

Abstract
Gramicidin A, a topical antibiotic made from alternating L and D amino acids, is characterized by its wide central pore; upon insertion into membranes, it forms channels that disrupts ion gradients. We present helical peptidomimetics with this characteristic wide central pore that have been designed to mimic gramicidin A channels. Mimetics were designed using molecular modeling focused on oligomers of heterochiral dipeptides of proline analogs, in particular azaproline (AzPro). Molecular Dynamics simulations in water confirmed the stability of the designed helices. A sixteen-residue Formyl-(AzPro-Pro)8-NHCH2CH2OH helix was synthesized as well as a full thirty-two residue Cbz-(AzPro-Pro)16-OtBu channels. No liposomal lysis activity was observed suggesting lack of channel formation, possibly due to inappropriate hydrogen-bonding interactions in the membrane. These peptidomimetics also did not hemolyze red blood cells, unlike gramicidin A.
Graphical Abstract

Introduction –
The design of peptide mimetics is a common strategy in medicinal chemistry due to the significant role that peptides/proteins have in molecular recognition in biology as well as their use in the improvement of ADME properties of pharmaceuticals. Organic non-peptides are used to mimic structural motifs for peptide molecular recognition as well as provide potential therapeutics for diseases. While organic scaffolds have been designed for various different types of secondary structures such as the α-helix [1, 2], β-sheet [3–6] as well as less probable structures [7, 8], mimetics for heterochiral secondary structures have yet to be described. Such structural motifs with alternating chirality and unique biological properties are found in nature; Gramicidin A, B and C contain heterochiral helical secondary structures with a wide central pore [9]. This helical motif was first described by Profs. G. N. Ramachandran and R. Chandrasekaran at the Second American Peptide Symposium held in Cleveland in 1968 [10] and later in 1972 [11]. Their structural analysis was based on the alternating L & D chiralities of amino acids found in gramicidins. Their postulated L,D-helix had 3.2 residues per turn and a rise of 1.55 Å per residue, and is now commonly referred to as a β6.3-helix, or LD4-helix [11]. The carbonyl oxygens face the center of the helix and are alternatively pointed towards the amino- and carboxy-terminals generating a cation-selective channel in lipid bilayers. To quote Ramachandran & Chandrasekaran, “the LD4-helix has the right size of cavity to bind ions like Na+ and K+, and thus appears to be the most probable helical structure for ion-transport peptides” [12–15].
Gramicidins were first isolated by Dubos in 1939 [16–18] for their bactericidal properties against gram-positive bacteria, and are still used topically as antibiotics. Due to their channel-forming properties in membranes, they show toxicity to human cells (hemolysis of red blood cells, for example), but have provided a model for antibacterial peptide research [19]. The amino acid sequence of gramicidin A (Figure 1a) including the alternative D,L chirality was determined by Sarges and Witkop in 1964 [20–22].
Figure 1.

a) Sequence structure of Gramicidin A monomer. R = Trp; B, R = Phe; and C, R = Tyr. b) Conformational states of gramicidin A monomers and dimers in phospholipid membranes [19].
Many biophysical and modeling studies on the structure of gramicidin A have been published with considerable controversy. Both a double-stranded helix and head-to-head dimer have been found depending on experimental conditions (Figure 1b), and the topology of the preferred structure has been hotly debated. It turned out that both the head-to-head dimer and double-stranded helix conformers are present in membrane environments [19], as is the monomer. Urry et al. prepared a covalently linked head-to-head dimer that functioned as a cation-selective channel [23] supporting the conjecture of Ramachandran and Chandrasekaran of the role of β6.3-helix (LD4-helix) in biological activity. The structure of the head-to-head dimer was later confirmed by solid-state NMR [24, 25]. Another possible channel structure formed by a double-stranded helix arises from the crystal structure of gramicidin (Figure 2) with sodium iodide [15]. The inherent conformational flexibility of gramicidins allows for multiple conformations to exist, with the preferred structure being dictated by experimental conditions (Figure 3). Development of a stable peptidomimetic of an L,D-helix (β6.3-helix) may lead a semi-rigid scaffold useful to probe channel biophysics as well as enhance the search for novel antibacterials. For example, the role of indole N-H bonding in gramicidin channel formation appears critical [30], but the mechanism remains elusive [31, 32]. Are tryptophan residues essential for channel formation and antibiotic activity? Previously, it was assumed that the transitions between on and off conductance states represented Urry et al. postulated that “monomers form in the single-stranded, β-helical conformation at the interface, transiently dip into the lipid layer and remain there only if a second monomer is appropriately encountered and a lipid spanning head-to-head dimeric channel results” [33]. Based on recent solid-state NMR experiments, Jones et al. proposed that gramicidin helices undergo a conformational transition near the permeation pathway which gates the channel formation [34]. Mo et al. found a series of kinked structures with a large degree of conformational heterogeneity. The C-terminal domain was rigid with a well-defined orientation in the bilayer interfacial region. On the other hand the N-terminal domain, although appearing to be conformationally rigid within the hydrophobic core of the bilayer, adopted a myriad of orientations relative to the bilayer normal [35] Hydrophobic mismatch (defined as the difference in length between the lipid hydrophobic thickness and the peptide hydrophobic region) is thought to be responsible for altering the lipid/protein dynamics of gramicidin [36, 37].
Figure 2.

Na+-ion positions (van der Waals surfaces) of the gD–NaI complex [15].
Figure 3.

Multiple conformations determined by NMR and crystallography [25]. A = Solid-state NMR-derived structure of gA from a lipid bilayer environment: single-stranded, right-handed, and 6.5 residues per turn; PDB = 1MAG [26]. B = X-ray crystallographic structure of crystals prepared from Cs+/MeOH solution: double-stranded, right-handed, and 7.2 residues per turn; PDB = 1AV2 [27]. C = A solution NMR structure from an SDS micellar environment: single-stranded, right-handed, and 6.3 residues per turn; PDB = 1GRM [28]. D = An X-ray crystallographic structure of crystals prepared from benzene/methanol solution: double-stranded, left-handed, and 5.6 residues per turn; PDB = 1ALZ [29]
Since gramicidin has become a prototypical membrane channel, numerous potential applications of gramicidin and its analogs have emerged. Gramicidin channels can also serve as molecular force probes for studying membrane physical properties, due to their unique ability to discriminate between changes in monolayer curvature and bilayer elastic moduli, and the consequences hereafter [38]. The mechanism of antimicrobial activity of gramicidins are still under investigation. Gramicidin A causes membrane permeabilization and induces formation of hydroxyl radicals; The latter may be the underlying mechanism of lethal activity against Staphylococcus aureus [39]. Solubilizing/stabilizing gramicidin by chemical modification enhances its antibacterial activity [40, 41] and reduces hemolysis as well as enhances activity in selective mitochondrial uncoupling [42, 43].
Simplified channels to facilitate ion movement across membranes have been investigated by Gokel [44] and others. Others have generated compounds that mimic the pore-forming properties of gramicidin A through self-assembly of smaller fragments [45, 46]. To our knowledge, however, no stable peptidomimetic of the heterochiral helix formed by gramicidin A itself has been disclosed.
Approach for Design
This study used molecular dynamics and molecular modeling to explore possible peptidomimetic designs for a stable oligomer of heterochiral dipeptides. The use of proline and proline analogs as the scaffold was a primary focus of this study. Oligomers of proline heterochiral dimers are excellent candidates because the dipeptides stabilize and initiate reverse turns [47, 48]; a succession of turns generate helices. Multiple dimers were tested and dimers of proline and azaproline (AzPro) were chosen as azaproline was known to stabilize cis-amide bonds in short peptides [49] In addition, the presence of the nitrogen replacing the α-carbon allows pyramidal inversion of the sp3-hybridized nitrogen costing only 5–10 kcal/mol, corresponding to interconversion between the two mirror image (R,S) forms [51]; this stereodynamic nitrogen allows conformational adjustment to environmental conditions. The dimer synthon is relatively easy to synthesize, and allows for the generation of repeating oligomeric motifs for probing molecular recognition. In addition, synthetic modification of the proline ring to orient different functional groups has been well documented [52], thus, it would be relatively simple to incorporate functional groups onto semi-rigid cyclic amino acids, such as proline or its homolog pipecolic acid and their analogs, for surface modulation of the peptide helices. Modular heterochiral dipeptides with different side chains appended to the cyclic ring would provide a toolkit for recognition studies. Simple modification of dipeptide units would allow for generation of multiple differing structures to probe for molecular-recognition motifs. It was found that oligomers of proline-azaproline [53, 54] dimers (Pro-AzPro)n generated stable mimetics of the heterochiral β6.3-helical secondary structure seen in gramicidins (Figure 4c and d).
Figure 4:
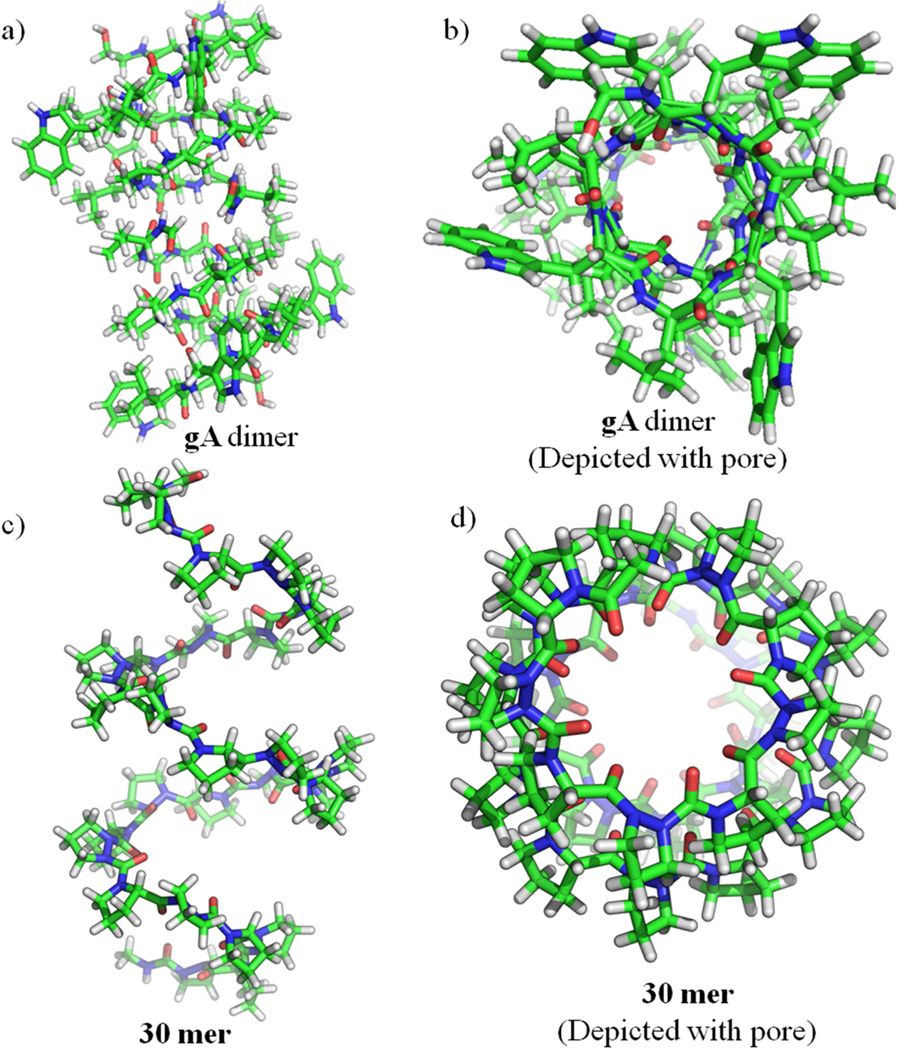
a) Structures of Gramicidin A (gA) Head-to-Head Dimer Helices (PDB ID: 1NRM [50]); side and top view, b) gA Depicted with pore, c) Helical structure of Pro-AzPro 30-residue helix (30 mer) from molecular modeling; side and top view, d) 30 mer Depicted with pore.
Computational Approach
Conformational Screening of Candidate Structures
Candidate dimers were built using Maestro (Schrodinger, Version 9.3) and to generate extended structures with total length of 30 residues. Conformer searching was done using the OpenEye OMEGA software package (version 2.5.1.4) by generating 20,000 unique conformers of the extended structure and the 10 lowest-energy conformers were output and minimized using SZYBKI (version 1.7.0) using the Poisson-Boltzmann Implicit Solvation Model. Energies for conformer stability comparisons were computed using parameters from the MMFF94 force field. The conformers were then visually evaluated, and if 7 out of the 10 conformers were similar to gramicidin A (helical with a pore and all carbonyls pointing towards the middle), the most stable conformer was used to carry out MD. This procedure is summarized in Figure 5.
Figure 5:

Flowchart of the procedure for testing candidate dimers for helical quality.
Molecular Dynamics Simulation of Helical Structures
To further determine if the helical structures generated by the OpenEye suite were structurally stable in aqueous conditions, the most stable conformer was passed into the SYBYL molecular dynamics engine using the MMFF94 Force Field [55]. For each candidate, the most stable helical conformer was solvated in a sphere of water with a diameter of 35 Å. The structure was then minimized using a steepest descent algorithm in explicit solvent with periodic boundary conditions. MD simulation using under NVT conditions at a constant temperature of 310 °K was run for 1 nanosecond with 1 femtosecond steps. The structure was then minimized using steepest descent once again at 310 °K. Fitting to any template helices, like Gramicidin A, for RMSD and flexibility information was done via alpha-carbon atom fit, or using the nitrogen atom of AzPro analogous to the alpha carbon in the helix residues. All final energy values were obtained using the final minimized structure and normalized by dividing by the molecular weight of the helix to allow comparison. Information about the helical character and helical pore size was obtained using PyMOL [56] and the python analysis library. Backbone analysis of the helix (Figure 6) demonstrates that both Φ and Ψ of the proline and azaproline residues are highly constrained over the course of the simulation. Both residues exist in a single state throughout, and very little variation is seen in either backbone dihedral angles. This provided initial evidence that the Pro-AzPro helical dimer would maintain a constrained helical character. However, it is important to note that this highly constrained helix was observed in simulations in water, and not necessarily indicative of the structure the helix would adopt once it was within the membrane. Furthermore, these distribution plots are derived from short simulation times.
Figure 6.

Torsion distribution plots computed from molecular dynamics simulations of the AzPro-Pro oligomer. a) Torsion angles of all proline residues found in the oligomer. b) Backbone torsions of all azaproline residues found in the oligomer. The Φ angle for azaproline was computed using the α-nitrogen in place of the α-carbon.
Visualization of the simulation provides further insight into the physical basis of the torsional shape. Initial intuition would suggest that the peptide would in fact exist in equilibrium with equal tendencies to be in either the helical conformation or an unfolded non-helical shape. This is suggested by the fact that unlike naturally occurring peptide helices, there are no hydrogen bonding sites on the backbone of the Pro-AzPro helix since neither amino acid has the necessary hydrogen bond donor-accepter combination. However, some helical structure might occur due to the torsional forces on each proline dimer as has been discussed in the literature [53, 54]. However, it was observed in the simulations that the stable helix shape was maintained due to the presence of polar solvent bridging the turns of the helix. The presence of a polar water solvent created hydrogen bonding network pathway that connected the carbonyl groups of each successive turn in the helix to one another, further stabilizing the helical shape of the peptide.
To further probe this polar-solvent bridging hypothesis, an ethanolamine solute was added to the peptide-solvent construct and simulated for an additional 50ns. Ethanolamine was chosen due to it also having both a hydrogen bond acceptor and a hydrogen bond donor on the molecule, and had a long enough carbon chain that it would be easily visible bridging the turns of the helix. The helical structure was maintained throughout the simulation, and by the end of the simulation the ethanolamine was observed bridging the helical turns (Figure 7). However, it is important to note that while it aided bridging the turns, it was not obstructing the pore, providing support for that the solvent-bridging hypothesis allows for stable pore formation.
Figure 7.

a) Ac-Pro-AzPro-Ac helical structure with ethanolamine embedded demonstrating hydrogen bond bridging between turns. b) Top view demonstrating an unoccluded pore.
Further insight into the ion binding properties of analogous peptides was sought using molecular simulation. 50 ns MD simulations were performed with SYBYL in aqueous conditions containing both Ca2+ and Cl- ions. Channel structure was maintained over the course of the simulation (Figure 8), as shown by the root-mean-square deviations (RMSD) and root-mean-square fluctuations per cyclopeptide (RMSF). 50 ns of simulation was sufficient for equilibrium sampling as indicated by the rapid entry of both water and cations into the nanotube, generally within the first nanosecond. Strong selectivity for cations is attributed to the negatively charged carbonyl oxygen atoms facing inside the channel.
Figure 8:

a) Helical structure of Ac-(Pro-AzPro)8-Ac (16 mer-CaCl2) helix (trapped with calcium ions from molecular modeling; side view and b) top view.
Synthesis of Gramicidin A-Channel Peptidomimetics
Incorporation of an aza-amino acid residue into the peptide chain requires a combination of hydrazine and peptide chemistry by adding a protected hydrazine to an isocyanate derivative of the peptide N-terminal. Unfortunately, it is not applicable when Pro occupies the N-terminal position as in many of our cases. Andre et al. [57] used triphosgene as the carbonylating reagent of a protected hydrazide and it is a mild, easy to handle, and an efficient carbonylating agent for the azapeptide synthesis. Alhough this method works well, in general, but the activated species can only be prepared in situ (under N2 at −10 °C), and formation of considerable amounts of side products, such as diazatides.were observed. More recently, synthetic routes to azapeptides have been investigated by Zhang et al. [50, 54] using a liquid-phase approach. Utilizing this synthetic strategy, we have synthesized a wide variety of Pro-AzPro-containing gramicidin A analogs including a full length 32-residue peptide listed in Table 1.
Table 1 :
List of synthesized AzPro-Pro peptide analogs, gramicidin A (gA) and their molecular weights.
| S. No. | Sequence | Compound code | Reaction code | Molecular weight |
|---|---|---|---|---|
| 1 | Cbz-(AzPro-Pro)8-NHCH2CH2OH | 1 | NDR-487 | 1756.99 |
| 2 | Ac-(AzPro-Pro)8-NHCH2CH2OH | 2 | NDR-488 | 1664.90 |
| 3 | Formyl-(AzPro-Pro)8-NHCH2CH2OH | 3 | NDR-363 | 1650.87 |
| 4 | H-(AzPro-Pro)8-NHCH2CH2OH | 4 | NDR-16MER | 1622.86 |
| 5 | Formyl-(AzPro-Pro)7-NHCH2CH2OH | 5 | NDR-14MER | 1455.65 |
| 6 | Formyl-(AzPro-Pro)6-NHCH2CH2OH | 6 | NDR-12MER | 1260.43 |
| 7 | Formyl-(AzPro-Pro)5-NHCH2CH2OH | 7 | NDR-10MER | 1065.21 |
| 8 | Cbz-(AzPro-Pro)8-OtBu | 8 | NDR-350 | 1770.03 |
| 9 | H-(Pro-AzPro)8-H | 9 | NDR-489 | 1533.77 |
| 10 | Boc-(Pro-AzPro)8-Cbz | 10 | NDR-409 | 1770.03 |
| 11 | Cbz-(AzPro-Pro)16-OtBu | 11 | NDR-353 | 3331.81 |
| 12 | Boc-(Pro-AzPro)8-OCOMe | 12 | NDR-477 | 1693.93 |
| 13 | Cbz-(AzPro-Pro)8-(N-Methyl AzPro) | 13 | NDR-354 | 1782.05 |
| 14 | Cbz-(AzPro-Pro)8-OH | 14 | NDR-451 | 1713.93 |
| 15 | HCl.H-(AzPro-Pro)8-OtBu | 15 | NDR-452 | 1672.36 |
| 16 | HCl. H-(Pro-AzPro)8-Cbz | 16 | NDR-478 | 1706.37 |
| 17 | Boc-(Pro-AzPro)8-H.HCl | 17 | NDR-479 | 1770.03 |
| 18 | Cbz-(AzPro-Pro)4-OtBu | 18 | NDR-347 | 989.14 |
| 19 | Cbz-(AzPro-Pro)4-OH | 19 | NDR-348 | 933.04 |
| 20 | H-(AzPro-Pro)4-OtBu | 20 | NDR-349 | 855.01 |
| 21 | Boc-(Pro-AzPro)4-Cbz | 21 | NDR-403 | 989.14 |
| 22 | Boc-(Pro-AzPro)4-H | 22 | NDR-375 | 855.01 |
| 23 | HCl.H-(Pro-AzPro)4-Cbz | 23 | NDR-476 | 925.49 |
| 24 | Formyl-L-Val-Gly-L-Ala-D-Leu-L-Ala-D-Val-L-Val-D-Val-L-Trp-D-Leu-L-Trp-DLeu-L-Trp-D-Leu-L-Trp-NHCH2CH2OH (Gramicidin A) | 24 | gA | 1882.29 |
Initially, we synthesized the azaproline peptide (Scheme-1) starting from Bochydrazine (25) which was acylated by carbobenzoxychloride (Cbz-Cl) to afford Boc-NH-NH-Cbz (26) with an excellent yield. Then the compound 26 was reacted with NaH in DMF and subsequent treatment by 1,3-dibromopropane furnished Boc-AzPro-OBzl (27). Boc group was deprotected by using 20% trifluoroacetic acid in dichloromethane to give Cbz-N,N’-propylhydrazine (28) quantitatively, which was activated with triphosgene at −20 °C in tetrahydrofuran (THF), and then treated with H-Pro-OtBu to give the Cbz-AzPro-Pro-OtBu dipeptide (29) in good yield. Removal of Cbz by hydrogenation gave H-AzPro-Pro-OtBu (30), which was acylated with Fmoc-Cl in presence of a catalytic amount of DMAP to afford Fmoc-AzPro-Pro-OtBu (31); then OtBu ester group was deprotected using 30% trifluroacitic acid in dichloromethane furnished Fmoc-AzPro-Pro-OH (32) acid quantitatively and then we initiated solid phase peptide synthesis for making AzPro-Pro- oligomers. Fmoc-AzPro-Pro-OH (32) was loaded into 2-chlorotrityl resin in DCM by using DIEA (see supplementary material S1.4, page S6). Then N-terminal Fmoc group was deprotected using 20% piperidine in DMF, but α-dehydrogenated azaproline was observed instead, which was further conformed by treating compound 31 with 20% piperidine in DMF, offered exclusively eliminated product (see supplementary material S1.4, page S6). The free azaproline peptides undergo α-elimination to give pyrazolidine derivatives even without any base, this spontaneous elimination was examined in all unprotected azaproline peptides.
Scheme 1.

Synthesis of protected Fmoc-AzPro-Pro-OtBu dipeptide.
Finally, we chose the other option, solution phase synthesis, dipeptide 29 was made into two parts; first, the OtBu ester group was deprotected with trifluoroacetic acidto obtain Cbz-AzPro-Pro-OH (34) quantitatively. Removal of Cbz by hydrogenation of a second sample gave H-AzPro-Pro-OtBu (30). Cbz-(AzPro-Pro)2-OtBu (35) tetrapeptide was prepared by using HATU peptide coupling protocol (Scheme-2). Using a similar synthetic strategy, Cbz-(AzPro-Pro)4-OtBu octamer (18), Cbz-(AzPro-Pro)8-OtBu hexadecamer (10), and 32-mer Cbz-(AzPro-Pro)16-OtBu (11) were prepared.
Scheme 2.

Solution phase synthesis of Cbz-(AzPro-Pro)n-OtBu oligomeric peptides.
Peptide Cbz-(AzPro-Pro)8-OtBu hexadecamer (10), the Cbz protecting group was deprotected by catalytic hydrogenation with masking of the deprotected azaproline with HCl. Further formylated used formic acid and acetic anhydride. Then N-terminal proline-OtBu was deprotected using 4N HCl in 1,4-dioxane at 50° C; in addition to the cleavage of the OtBu ester, the formyl group also cleaved and dimer-shortened fragments were generated as shown in Scheme 3. We further formylated the mixture of dimer-shortened peptides furnished mixture of compounds 39, 40, 41 and 42 (see the detailed LCMS data in supporting supplementary material S8, page S32), followed by coupling with ethanolamine in the presence of EDCL to generate dimer-fragmented gramicidin A analogs (see the detailed LCMS data supplementary material S8, page S33). All of them were well separable by HPLC and isolated major fractions are decamer, dodecamer, tetradecamer and hexadecamer (7, 6, 5 and 3 respectively). No dimer shortened fragments were generated using 4N HCl in 1,4-dioxane at 22° C, but the reaction was rather slow. Selectively OtBu ester was cleaved quantitatively under 50% TFA/DCM conditions; insertion of ethanolamine produces a formylated gA analog as a single compound as shown in Scheme 4.
Scheme 3.

Generation of a set of formyl-(AzPro-Pro)-oligomers by limited acid hydrolysis in 4N HCl/dioxane followed by addition of C–terminal ethanolamine to form gA peptidomimetics of different lengths (10, 12, 14 & 16).
Scheme 4.

Synthesis of gramicidin A analogous peptides.
These formyl protected gA peptidomimetics of different lengths (10, 12, 14 & 16) were unstable, undergoing auto deformylation with time and labile with temperature also. The peptides are stable upon replacing formyl with acetyl or carboxybenzyl at the N-terminus of AzPro residues. Peptide Cbz-(AzPro-Pro)8-OtBu hexadecamer (17) had its Cbz protecting group cleaved under catalytic hydrogenation and further acetylated using acetic anhydride to furnish the acetylated hexadecamer OtBu ester (42). C-terminal proline-OtBu was deprotected quantitatively with 50% TFA/DCM followed by coupling with ethanolamine using the standard HATU method gave gA analogous peptide 2 in high yield (Scheme 4). Deprotection of C-terminal proline-OtBu ester of peptide 10 generated the hexadecamer free acid 14, followed by insertion of ethanolamine produce the carboxybenzylated gA peptide analog 1 and coupling with N-methyl AzPro generated peptide 13 as represented in Scheme 4.
The other sequence where proline is present at the N-terminus and azaproline at the C-terminus of β6.3-helical peptdiomimetics is represented in Scheme 5. Due to resonance, azaproline amine in H-AzPro-Cbz (45) is a poorer nucleophile than proline, We attempted many methods (see supplementary material S1.1, page No. S3) to couple 45 with Boc-Pro-OH. Finally, Dipeptide 46 was obtained in excellent yield by HATU coupling of Boc-Pro-OH and Cbz-AzPro-H (45) which was obtained by selective Boc deprotection using 50% TFA/DCM. Then dipeptide 46 was divided into two parts, the Boc group was deprotected with TFA to obtain TFA.H-Pro-AzPro-Cbz (48) quantitatively, removal of Cbz by hydrogenation from the other part gave Boc-Pro-AzPro-H. We were unsuccessful in connecting two fragments with a urea bond using 1,1’-carbonyldiimidazole. After several attempts (see supplementary material S1.2, page No. S4), we succeeded in making Boc-(Pro-AzPro)2-Cbz tetrapeptide (49) using triphosgene and N-methylmorpholine (NMM) in THF (Scheme 5). Using a similar synthetic strategy, Boc-(Pro-AzPro)4-Cbz octamer (21), hexadecamer 10 (Boc-(Pro-AzPro)8-Cbz) and unprotected hexadecamer 9 were generated.
Scheme 5.

Synthesis of Boc-(Pro-Aza)8-Cbz gramicidin analogous peptide.
Circular Dichroism studies:
The conformational states of gA and Pro-AzPro peptides were studied by circular dichroism spectroscopic using a JASCO CD spectrometer. All samples were prepared in PBS buffer and methanol at a concentration of 100 μM and a quartz cuvette with a 0.1 mm path length was used for the wavelength scans. The CD spectra were recorded from 190 to 260 nm in MeOH and 200–260 nm range in PBS buffer at 25 °C, with a spectrometer bandwidth of 1 nm and average of five scans.
The CD spectrum of any molecule under given conditions provides the sum of the spectra of all the individual conformers present in solution weighted by their relative abundance. The CD spectrum of purified gramicidin A (gA) in methanol displays two strong negative absorption bands at 210 nm and 228 nm as in shown in Figure 9a. These absorption bands can be attributed to the various forms of intertwined double helical species present in this solvent [58, 59] and a distinctive positive band at 223 nm thought to be monomeric state [60–62]. Figure 9a represents the CD spectra of all 16-mer gA analogous peptides; (1-3, 8, and 10) show positive absorption band attributed at 223 (θ max at 223 nm) that are similar to that of gA monomeric n→π∗ transition (θ max at 223 nm) that is indicative of structural persistence. Indeed the full length 32-mer (11) shows a similar pattern. CD spectra of the series of dimer-shortened fragmented peptide analogs are plotted in Figure 9b and shows θmax at 221 nm for hexadecamer 4, 218 nm for tetradecamer 5, 216 nm for dodecamer and 214 nm for decamer which clearly supports structural deviation from beta helix in 16mer (4) to reverse turn in the 10mer (7). Figure 9c represents the comparison of CD spectra of a series of 32mer to dipeptides Cbz-(AzPro-Pro)n-OtBu (n = 16(11), 8 (8), 4(18), 2(29) 1(35)), reveals that 32mer and 16mer adopt a β6.3-helical structure in methanol, whereas the octamer is blue shifted to 214 nm while the tetrapeptide and dipeptide CD spectra (θmax at 211 nm) support a reverse-turn conformation. Similar CD pattern (Figure 9d) were observed in the Boc-(Pro-AzPro)n-Cbz (n = 8(10), 4(21), 2(49), and 1(46)) series as well.
Figure 9:

CD spectrum comparision of gA and its analogous peptides in methanol (100 μM) at 25 °C. a) gramicidin A (gA) and its analogous peptides (1-3, 8, and 10) hexadecamers and 11 is the full-length 32mer. b) gA, its analog 3 and dimer shortened peptides (39–41). c) gA and Cbz-(AzPro-Pro)n-OtBu (n = 16(11), 8 (8), 4(18), 2(29) 1(35)) analogous peptides. d) gA and Boc-(AzPro-Pro)n-Cbz (n = 8 (10), 4(21), 2(49), 1(46)) analogous peptides.
Figure 10a represents the CD spectrum of gA (dissolved in 1:1 of PBS buffer in ethanol) and peptide analogous (1-3, 8, and 10) including 32-mer (11) in PBS buffer shows distinctive positive absorption band attributed at 223 (θ max at 223 nm) that are similar to that of gA monomeric n→π∗ transition (θ max at 223 nm) which is indicative of structural persistence. CD spectra of the series of dimer-shortened fragmented peptide analogs in PBS buffer are plotted in Figure 10b shows θmax at 221 nm for hexadecamer (3), 218 nm for tetradecamer (5), 216 nm for dodecamer (6) and 214 nm for decamer (7) which clearly supports structural deviation from beta helix in 16mer (4) to reverse turn in the 10-mer (7).
Figure 10:

Comparison of far-UV CD spectrum of gramicidin A (gA) and its analogous peptides in PBS buffer (100 μM) at 25 °C . a) gramicidin A (gA) and its analogous hexadecamers (1-3) and 11 is the full-length 32mer. b) gA, 2, 3, 11 and dimer shortened peptides (5–7).
Hemolysis of erythrocytes (red blood cells, RBCs) –
One limitation restricting the use of gramicidin A as an antibiotic is its toxicity with human membranes as exemplified by lysis of erythrocytes. The peptidomimetic analogs were tested for RBC hemolysis by direct comparison with gramicidin A (Figure 11). As shown, the peptidomimetic analogs demonstrated little, if any, dose-dependent hemolysis as compared with authentic gramicidin A (gA) with a reported HC50 = 5 μM [63]. Mao et al. have demonstrated that a gramicidin A analog having lactam-bridged between the side chains of the fourth lysine and tenth glutamic acid residues stabilize the β6.3-helical conformation while significantly eliminating most antibacterial activity as well as RBC hemolysis [41]. This suggests that the stabilized helix may not be the bioactive conformation, rationalizing our results.
Figure 11:

RBC hemolysis comparison: a) gA and its hexadecameric analogous peptides. b) gA and shorter peptide analogs.
Binding assays of peptides on immobilized liposomes by surface plasmon resonance (SPR) spectroscopy:
We used surface plasmon resonance (Reichert) to (i) obtain more information on the modes of binding of peptides with liposomes, and ii) to determine the binding kinetics and affinity of liposomes with analogous peptides. SPR binding experiments were conducted using a Reichart SR7100DC SPR instrument. HEPES buffer (10 mM HEPES, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid; 100 mM KCl at pH 7.4) was used as a running buffer for the SPR assay. Carboxyl groups on a dextran-coated chip surface activated with N-hydroxysuccinimide, followed by covalent bonding of the ligands to the chip surface via amide linkages with 5-hydroxy sphingosine lipid and excess activated carboxyls were blocked with ethanolamine. Reference surface prepared in the same manner, except that all carboxyls are blocked and no ligand is added. A liposome solution (5 mg·mL−1) in PBS at a flow rate of 25 μL·min−1 was used to form anti-LPS-liposome complexes on the chip surface and coverage of the chip surface by the liposome complexes was assessed by a protein standard (BSA). During analysis, peptides in PBS solutions were passed over the immobilized liposome complexes at a rate of 25 μL min−1 and the final concentration of bound ligand, expressed in response units (RU), which is calculated by subtracting the reference RU from the ligand RU. The chip surface is regenerated by removal of analyte with a 20 mM CHAP solution followed by immobilizing with lipososomes. The changes of SPR signal as a function of time deliberates the accumulation of adsorbed peptide on the immobilized liposome. The maximum RU with each analyte demonstrates the level of interaction, and reflects comparative binding affinity.
All hexadecamer peptides (1, 2, 8 and 10) were introduced to the POPC lipid systems at 100 μM concentration and showed some binding response. We did not succeed with gramicidin A, because it was solubilised in DMSO and precipitated out of solution upon diluting with assay buffer. The sensorgrams of each peptide (1, 2, 8 and 10) binding at difference concentrations from 1000, 500, 250, 100, 50, 25 and 10 μM to the POPC lipid system are shown in Figure 12, which indicate that the initial binding of each peptide to the model membrane is rapid and the response was also proportional to peptide concentration. At above 100 μM concentration, the peptides dissociated very little after the introduction of peptides (Figure 12), suggesting that these peptides have just associated with lipid vesicles instead of rupturing the liposomes. The maximum response of each concentration plotted with peptide concentration to determine the equilibrium dissociation constant (KD) and Rmax for analogous peptides 1, 2, 8 and 10 as shown in Figure 12.
Figure 12:

Surface-plasmon resonance sensorgrams of the interaction of liposomes with peptide analogs. Dilution-series binding and dose-response plot of the interaction with POPC liposomes. a), b) Peptide 1; c), d) Peptide 2; e), f) Peptide 8 and g), h) Peptide 10.
Peptide 1 and 10 showed higher affinity binding with a KD of 3.7 × 10−4 M, 4.4 × 10−4 M respectively. A weaker affinity was observed for peptide 2 (6.1 × 10−4) and 8 (2.58 × 10−3). We therefore conclude that these peptides have a low affinity for POPC liposomes. A weaker affinity interaction could be due to the peptide not making a channels within the liposome membrane.
Liposomal assay of channel formation -
Liposome preparation and efflux was followed according to Saito et al. [64] 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids) composition of liposomes was used for all experiments. 5 mg of lipid was dissolved in chloroform (1 mL), and evaporated chloroform under N2 gas, and residual chloroform was removed using freeze dryer for 2 h, dissolved in 0.5 mL diethyl ether and hydrated in 0.5 mL HEPES buffer (10 mM HEPES, 100 mM KCl, pH 7.4, containing carboxyfluorescein (CF) - 50 mM). The azeotropic mixture was sonicated for 30s in a bath sonicator (1,600 W) for five cycles and removed residual ether under reduced pressure. Liposomes were downsized using 200 nm polycarbonate filters (Nucleopore, Pleasanton, California) by passes 11 times in a hand-held, small-scale extruder (Avestin Europe GmbH, Mannheim, Germany) and separated from unincorporated fluorescein compounds by Sephacryl S-300 (Sigma) chromatography. Liposome size was determined by dynamic light scattering method using an N4MD submicron particle analyser (Coulter, Hialeaha, Florida) and CF vesicles were used at 2.2 × 1010 vesicles per ml.
Using a monodisperse preparation of liposomes, we undertook the quantitative study of gA-peptidomimetic pore formation. In these liposomes the CF at quenching concentrations; dequenching can be resulted by release of CF from liposome vesicles of this size upon rupturing. The kinetics of pore activation could be monitored experimentally by the dequenching rate. Gramicidin A and a series of (AzPro-Pro)n peptide analogs, were tested to determine the impact of channel length on release of CF. In all the peptides and gA were tested at 20 μM concentration and peptide solution was mixed with vesicle for 3 min of equilibration. Figure 13a represents the POPC liposome leakage comparison of gramicidin A with hexadecamer analogs at 20 μM concentration. Data reveals that gA causes 100% leakage with POPC liposomes; however, none of the analogs damaged POPC vesicles even at higher concentrations (Figure 13b). No liposome leakage was observed with the shortened peptides as in Figure 13c. The DOPC liposome leakage comparison of gramicidin A with hexadecamer analogs at 20 μM concentration of peptide analogs are plotted in Figure 13d. gA causes 100% leakage with DOPC as well; however, none of the hexadecamer analogous peptides creates channels to liposome except peptide 1 where only 13% liposome leakage (Figure 13d) was observed.
Figure 13:

Gramicidin A and its analogous peptide-caused lysis of a), b), c) POPC, and d) DOPC liposomes.
Antibacterial data analysis:
The assessment of the antibacterial activity of azaproline-containing peptides and gA was performed using a standard minimal inhibitory concentration (MIC) test against Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, and Staphylococcus aureu. CO-ADD (The Community for Antimicrobial Drug Discovery), The University of Queensland (Australia) screened the antimicrobial activity. Colistin and vancomycin were used as positive inhibitor standards for gram-negative and gram-positive bacteria, respectively. All bacterial strain were cultured in cation-adjusted Muller Hinton broth (CAMHB) at 37 °C for 12 h. Cultured samples were diluted 40-fold in fresh broth and incubated at 37 °C for 1.5–3 h. The resultant mid-log phase cultures were diluted (CFU/mL mesured by OD600), then 45 μL was added to each well of the compound-containing plates, giving a cell density of 5×105 CFU/mL and a final compound concentration of 32 μg/mL.. Then all the plates were left standing at 37° C for 18 h.
Growth inhibition was determined based on the amount of light scattered by the culture (by reading the optical density at 600 nm, OD600), using a Tecan M1000 Pro monochromator plate reader. Bacterial growth inhibition percentage was calculated for each well, using the negative control (media only) and positive control (bacteria without inhibitors) on the same plate as references. Z-score determine the significance of the inhibition values, which was calculated using the average and standard deviation of the sample wells (no controls) on the same plate. Samples with inhibition value above 80% and abs (Z-Score) above 2.5 were classed as actives and 20–80% as partial actives.
Initially, the compound Gramicidin complex (Cat BIA-G1592–1, bioaustralis fine chemicals) was subjected to whole cell growth inhibition assay as a 12-point dose response to determine the minimum inhibitory concentration (MIC) against. Two plate types were compared, non-binding surface (NBS) 96-well plates and polystyrene (PS) 96-well plates, assay was performed in duplicates (n=2). The compound tested was found to be active only against Staphylococcus aureus (MRSA) with MIC 50–25 μg/mL in polystyrene (PS) plates (Table 2). The compound was not active in the non- binding surface plates (NBS).
Table 2:
Minimum inhibition concentrations (MIC) at 50 μg/mL of reference compounds (Vancomycin, Colistin and Gramicidin) against four bacterial pathogens.
| Compound name | E. coli | K. pneumoniae | A. baumannii | S. aureus | ||||
|---|---|---|---|---|---|---|---|---|
| FDA Control | MDR | Type | MRSA | |||||
| NBS plate | PS plate | NBS plate | PS plate | NBS plate | PS plate | NBS plate | PS plate | |
| Vancomycin | >32 | >32 | >32 | >32 | >32 | >32 | 2 | 2 |
| Vancomycin | >32 | >32 | >32 | >32 | >32 | >32 | 2 | 2 |
| Colistin | 0.016 | 0.125 | 0.06 | 0.25 | 0.125 | 1 | >32 | >32 |
| Colistin | 0.016 | 0.125 | 0.06 | 0.25 | 0.06 | 1 | >32 | >32 |
| Gramicidin | >50 | >50 | >50 | >50 | >50 | >50 | >50 | 50 |
| Gramicidin | >50 | >50 | >50 | >50 | >50 | >50 | >50 | 25 |
Later we have isolated gramicidin A from the complex by HPCL 33–95% acetonitrile gradient over 30 minutes and characterized it by LCMS. The results, listed in Figure 14, show activity for purified gA (17 μM) and its peptide analogs at 32 μg/mL which is 15–17 μM concentration depending on the molecular weight of hexadecamers. Data suggest that gA is much less active against S. aureus is 2.5 μM reported by Wang et al. [40, 65]. Comparing the activity of Pro-AzPro peptides with earlier literature gA activity reveals that there is tremendous loss in activity of the analogs against S. aureus.
Figure 14:

Inhibition percentage of gA (17 μM) and its analogs against five bacterial pathogens.
Data reveals that gA peptide analogue 1 bearing Cbz group at N-terminus inhibits 38% of A. baumannii growth selectively, whereas 1, 9, 13 and 22 are selective with S. aureus. However, the full length 32mer (11) and peptides 2, 4 and 14 display a considerable loss of activity. Hexadecameric peptides 16 and 17 as amine hydrochloride salts showed some activity with both S. aureus and A. baumannii and surprisingly, octamers 18, 19, 22 and 23 showed little inhibition.
Shortening the peptide length of Pro-AzPro oligomers (5, 6, 14 and 20) from hexadecamer in L&D-helical structure largely abolishes all activity with any of the five bacterial strains. The exact process of the membrane disruption is not completely understood [66]. This study demonstrates the presence of β6.3-helical structure alone in the gA analogs cannot be used for the prediction of potential antimicrobial activity and may indicate a more complicated mechanism.
Antifungal data analysis:
Antifungal activity was measured by a quantitative microspectrophotometric assay. Fungi strains cultured for 3 days on Yeast Extract-Peptone Dextrose (YPD) agar at 30 °C. Cultured yeast suspension of 1–5 million cells/mL (as determined by OD530) was prepared from five colonies. These stock suspensions were diluted with Yeast Nitrogen Base (YNB) broth to a make final concentration of 2.5 ×103 CFU/mL. Then, 45 μL of the fungi suspension was added to each well of the compound-containing plates, giving a final concentration of 32 μg/mL for the tested samples and incubated plates at 35 °C for 24 h without shaking. Fluconazole was used as a positive fungal inhibitor standard for C. albicans and C. neoformans.
Growth inhibition of C. albicans was determined based on the amount of light scattered by the culture at 530 nm (by reading the optical density at 530 nm, OD530 and C. neoformans growth inhibition was determined by measuring the difference in the amount of light scattered between 600 and 570 nm (OD600–570), after the addition of resazurin (0.001% final concentration) and incubation at 35° C for an additional 2 h. Analysis of data reveals that the peptides inhibit the growth of C. albicans is found to be not more than 10% (Figure 15) including gA. Surprisingly, these peptides enhance C. neoformans growth (Figure 15).
Figure 15:

Inhibition percentage (C. albicans = blue bars) of gramicidin A and its analogous peptides against two fungal pathogens. Note that growth of C. neoformans was enhanced (red bars)
Discussion and Conclusion
In conclusion, the biomimetic syntheses of gA analogs including full length 32-residue channel peptide 11 was successfully demonstrated. Synthetic azaproline (AzPro) allowed generation of several gA analogs having L-Pro-AzPro and AzPro-L-Pro repeating units. The conservation of β6.3-helical character was confirmed for all 16-residue peptides, full length 32-residue peptide (11), and gA using circular dichroism. No ability to lyse liposomes was established suggesting a lack of channel formation, nor did these peptidomimetics hemolyze red blood cells, unlike gramicidin A. SPR binding assay study and liposome leakage experiment showed that analogous peptides just associated with lipid liposomes vesicles instead of rupturing them. Analysis of the antimicrobial activity of the 19 peptides revealed a lowered bactericidal effect for peptide analogs compared with gA. Apparently, modification of the gA with azaproline and proline residue is counterproductive with respect to antibacterial activity even when the β6.3-helical character is preserved. 16-mers having AzPro-Pro repeating unit (1, 8, 41, 13 and 9) showed highest activity, surprisingly drastic loss in activity of the 32-mer (11), perhaps due to a length that was greater than the membranes width. No antifungal activity was observed for any of these analogs including gA. We hypothesize that introduction of lysine residues at the N-terminus region of the peptides should be explored in future development of gA-based peptidomimetic antibiotics.
Experimental Section
1). Chemistry General.
All the reactions were performed in oven-dried apparatus and were stirred using magnetic stirbars. Starting materials, reagents, and solvents were purchased from commercial vendors unless otherwise noted. Chromatography grade ethylacetate, dichloromethane, acetonitrile, and hexanes were obtained from Sigma-Aldrich. Column chromatography was performed on silica gel (100–200 mess) purchased from Sorbent Technologies. TLC was carried out on Analtech 200 microns silica-gel coated plastic-fiber sheets. All reactions were monitored by thin layer chromatography (TLC) carried out on Merck silica-gel plates (0.25 mm thick, 60F254), visualized by using UV (254 nm) or dyes such as ninhydrin, KMnO4, p-anisaldehyde or CAM (ceric ammonium molybdate). High-performance liquid chromatography (HPLC) was carried out on GILSON GX-281 using Waters C18 5μM, 4.6 × 50mm and Waters Prep C18 5μM, 19 × 150mm reverse phase columns. The mobile phases used were A: H2O with 0.05% TFA, B: CH3CN with 0.05% TFA using a solvent gradient of A-B over 30 min with a flow rate of 14.8 mL/min, with detection at 220 and 254 nm UV detectors. Purity assessment and mass spectra (MS) data were obtained using a Hewlett-Packard HPLC/MSD using electrospray ionization (ESI) for detection. 1H and 13C NMR spectra were measured on a Varian 400 MHz NMR instrument. Chemical shifts are expressed in parts per million (ppm) from the residual of nondeuterated solvents as internal standard. (1H NMR: TMS = 0.00 ppm, CDCl3 δ = 7.26 ppm, DMSO-d6 δ = 2.50 ppm, D2O: δ = 4.79 ppm; 13C NMR (APT): TMS δ = 0.00 ppm, CDCl3 δ = 77.16 ppm, DMSO-d6 δ = 39.52 ppm). Coupling constants (J) are given in hertz (Hz). The following abbreviations were used to express the multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; p = pentet; quin = quintet; sep = septet; hept = heptet; m = multiplet; dd = doublet of doublets; dt = doublet of triplet; td = triplet of doublet; m = multiplet; bs = broad singlet. All compounds used for CD, liposome lysis SPR studies and biological assays were >95% pure based on NMR and LC-MS by UV absorbance at 210 nm and 254 nm wavelengths.
2). General Procedure for Azaproline Synthesis:
2a). Synthesis of Boc-NH-NH-Cbz (26):
To a cold solution (0° C) of tert-butyl hydrazinecarboxylate hydrochloride (7.1 g, 53.8 mmol) in and N-methylmorpholine (17.7 mL, 161 mmol) in tetrahydrofuran (100 mL) was added benzyl chloroformate (7.7 g, 53.8 mmol) and slowly brought to room temperature and stirred for 10 h. Tetrahydrofuran was removed under reduced pressure and the resulting viscous solution was diluted with water (40 mL) and thoroughly extracted with ethyl acetate (50×3 mL). The combined organic extracts were washed with 1 N HCl (25 mL), saturated aqueous sodium bicarbonate (NaHCO3) (25 mL) and dried over anhydrous sodium sulphate (Na2SO4) and concentrated to give a residue, which was purified by by silica gel column chromatography (EtOAc:Hexane – 2:5) to give a white solid (13.8 g, yield: 96%). 1H NMR (400 MHz, CDCl3) δ ppm: 7.44 – 7.27 (m, 5 H), 6.48 (bs, 1 H), 6.29 (bs, 1 H), 5.18 (s, 1H), 1.46 (bs, 9 H). MS (ESI): found: [M + Na]+, 289.2.
2b). Boc-AzPro-Cbz (24):
55% Sodium hydride (2.1 g, 52.6 mmol) was taken into 250 mL round bottom flask under nitrogen atmosphere and dry hexane (25 mL) was added and stirred for 5 min, then removed hexane with syringe. Repeated this process twice to activate the sodium hydride from paraffin. Then Boc-NH-NH-Cbz (7 g, 26.3 mmol) in dry N,N-dimethylformamide (75 mL) was added to the activated sodium hydride and stirred at 0° C for 10 min. Then 1,3-dibromopropane (2.65 mL, 26.3 mmol) was added dropwise over 10 min, slowly brought to room temperature and stirred for 3 h. Reaction was quenched with 1N HCl and N,N-dimethylformamide was removed under reduced pressure and the resulting viscous solution was diluted with water (20 mL) and thoroughly extracted with ethyl acetate (40×3 mL), then dried over anhydrous sodium sulphate (Na2SO4) and concentrated to give a residue, which was purified by by silica gel column chromatography (EtOAc:Hexane – 3:5) as a viscous oil (7.13 g, yield: 89%). 1H NMR (400 MHz, CDCl3) δ ppm: 7.42 – 7.27 (m, 5 H), 5.26 (d, J = 11.9 Hz, 1 H), 5.11 (d, J = 11.8 Hz, 1 H), 4.02 – 3.82 (m, 2 H), 3.34 – 3.26 (m, 1 H), 3.26 – 3.09 (m, 1 H), 2.04 (quin, J = 7.1 Hz, 2 H), 1.42 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ ppm: 136.2, 128.4, 128.0, 127.9, 81.5, 77.3, 77.0, 76.7, 67.7, 46.7, 46.2, 28.1, 25.6. MS (ESI): found: [M + Na]+, 329.3.
2c). TFA-AzPro-Cbz (28):
Boc-AzPro-Cbz (5 g, 16.3 mmol) was dissolved in 15% trifluoroacetic acid in CH2Cl2 (50 mL) and stirred at 0° C for 3 hours. Solvent was removed by rotary evaporation, and the residue washed with hexane (3 × 10 mL) for removal of a trace amount of trifluoroacetic acid. The residue was dried in vacuo over a KOH trap to obtain the desired Boc deprotected amine salt as a viscous oil (5.56 g, yield: 100%) and used without further purification. MS (ESI): found: [M + Na]+, 229.3
2d). Cbz-AzPro-Pro-OtBu (29):
To a cold solution (−30 °C) of Boc-AzPro-H.TFA (4 g, 12.6 mmol) in dry tetrahydrofuran (THF) (50 mL) was added N-methylmorpholine (1.04 mL, 9.3 mmol) and triphosgene (1.3 g, 4.4 mmol) then stirred for 30 min. Then a solution of proline t-butyl ester hydrochloride (2.15 g, 12.6 mmol) in N,N-dimethylformamide (8 mL) was added to the flask followed by N-methylmorpholine (1.04 mL, 9.3 mmol) and the reaction mixture was slowly brought to room temperature and stirred till the starting material was consumed completely. N,N-dimethylformamide and tetrhydrofuran were removed under reduced pressure and the resulting viscous solution was diluted with water (10 mL) and thoroughly extracted with ethyl acetate (20×3 mL). The combined organic extracts were washed with 1 N HCl (15 mL), saturated aqueous sodium bicarbonate (NaHCO3) (15 mL) and dried over anhydrous sodium sulphate (Na2SO4) and concentrated to give a residue, which was purified by silica gel column chromatography (EtOAc : Hexane – 2:3) as a viscous oil (4.1 g, yield: 82%). 1H NMR (400 MHz, CDCl3) δ ppm: 7.39 – 7.28 (m, 5 H), 5.22 (d, J = 12.0 Hz, 1 H), 5.15 (d, J = 12.0 Hz, 1 H), 4.48 – 4.40 (m, 1 H), 3.81 – 3.71 (m, 1 H), 3.71 – 3.60 (m, 2 H), 3.51 (td, J = 6.4, 10.4 Hz, 2 H), 3.41 – 3.23 (m, 1 H), 2.20 – 2.09 (m, 1 H), 2.04 (quin, J = 7.2 Hz, 2 H), 1.95 – 1.76 (m, 3 H), 1.44 (s, 9 H). MS (ESI): found: [M + Na]+, 426.4.
3). HATU peptide coupling protocol for azaproline coupling:
To a solution (0° C) of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (1.3 mmol) in N,N-dimethylformamide (10 mL), N,N-diisopropylethylamine (3 mmol) was added and stirred for 15 min. Then added azaproline fragment (1 mmol) in N,N-dimethylformamide (4 mL) followed by DIEA (2 equivalents) and stirred it for 7–8 h at 4 °C., and the reaction mixture stirred at room temperature till the starting material was consumed completely. N,N-dimethylformamide was removed under reduced pressure and the resulting viscous solution was diluted with water (5 mL) and thoroughly extracted with ethyl acetate (15 mL). The combined organic extracts were washed with 1 N HCl (5 mL), saturated aqueous sodium bicarbonate (NaHCO3) (5 mL) and dried over anhydrous sodium sulphate (Na2SO4) and concentrated to give a residue, which was purified by silica gel (100–200 mesh) flash column chromatograph.
4). General procedure for triphosgene coupling protocol:
To a cold solution (−30 °C) of Boc-AzPro-H (1.0 mmol) in dry tetrahydrofuran (THF) (5 mL) N-methylmorpholine (1.2 mmol) and triphosgene (0.35 mmol) were added and stirred for 30 min. Then a solution of amino acid ester hydrochloride (1 mmol) in N,N-dimethylformamide (2 mL) flask was added, followed by N-methylmorpholine (2.2 mmol) and the reaction mixture was slowly brought to room temperature and stirred till the starting material was consumed completely. N,N-dimethylformamide and dichloromethane were removed under reduced pressure and the resulting viscous solution was diluted with water (5 mL) and thoroughly extracted with ethyl acetate (15 mL). The combined organic extracts were washed with 1 N HCl (5 mL), saturated aqueous sodium bicarbonate (NaHCO3) (5 mL) and dried over anhydrous sodium sulphate (Na2SO4) and concentrated to give a residue, which was purified by silica gel (100–200 mesh) flash column chromatograph.
5). General Procedure - Acidic cleavage of N-tert-butyloxycarbonyl (Boc) using trifluoroacetic acid:
The protected peptide (1.0 mmol) was dissolved in 15% trifluoroacetic acid in CH2Cl2 (5 mL) and stirred at 0° C for 3 hours. Solvent was removed by rotary evaporation and the residue washed with hexane (3 × 10 mL) for removal of a trace amount of trifluoroacetic acid. The residue was dried in vacuo over a KOH trap and used without further purification.
6). General Procedure - Acidic cleavage of OtButylester (OtBu) protecting group using trifluoroacetic acid:
The protected peptide (1.0 mmol) was dissolved in 30% trifluoroacetic acid in CH2Cl2 (5 mL) and stirred at 0° C for 3 hours. Solvent was removed by rotary evaporation and the residue washed with hexane and evaporated (3 × 10 mL) for removal of trace amounts of trifluoroacetic acid. The residue was dried in vacuo over a KOH trap and used without further purification.
Supplementary Material
Highlights.
Biomimetic of gramacidin A (gA) analogs including full length 32-residue channel peptide was successfully demonstrated.
Molecular dynamics simulations and Circular Dichroism studies confirmed the structural persistence of gA and designed peptide mimetics.
Synthesised peptide analogs did not hemolyze red blood cells, unlike gA.
Analysis of the antimicrobial activity of the 19 non-hemolytic peptides revealed, 16-mers having AzPro-Pro repeating unit (1, 8, 41, 13 and 9) showed highest activity.
Acknowledgements
The authors thank the Division of Biology and Biological Sciences at Washington University for a graduate fellowship (SS), the Washington University Career Center for an Undergradate Summer Internship Award (SS), a gift from Suzanne and Garland Marshall to the Department of Chemistry to support undergraduate research, and the Department of Biochemistry and Molecular Biophysics, WUSM for research support. Antimicrobial screening was performed by CO-ADD (The Community for Antimicrobial Drug Discovery), funded by the Wellcome Trust (UK) and The University of Queensland (Australia). We are grateful to Prof. Carl Frieden for providing access to his CD facility.
Abbreviation
- AzPro
azaproline
- MD
molecular dynamic
- MMFF
Merck molecular force field
- NVT
constant temperature, constant volume
- RMSD
root-mean-square deviation
- ADME
absorption, distribution, metabolism, and excretion
- MIC
minimal inhibitory concentration
- NMR
nuclear magnetic resonance spectroscopy
- PDB
protein data bank
- Cs
Caesium
- SDS
Sodium dodecyl sulfate
- DIEA
diisopropylethylamine
- Boc
tert-butyloxycarbonyl
- Cbz-Cl
carbobenzoxychloride
- Cbz
carboxybenzyl
- Fmoc-Cl
chloroformic acid 9H-fluoren-9-ylmethyl ester
- DMAP
4-dimethylaminopyridine
- NaH
Sodium hydride
- DMF
N,Ndimethylformamide
- OtBu
ortho-tert-butyl
- LiOH
lithium hydroxide
- MeOH
methanol
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- DCM
dichloromethane
- EtOAc
ethylacetate
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- BSA
bovine serum albumin
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- KCl
Potassium chloride
- HCl
hydrochloric acid
- TFA
trifluoroacetic acid
- HPLC
high performance liquid chromatography
- SPR
surface plasmon resonance
Footnotes
Supplementary Material
Detailed experimental procedures, purity and spectral data of compounds, PDB coordinates in machine readable format for computational models, PDB reference IDs of gramicidin A, RBC hemolysis and antimicrobial activity data.
Predicted Models: The PDB coordinates of the predicted helical structure Ac-(Pro-AzPro)14-COOMe (30 mer) and Ac-(Pro-AzPro)8-Ac (16 mer-CaCl2) complex in water. “Authors will release the atomic coordinates upon article publication.”
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cabezas E, Satterthwait AC, The hydrogen bond mimic approach: solid-phase synthesis of a peptide stabilized as an α-helix with a hydrazone link, J. Am. Chem. Soc, 121 (1999) 3862–3875. [Google Scholar]
- [2].Chapman RN, Dimartino G, Arora PS, A highly stable short α-helix constrained by a main-chain hydrogen-bond surrogate, J. Am. Chem. Soc, 126 (2004) 12252–12253. [DOI] [PubMed] [Google Scholar]
- [3].Stanger HE, Gellman SH, Rules for antiparallel β-sheet design: D-Pro-Gly is superior to L-Asn-Gly for β-hairpin Nucleation, J. Am. Chem. Soc, 120 (1998) 4236–4237. [Google Scholar]
- [4].Schneider JP, Kelly JW, Templates that induce α-helical, β-sheet, and loop conformations, Chem. Rev, 95 (1995) 2169–2187. [Google Scholar]
- [5].Kemp DS, Bowen BR, Muendel CC, Synthesis and conformational analysis of epindolidione-derived peptide models for β-sheet formation, J. Org. Chem, 55 (1990) 4650–4657. [Google Scholar]
- [6].Fisk JD, Gellman SH, A parallel β-sheet model system that folds in water, J. Am. Chem. Soc, 123 (2000) 343–344. [DOI] [PubMed] [Google Scholar]
- [7].Reddy DN, Thirupathi R, Prabhakaran EN, Accessing the disallowed conformations of peptides employing amide-to-imidate modification, Chem. Commun, 47 (2011) 9417–9419. [DOI] [PubMed] [Google Scholar]
- [8].Tumminakatti S, Reddy DN, Prabhakaran EN, Exploring the consequences of a representative “disallowed” conformation of Aib on a 310-helical fold, Biopolymers 104 (2015) 21–36. [DOI] [PubMed] [Google Scholar]
- [9].Ketchem RR, Lee K-C, Huo S, Cross TA, Macromolecular structural elucidation with solid-state NMR-derived orientational constraints, J. Biomol. NMR, 8(1996) 1–14. [DOI] [PubMed] [Google Scholar]
- [10].Ramachandran GN, Chandrasekaran R, Studies on dipeptide conformation and on peptides with sequences of alternating L and D residues with special reference to antibiotic and ion transport peptides, Prog. Peptides Res 2(1972) 195–215. [Google Scholar]
- [11].Ramachandran GN, Chandrasekaran R, Conformation of peptide chains containing both L- & D-residues: Part I - helical structures with alternating L- & D-residues with special reference to the LD-ribbon & the LD-helix, Indian J. Biochem. Biophys, 9 (1972) 1–11. [PubMed] [Google Scholar]
- [12].Bystrov VF, Arseniev AS, Diversity of the gramicidin a spatial structure: two-dimensional 1H NMR study in solution, Tetrahedron, 44 (1988) 925–940. [Google Scholar]
- [13].Chen Y, Tucker A, Wallace BA, Solution structure of a parallel left-handed double-helical gramicidin-A determined by 2D 1H NMR, J. Mol. Biol, 264 (1996) 757–769. [DOI] [PubMed] [Google Scholar]
- [14].Doyle DA, Wallace BA, Shifting the equilibrium mixture of gramicidin double helices toward a single conformation with multivalent cationic salts, Biophys. J, 75 (1998) 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Olczak A, Glowka ML, Szczesio M, Bojarska J, Wawrzak Z, Duax WL, The first crystal structure of a gramicidin complex with sodium: high-resolution study of a nonstoichiometric gramicidin D-NaI complex, Acta Crystallogr. D Biol. Crystallogr, 66 (2010) 874–880. [DOI] [PubMed] [Google Scholar]
- [16].Dubos RJ, Studies on a bactericidal agent extracted from a soil bacillus : ii. Protective effect of the bactericidal agent against experimental pneumococcus infections in mice, J. Exp. Med, 70 (1939) 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dubos RJ, Studies on a bactericidal agent extracted from a soil Bacillus : I. Preparation of the agent. Its activity in vitro, J. Exp. Med, 70 (1939) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dubos RJ, Cattaneo C, Studies on a bactericidal agent extracted from a soil bacillus : iii. preparation and activity of a protein-free fraction, J. Exp. Med, 70 (1939) 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dzikovski BG, Borbat PP, Freed JH, Channel and nonchannel forms of spin-labeled gramicidin in membranes and their equilibria, J. Phys. Chem. B, 115 (2011) 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sarges R, Witkop B, Gramicidin. Vii. The structure of valine- and isoleucine-gramicidin B, J. Am. Chem. Soc, 87 (1965) 2027–2030 [DOI] [PubMed] [Google Scholar]
- [21].Sarges R, Witkop B, Gramicidin A.Vi. The synthesis of valine- and isoleucine-gramicidin A, J. Am. Chem. Soc, 87 (1965) 2020–2027. [DOI] [PubMed] [Google Scholar]
- [22].Sarges R, Witkop B, Gramicidin AV The structure of valine- and isoleucine-gramicidin A, J. Am. Chem. Soc, 87 (1965) 2011–2020. [DOI] [PubMed] [Google Scholar]
- [23].Urry DW, Goodall MC, Glickson JD, Mayers DF, The gramicidin A transmembrane channel: characteristics of head-to-head dimerized (L,D) helices, Proc. Natl. Acad. Sci. U. S. A, 68 (1971) 1907–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cross TA, Arseniev A, Cornell BA, Davis JH, Killian JA, Koeppe RE, Nicholson LK, Separovic F, Wallace BA, Gramicidin channel controversy--revisited, Nat. Struct. Biol, 6 (1999) 610–611; discussion 611–612. [DOI] [PubMed] [Google Scholar]
- [25].Kovacs F, Quine J, Cross TA, Validation of the single-stranded channel conformation of gramicidin A by solid-state NMR, Proc. Natl. Acad. Sci. U. S. A, 96 (1999) 7910–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ketchem RR, Lee KC, Huo S, Cross TA, Macromolecular structural elucidation with solid-state NMR-derived orientational constraints, J. Biomol. NMR, 8 (1996) 1–14. [DOI] [PubMed] [Google Scholar]
- [27].Burkhart BM, Li N, Langs DA, Pangborn WA, Duax WL, The conducting form of gramicidin A is a right-handed double-stranded double helix, Proc. Natl. Acad. Sci. U. S. A, 95 (1998) 12950–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lomize AL, Orekhov V, Arsen’ev AS, Refinement of the spatial structure of the gramicidin A ion channel. Bioorg. Khim 18(1992), 182–200. [PubMed] [Google Scholar]
- [29].Burkhart BM, Gassman RM, Langs DA, Pangborn WA, Duax WL, Heterodimer formation and crystal nucleation of gramicidin D, Biophys. J, 75 (1998) 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chaudhuri A, Haldar S, Sun H, Koeppe RE 2nd, A. Chattopadhyay, Importance of indole N-H hydrogen bonding in the organization and dynamics of gramicidin channels, Biochim. Biophys. Acta, 1838 (2014) 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gu H, Lum K, Kim JH, Greathouse DV, Andersen OS, Koeppe RE, The membrane interface dictates different anchor roles for “inner pair” and “outer pair” tryptophan indole rings in gramicidin A channels, Biochemistry, 50 (2011) 4855–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Haldar S, Chaudhuri A, Gu H, Koeppe RE, Kombrabail M, Krishnamoorthy G, Chattopadhyay A, Membrane organization and dynamics of “inner pair” and “outer pair” tryptophan residues in gramicidin channels, J. Phys. Chem. B, 116 (2012) 11056–11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Urry DW, Alonso-Romanowski S, Venkatachalam CM, Trapane TL, Prasad KU, The source of the dispersity of gramicidin A single-channel conductances. The L X Leu5-gramicidin A analog, Biophys. J, 46 (1984) 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jones TL, Fu R, Nielson F, Cross TA, Busath DD, Gramicidin channels are internally gated, Biophys. J, 98 (2010) 1486–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mo Y, Cross TA, Nerdal W, Structural restraints and heterogeneous orientation of the gramicidin A channel closed state in lipid bilayers, Biophys. J, 86 (2004) 2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yoo J, Cui Q, Membrane-mediated protein-protein interactions and connection to elastic models: a coarse-grained simulation analysis of gramicidin A association, Biophys. J, 104 (2013) 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Basu I, Chattopadhyay A, Mukhopadhyay C, Ion channel stability of Gramicidin A in lipid bilayers: effect of hydrophobic mismatch, Biochim. Biophys. Acta, 1838 (2014) 328–338. [DOI] [PubMed] [Google Scholar]
- [38].Lundbaek JA, Collingwood SA, Ingolfsson HI, Kapoor R, Andersen OS, Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes, Journal of the Royal Society, Interface / the Royal Society, 7 (2010) 373–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liou JW, Hung YJ, Yang CH, Chen YC, The antimicrobial activity of gramicidin A is associated with hydroxyl radical formation, PLoS One, 10 (2015) e0117065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang F, Qin L, Pace CJ, Wong P, Malonis R, Gao J, Solubilized gramicidin A as potential systemic antibiotics, ChemBioChem, 13 (2012) 51–55. [DOI] [PubMed] [Google Scholar]
- [41].Mao J, Kuranaga T, Hamamoto H, Sekimizu K, Inoue M, Rational design, synthesis, and biological evaluation of lactam-bridged gramicidin A analogues: discovery of a low-hemolytic antibacterial peptide, ChemMedChem, 10 (2015) 540–545. [DOI] [PubMed] [Google Scholar]
- [42].Sorochkina AI, Plotnikov EY, Rokitskaya TI, Kovalchuk SI, Kotova EA, Sychev SV, Zorov DB, Antonenko YN, N-terminally glutamate-substituted analogue of gramicidin A as protonophore and selective mitochondrial uncoupler, PLoS One, 7 (2012) e41919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Silachev DN, Khailova LS, Babenko VA, Gulyaev MV, Kovalchuk SI, Zorova LD, Plotnikov EY, Antonenko YN, Zorov DB, Neuroprotective effect of glutamate-substituted analog of gramicidin A is mediated by the uncoupling of mitochondria, Biochim. Biophys. Acta, 1840 (2014) 3434–3442. [DOI] [PubMed] [Google Scholar]
- [44].Gokel GW, Negin S, Synthetic ion channels: from pores to biological applications, Acc. Chem. Res, 46 (2013) 2824–2833. [DOI] [PubMed] [Google Scholar]
- [45].Xin P, Zhu P, Su P, Hou JL, Li ZT, Hydrogen-bonded helical hydrazide oligomers and polymer that mimic the ion transport of gramicidin A, J. Am. Chem. Soc, 136 (2014) 13078–13081. [DOI] [PubMed] [Google Scholar]
- [46].Barboiu M, Le Duc Y, Gilles A, Cazade PA, Michau M, Marie Legrand Y, van der Lee A, Coasne B, Parvizi P, Post J, Fyles T, An artificial primitive mimic of the gramicidin A channel, Nat. Commun, 5 (2014) 4142. [DOI] [PubMed] [Google Scholar]
- [47].Takeuchi Y, Marshall GR, Conformational analysis of reverse-turn constraints by N-methylation and N-hydroxylation of amide bonds in peptides and non-peptide mimetics, J. Am. Chem. Soc, 120 (1998) 5363–5372. [Google Scholar]
- [48].Chalmers DK, Marshall GR, Pro-D-NMe-amino acid and D-Pro-NMe-amino acid - simple, efficient reverse-turn constraints (Vol 117, Pg 5927, 1995) (Additions & Corrections), J. Am. Chem. Soc, 118 (1996) 1579. [Google Scholar]
- [49].Townsley LE, The three-dimensional structure of gramicidin analogs in micellar environments determined using two-dimensional nuclear magnetic resonance spectroscopic techniques. Ph.D. Dissertation, University of Arkansas, Fayetteville, AR, USA, 2000. [Google Scholar]
- [50].Zhang WJ, Berglund A, Kao JL, Couty JP, Gershengorn MC, Marshall GR, Impact of azaproline on amide cis-trans isomerism: conformational analyses and NMR studies of model peptides including TRH analogues, J. Am. Chem. Soc, 125 (2003) 1221–1235. [DOI] [PubMed] [Google Scholar]
- [51].Zhang Y, Malamakal RM, Chenoweth DM, A single stereodynamic center modulates the rate of self-assembly in a biomolecular system. Angew. Chem. Int. Ed. Engl 2015, 54, 10826–10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Calaza MI, Cativiela C, Stereoselective synthesis of quaternary proline analogues, Eur. J. Org. Chem, 2008 (2008) 3427–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Che Y, Marshall GR, Impact of azaproline on peptide conformation, J. Org. Chem, 69 (2004) 9030–9042. [DOI] [PubMed] [Google Scholar]
- [54].Che Y, Marshall GR, Impact of cis-proline analogs on peptide conformation, Biopolymers, 81 (2006) 392–406. [DOI] [PubMed] [Google Scholar]
- [55].Halgren TA, Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94, J. Comput. Chem, 17 (1996) 490–519. [Google Scholar]
- [56].DeLano WL, The PyMOL molecular graphics system, version 1.6, Schrödinger, LLC, 2002. [Google Scholar]
- [57].Andre F, Marraud M, Tsouloufis T, Tzartos SJ, Boussard G, Triphosgene: an efficient carbonylating agent for liquid and solid-phase aza-peptide synthesis. Application to the synthesis of two aza-analogues of the AChR MIR decapeptide, J. Pept. Sci, 3 (1997) 429–441. [DOI] [PubMed] [Google Scholar]
- [58].Veatch WR, Fossel ET, Blout ER, Conformation of gramicidin A, Biochemistry, 13 (1974) 5249–5256. [DOI] [PubMed] [Google Scholar]
- [59].Chen Y, Wallace BA, Solvent effects on the conformation and far UV CD spectra of gramicidin, Biopolymers, 42 (1997) 771–781. [DOI] [PubMed] [Google Scholar]
- [60].Urry DW, Mayers DF, Haider J, Spectroscopic studies on the conformation of gramicidin A’. Evidence for a new helical conformation, Biochemistry, 11 (1972) 487–493. [DOI] [PubMed] [Google Scholar]
- [61].Bouchard M, Benjamin DR, Tito P, Robinson CV, Dobson CM, Solvent effects on the conformation of the transmembrane peptide gramicidin A: insights from electrospray ionization mass spectrometry, Biophys. J, 78 (2000) 1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Killian JA, Gramicidin and gramicidin-lipid interactions, Biochim. Biophys. Acta, 1113 (1992) 391–425. [DOI] [PubMed] [Google Scholar]
- [63].Wang F, Qin L, Pace CJ, Wong P, Malonis R, Gao J, Solubilized gramicidin A as potential systemic antibiotics, ChemBioChem, 13 (2012) 51–55. [DOI] [PubMed] [Google Scholar]
- [64].Saito M, Korsmeyer SJ, Schlesinger PH, BAX-dependent transport of cytochrome c reconstituted in pure liposomes, Nat. Cell Biol, 2 (2000) 553–555. [DOI] [PubMed] [Google Scholar]
- [65].Wang F, Qin L, Wong P, Gao J, Effects of lysine methylation on gramicidin A channel folding in lipid membranes, Biopolymers, 100 (2013) 656–661. [DOI] [PubMed] [Google Scholar]
- [66].Prenner EJ, Lewis RNAH, McElhaney RN, The interaction of the antimicrobial peptide gramicidin S with lipid bilayer model and biological membranes, Biochim. Biophy. Acta - Biomem, 1462 (1999) 201–221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.