

Abstract
Objectives:
Autosomal dominant (ADPKD) and recessive (ARPKD) polycystic kidney diseases are the most common hepatorenal fibrocystic diseases (ciliopathies). Characteristics of liver disease of these disorders are quite different. All of the patients with ARPKD have congenital hepatic fibrosis (CHF) often complicated by portal hypertension. In contrast, typical liver involvement in ADPKD is polycystic liver disease, although rare atypical cases with CHF are reported. Our goal was to describe the characteristics of CHF in ADPKD.
Patients and Methods:
As a part of an intramural study of the National Institutes of Health on ciliopathies (www.clinicaltrials.gov, trial NCT00068224), we evaluated 8 patients from 3 ADPKD families with CHF. We present their clinical, biochemical, imaging, and PKD1 and PKHD1 sequencing results. In addition, we tabulate the characteristics of 15 previously reported patients with ADPKD-CHF from 11 families.
Results:
In all of the 19 patients with ADPKD-CHF (9 boys, 10 girls), portal hypertension was the main manifestation of CHF; hepatocelllular function was preserved and liver enzymes were largely normal. In all of the 14 families, CHF was not inherited vertically, that is the parents of the index cases had PKD but did not have CHF-suggesting modifier gene(s). Our 3 families had pathogenic mutations in PKD1; sequencing of the PKHD1 gene as a potential modifier did not reveal any mutations.
Conclusions:
Characteristics of CHF in ADPKD are similar to CHF in ARPKD. ADPKD-CHF is caused by PKD1 mutations, with probable contribution from modifying gene(s). Given that both boys and girls are affected, these modifier(s) are likely located on autosomal chromosome(s) and less likely X-linked.
Keywords: autosomal dominant polycystic kidney disease, autosomal recessive polycystic kidney disease, congenital hepatic fibrosis, polycystic liver disease, portal hypertension
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common inherited diseases in man with a frequency of 1 in 500 to 1000 (1). It belongs to the group of hepatorenal fibrocystic diseases referred to as “ciliopathies” after the recent discovery that fibrocystic disease proteins localize to the primary cilia. ADPKD is characterized by progressive cystic degeneration of the kidneys resulting in hypertension, renal pain, and renal insufficiency; approximately 50% of individuals with ADPKD have end-stage renal disease by age 60 years (1). Extra-renal manifestations of ADPKD include polycystic liver disease (PLD), intracranial aneurysms, dilatation of the aortic root, mitral valve prolapse and cysts in the pancreas, arachnoid membrane, and seminal vesicles (1,2). Typically, congenital hepatic fibrosis (CHF) and portal hypertension (PH) do not occur in ADPKD (3). ADPKD is genetically heterogeneous, with 2 genes identified: PKD1 encoding polycystin-1 and PKD2 encoding polycystin-2 (4,5). Approximately 85% of ADPKD is caused by PKD1 and 15% by PKD2 mutations. Patients with mutations in PKD1 and PKD2 have similar liver involvement in the form of PLD, but PKD1 mutations cause more severe kidney disease and are associated with earlier onset of end-stage renal disease compared with PKD2 mutations (54.3 years for PKD1, 74.0 years for PKD2) (6,7).
Autosomal recessive polycystic kidney disease (ARPKD) is the most common childhood-onset PKD with a frequency of approximately 1 in 20,000 (8–10). By definition, all of the patients with ADPKD have CHF, complicated by PH in most cases. ARPKD is caused by mutations in the PKHD1 gene that encodes fibrocystin.
Here we report 8 patients from 3 families with ADPKD, in which some individuals with PKD had CHF complicated by PH. We present the DNA sequencing results of PKD1 and PKHD1 genes and the clinical, biochemical, and ultrasound (USG) and magnetic resonance imaging (MRI) findings. In addition, we tabulate the previously reported patients with ADPKD with CHF complicated by PH and discuss potential explanations for this variation in the liver manifestations of ADPKD.
PATIENTS AND METHODS
Patients
All of the patients were enrolled in the protocol Clinical Investigations into the Kidney and Liver Disease in Autosomal Recessive Polycystic Kidney Disease/Congenital Hepatic Fibrosis and other Ciliopathies (www.clinicaltrials.gov, trial NCT00068224), approved by the NHGRI institutional review board. Patients or their parents gave written informed consent.
Sequencing and Analysis
All of the coding exons of PKHD1 and PKD1 and their intronic boundaries were sequenced using a Beckman CEQ 8000 system (Beckman Coulter Inc, Fullerton, CA). DNA variant analyses were performed using Sequencher (GeneCodes, Ann Arbor, MI).
Imaging Studies
USG evaluations were performed using standard (4 MHz) and high-resolution (7 MHz) ultrasonographic probes (AVI Sequoia Inc, Mountain View, CA). MRI was performed on a 1.5-T machine (Philips Medical Systems, NA, Bothell, WA; General Electric Healthcare, Waukesha, WI). Kidney volumes were calculated from MRI images (11,12) at the Image Processing Center of the National Institutes of Health (NIH).
Literature Review
To find all of the previous reports of ADPKD cases with CHF, we searched the medical literature using PubMed Database (National Center for Biotechnology Information) using multiple key words including “CHF,” “PH,” “Caroli’s,” “ADPKD,” and “liver.” In addition, we reviewed the references of these publications. We included only the reports with clear documentation of the dominant nature of the inheritance of PKD.
RESULTS
Family 1
In family 1 (Fig. 1), 6 individuals including all of the 3 children (patients III-1, III-2, and III-3), their father (II-3), paternal aunt (II-2), and paternal grandmother (I-2) had typical renal findings of ADPKD. Two of the children (III-1 and III-3) had CHF complicated by PH, whereas 4 other family members with ADPKD had normal-sized spleens. The father (II-3) was diagnosed with ADPKD at age 34 and received a renal allograft at age 47. USG at NIH at age 49 revealed multiple small liver cysts and a normal-sized spleen. Liver enzymes and synthetic function were normal (Table 1).
FIGURE 1.
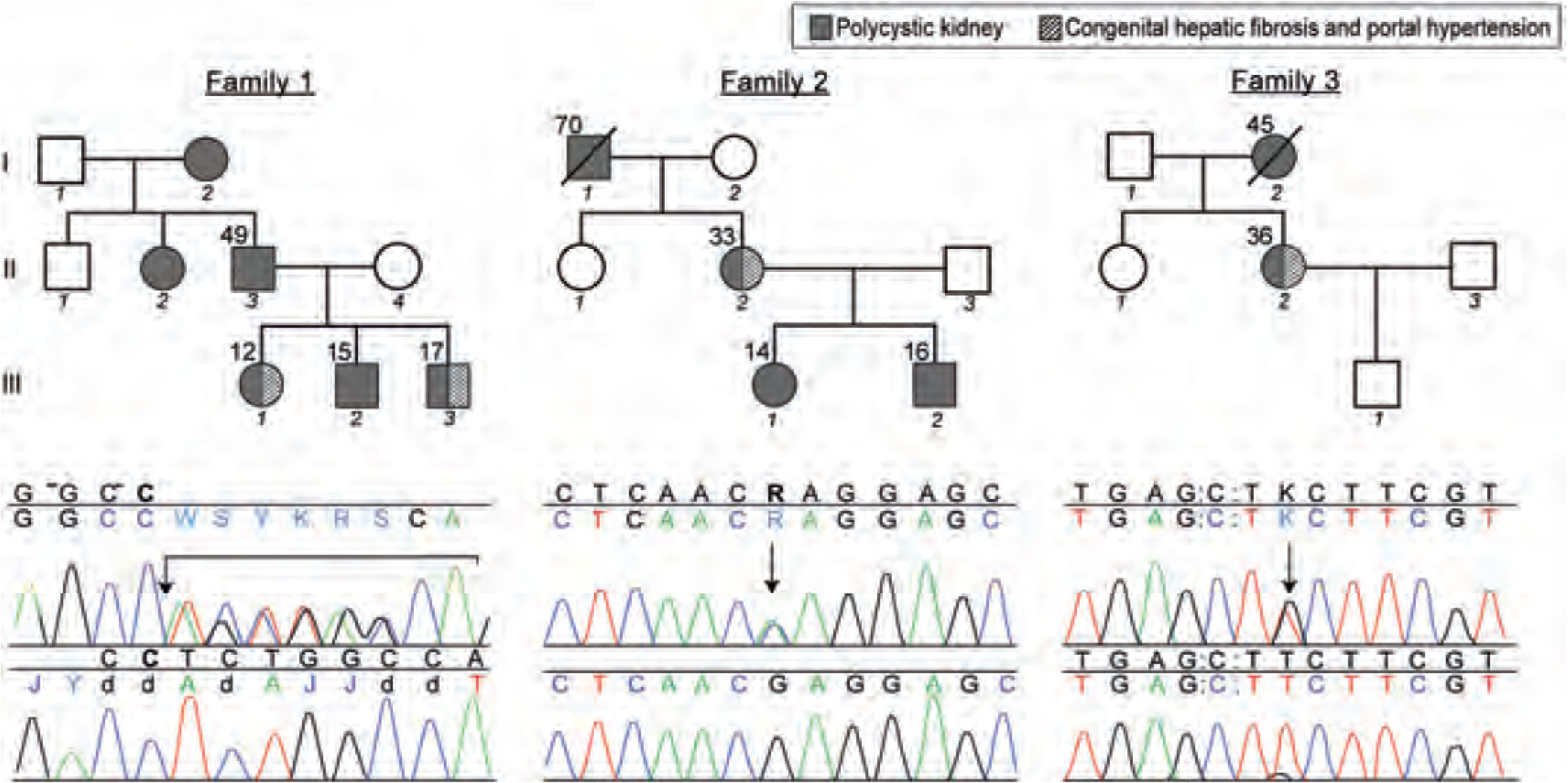
Pedigrees and DNA sequences displaying mutations in the PKD1 gene. Upper panel shows mutations in comparison with the normal control sequence (lower panel). The Sequencher software assigns letters other than A,T,C, or G to indicate heterozygous states. In family 1, the father (II-3) and all of the 3 children (III-1, III-2, and III-3) displayed a 7-base-pair deletion at position g.17554 (arrow) in exon 5, resulting in a frameshift. In family 2, the mother (II-2) and both the children (III-1 and III-2) had a missense mutation at position g.425323 (arrow) in exon 61, replacing a conserved serine with arginine. In family 3, patient II-2 had a missense mutation at position g.26918 (arrow) in exon 15, replacing a conserved phenylalanine with cysteine.
TABLE 1.
Liver-and kidney-related laboratory results of presented patients
| Family no. | Patient no. | PT, s | Albumin, g/dL | Total bilirubin, mg/dL | Ammonia, μmol/L | ALT, U/L | AST, U/L | AP, U/L | GGT, U/L | Creatinine clearance, mL · min−1 · 1.73 m−2 | Cystatin C, mg/L | WBC counts, K/μL | Hb, g/dL | PLT counts, K/μL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | II-3 | 12.2 | 4.1 | 0.9 | 20 | 20 | 19 | 134 | 24 | Transplant | Transplant | 3.5 | 14.1 | 147 |
| 1 | III-1 | 15.7 | 4 | 1 | 58 | 32 | 31 | 199 | 27 | 143 | 0.55 | 5.36 | 12 | 229 |
| 1 | III-2 | 15.1 | 3.8 | 0.6 | 28 | 20 | 19 | 228 | 15 | 105 | 0.58 | 4.29 | 12.2 | 265 |
| 1 | III-3 | 15.3 | 4.1 | 0.5 | 26 | 24 | 16 | 124 | 28 | 121 | 0.68 | 3.15 | 15.4 | 115 |
| 2 | II-2 | 13.5 | 3.7 | 0.5 | 47 | 30 | 46 | 57 | 26 | 105 | 0.92 | 3.2 | 12 | 87 |
| 2 | III-1 | 13.1 | 4 | 0.7 | NA | 18 | 19 | 67 | 17 | 118 | 0.59 | 6.8 | 14.8 | 239 |
| 2 | III-2 | NA | 4.4 | 1.7 | NA | 15 | 21 | 165 | 10 | NA | 0.93 | 4.5 | 15.3 | 223 |
| 3 | II-2 | 16.8 | 3.4 | 1.2 | 77 | 12 | 19 | 61 | 21 | 53 | 1.84 | 2.77 | 11 | 64 |
| Normal values | 11.6–15.2 | 3.7–4.7 | 0.1–1.0 | 11–32 | 6–41 | 9–34 | 37–375* | 7–38 | 90–125 | 0.55–1.03 | 3.3–9.6 | 12.7–16.7 | 154–345 |
ALT = Alanine aminotransferase; AP = alkaline phosphatase; AST = aspartate aminotransferase; GGT = γ-glutamyltransferase; Hb = hemoglobin; PLT = platelets; PT = prothrombin time; WBC = white blood cell.
Normals vary by age within this range.
The youngest of the children (III-1) first came to medical attention when she presented with esophageal variceal bleeding at age 4 years (Table 2) (13–19). The spleen was enlarged. Liver biopsy showed CHF. She was started on propanolol and subsequently underwent distal splenorenal shunt placement, which resulted in reduction of spleen size and improved growth. NIH evaluation at age 12 revealed a healthy-appearing female with normal growth. Liver was palpable 6 cm at the xiphoid but non-palpable at right costal margin, with moderately increased consistency and smooth surface. Spleen was not palpable. USG revealed a moderately hyperechogenic liver with coarsened echo-texture. There were extensive collaterals. Kidney imaging showed multiple macrocysts in both kidneys consistent with ADPKD (Fig. 2A). Liver and spleen volumes, calculated based on MRI imaging, were increased at 1254 and 288 mL, respectively. Liver enzymes were normal and synthetic function of the liver was intact (Table 1).
TABLE 2.
Present and reported patients with congenital hepatic fibrosis and autosomal dominant polycystic kidney disease
| References | Family (patient) | Sex | Age at evaluation, y | DNA analysis | Family history | Family members with ADPKD without CHF/PH | Liver biopsy (age) | Splenomegaly (age) | WBC count, K/μL | PLT count, K/μL | Esophageal varices | Variceal bleeding (age) | Liver cysts | Liver synthetic function | Liver enzymes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Present report | Family 1 (III-1) | F | 12 | c. 856_862delTCTGGCC p. Ala286fs2X in PKD1, no mutation in PKHD1 | ADPKD | Father, paternal aunt, paternal grandmother, and brother | CHF (33) | Yes | 5.36 | 229 | Yes | Yes (4) | No | Normal | Normal |
| Family 1 (III-3) | M | 17 | NR | Yes | 3.15 | 115 | Yes, grade 1 | No | No | Normal | Normal | ||||
| Family 2 (II-2) | F | 33 | g.31944G>A p.Glu2771Lys in PKD1 No mutation in PKHD1 | ADPKD | Father, daughter, and son | CHF (1) | Yes | 3.2 | 87 | Yes, grade 3 | No | Cysts in right posterior | Normal | Normal | |
| Family 3 (II-2) | F | 36 | g.26918T>G p.Phe2132Cys in PKD1 no mutation in PKHD1 | ADPKD | Mother | CHF (33) | Yes | 2.77 | 64 | No | No | Multiple small cysts | Partially impaired | Normal | |
| Tazelaar et al (19) | Index case | F | 19 | NR | ADPKD | Mother, maternal aunt, and second cousin | CHF (19) | Yes (5) | NR | NR | Yes | Yes | No | NR | NR |
| Lee et al (18) | Index case | F | 7 | NR | ADPKD | Father (no CHF on biopsy), paternal grandmother (no splenomegaly on autopsy at age 51) | CHF(7) | Yes, splenorenal shunt | NR | NR | Yes | Yes, recurrent | NR | Normal | Normal |
| Brother | M | 14 | NR | Yes | NR | NR | NR | No | No | NR | NR | ||||
| De Vos et al (17) | Family 1 (III-1) | M | 7 | NR | ADPKD | Mother (no CHF on biopsy) | CHF (7) | Yes, splenectomy, portocaval shunt at age 7 | NR | Reduced | NR | No | NR | NR | NR |
| Family 1 (III-3 | M | 3 | CHF | Yes, portocaval shunt | 2.8 | 43 | Yes | No | No | NR | Mildly elevated | ||||
| Family 2 (II-3) | M | 12 | NR | ADPKD | Mother, sister, and 2 brothers | CHF | Yes (10), portocaval shunt | NR | 21 | Yes | Yes | Dilated CBD, intrahepatic cysts in communication with biliary tree | Normal | Mildly elevated | |
| Family 2 (II-5) | M | 7 | ADPKD | CHF | Yes, distal splenorenal shunt | NR | 97 | Yes | No | Multiple small cysts | Normal | Mildly elevated | |||
| Matsuda et al (16) | Index case | M | 33 | NR | ADPKD | Father (no CHF on biopsy), 2 paternal uncles | CHF, no von Meyenberg complexes (33) | Yes (17) | NR | NR | NR | NR | No | Normal | NR |
| Cobben et al (15) | Family PK2 (III-3) | F | 29 | Linkage to chromosome 16 | ADPKD | 2 sisters | CHF (8) | Yes splenectomy at age 8 | NR | NR | Yes | Yes (12) | No | NR | NR |
| Family PK2 (III-4) | F | 27 | CHF (16) | Yes | NR | NR | Yes | Yes (16) | No | NR | NR | ||||
| Family PK67 (III-1) | M | 29 | Linkage to chromosome 16 | ADPKD | Mother and sister | CHF (3) | Yes | Reduced | Reduced | No | No | No | NR | NR | |
| Family PK67 (III-2) | F | 27 | CHF (18) | Yes (birth) | NR | NR | Yes | No | No | NR | NR | ||||
| Family PK11 (III-7) | M | 19 | Linkage to chromosome 16 | ADPKD | Mother, 2 maternal aunts, and maternal grandmother | CHF (11) | Yes (11) | Reduced | Reduced | Yes | No | No | NR | NR | |
| Lipschitz et al (14) | Index case | F | 19 | NR | ADPKD | Mother maternal grandfather, 2 sisters, and a brother | CHF (14) | Yes (4) | 2.8 | 62 | Yes | Yes | No | Normal | Normal |
| Kanaheswari et al (13) | Index case | F | 6 | NR | ADPKD | Father | CHF (6) | Yes | 6.6 | 67 | NR | No | No | Normal | Normal |
ADPKD = autosomal dominant polycystic kidney disease; CHF = congenital hepatic fibrosis; F = female; M = male; NR = not reported; PH = portal hypertension.
FIGURE 2.
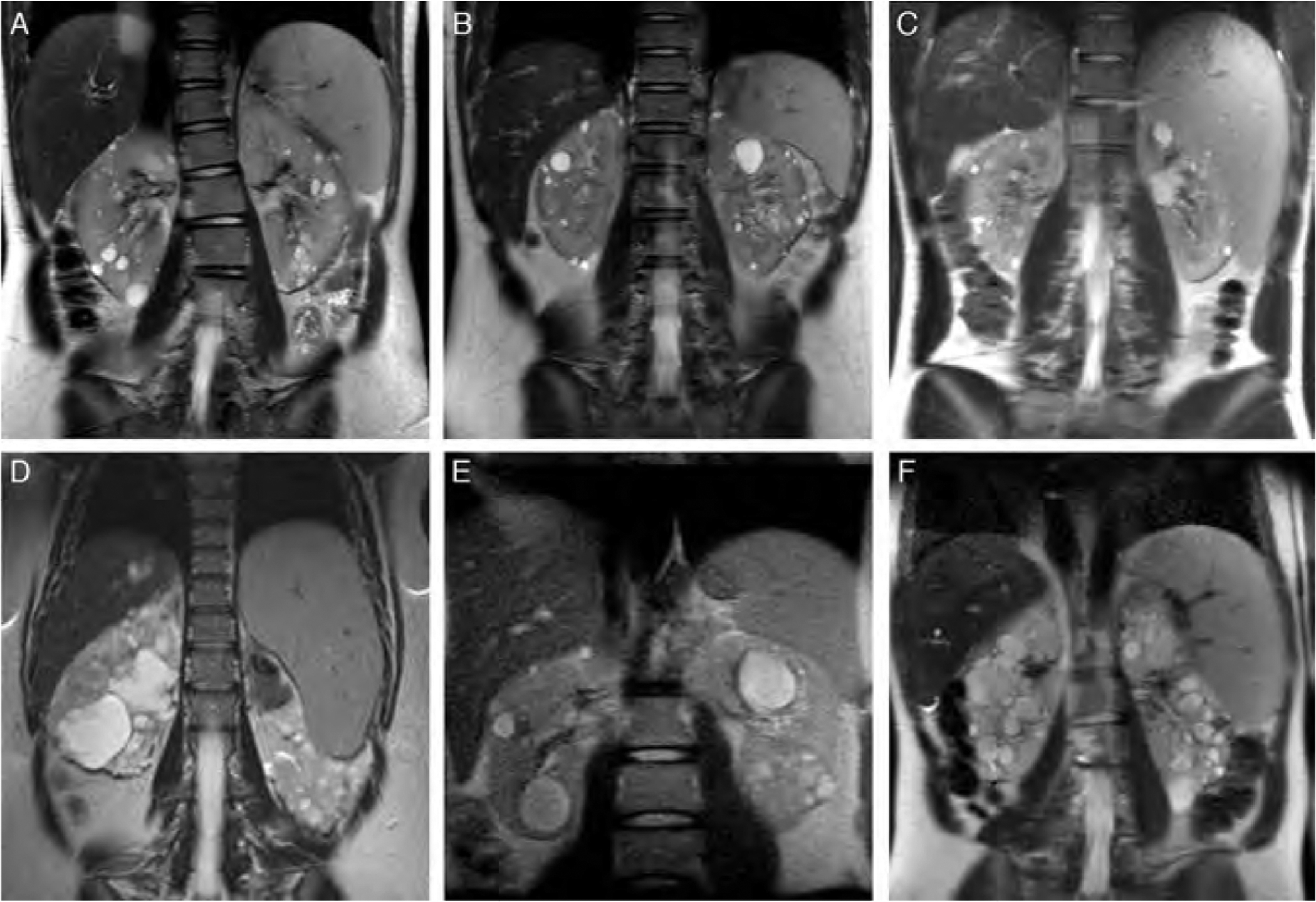
Abdominal MRI images displaying kidney, liver, and spleen findings. All of the patients displayed polycystic kidneys. (A), (B), and (C) belong to the 3 siblings in family 1. Patient III-1, who underwent splenorenal shunt placement, had a mildly enlarged spleen (A). Patient III-2 had a normal-sized spleen (B). Patient III-3 had an enlarged spleen (C). In family 2, patient II-2 had an enlarged spleen (D), whereas her daughter (III-1) had a normal-sized spleen at age 16 (E). In family 3, patient II-2 also displayed splenomegaly (F).
The 12-year-old brother (III-2) of case III-1 also had multiple macrocysts on both kidneys (Fig. 2B). On USG, the liver was mildly hyperechogenic. Spleen volume, calculated based on MRI imaging, was at the upper end of normal at 258 mL (Fig. 2B). Serum chemistries were normal (Table 1).
The NIH evaluation of the 14-year-old boy (III-3) revealed a healthy-appearing teenager with normal growth. He had multiple cysts in both kidneys (Fig. 2C). On USG, liver was hyperechogenic. MRI-based liver and spleen volumes were increased at 1899 and 931 mL, respectively (20–22) (Fig. 2C). Liver enzymes and synthetic functions were normal (Table 1).
All of the coding exons of the PKD1 and PKHD1 genes were sequenced in the father and the 3 children. All of the 4 patients were heterozygous for a 7-bp deletion at position g.17554 in exon 5 of the PKD1 gene (Fig. 1, family 1). Sequencing of the 66 coding exons of the PKHD1 gene revealed no mutations.
Family 2
In Family 2 (Fig. 1), 4 individuals including the mother (II-2), both of her children (III-1, III-2), and the maternal grandfather (I-1) had typical renal findings of ADPKD. The mother had CHF complicated by PH, whereas 3 other individuals with ADPKD had normal-sized spleens (Table 2, Fig. 2D and E).
The mother (II-2) presented with splenomegaly at age 6 months. Liver biopsy was consistent with CHF. She developed esophageal varices that required banding throughout childhood and into adulthood. In addition, she had 3 episodes of cholangitis. The NIH evaluation at age 33 revealed a distended abdomen. The liver was palpable 13 cm below the xiphoid with firm consistency. The spleen was 12 cm palpable below the left costal margin. Based on MRI images, liver and spleen volumes were markedly increased at 2310 and 908 mL, respectively (20–22). USG showed increased liver echogenicity with 3 cysts in the posterior right lobe; on magnetic resonance cholangiopancreatography (MRCP) these cysts were in continuity with the bile tree. The kidneys were markedly enlarged with multiple macrocysts (Fig. 2D). Liver function tests were normal. The patient had thrombocytopenia and leukopenia due to hypersplenism (Table 1).
Her 16-year-old daughter (III-1) (Fig. 2E) and 14-year-old son (III-2) both had multiple renal cysts. Their liver ultrasound pattern, biliary system, and spleen size were normal, and serum chemistries were unremarkable (Table 1). The mother and both children had a missense mutation at position g425323 in exon 61 of the PKD1 gene, replacing a conserved serine with arginine (Fig. 1, family 2). Sequencing of the PKHD1 gene did not reveal any mutations.
Family 3
In this family, the proband (II-2), a 36-year-old female with ADPKD, was diagnosed with CHF at age 33, based on a liver biopsy prompted by splenomegaly (Table 2, Fig. 2F). Her mother died of renal complications of ADPKD; she did not have PH. At the NIH Clinical Center, USG of patient II-2 showed moderately echogenic liver with coarsening of the echotexture and several small cysts. MRCP showed no dilatation of the bile ducts. There was extensive collateral formation. Based on MRI imaging, the liver and spleen volumes were increased at 1687 and 1090 mL, respectively (20,22). The kidneys contained multiple cysts. Leukocyte and platelet counts were low due to hypersplenism (Table 1). The patient had mild postprandial hyperammonemia; other liver function tests and liver enzymes were normal. She had a missense mutation at position g.26918 in exon 15 of the PKD1 gene, replacing a conserved phenylalanine with cysteine (Fig. 1, family 3). Sequencing of the PKHD1 gene did not reveal any mutations.
Literature Review
In Table 2, we listed the characteristics of liver disease and family history of 19 patients with ADPKD associated with CHF complicated by PH, from 14 ADPKD families, reported in 7 publications between 1984 and 2010 (13–19). Other reports were reviewed but not included because the evidence for autosomal dominant inheritance of PKD was not convincing (23–27). All of the 19 patients had family histories of ADPKD. There were 9 boys and 10 girls. Age at the time of evaluation ranged from 3 to 36 years (mean ± SD, 18.7 ± 10.5) (Table 2). All of the 19 patients had splenomegaly; in many patients, an enlarged spleen was first noted at birth or in early childhood. Five patients had portosystemic shunt placement and 2 had splenectomy. Histopathological evaluation of the liver showed CHF in all of the 17 patients who underwent liver biopsy (Table 2). Age at the time of the liver biopsy ranged from 1 to 33 years (14.9 ± 11.1) in 14 patients for whom this information was available. Thirteen of the 15 patients evaluated had esophageal varices; 7 had bleeding from esophageal varices, the youngest at age 4 years (Table 2). The majority of the patients had decreased platelet and white blood cell counts due to hypersplenism. Synthetic function of the liver was preserved and liver enzymes were normal or midly elevated (Table 2). In all of the 14 families, there were one or more family members with ADPKD but without CHF/PH; 2 fathers and a mother with ADPKD from 3 families had liver biopsies that did not show CHF (Table 2).
DISCUSSION
Although both ADPKD and ARPKD are associated with liver involvement, the characteristics of liver disease in these 2 types of PKD are quite different. The liver disease of ARPKD involves CHF that is often complicated by PH (28,29) and its consequent hypersplenism and esophageal varices. Many patients with ARPKD also have cystic dilatations of the intrahepatic bile ducts that are continuous with the biliary system; a combination referred to as Caroli syndrome (30,31). In contrast, the liver cysts of ADPKD originate from biliary microhamartomas (von Meyenburg complexes) that are embedded in fibrous tissue; hence, they are not in continuity with the intrahepatic biliary tree (32–34). Imaging of the intrahepatic biliary system, preferably performed using MRCP because endoscopic pancreatiocholangiography increases the risk for cholangitis, is useful in the differential diagnosis (33). In ADPKD, enlarged liver cysts cause complications that are largely due to a mass effect, including chronic upper abdominal pain and distension, early satiety, nausea and dyspnea, and rare cases of hepatic venous outflow obstruction (2,35,36). PH caused by CHF is not typical for ADPKD.
The proteins encoded by the PKD genes (polycystin-1, polycystin-2, and fibrocystin) localize to the primary cilia. Intact cilia-based signaling via the PKD proteins is required for normal development of the portobiliary system (37,38) and renal tubules (39). Dysfunction of cholangiocyte cilia results in defective remodeling of the developing biliary system that is referred to as “ductal plate malformation” (DPM) (28,40,41). DPM is the main pathology that underlies the liver disease in ciliopathies (28). It is characterized by retention of excessive numbers of primitive bile duct remnants in their original, peripheral, interrupted ring-like position. Depending on the level of the affected portobiliary tree, DPM results in a spectrum of abnormalities ranging from CHF (microscopic bile ducts), to CHF/CS (microscopic and medium-size bile ducts), to Caroli disease (CD) (medium and large bile ducts) (40). Liver biopsy in CHF shows abnormal portal tracts with an excessive number of abnormally shaped embryonic bile ducts, abnormal portal vein, and periportal fibrosis without inflammation (29). The severity ofDPM and the level of the portobiliary tree affected by DPM vary within and among individual ciliopathies. The isolated liver cysts in the PLD associated with ADPKD probably represent DPM affecting the most peripheral end of the biliary system (40,42).
Typically, kidney and liver disease of ADPKD becomes symptomatic in adulthood. However, many patients with ADPKD-CHF presented with PH at birth or in early childhood (Table 2). Out of the 13 patients for whom age at the time of liver biopsy was available, 9 were younger than 18 years, including patients at ages 1 and 3 years (Table 2). Three of the 7 patients with esophageal variceal bleeding were at ages 4, 12, and 16 years at the time of the bleed. Histopathological and other clinical characteristics of CHF in ADPKD were similar to those of CHF in ARPKD (Table 2). Thrombocytopenia and neutropenia caused by hypersplenism were common. Synthetic function of the liver was preserved and liver enzymes were normal or only mildly elevated (Table 2).
The presence of CHF in these rare ADPKD families raises the question whether the PKD in these families is caused by one of the known genes of ADPKD (PKD1 or PKD2 that typically cause ADPKD associated with PLD) or by another yet to be identified gene(s). The first indirect molecular data on this came from the linkage of ADPKD-CHF to chromosome 16, where PKD1 gene resides (15) (Table 2). By documenting that all of the affected individuals in our 3 families had pathogenic mutations in PKD1, we show for the first time that the PKD in families with ADPKD-CHF is due to PKD1 mutations (Table 2). Given that only some affected individuals develop CHF, despite the fact that all of them (those with PKD and typical liver disease in the form of PLD and those with PKD and CHF) carry the same mutation in PKD1, suggests that the atypical nature of liver disease in these families is not explainable by the location or type of the PKD1 mutation. In all of the 14 families with ADPKD-CHF, the parents of the index cases had PKD but did not have CHF; liver biopsies of 2 fathers and a mother with PKD did not show CHF (Table 2).
There were several families in which several siblings had ADPKD with CHF (Table 2). This suggests contribution of modifier mutation(s) in other gene(s). Given that CHF is observed in both boys (9) and girls (10) with ADPKD, these modifier genes are probably located on autosomal chromosomes and less likely X-linked. PKHD1 and other ciliopathy genes are likely candidates for such modifiers. We sequenced PKHD1 in our 3 families and did not identify any pathogenic mutations. However, it remains possible that a variant in the noncoding parts of PKHD1 or a single-nucleotide polymorphism can be contributing.
In summary, CHF complicated by PH is a rare but potentially life-threatening complication of ADPKD. Increased liver echogene-city on USG, decreased platelet count, enlarged left lobe of liver, or enlarged spleen should alert physicians to this possibility. Upon diagnosis of an index case, other family members, especially siblings, should be evaluated for CHF/PH. We recommend abdominal ultrasound and platelet count on siblings at the time of diagnosis of the proband. Repeat of these tests every 2 years would be warranted especially early in life, as some patients may progress slower than others. Abnormalities in these tests should prompt further work-up. Early diagnosis of CHF/PH in other family members with ADPKD can be lifesaving with appropriate monitoring and treatment of esophageal varices.
Acknowledgments:
We thank the patients with ADPKD and families who generously participated in this investigation.
This study was supported by the Intramural Research Program of the National Human Genome Research Institute and the NIH Clinical Center.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med 2009;60:321–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: GeneReviews at GeneTests: Medical Genetics Information Resource. University of Washington, Seattle: 1997–2008. Available at http://www.genetests.org.AccessedNovember 10, 2010. [PubMed] [Google Scholar]
- 3.Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol 2005;100:2569–82. [DOI] [PubMed] [Google Scholar]
- 4.Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 1995;10:151–60. [DOI] [PubMed] [Google Scholar]
- 5.Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996;272:1339–42. [DOI] [PubMed] [Google Scholar]
- 6.Hateboer N, v Dijk MA, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 study group. Lancet 1999;353:103–7. [DOI] [PubMed] [Google Scholar]
- 7.Gartzke J, Majewski E. Aromatic carboxylic acids in urine. II. A quantitative method for the determination of (5-hydroxy-3-indolyl)-acetic acid and vanillinmandelic acid from urine, and some problems of methodology (author’s transl). Z Klin Chem Klin Biochem 1972;10:108–11. [PubMed] [Google Scholar]
- 8.Zerres K, Rudnik-Schoneborn S, Senderek J, et al. Autosomal recessive polycystic kidney disease (ARPKD). J Nephrol 2003;16:453–8. [PubMed] [Google Scholar]
- 9.Gunay-Aygun M, Font-Montgomery E, Lukose L, et al. Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol 2010;5:972–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunay-Aygun M, Avner ED, Bacallao RL, et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: summary statement of a first National Institutes of Health/Office of Rare Diseases conference. J Pediatr 2006;149:159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med 2006;354:2122–30. [DOI] [PubMed] [Google Scholar]
- 12.Cheong B, Muthupillai R, Rubin MF, et al. Normal values for renal length and volume as measured by magnetic resonance imaging. Clin J Am Soc Nephrol 2007;2:38–45. [DOI] [PubMed] [Google Scholar]
- 13.Kanaheswari Y, Hamzaini AH, Wong SW. Congenital hepatic fibrosis in a child with autosomal dominant polycystic kidney disease. Med J Malaysia 2008;63:251–3. [PubMed] [Google Scholar]
- 14.Lipschitz B, Berdon WE, Defelice AR, et al. Association of congenital hepatic fibrosis with autosomal dominant polycystic kidney disease. Report of a family with review of literature. Pediatr Radiol 1993;23:131–3. [DOI] [PubMed] [Google Scholar]
- 15.Cobben JM, Breuning MH, Schoots C, et al. Congenital hepatic fibrosis in autosomal-dominant polycystic kidney disease. Kidney Int 1990;38:880–5. [DOI] [PubMed] [Google Scholar]
- 16.Matsuda O, Ideura T, Shinoda T, et al. Polycystic kidney of autosomal dominant inheritance, polycystic liver and congenital hepatic fibrosis in a single kindred. Am J Nephrol 1990;10:237–41. [DOI] [PubMed] [Google Scholar]
- 17.De Vos M, Barbier F, Cuvelier C. Congenital hepatic fibrosis. J Hepatol 1988;6:222–8. [DOI] [PubMed] [Google Scholar]
- 18.Lee FI, Paes AR. Congenital hepatic fibrosis and adult-type autosomal dominant polycystic kidney disease in a child. Postgrad Med J 1985;61:641–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tazelaar HD, Payne JA, Patel NS. Congenital hepatic fibrosis and asymptomatic familial adult-type polycystic kidney disease in a 19-year-old woman. Gastroenterology 1984;86:757–60. [PubMed] [Google Scholar]
- 20.Watanabe Y, Todani T, Noda T, et al. Standard splenic volume in children and young adults measured from CT images. Surg Today 1997;27:726–8. [DOI] [PubMed] [Google Scholar]
- 21.Coppoletta JM, Wolbach SB. Body length and organ weights of infants and children: a study of the body length and normal weights of the more important vital organs of the body between birth and twelve years of age. Am J Pathol 1933;9:55–70. [PMC free article] [PubMed] [Google Scholar]
- 22.Yu HC, You H, Lee H, et al. Estimation of standard liver volume for liver transplantation in the Korean population. Liver Transpl 2004;10:779–83. [DOI] [PubMed] [Google Scholar]
- 23.Gaisford W, Bloor K. Congenital polycystic disease of kidneys and liver. Portal hypertension: portacaval anastomosis. Proc R Soc Med 1968;61:304. [PMC free article] [PubMed] [Google Scholar]
- 24.Longmire WP Jr, Mandiola SA, Gordon HE. Congenital cystic disease of the liver and biliary system. Ann Surg 1971;174:711–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradford WD, Bradford JW, Porter FS, et al. Cystic disease of liver and kidney with portal hypertension. A cause of sudden unexpected hematemesis. Clin Pediatr (Phila) 1968;7:299–306. [DOI] [PubMed] [Google Scholar]
- 26.Manes JL, Kissane JM, Valdes AJ. Congenital hepatic fibrosis, liver cell carcinoma and adult polycystic kidneys. Cancer 1977;39:2619–23. [DOI] [PubMed] [Google Scholar]
- 27.Mindikoglu AL, Regev A, O’Sullivan MJ, et al. Multiple normal deliveries in a woman with severe portal hypertension due to congenital hepatic fibrosis: the importance of preserved hepatocellular function. Am J Gastroenterol 2005;100:2359–61. [DOI] [PubMed] [Google Scholar]
- 28.Gunay-Aygun M Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet 2009;151C:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kerr DN, Okonkwo S, Choa RG. Congenital hepatic fibrosis: the long-term prognosis. Gut 1978;19:514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caroli J Diseases of intrahepatic bile ducts. Isr J Med Sci 1968;4: 21–35. [PubMed] [Google Scholar]
- 31.Kerkar N, Norton K, Suchy FJ. The hepatic fibrocystic diseases. Clin Liver Dis 2006;10:55–71. [DOI] [PubMed] [Google Scholar]
- 32.Tahvanainen E, Tahvanainen P, Kaariainen H, et al. Polycystic liver and kidney diseases. Ann Med 2005;37:546–55. [DOI] [PubMed] [Google Scholar]
- 33.Morgan DE, Lockhart ME, Canon CL, et al. Polycystic liver disease: multimodality imaging for complications and transplant evaluation. Radiographics 2006;26:1655–68. [DOI] [PubMed] [Google Scholar]
- 34.Caroli J Diseases of the intrahepatic biliary tree. Clin Gastroenterol 1973;2:147–61. [PubMed] [Google Scholar]
- 35.Chauveau D, Grunfeld JP, Durand F, et al. Ascites in a polycystic patient. Nephrol Dial Transplant 1997;12:228–30. [DOI] [PubMed] [Google Scholar]
- 36.Uddin W, Ramage JK, Portmann B, et al. Hepatic venous outflow obstruction in patients with polycystic liver disease: pathogenesis and treatment. Gut 1995;36:142–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masyuk T, Masyuk A, LaRusso N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Curr Opin Gastroenterol 2009;25:265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masyuk AI, Masyuk TV, Splinter PL, et al. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology 2006;131:911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol 2007;18:1381–8. [DOI] [PubMed] [Google Scholar]
- 40.Desmet VJ. Ludwig symposium on biliary disorders: part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc 1998;73: 80–9. [DOI] [PubMed] [Google Scholar]
- 41.Gunay-Aygun M, Heller T, Gahl WA. Congenital hepatic fibrosis overview. In: GeneReviews at GeneTests: Medical Genetics Information Resource. University of Washington, Seattle; 1997–2008. Available at http://www.genetests.org. AccessedNovember 10, 2010. [Google Scholar]
- 42.Desmet VJ. What is congenital hepatic fibrosis? Histopathology 1992;20:465–77. [DOI] [PubMed] [Google Scholar]