifs function as both recombination and mutation hotspots [32]. As the target sequence is cleaved and recombined, mutations accumulate due to imperfect repair and these mismatches can inhibit future binding of PRDM9 [34]. Alleles lacking PRDM9 motifs are under positive selection, because the lack of recombination allows them to be inherited more frequently [35,36]. The mutational processes at play lead to both rapid evolution of the PRDM9 zinc finger binding domain and meiotic recombination hotspots across the genome.
Nonallelic homologous recombination (NAHR) can result in SV hotspots and occurs by ectopic recombination at or near repetitive sequences in the genome during meiosis [37]. For example, chromosome 17p contains 1.7 kb segmental duplications flanking the dosage sensitive PMP22 gene. Homologous recombination between the incorrect (98.7% identical) segmental duplications during meiosis results in either duplication or deletion of PMP22, leading to Charcot-Marie-Tooth (CMT1A) or Hereditary Neuropathy with Liability to Pressure Palsies (HNPP), respectively (Figure 2B) [38]. The rate of NAHR can be significantly higher than that of SNVs, with CMT1A duplication and HNPP deletion events occurring at a frequency of ~2–4× 10−5 per every male meiosis [39]. In addition to segmental duplications, transposable elements may facilitate NAHR in some regions of the genome, including the VHL and SPAST loci [40].
Centromeric Rearrangements
Centromeric DNA can be a hotspot for rearrangements because it largely comprises satellite repeat DNAs and transposable elements [41]. Heterochromatin marks on centromeres are important for maintaining centromere integrity and preventing rearrangements [42]. Centromeric rearrangements can involve entire chromosome arms, affecting the dosage of many genes, and can act as a precursor to aneuploidy in cancer [43,44]. Translocations occurring at the centromere are common across almost all tumor types due to the high amount of homology between centromeric loci of many human chromosomes [45,46]. Chromosome mis-segregation and errors in DNA replication can lead to chromosome breakage and result in unbalanced events, such as those occurring in squamous cell carcinomas [47]. The large scale of centromeric mutations can result in the many gene dosage alterations that are typically seen in cancer.
Telomeres and Subtelomeric Regions
Telomeres have an important role in protecting chromosome ends from erosion over many cellular divisions. Shortened telomeres trigger cell cycle arrest and apoptosis. Apoptosis prevents the potential joining of chromosomal ends (chromosome fusion) in senescent cells or aberrantly proliferating pretumor cells [48]. Cells that do not initiate apoptosis enter a state of telomere crisis, which is characterized by frequent telomere fusions, large SVs, chromothripsis, kataegis, and tetraploidy [49,50]. Telomere crisis is seen in many cancers, including chronic lymphocytic leukemia, breast cancer, colorectal adenomas, and gliomas.
Subtelomeric regions are gene-rich sites of frequent meiotic recombination [51]. Subtelomeric sequences are highly polymorphic in copy number, and their rearrangement contributes significantly to intellectual disability, autism, and birth defects [51,52]. The olfactory receptor gene family resides within subtelomeric loci, and variants due to recombination and mutation are common in human olfactory receptor genes [53].
Replication Timing
The first analysis of DNA replication timing established three timepoints of S phase: early; middle; and late [54]. Actively transcribed gene-rich domains in the nuclear interior replicate early, whereas heterochromatin domains comprising centromeric and telomeric repeats, which are largely located in the nuclear periphery, replicate later [55]. Increased mutation rates are correlated with these late-replicating regions [56]. For an in-depth review of the mutational landscape of replication timing, see [57].
The rate of point mutations along chromosomes correlates tightly with replication timing [58]. Late-replicating regions contain a two- and sixfold increase in transition and transversion SNV mutations, respectively, compared with early-replicating loci [58]. Factors contributing to increased mutations in late-replicating regions include replication fork stress and induction of DNA damage response pathways [57]. Other mutation types are correlated with differences in replication timing. For example, SVs mediated by NHEJ are twice more enriched in late-replicating regions, whereas SVs driven by NAHR are four times more enriched in early replicating regions (Figure 3A) [59].
Figure 3. Mutations during DNA Replication.

(A) Regions replicated during early S phase generally comprise euchromatic DNA and are localized towards the center of the nucleus. Regions replicated during late S phase are primarily heterochromatic and localized near the nuclear periphery. Replication timing is correlated with mutation type. Structural variants (SVs) mediated by nonallelic homologous recombination (NAHR) primarily occur during early S phase. Structural variants mediated by nonhomologous end-joining (NHEJ) as well as single-base transitions and transversions occur more frequently during late S phase. (B) Transcriptionally active G nucleotide-rich repeat tracts can result in secondary structure-forming G-quadruplexes and R-loops. The stabilization of transcription forks at these loci poses a threat to genomic integrity in two ways. First, single-stranded DNA at R-loops is exposed to apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) family enzymes, allowing C:U mutations on the free strand (indicated as red asterisks). Second, if R-loops are not resolved before replication, the transcription and replication forks may collide, causing replication fork collapse and potentially triggering SV formation. Abbreviations: DNAP, DNA polymerase; RNAP, RNA polymerase.
In cancer cells, late-replicating regions have a two to three times higher point mutation rate [60] and a loss of DNA methylation [61]. In prostate and breast cancer, late-replicating regions are correlated with cis-chromosomal rearrangements, whereas early replicating regions contain more trans-chromosomal rearrangements and tandem duplications [61,62]. Collectively, these results exhibit a relationship between replication timing, point mutations, and structural variants, which can contribute to differences in mutation rate across the genome.
Common Fragile Sites
Common fragile sites (CFSs) were first described in 1984 as sites that formed visible gaps and breaks on human metaphase chromosomes following inhibition of DNA synthesis [63]. After sequencing of the human genome, CFSs were found to contain long stretches of AT microsatellite sequences that are prone to secondary structure and double-strand breaks (DSBs), making them susceptible to SV formation [64]. CFSs are late replicating, enriched in large genes, have a low concentration of replication origins, and display cell-type specificity [65,66].
CFSs are prone to mutation through a variety of mechanisms. Through recurrent DSBs, CFSs can lead to structural variants in nearby loci and within topologically associated domains [67,68]. Additionally, instability of CFS sequences can lead to the deletion of oncogenes by an NHEJ-independent mechanism [69]. A model for CFS instability and SV hotspots involves transcription-dependent double fork failure [66]. In this scenario, late-replicating genes interfere with actively transcribed regions. The replication fork and transcription machinery collide, create DSBs, and may be improperly repaired resulting in SVs [66]. This phenomenon is more common in genes with large transcription units that are >500 kb in length, such as LSAMP and AUTS2 [66].
DNA Structures Prone to Variation
AT-Rich and Hairpin-Forming Palindromes
Palindromic sequences can lead to unusual DNA conformations and genomic instability [70,71]. Palindromic AT-rich repeats (PATRRs) form opposing hairpins in DNA, creating a four-way Holliday junction that resembles a cruciform. Upon cleavage, the resolution of the cruciform structures can result in translocations, some of which are recurrent [72]. One such PATRR-mediated translocation results in a supernumerary chromosome containing genetic material from chromosomes 11 and 22, and can lead to Emanuel syndrome, which is characterized by developmental delay and hypotonia in infants [73]. Another translocation between PATRRs on chromosomes 17 and 22 disrupts the NF1 gene and manifests as neurofibromatosis type 1, characterized by patches of darkened skin and benign tumors [74].
Hairpin-forming palindromes throughout the genome can also be nucleation sites for APOBEC-mediated mutations [75]. Some recurrent hotspots of APOBEC mutation occur outside of hairpins in oncogenes or tumor suppressors, and appear to be under positive selection for cellular growth advantages; these are important hotspots for understanding tumor evolution [75]. Other documented APOBEC hotspots may be functionally insignificant and simply occur because they are located within hairpin-forming palindromes that serve as optimal APOBEC substrates [75,76]. Therefore, mutation hotspots in cancer due to the action of APOBEC family proteins may not reflect tumor evolutionary processes.
G-Quadruplexes and R Loops
G-quadruplexes are long stretches of G nucleotide-rich sequences that can self-stack and act as common sites of replication stress and fork stalling. G-quadruplexes are enriched in promoter regions and have been shown to act as docking sites for transcription factors [77,78]. Given their involvement in both active transcription and replication stress, G-quadruplexes serve as sites where transcription and replication forks may collide [79,80].
R-loops are structures where the RNA remains hybridized with the transcribed template DNA strand, exposing the bare nontemplate DNA strand; these three-stranded structures are associated with nontemplate strand G-quadruplexes [81]. R-loops provide a link between late-replicating large transcription units and SV formation due to CFSs [66]. Stabilized R-loops located in late-replicating regions can promote convergence of transcription and replication forks, leading to DSBs and SV formation [66] (Figure 3B). Interestingly, due to cell- and tissue-specific transcriptional programs, these SV-prone fragile sites are likely to differ among species as well as tissues.
Chromatin and Mutational Processes
The 3D organization of DNA has an important and emerging role in the repair of DSBs via HR, and may influence NAHR [82]. DSBs are preferentially repaired using a homologous DNA template in close special proximity, with similar replication timing, and within overlapping chromosome territories [83]. One key example is the BCR-ABL fusion protein (Box 1); whereby the BCR and ABL genes are replicated at the same time, and are located near each other in the nucleus [84]. Both yeast and human studies show that proximal genomic regions recombine more efficiently than distal genomic regions, and this can have a role in either facilitating or preventing NAHR [83,85]. Chromosome loop anchors are responsible for bringing two loci into close spatial proximity [86]. Anchor binding sites may be cut by topoisomerases to rearrange DNA topology, and mutations at topoisomerase binding sites may result in ectopic recombination between chromosome loop anchors and chromosomal rearrangements [87]. These studies highlight how DNA proximity can affect homologous recombination and may prevent or facilitate NAHR.
Focal Mutation Hotspots
Chromothripsis
The first documented chromothriptic event showed that a single complex genomic rearrangement can drive cancer progression by simultaneously deleting tumor suppressors and duplicating oncogenes [15]. This event resulted in 42 somatic rearrangements all on chromosome 4q, but a scarcity of rearrangements elsewhere. Chromothripsis encompasses complex clusters of rearrangements, including duplications, deletions, and inversions, that are proposed to occur in a single chromosome-shattering event. Chromothripsis is highly prevalent in some cancers, occurring in >50% of liposarcomas, osteosarcomas, and glioblastomas [88]. The mechanisms that result in chromothripsis are only now becoming clear. Micronuclei formation is a precursor to chromothripsis, and can result from telomere fusion, chromosomal bridges, unrepaired DSBs, and nuclear envelope collapse [89–91]. The breakage and improper repair of chromosomal bridges can also lead to chromothripsis and has been attributed to the cytoplasmic exonuclease TREX1 [91,92]. It is difficult to distinguish which structural variants from a chromothriptic event are oncogenic. As mentioned earlier, micronuclei harboring amplified oncogenes are one potential contributor to positive selection in tumorigenesis (Box 1). To further understand the functional consequences of chromothripsis, a micronuclei-based model system can now be used [93].
Kataegis
In addition to large-scale chromosomal rearrangements, tumors also harbor clusters of point mutations termed mutation showers or kataegis [94]. Some of these SNVs cluster at TpC dinucleotides that are adjacent to somatic rearrangements, and can be hundreds to thousands of times more common than the background mutation rate [95]. Overexpression of APOBEC enzymes is associated with mutation, genomic damage, and cancer progression (cancer signatures SBS13 and possibly DBS11 [17]) [10]. This family of enzymes mutates TpC dinucleotides, suggesting that they have a role in kataegic foci formation [96,97]. Kataegic foci were first observed in cancer genomes as clustered mutations commonly found at sites of DSBs, DNA repair, and structural variation [95,97–99].
Structural variation and kataegis can co-occur focally in human cancer, suggesting that they are mechanistically linked [96,97,100,101]. Break-induced replication (BIR), a process that repairs single-ended DNA breaks by copying homologous templates, can give rise to kataegic foci [98,101]. During this repair process, single-stranded DNA is made vulnerable to APOBEC cytosine deamination, creating many de novo SNV lesions in the replicated strand (Figure 4A) [102]. Another example involves telomere crisis and chromothripsis [90]. Telomere crisis may induce chromothriptic events surrounded by APOBEC3B-mediated kataegic foci [91].
Figure 4. Replication-Based Repair Mechanisms Are Associated with Clustered Mutations.
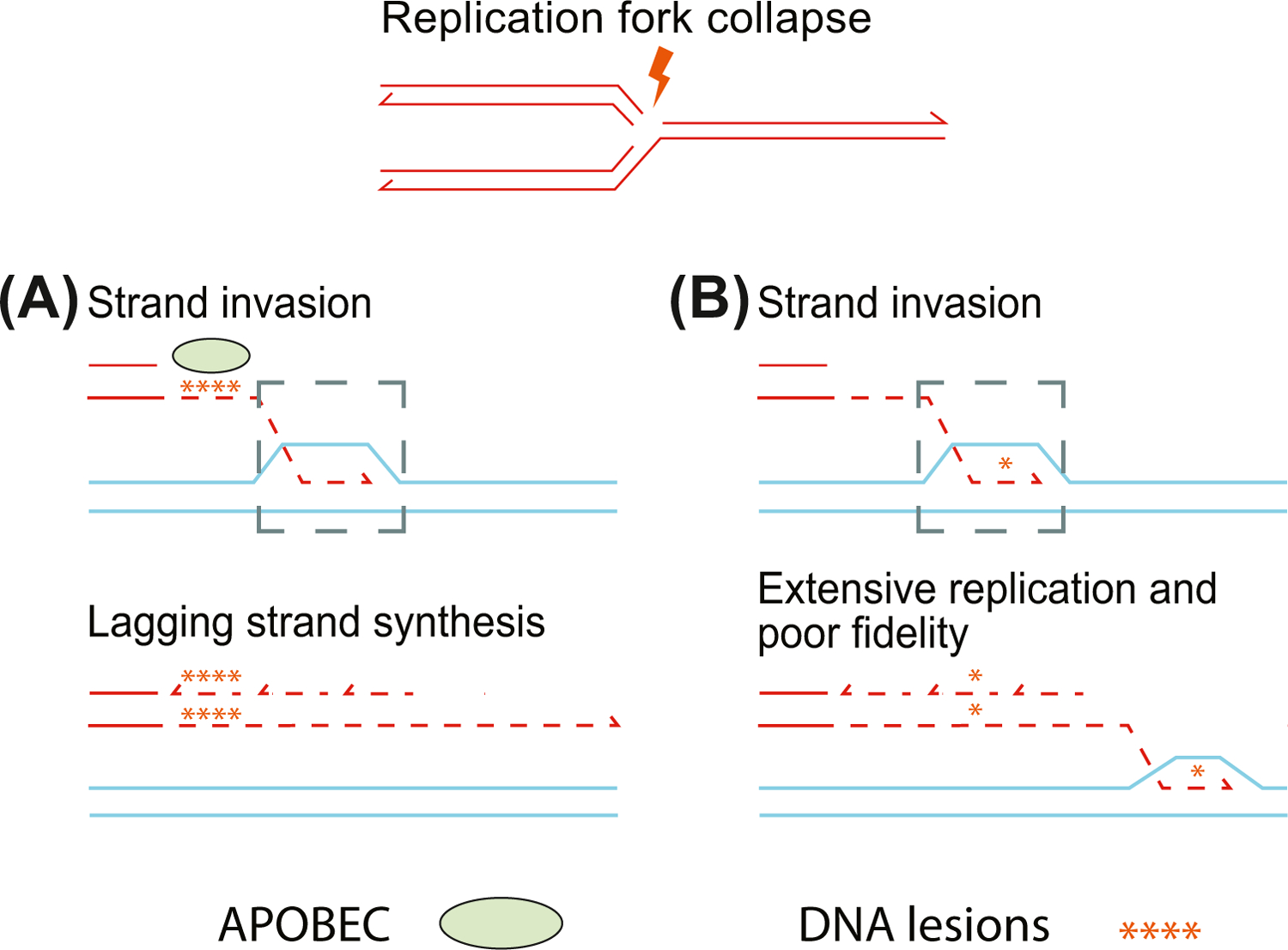
(A) Repair pathways that expose single-stranded DNA are subject to apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC)-mediated hypermutation. Following replication fork collapse, mutations are primarily focused on exposed single-stranded DNA. In this break-induced replication repair model, the leading strand from the damaged chromosome invades the homologous region of its mate and forms a displacement loop (gray dashed-line box). This single-stranded DNA is exposed to lesions from APOBEC-mediated deamination. These lesions are retained as the lagging strand of the broken chromosome is synthesized. (B) Replication-based repair mechanisms use the homologous chromosome as a template for repair. This creates localized regions of conservative replication leading to accumulation of variants in the homologous region. The displacement loop created by strand invasion can proceed for regions up to ~1 MB, and this process introduces errors or mutations that are not efficiently repaired.
Long Tracts of Mutations Surround BIR-Mediated SVs
De novo, nonrecurrent SVs have been shown to harbor clusters of indels and polymerase slippage events close to their breakpoint junctions [103], but mutations can also extend up to 1 Mb away from the breakpoint [104]. Replication-based repair mechanisms could explain these mutation clusters because the displacement loop and synthesis results in conservative inheritance of new DNA (as opposed to traditional semiconservative inheritance), and can lead to a 1000-fold increase in mutations compared with normal replication (Figure 4B) [104,105]. Indels around de novo, nonrecurrent SVs are likely the result of polymerase slippage and occur within poly(A) tracts and tandem duplications [103]. These findings point to multiple mechanisms leading to higher mutation rates accompanying SV formation.
Concluding Remarks
Mutation hotspots are highly dependent on DNA sequence and structure and are subject to selection. Mutations can occur due to cellular processes, such as DNA replication and repair, meiotic recombination, and immunoglobulin specification. Mutations are not always under positive selective pressure, because some are simply passenger mutations resulting from a multimutational event. Here, we reviewed many of the known and recently identified mutational hotspots and the mechanisms of their formation. However, the mechanistic relationships of certain mutational signatures found in cancers currently remain unclear [17]. The investigation of mutation hotspots in cancer has also resulted in novel therapeutic approaches (see Box 1). As technology advances and more large-scale sequencing studies are completed, we will gain further insight into hotspots of genetic variation and their frequency across the human population and across diverse tissues, as well as how these processes contribute to disease (see Outstanding Questions).
Outstanding Questions.
How do genomic differences between two individuals influence mutational hotspots?
Do specific defects DNA repair results in mutation hotspots that can be tumorigenic in specif tissues?
What are the mechanisms underlying all patterns of somatic and germline mutations?
How does the epigenetic landscape influence focal mutation mechanisms and rates?
How do mutational hotspots influence healthy genotypic and phenotypic variation?
Highlights.
Genetic mutations are influenced by sequence context, structure, and genomic features.
Mechanisms responsible for many mutational hotspots have been identified.
Hotspots are largely related to loci prone to mutation during replication or DNA repair.
Selection leads to recurrent mutations in somatic tissues that can be exploited for therapeutic purposes.
Acknowledgments
We thank the members of the Beck lab and colleagues at the Jackson Laboratory for Genomic Medicine for reading and editing the review; in particular we appreciated input from Francesca Menghi and Samirkumar B. Amin. This work was supported in part by the National Institute of General Medical Sciences grants R00GM120453, R35GM133600, and startup funds from the University of Connecticut Health and the Jackson Laboratory for Genomic Medicine to C.R.B.
Glossary
- Apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC)
family of cytidine deaminases that modify single-stranded DNA at the C of TpC dinucleotides converting them to uracil. APOBEC3B and others are implicated in mutagenesis in cancer
- Apoptosis
type of programmed cell death triggered by either intrinsic or extrinsic pathways, such as DNA damage response or tumor necrosis factor receptor binding, respectively
- Chromothripsis
a single event that results in up to hundreds of genomic rearrangements in a localized area
- Copy number variation (amplification/deletion)
structural alteration of the genome resulting in differences in the number of copies of a gene or locus between individuals in a population
- CpG Islands
~300–3000-base pair loci largely consisting of CpG dinucleotides
- Driver mutations
somatically acquired mutations that facilitate clonal cellular growth
- Extrachromosomal circular DNA
circular DNAs that originate from normal chromosomal DNA during tumorigenesis. Also known as double minutes, these DNAs are common in neurological and other tumors, and often harbor amplified copies of oncogenes
- Gene conversion
exchange between two similar DNA sequences where a donor sequence is used to replace an acceptor sequence
- Holliday junction
cross-shaped DNA structure formed between two homologous DNA duplexes during recombination
- Kataegis
clusters of simultaneously occurring nucleotide substitutions within a 1-kb window that are commonly associated with structural variants
- Meiotic recombination
in meiosis, the formation of DSBs allows DNA to be exchanged between homologous regions of both parental chromosomes
- Microsatellite
a one to six-base pair DNA sequence that is usually tandemly repeated up to 50 times
- Microsatellite instability
change in the number of repeats in a microsatellite
- Mutation
permanent change in a genome at the nucleotide level
- Mutational signatures
characteristic somatic mutation patterns that result from distinct mutational processes, often occurring in cancers with a specific genetic, chemical, or environmental insult
- Nonallelic homologous recombination
process in which two highly similar sequences that are not alleles, such as segmental duplications, serve as an incorrect template for homologous recombination
- Polymerase slippage (slipped strand mispairing)
indels commonly form in regions that are difficult to replicate, such as repeats, which are prone to polymerase displacement and re-annealing at nearby, ectopic locations
- Satellite repeat DNAs
highly repetitive segments found in centromeres and telomeres of eukaryotes
- Segmental duplication (low copy repeat)
loci >1 kb in length with >90% sequence similarity
- Selection (positive, negative, and neutral)
mutations either tend to be maintained and/or enriched over several cell divisions (i.e., positive selection) or lost over time (i.e., negative selection). Mutations that do not impact fitness are not under selection and evolve through neutral evolution or genetic drift
- Spontaneous deamination
loss of an amino group from a cytosine or 5-methylcytosine. These modified cytosines become uracil or thymine respectively
- Subtelomeric regions
the repeat-rich ~500-kb DNA segments adjacent to telomeric repeats and DNA that uniquely map to a chromosome arm
- Tandem repeats
segment of DNA containing one or more nucleotides repeated without interruption
- Telomere crisis
state of significantly heightened chromosomal fusions and instability, resulting from the failure of a cell to apoptose upon the natural shortening of telomeric repeats
- Topologically associating domains
loci that physically interact with each other in 3D space
- Transposable elements
segments of DNA that comprise almost half of the human genome and, at one point in time, could move from one place to another
- Variation
mutations that distinguish between individuals within a population of organisms; these can be new, rare, or common
- Whole-genome duplication
a single event where the entire genome of a cell is duplicated, resulting in polyploidy
References
- 1.Gusella JF et al. (1983) A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–238 [DOI] [PubMed] [Google Scholar]
- 2.Andrew SE et al. (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet 4, 398–403 [DOI] [PubMed] [Google Scholar]
- 3.Falush D et al. (2001) Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial underascertainment of late-onset cases. Am. J. Hum. Genet 68, 373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacDonald ME et al. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 5.Michaelson JJ et al. (2012) Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shendure J and Akey JM (2015) The origins, determinants, and consequences of human mutations. Science 349, 1478–1483 [DOI] [PubMed] [Google Scholar]
- 7.Sasani TA et al. (2019) Large, three-generation human families reveal post-zygotic mosaicism and variability in germline mutation accumulation. eLife 8, e46922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coulondre C et al. (1978) Molecular basis of base substitution hotspots in Escherichia coli. Nature 274, 775–780 [DOI] [PubMed] [Google Scholar]
- 9.Muramatsu M et al. (2000) Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563 [DOI] [PubMed] [Google Scholar]
- 10.Burns MB et al. (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montgomery SB et al. (2013) The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res 23, 749–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin Y et al. (2018) Architecture of polymorphisms in the human genome reveals functionally important and positively selected variants in immune response and drug transporter genes. Hum. Genomics 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins RL et al. (2020) A structural variation reference for medical and population genetics. Nature 581, 444–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conrad DF et al. (2010) Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephens PJ et al. (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bielski CM et al. (2018) Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet 50, 1189–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrov LB et al. (2020) The repertoire of mutational signatures in human cancer. Nature 578, 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duret L and Galtier N (2009) Biased gene conversion and the evolution of mammalian genomic landscapes. Annu. Rev. Genomics Hum. Genet 10, 285–311 [DOI] [PubMed] [Google Scholar]
- 19.Nachman MW and Crowell SL (2000) Estimate of the mutation rate per nucleotide in humans. Genetics 156, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeziorska DM et al. (2017) DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. U. S. A 114, E7526–E7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomso DJ and Bell DA (2003) Sequence context at human single nucleotide polymorphisms: overrepresentation of CpG dinucleotide at polymorphic sites and suppression of variation in CpG islands. J. Mol. Biol 327, 303–308 [DOI] [PubMed] [Google Scholar]
- 22.Xia J et al. (2012) Investigating the relationship of DNA methylation with mutation rate and allele frequency in the human genome. BMC Genomics 13, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glémin S et al. (2015) Quantification of GC-biased gene conversion in the human genome. Genome Res 25, 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutta R et al. (2018) 1000 human genomes carry widespread signatures of GC biased gene conversion. BMC Genomics 19, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lachance J and Tishkoff SA (2014) Biased gene conversion skews allele frequencies in human populations, increasing the disease burden of recessive alleles. Am. J. Hum. Genet 95, 408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lander ES et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921 [DOI] [PubMed] [Google Scholar]
- 27.Verkerk AJ et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 [DOI] [PubMed] [Google Scholar]
- 28.La Spada AR et al. (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79 [DOI] [PubMed] [Google Scholar]
- 29.Christopher J et al. (2019) Quantifying microsatellite mutation rates from intestinal stem cell dynamics in msh2-deficient murine epithelium. Genetics 212, 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lang GI et al. (2013) Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast. G3 GenesGenomesGenetics 3, 1453–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baudat F et al. (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berg IL et al. (2010) PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet 42, 859–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keeney S et al. (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384 [DOI] [PubMed] [Google Scholar]
- 34.Schwartz JJ et al. (2014) Primate evolution of the recombination regulator PRDM9. Nat. Commun 5, 4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeffreys AJ and Neumann R (2002) Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat. Genet 31, 267–271 [DOI] [PubMed] [Google Scholar]
- 36.Boulton A et al. (1997) The hotspot conversion paradox and the evolution of meioticrecombination. Proc. Natl. Acad. Sci. U. S. A 94, 8058–8063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stankiewicz P and Lupski JR (2002) Genome architecture, rearrangements and genomic disorders. Trends Genet 18, 74–82 [DOI] [PubMed] [Google Scholar]
- 38.Lupski JR et al. (1991) DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219–232 [DOI] [PubMed] [Google Scholar]
- 39.Turner DJ et al. (2008) Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet 40, 90–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song X et al. (2018) Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res 28, 1228–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warburton PE et al. (1993) Nonrandom localization of recombination events in human alpha satellite repeat unit variants: implications for higher-order structural characteristics within centromeric heterochromatin. Mol. Cell. Biol 13, 6520–6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu G-L et al. (1999) Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191 [DOI] [PubMed] [Google Scholar]
- 43.Ben-David U and Amon A (2020) Context is everything: aneuploidy in cancer. Nat. Rev. Genet 21, 44–62 [DOI] [PubMed] [Google Scholar]
- 44.Ribeiro IP et al. (2020) Chromosomal breakpoints in a cohort of head and neck squamous cell carcinoma patients. Genomics 112, 297–303 [DOI] [PubMed] [Google Scholar]
- 45.Kim T-M et al. (2013) Functional genomic analysis of chromosomal aberrations in a compendium of 8000 cancer genomes. Genome Res 23, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barra V and Fachinetti D (2018) The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun 9, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martínez JG et al. (2012) Localization of centromeric breaks in head and neck squamous cell carcinoma. Cancer Genet 205, 622–629 [DOI] [PubMed] [Google Scholar]
- 48.Harley CB et al. (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 [DOI] [PubMed] [Google Scholar]
- 49.Maciejowski J and de Lange T (2017) Telomeres in cancer: tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol 18, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayashi MT et al. (2012) A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol 19, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown WRA et al. (1990) Structure and polymorphism of human telomere-associated DNA. Cell 63, 119–132 [DOI] [PubMed] [Google Scholar]
- 52.Luo Y et al. (2011) Diverse mutational mechanisms cause pathogenic subtelomeric rearrangements. Hum. Mol. Genet 20, 3769–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trask BJ et al. (1998) Members of the olfactory receptor gene family are contained in large blocks of DNA duplicated polymorphically near the ends of human chromosomes. Hum. Mol. Genet 7, 13–26 [DOI] [PubMed] [Google Scholar]
- 54.Stubblefield E (1975) Analysis of the replication pattern of Chinese hamster chromosomes using 5-bromodeoxyuridine suppression of 33258 Hoechst fluorescence. Chromosoma 53, 209–221 [DOI] [PubMed] [Google Scholar]
- 55.Kim S-M et al. (2003) Early-replicating heterochromatin. Genes Dev 17, 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yehuda Y et al. (2018) Germline DNA replication timing shapes mammalian genome composition. Nucleic Acids Res 46, 8299–8310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaboriaud J and Wu P-YJ (2019) Insights into the link between the organization of DNA replication and the mutational landscape. Genes 10, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koren A et al. (2014) Genetic variation in human DNA replication timing. Cell 159, 1015–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koren A et al. (2012) Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet 91, 1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woo YH and Li W-H (2012) DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat. Commun 3, 1004. [DOI] [PubMed] [Google Scholar]
- 61.Du Q et al. (2019) Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun 10, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y et al. (2020) Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glover TW et al. (1984) DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet 67, 136–142 [DOI] [PubMed] [Google Scholar]
- 64.Zhang H and Freudenreich CH (2007) An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 27, 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glover TW et al. (2017) Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 17, 489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilson TE et al. (2015) Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res 25, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei P-C et al. (2016) Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell 164, 644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarni D et al. (2020) 3D genome organization contributes to genome instability at fragile sites. Nat. Commun 11, 3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yunis JJ (1984) Fragile sites and predisposition to leukemia and lymphoma. Cancer Genet. Cytogenet 12, 85–88 [DOI] [PubMed] [Google Scholar]
- 70.Kato T et al. (2012) Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev 22, 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nasar F et al. (2000) Long palindromic sequences induce double-strand breaks during meiosis in yeast. Mol. Cell. Biol 20, 3449–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inagaki H et al. (2013) Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat. Commun 4, 1592. [DOI] [PubMed] [Google Scholar]
- 73.Kurahashi H and Emanuel BS (2001) Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum. Mol. Genet 10, 2605–2617 [DOI] [PubMed] [Google Scholar]
- 74.Kurahashi H et al. (2003) The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am. J. Hum. Genet 72, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buisson R et al. (2019) Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 364, eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carter H (2019) Mutation hotspots may not be drug targets. Science 364, 1228–1229 [DOI] [PubMed] [Google Scholar]
- 77.Huppert JL and Balasubramanian S (2007) G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res 35, 406–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raiber E-A et al. (2012) A non-canonical DNA structure is a binding motif for the transcription factor SP1 in vitro. Nucleic Acids Res 40, 1499–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.French S (1992) Consequences of replication fork movement through transcription units in vivo. Science 258, 1362–1365 [DOI] [PubMed] [Google Scholar]
- 80.Hamperl S et al. (2017) Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170, 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Belotserkovskii BP et al. (2018) R-loop generation during transcription: formation, processing and cellular outcomes. DNA Repair 71, 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De S and Michor F (2011) DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat. Biotechnol 29, 1103–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Agmon N et al. (2013) Effect of nuclear architecture on the efficiency of double-strand break repair. Nat. Cell Biol 15, 694–699 [DOI] [PubMed] [Google Scholar]
- 84.Lukásová E et al. (1997) Localisation and distance between ABL and BCR genes in interphase nuclei of bone marrow cells of control donors and patients with chronic myeloid leukaemia. Hum. Genet 100, 525–535 [DOI] [PubMed] [Google Scholar]
- 85.Girelli G et al. (2020) GPSeq reveals the radial organization of chromatin in the cell nucleus. Nat. Biotechnol 38, 1184–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rao SSP et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Canela A et al. (2017) Genome organization drives chromosome fragility. Cell 170, 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cortés-Ciriano I et al. (2020) Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet 52, 331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang C-Z et al. (2015) Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maciejowski J et al. (2015) Chromothripsis and kataegis induced by telomere crisis. Cell 163, 1641–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maciejowski J et al. (2020) APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet 52, 884–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Umbreit NT et al. (2020) Mechanisms generating cancer genome complexity from a single cell division error. Science 368, eaba0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kneissig M et al. (2019) Micronuclei-based model system reveals functional consequences of chromothripsis in human cells. eLife 8, e50292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Campbell PJ et al. (2020) Pan-cancer analysis of whole genomes. Nature 578, 82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nik-Zainal S et al. (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roberts SA et al. (2013) An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet 45, 970–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burns MB et al. (2013) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet 45, 977–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sakofsky CJ et al. (2014) Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep 7, 1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alexandrov LB et al. (2013) Signatures of mutational processes in human cancer. Nature 500, 415–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sakofsky CJ et al. (2019) Repair of multiple simultaneous double-strand breaks causes bursts of genome-wide clustered hypermutation. PloS Biol 17, e3000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roberts SA et al. (2012) Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 46, 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Elango R et al. (2019) Repair of base damage within break-induced replication intermediates promotes kataegis associated with chromosome rearrangements. Nucleic Acids Res 47, 9666–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carvalho CMB et al. (2013) Replicative mechanisms for CNV formation are error prone. Nat. Genet 45, 1319–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Beck CR et al. (2019) Megabase length hypermutation accompanies human structural variation at 17p11.2. Cell 176, 1310–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Saini N et al. (2013) Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nielsen R (2005) Molecular signatures of natural selection. Annu. Rev. Genet 39, 197–218 [DOI] [PubMed] [Google Scholar]
- 107.Perry GH et al. (2007) Diet and the evolution of human amylase gene copy number variation. Nat. Genet 39, 1256–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martincorena I et al. (2017) Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lawrence MS et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rheinbay E et al. (2020) Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stratton MR et al. (2009) The cancer genome. Nature 458, 719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim H et al. (2020) Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet 52, 891–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Loh P-R et al. (2018) Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Imielinski M et al. (2017) Insertions and deletions target lineage-defining genes in human cancers. Cell 168, 460–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Druker BJ et al. (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med 344, 1031–1037 [DOI] [PubMed] [Google Scholar]
- 116.Forbes SA et al. (2008) The catalogue of somatic mutations in cancer (COSMIC). Curr. Protoc. Hum. Genet 10, 10–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kong-Beltran M et al. (2006) Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 66, 283–289 [DOI] [PubMed] [Google Scholar]
- 118.Bollag G et al. (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wolf J et al. (2020) Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med 383, 944–957 [DOI] [PubMed] [Google Scholar]
- 120.Canon J et al. (2019) The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 [DOI] [PubMed] [Google Scholar]
- 121.Tonegawa S (1983) Somatic generation of antibody diversity. Nature 302, 575–581 [DOI] [PubMed] [Google Scholar]
- 122.Oettinger MA et al. (1990) RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248, 1517–1523 [DOI] [PubMed] [Google Scholar]
- 123.Lieber MR (1992) The mechanism of V(D)J recombination: a balance of diversity, specificity, and stability. Cell 70, 873–876 [DOI] [PubMed] [Google Scholar]
- 124.Weigert MG et al. (1970) Variability in the lambda light chain sequences of mouse antibody. Nature 228, 1045–1047 [DOI] [PubMed] [Google Scholar]
- 125.Petersen-Mahrt SK et al. (2002) AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418, 99–103 [DOI] [PubMed] [Google Scholar]
- 126.Kleinstein SH et al. (2003) Estimating hypermutation rates from clonal tree data. J. Immunol 171, 4639–4649 [DOI] [PubMed] [Google Scholar]