

Abstract
Mix-and-inject serial crystallography is an emerging technique that utilizes X-ray free-electron lasers (XFELs) and microcrystalline samples to capture atomically detailed snapshots of biomolecules as they function. Early experiments have yielded exciting results; however, there are limited options to characterize reactions in crystallo in advance of the beamtime. Complementary measurements are needed to identify the best conditions and timescales for observing structural intermediates. Here, we describe the interface of XFEL compatible mixing injectors with rapid freeze-quenching and X-band EPR spectroscopy, permitting characterization of reactions in crystals under the same conditions as an XFEL experiment. We demonstrate this technology by tracking the reaction of azide with microcrystalline myoglobin, using only a fraction of the sample required for a mix-and-inject experiment. This spectroscopic method enables optimization of sample and mixer conditions to maximize the populations of intermediate states, eliminating the guesswork of current mix-and-inject experiments.
Graphical Abstract
Many biological mysteries are hidden in the transient states of enzyme-ligand interactions. These short-lived structures hold valuable information about key functional mechanisms and may offer novel drug targets to combat disease. Structural enzymology would be revolutionized by a technique that routinely captures the atomically detailed structure of these intermediates. The emerging technique of mix-and-inject serial crystallography (MISC) using X-ray free-electron lasers (XFELs) offers the possibility to capture crystal structures of reaction intermediates with atomic detail;1–4 however, further development is necessary to make the technique routine.
Enzymes often maintain their functionality in crystals, presenting the opportunity to measure high-resolution structures of reaction intermediates. However, to resolve an intermediate’s structure, a significant fraction of the enzymes within the crystal must populate that particular state during the measurement. To accomplish this in ligand-initiated reactions, the ligand must be soaked into the crystal on a timescale that is short compared to the lifetime of the intermediate. The large crystals used in traditional crystallography preclude this, as the soaking time is too long. Mix-and-Inject Serial Crystallography overcomes this barrier by exploiting the high brightness of XFELs to acquire data from individual microcrystals. In these tiny crystals, soaking times can be very fast, making it possible to quickly initiate reactions and resolve millisecond-scale intermediates.5
In MISC, a specialized mixing injector initiates the reaction and delivers the reacting sample to the X-rays. Figure 1 shows a simplified schematic of the apparatus. Mixing injectors rely on Gas Dynamic Virtual Nozzles (GDVNs), a staple of XFEL sample delivery, which use a high-speed gas stream to propel a thin jet of liquid into the path of the X-ray beam.6 A mixing injector contains additional components upstream of the GDVN nozzle to rapidly combine microcrystals with a reactant.7–10 The reacting crystals age as they flow toward the nozzle before being jetted into the X-ray beam. Structures are captured at various timepoints during the reaction by changing the delay between the mixing region and intersection with the X-ray probe.
Figure 1.
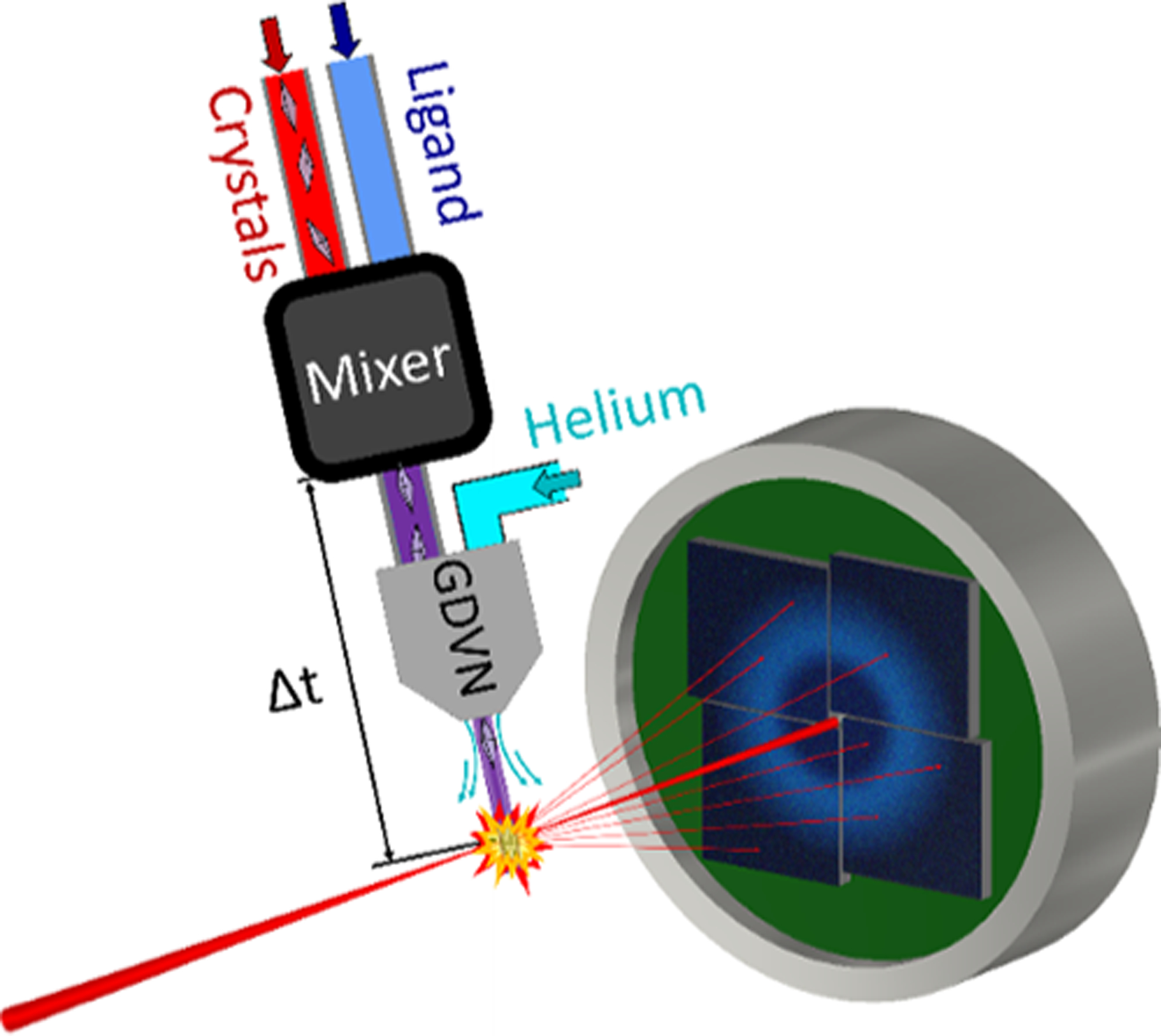
Schematic of a MISC experiment and mixing injector. Microcrystals and an activating ligand are combined in the mixer and subsequently propelled into the X-ray beam by the GDVN. Snapshot diffraction images are collected by the detector.
A typical MISC beamtime may only afford the time to collect a few time-resolved reaction points; therefore, to ensure that the target intermediates are captured, viable conditions must be identified in advance of the beamtime. Variables such as the crystallization conditions, choice of ligand, time delay after mixing, and flow parameters can determine the success or failure of an experiment because they profoundly affect the population of elusive reaction intermediates. However, there are currently limited options to assess these quantities in crystallo. Solution assays provide some guidance for optimizing conditions, but reaction rates in crystals and solutions can vary by several orders of magnitude.11–14 These differences may result from ligand diffusion times, interference from crystal contacts, limited active site accessibility, or buffer chemistry. Kinetics can also vary for different crystallization conditions for the same biomolecule. For example, Olmos et al. found that the build-up of intermediate states differs dramatically between two crystal forms of the same enzyme.3 There is a strong need for advanced characterization of reactions in the same microcrystal system that will be used in the MISC beamtime.
While previous experiments have characterized reactions in microcrystals using a stopped-flow mixer,11–14 the mixing methods and delay mechanisms associated with this technique differ significantly from those of current XFEL mixing injectors. Therefore, timescales may not transfer between these devices. In addition, the turbulence associated with stopped-flow mixing may damage fragile microcrystals, invalidating the results. For optimal success in observing transiently occurring populations, timescales must be characterized in the same crystallization and flow conditions used in the XFEL experiment, using the same mixing injectors. A new approach that combines in-lab spectroscopic measurements with XFEL mixing injectors achieves these goals, ensuring direct transferability of timescales between the spectroscopic and crystallographic measurements.
Here, we present a new method that combines rapid freeze quench (RFQ) electron paramagnetic resonance (EPR) with XFEL mixing injectors to quantify intermediate states in microcrystals under the same conditions as a mix-and-inject experiment. We demonstrate this method by tracking the reaction between myoglobin and azide for both aqueous and crystalline samples using our previously developed mixing injectors.9 For EPR-active systems, this technology allows mix-and-inject parameters to be optimized for the highest structural intermediate occupancy. It also provides a means to independently quantify and characterize intermediate populations, providing a cross-check for mix-and-inject results and giving additional details that are inaccessible via crystallography. Our method can be readily performed with any GDVN based mixing injector, ensuring broad applicability for future time-resolved mix-and-inject experiments at XFELs.
BACKGROUND
Electron paramagnetic resonance is a powerful tool for characterizing paramagnetic states, which often occur in enzymes incorporating transition metal ions. The most common type of EPR measurement, continuous-wave X-band EPR, is well suited for investigating many metal-loenzymes. X-band EPR can identify the type of metal in a metal center, along with its oxidation and spin state. It can also provide insights into the local environment of the metal center.15–17
EPR is an excellent technique to combine with XFEL mixers for in-lab characterization of mix-and-inject conditions. EPR spectrometers are relatively common, and measurement times are on the order of minutes. Each measurement consumes only small amounts of sample (~100–200 μL at concentrations on the μM–mM scale, depending on the sample16,17). In addition, EPR is commonly used in conjunction with Rapid Freeze-Quench (RFQ) to perform time-resolved mixing experiments.
In a standard RFQ experiment, a turbulent mixer combines a sample with a ligand before ejecting the mixture into a bath of liquid cryogen, commonly isopentane. The mixture rapidly freezes, quenching the reaction and locking in intermediate states.18–20 Reactions can be quenched in under 5 ms.19,20 After quenching, the sample is packed into an EPR tube using a chilled rod, and the tubes are then stored in a dewar until measurement. The frozen samples can be measured with a standard EPR setup since cryogenic temperatures are typically required to resolve spin states as a result of small energy differences between them.15
The small, fast jets produced by XFEL mixing injectors are well suited for fast freezing. It is well documented that using smaller jets results in shorter freezing times.21,22 XFEL mixing injectors produce jets that are ~10 μm in diameter, in contrast to typical quench freezing jets that have diameters of a few 100 microns. In addition, the jets from XFEL mixing injectors travel at a speed similar to conventional RFQ jets (~10 m/s), so the flight time from the tip of the nozzle to the cryogen is on the order of 1 ms. Consequently, the total quenching time for a sample produced by mixing injectors should be less than a few milliseconds. This is an order of magnitude faster than the shortest timepoint probed during a mix-and-inject experiment to date3 and should be adequate for most experiments.
Combining mixing injectors with RFQ presents a challenge: past work shows the impracticality of packing powder from fine jets.23 Alternative packing methods do exist and can be divided into three categories: cold centrifugation,23 flow packing,24,25 and quenching on cold metal wheels.26,27 Since the submilli-second quenching offered by the cold metal freezing is not required for the XFEL application, and many labs do not have a cryocompatible centrifuge, we chose to adapt the flow packing method. In this scheme, following quenching, the cryogen is pumped out through a filter at the bottom of an EPR tube, concentrating and trapping the frozen sample. Very fine frozen powders are easily packed with this method.
RFQ FLOW PACKING SYSTEM FOR USE WITH MIXING INJECTORS
We developed a custom setup and procedure to enable the use of mixing injectors with RFQ. This technology adapts the flow packing method to achieve fast quenching and efficient packing with robust temperature control.
Figure 2 shows an overview of the procedure. First, the reacting sample sprayed from a mixing injector is quenched in a small volume of cryogenic isopentane (Figure 2a). The isopentane containing the quenched sample is then transferred to our custom flow packing system, shown in Figure 2b). The flow packing system consists of a metal reservoir connected to a modified EPR tube which terminates in a Teflon filter. The entire flow packing system is maintained at ~160 K by a gas cryostat. After transferring the sample, the flow packing system is sealed off and pressurized by nitrogen gas-cooled in a copper coil to 160 K (Figure 2c). This pumps the isopentane out through the filter, leaving behind the concentrated quenched sample (Figure 2d). Low temperatures are maintained in both the flow packing system and the pressurization gas to ensure that the quenched sample is kept below its glass transition temperature throughout the flow packing process, preventing the reaction from continuing. (See the Supporting Information and Figures S1 and S2 for more details about the cryostat and flow packing system.)
Figure 2.
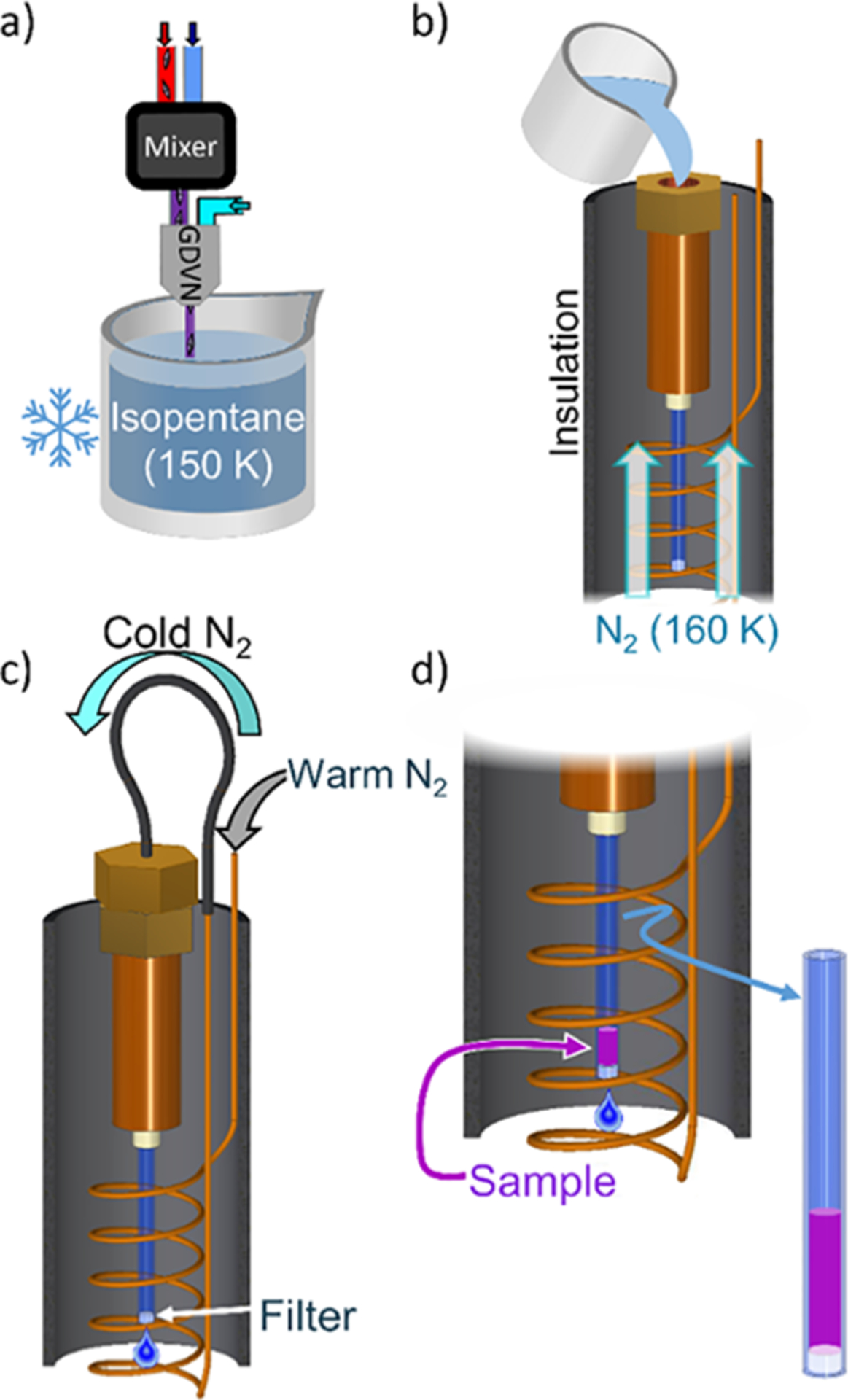
Cartoon of the RFQ process developed for mixing injectors. (a) Reacting sample is quenched in cryogenic isopentane. (b) Quenched sample is transferred to the cold flow packing system. (c) Flow packing system is pressurized by cold nitrogen gas. (d) Sample accumulates behind the filter as isopentane is drained.
METHODS
Sample Prep.
Horse heart myoglobin (Sigma-Aldrich) was batch crystallized as previously described.28 Briefly, solid ammonium sulfate was added to a pH 7.8, 100 mM phosphate buffer containing 60 mg/mL myoglobin, bringing the ammonium sulfate concentration to ~3.7 M. The crystallization protocol was optimized to reduce the free protein in solution. The final crystals were plate-like and had a broad size distribution. Typical crystals appear to be few microns thick and between 5 and 15 μm in length and width. (See the Supporting Information for full details; Figure S3 shows pictures of the crystals.)
Mixing Injectors and Sample Delivery.
Hydrodynamically focusing, diffusive mixing injectors7 were built using the framework described previously.9 Two slightly different designs were used. The injectors for solution samples had a 75 micron diameter mixing constriction and accessed timepoints from 16 to 88 ms. Unfortunately, samples collected from the 16 ms timepoint were lost due to a problem with the helium cryostat in the EPR spectrometer, and the shortest solution timepoint reported here is 32 ms. To avoid clogging when flowing the relatively large crystals, the mixing constriction was increased to 100 micron diameter for the crystalline samples. These mixers accessed timepoints from 32 to 250 ms. (See the Supporting Information for details of how timepoints are determined.)
Syringe pumps (Harvard Apparatus, Holliston, MA) supplied the injector with myoglobin and azide. A 3:1 azide:myoglobin flow rate ratio at a combined flow rate of 75 μL/min was used for both solution and crystal mixing experiments. (See Figure S4 for a schematic of the flow path.) After mixing, the final myoglobin and azide concentrations were 500 μM and 15 mM, respectively, for solution experiments, and ~150 μM and 114 mM for crystal experiments. Several batches of crystals were required, and their concentrations varied slightly.
Freeze-Quenching.
The reacting sample (150 μL) was jetted into ~13 mL of ~150 K isopentane while using a magnetic stir bar to ensure homogenous temperature. The sample was transferred to the flow packing system, which was sealed and pressured with ~160 K, 120 PSI nitrogen gas. A mass flow meter was used to monitor the flow rate of the pressurization gas to determine when all the isopentane had flowed through the filter. Flow packing was typically complete in less than 2 min. The reservoir was depressurized and the sample further compressed with a chilled aluminum rod. The EPR tube was disconnected from the reservoir and placed in a liquid nitrogen storage dewar to await measurement (See the Supporting Information and Figures S6–S15 for a detailed protocol and pictures).
Preparation of Reaction Standards for EPR Experiments.
In the buffer conditions chosen for this experiment, in the absence of azide, the myoglobin is in the high-spin iron state (Fe3+, S = 5/2).24 Addition of excess azide induces the low-spin state (Fe3+, S = 1/2). High- and low-spin standards for the myoglobin/azide reaction were prepared for both sample types. For the solution experiments, both spin standards contained 1 mM myoglobin. The low-spin solution standard included 20 mM azide. The microcrystalline standards both contained 600 μM myoglobin. The low-spin standard included 20 mM azide. After equilibration, all standard samples were dispensed into an EPR tube and slowly frozen in liquid nitrogen.
EPR Measurements.
EPR spectra were recorded at ACERT on a Bruker ElexSys E500 EPR spectrometer at 9.4 GHz, using an ESR910 liquid-helium cryostat (Oxford Instruments) maintained at 12 K. The spectrometer settings were as follows: modulation frequency, 100 kHz; modulation amplitude, 6 Gauss; microwave power, 0.02 mW. The field sweeps were calibrated with a BRUKER ER 035 Gauss meter, and the microwave frequency was monitored with a frequency counter. Data acquisition and manipulation were performed with Xepr software.
EPR Data Analysis.
The myoglobin/azide reaction is a well-characterized calibration standard for RFQ.19,20,24 Myoglobin begins in the high-spin state, binds to azide, and converts to the low-spin state. IHS Standard is the high-spin intensity of a sample with no azide, and ILS Standard is the low-spin intensity of a sample equilibrated with azide (see Figure 3a). These peaks are found at approximately 1100 Gauss and 3050 Gauss (g = 6.1 and 2.2), respectively.
Figure 3.
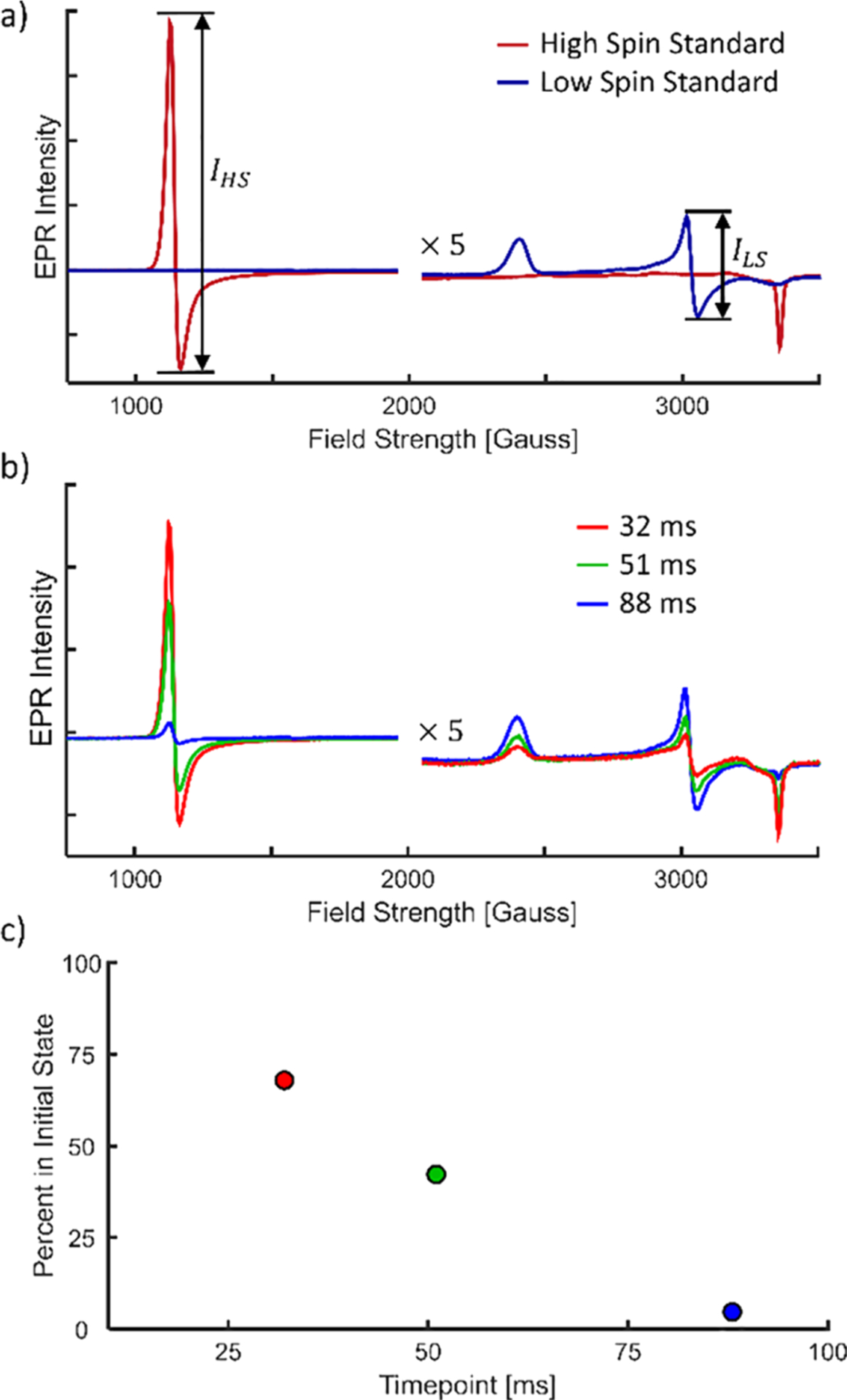
EPR data for the myoglobin and 15 mM azide reaction in the solution state. EPR intensities are provided in relative units. For high field strengths, EPR intensities are multiplied by 5 for ease of viewing. (a) EPR spectra of the high- and low-spin solution standards. Peak-to-peak intensities used in data analysis are illustrated. (b) Time-resolved EPR spectra of myoglobin/azide reaction. Curves are normalized by the total myoglobin concentration (see the Supporting Information and Figure S5 for details). (c) Percent of the initial state (FHS(t)) remaining as a function of time.
In time-resolved samples, the population fraction of high-spin (azide unbound) myoglobin, FHS(t), is determined from the EPR data as described previously:20,24
| (1) |
where
| (2) |
| (3) |
Here, IHS(t) and ILS(t) represent the EPR intensity of the high-and low-spin peaks in the time-resolved samples, respectively. Ri/f is a calibration factor representing the ratio of peak intensities at the initial and final states of the reaction.
RESULTS
EPR with Solution Samples.
As a test of the interface of the XFEL mixing injector with the RFQ apparatus, we prepared EPR compatible samples to measure the reaction of myoglobin in solution with 15 mM azide. Figure 3a shows the EPR spectra of the high- and low-spin solution standards. Figure 3b shows the time-resolved EPR spectra for 32, 51, and 88 ms after mixing. A clear trend emerges with time: the high-spin state decreases and the low-spin state increases. Figure 3c shows the percentage of myoglobin remaining unbound versus time, calculated with eq 1, showing the progression of the reaction.
EPR with Crystalline Samples.
Following the success with the solution reaction, the mixing injectors/RFQ/EPR system was then applied to monitor the reaction between myoglobin in microcrystals and 114 mM azide – an ~8× increase compared to the solution data. Calibration standards were prepared using crystalline myoglobin. Figure 4a shows EPR intensities for these standard samples. Subtle differences between the spectra for crystal and solution standards may reflect changes in buffer conditions or may result from crystal contacts. Figure 4b shows the time-resolved data for the reaction of microcrystals with azide. Two separate samples were prepared and measured for the 32, 88, and 250 ms timepoints. The replicates allowed us to assess reproducibility in the time-resolved data. Figure 4c shows the percentage of unbound myoglobin remaining over time. One solution data point, produced using the same mixing injector and 114 mM azide, is included for comparison.
Figure 4.

EPR data for the reaction between myoglobin microcrystals and 114 mM azide. EPR intensities are shown in relative units. (a) EPR spectra of the high- and low-spin crystal standards. (b) Time-resolved EPR spectra of the myoglobin/azide reaction normalized for myoglobin concentration (see the Supporting Information). The spectra for timepoints with duplicate measurements are averaged. (c) Percent of the initial state remaining as a function of time for the reaction in microcrystals (circles) and solution at the same azide concentration for comparison (black triangle). The 2× label indicates two overlapping datapoints at 250 ms.
DISCUSSION
Comparison of Solution and Crystal Data.
Neglecting all other differences between the solution and crystal measurements, the 8× higher azide concentration used in the microcrystal reaction should result in a more rapid reaction than in the solution sample. However, this was not the case; the reaction time in crystalline samples was about a factor of two slower. This discrepancy is largely due to the altered kinetics in crystals versus solution. (There is also a small contribution from the mixing injectors due to the ~2× slower mixing times in the crystal vs solution mixers. However, this difference is not nearly enough to account for the observed change in rate between solution and crystals.) To verify the dramatic change in reaction rates between solution and crystalline samples, one solution reaction timepoint, 32 ms, was acquired using the same mixing injector and the same 8× azide concentration as the crystal reaction data. This datapoint is shown as a black triangle in Figure 4c). After 32 ms, more of the proteins in the solution sample are bound to azide than after 88 ms in the crystalline sample.
The difference in reaction times for crystalline and solution samples has been documented11,13,14 and is not surprising. Whether this difference is the result of changes in buffer conditions, mixing methods, or the crystalline versus solution state is irrelevant: our results emphasize that one cannot assume that kinetics measured in solution will transfer to a mix-and-inject experiment. Characterizing the reactions in crystals is essential to the success of a MISC beamtime: reaction intermediates may appear at very different timepoints in crystals than in solution.
Sample Consumption.
Time-resolved EPR using RFQ and XFEL mixing injectors uses only a fraction of the sample required for the eventual MISC experiments. The former uses only ~90 μL of crystals per timepoint (including flushing the device ahead of time), while the latter consumes over 1 mL of sample per timepoint. Thus, if sufficient samples can be produced for a mix-and-inject experiment, consumption for a complementary RFQ experiment is not a barrier.
Measuring Kinetics in Crystals.
The main goal of a mixand-inject experiment is to capture the structure of intermediates; measurements of kinetic rates in the crystals are of lesser importance since they most likely differ from the rates in physiologically relevant solution conditions. In some cases where in crystallo rate constants are important, they can be obtained from this RFQ technique. However, diffusive mixing injectors are often not suitable for measuring diffusion-limited binding rate constants.
As can be seen in Figure 3c and Figure 4c, reactions initiated in these diffusive mixing injectors do not follow simple first-order kinetics because mixing continues for a significant portion of the reaction time. When studied with a device that mixes nearly instantly, such as a stopped-flow mixer, the first-order kinetics of the myoglobin/azide system yield an exponential decay of the unbound species. After near-instantaneous mixing to 15 mM azide, the myoglobin in solution should be 86% bound in 48 ms, as calculated from a previously measured rate constant.24 We observe this level of binding in soluble myoglobin after ~80 ms. The slower reaction rate is expected due to the noninstantaneous mixing.
Extension to Other Systems.
The combination of the mixing injectors, the custom RFQ technology, and EPR spectroscopy can be readily applied to a wide range of systems. EPR with RFQ has previously been used to study a wide variety of protein–substrate reactions in solution, including the inactivation of pyruvate formate-lyase by dioxygen,29 the interactions of metallo-β-lactamases with antibiotics,30 and the radical formation in cytochrome c oxidase.31 These systems and many others are excellent candidates for future mix-and-inject experiments and would greatly benefit from characterization with this new method. Though the myoglobin/azide reaction studied here is a simple binding step without transient intermediate states, this technique clearly and effectively captured changing populations during a reaction occurring in tens of milliseconds in a crystalline sample. When applied to EPR active systems that do access transient states, this technique could be applied to quantify the occupancy of intermediates and optimize timepoints and conditions for their structural characterization via MISC experiments.
The sample concentration under XFEL mixing conditions will likely be high enough to generate sufficient EPR signal strength for many systems. While the required sample concentration is system-specific, previous RFQ EPR studies of enzymatic reactions were successfully performed with final mixed sample concentrations ranging from ~25 to 500 μM.29–33 XFEL mix-and-inject experiments typically operate in a comparable range (~30–430 μM final mixed concentration).1–4 Therefore, it is likely that the sample concentrations used for a MISC beamtime will yield enough signal for interpretable EPR data. In cases where the signal strength is insufficient, the use of newly developed mixing injectors that can operate at significantly lower reactant-to-sample ratios10 could significantly boost the EPR signal by increasing the mixed sample concentration.
Some samples of interest may require a different analysis method than the two-peak ratio used here to characterize the populations of intermediate states in a reaction. Previous EPR RFQ studies have probed many reactions containing states which are either EPR-silent or have peaks that overlap with other intermediates. For these systems, alternative analysis methods, including synthesizing EPR curves from known intermediate traces,29,32 simulation of EPR signals,30 or tracking the EPR intensity over time,33 can be used successfully to quantify the populations of intermediate states. The packing efficiencies achieved here had a standard deviation of ~10% for crystals grown in the same batch; therefore, samples with EPR-silent states could be probed without normalization, with only small errors in EPR intensity (see the Supporting Information and Figure S5 for details).
Utility of Technique for Enhancing Mix-and-Inject Experiments.
The most straightforward application of this technique is to select the ideal timepoints for observing structural intermediates. However, this technique can also be employed to optimize crystal forms and buffer conditions for future MISC studies. Solvent channel size, active site accessibility, buffer chemistry, and effects of crystal contacts are just some of the parameters that can alter the reaction rates. Characterizing the effects of these parameters in advance of the XFEL beamtime will allow experimenters to select measurement conditions that maximize the populations of transient intermediate states.
Furthermore, the EPR data provide essential details about an enzyme’s active site that complement structural data. Information about metal oxidation and spin states may be difficult or impossible to determine from crystallography alone but are easily detectable by EPR. This approach adds rigor to the mix-and-inject data since an experimenter can compare transient state occupancy between the EPR and mix-and-inject results.
Recent progress in synchrotron serial crystallography experiments demonstrates the feasibility of performing microcrystal mixing experiments for some systems without an XFEL.34–36 The resulting increased availability of the MISC method underscores the need for complementary experiments, like these, which characterize the reactions of interest.
Finally, the combination of spectroscopy with mixing injectors is not limited to EPR. This proof-of-principle experiment demonstrates that reactions in microcrystals can be reliably monitored under mix-and-inject conditions. In the future, mixing injectors could be interfaced with other spectroscopies to report a broader range of reactions. Such characterization will dramatically improve the efficiency and information content of mix-and-inject experiments and will propel the technique to a wider utility.
CONCLUSIONS
XFEL mixing injectors were successfully interfaced with EPR spectroscopy using a custom RFQ setup to track the populations of two chemical states during the myoglobin/azide reaction, both in crystalline and solubilized proteins. The measured difference between crystal and solution reaction rates emphasizes the need for in crystallo characterization to optimize sample and mixing parameters in advance of MISC beamtimes. This technique, as well as variations where mixing injectors are interfaced with other types of spectroscopy, permits reaction characterization in microcrystals using the same experimental setup as in a mix-and-inject experiment, thus increasing the efficiency of these challenging measurements.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Ilme Schlichting for advice and input on myoglobin crystal preparation. The authors also thank Chris Pollock for useful discussions about RFQ. EPR data were collected at ACERT, which is supported by NIH grant P41GM103521. The authors thank Nathan Ellis, Jeff Koski, and Rob Page for useful discussion and assistance in fabricating the flow packing system. Mixers were partially fabricated at Cornell Center for Materials Research Shared Facilities, which are supported through the NSF MRSEC program (DMR-1719875). This work was funded by the NSF Science and Technology Center Grant No. 1231306. K.A.Z. was supported by the Cornell Molecular Biophysics Training Program (NIH T32-GM008267).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c02569.
Additional information on the cryostat and flow packing system, EPR data normalization, and full parts list and RFQ protocol (PDF)
Technical drawings for the cryostat and flow packing system (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c02569
The authors declare no competing financial interest.
Contributor Information
George D. Calvey, School of Applied and Engineering Physics, Cornell University, Ithaca, New York 14853, United States
Andrea M. Katz, School of Applied and Engineering Physics, Cornell University, Ithaca, New York 14853, United States
Kara A. Zielinski, School of Applied and Engineering Physics, Cornell University, Ithaca, New York 14853, United States
Boris Dzikovski, Department of Chemistry and Chemical Biology and National Biomedical Center for Advanced ESR Technology, Cornell University, Ithaca, New York 14853, United States.
Lois Pollack, School of Applied and Engineering Physics, Cornell University, Ithaca, New York 14853, United States.
REFERENCES
- (1).Kupitz C; Olmos JL Jr.; Holl M; Tremblay L; Pande K; Pandey S; Oberthür D; Hunter M; Liang M; Aquila A; Tenboer J; Calvey G; Katz A; Chen Y; Wiedorn MO; Knoska J; Meents A; Majriani V; Norwood T; Poudyal I; Grant T; Miller MD; Xu W; Tolstikova A; Morgan A; Metz M; Martin-Gracia J; Zook JD; Roy-Chowdhury S; Coe J; Nagaratnam N; Meza D; Fromme R; Basu S; Frank M; White T; Barty A; Bajt S; Yefanov O; Chapman HN; Zatsepin N; Nelson G; Weierstall U; Spence J; Schwander P; Pollack L; Fromme P; Ourmazd A; Phillips GN; Schmidt M Struct. Dyn 2017, 4, No. 044003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stagno JR; Liu Y; Bhandari YR; Conrad CE; Panja S; Swain M; Fan L; Nelson G; Li C; Wendel DR; White TA; Coe JD; Wiedorn MO; Knoska J; Oberthuer D; Tuckey RA; Yu P; Dyba M; Tarasov SG; Weierstall U; Grant TD; Schwieters CD; Zhang J; Ferré-D’Amaré AR; Fromme P; Draper DE; Liang M; Hunter MS; Boutet S; Tan K; Zuo X; Ji X; Barty A; Zatsepin NA; Chapman HN; Spence JCH; Woodson SA; Wang Y-X Nature 2017, 541, 242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Olmos JL Jr.; Pandey S; Martin-Garcia JM; Calvey G; Katz A; Knoska J; Kupitz C; Hunter MS; Liang M; Oberthuer D; Yefanov O; Wiedorn M; Heyman M; Holl M; Pande K; Barty A; Miller MD; Stern S; Roy-Chowdhury S; Coe J; Nagaratnam N; Zook J; Verburgt J; Norwood T; Poudyal I; Xu D; Koglin J; Seaberg MH; Zhao Y; Bajt S; Grant T; Mariani V; Nelson G; Subramanian G; Bae E; Fromme R; Fung R; Schwander P; Frank M; White TA; Weierstall U; Zatsepin N; Spence J; Fromme P; Chapman HN; Pollack L; Tremblay L; Ourmazd A; Phillips GN; Schmidt M BMC Biol 2018, 16, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dasgupta M; Budday D; de Oliveira SHP; Madzelan P; Marchany-Rivera D; Seravalli J; Hayes B; Sierra RG; Boutet S; Hunter MS; Alonso-Mori R; Batyuk A; Wierman J; Lyubimov A; Brewster AS; Sauter NK; Applegate GA; Tiwari VK; Berkowitz DB; Thompson MC; Cohen AE; Fraser JS; Wall ME; van den Bedem H; Wilson MA Proc. Natl. Acad. Sci. U. S. A 2019, 116, 25634–25640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schmidt M Adv. Condens. Matter Phys 2013, 2013, 1–10. [Google Scholar]
- (6).DePonte DP; Weierstall U; Schmidt K; Warner J; Starodub D; Spence JCH; Doak RB J. Phys. D: Appl. Phys 2008, 41, 1–12. [Google Scholar]
- (7).Calvey GD; Katz AM; Schaffer CB; Pollack L Struct. Dyn 2016, 3, No. 054301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Oberthuer D; Knoška J; Wiedorn MO; Beyerlein KR; Bushnell DA; Kovaleva EG; Heymann M; Gumprecht L; Kirian RA; Barty A; Mariani V; Tolstikova A; Adriano L; Awel S; Barthelmess M; Dörner K; Xavier PL; Yefanov O; James DR; Nelson G; Wang D; Calvey G; Chen Y; Schmidt A; Szczepek M; Frielingsdorf S; Lenz O; Snell E; Robinson PJ; Šarler B; Belšak G; Maček M; Wilde F; Aquila A; Boutet S; Liang M; Hunter MS; Scheerer P; Lipscomb JD; Weierstall U; Kornberg RD; Spence JCH; Pollack L; Chapman HN; Bajt S Sci. Rep 2017, 7, 44628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Calvey GD; Katz AM; Pollack L Anal. Chem 2019, 91, 7139–7144. [DOI] [PubMed] [Google Scholar]
- (10).Knoška J; Adriano L; Awel S; Beyerlein KR; Yefanov O; Oberthuer D; Murillo GEP; Roth N; Sarrou I; Villanueva-perez P; Wiedorn MO; Wilde F; Chapman HN; Heymann M Nat. Commun 2020, 11, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chance B; Ravilly A; Rumen NJ Mol. Biol 1966, 17, 525–534. [DOI] [PubMed] [Google Scholar]
- (12).Rupley JA; Gates VJ Mol. Biol 1968, 35, 477–481. [DOI] [PubMed] [Google Scholar]
- (13).Chance B; Ravilly AJ Mol. Biol 1966, 21, 195–198. [DOI] [PubMed] [Google Scholar]
- (14).Theorell H; Chance B; Yonetani TJ Mol. Biol 1966, 17, 513–524. [DOI] [PubMed] [Google Scholar]
- (15).Hagen WR Dalton Trans 2006, 4415–4434. [DOI] [PubMed] [Google Scholar]
- (16).Cammack R Electron Paramagnetic Resonance Spectroscopy of Metalloproteins. In Methods in Molecular Biology; Jones C, Mulloy B, Thomas AH, Eds.; 2019; 1876, pp. 197–211. DOI: 10.1007/978-1-4939-8864-8_13. [DOI] [PubMed] [Google Scholar]
- (17).More C; Belle V; Asso M; Fournel A; Roger G; Guigliarelli B; Bertrand P Biospectroscopy 1999, 5, 3–18. [DOI] [PubMed] [Google Scholar]
- (18).Bray RC Biochem. J 1961, 81, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ballou DP; Palmer GA Anal. Chem 1974, 46, 1248–1253. [Google Scholar]
- (20).Pievo R; Angerstein B; Fielding AJ; Koch C; Feussner I; Bennati M ChemPhysChem 2013, 14, 4094–4101. [DOI] [PubMed] [Google Scholar]
- (21).Palmer G; Bray RC; Beinert HJ Biol. Chem 1964, 239, 2657–2666. [PubMed] [Google Scholar]
- (22).Ballou DP Methods Enzymol 1978, 54, 85–93. [DOI] [PubMed] [Google Scholar]
- (23).Cherepanov AV; De Vries S Biochim. Biophys. Acta 2004, 1656, 1–31. [DOI] [PubMed] [Google Scholar]
- (24).Nami F; Gast P; Groenen EJ J. Appl. Magn. Reson 2016, 47, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsai AL; Berka V; Kulmacz RJ; Wu G; Palmer G Anal. Biochem 1998, 264, 165–171. [DOI] [PubMed] [Google Scholar]
- (26).Lin Y; Gerfen GJ; Rousseau DL; Yeh S-R Anal. Chem 2004, 75, 5381–5386. [DOI] [PubMed] [Google Scholar]
- (27).Tanaka M; Matsuura K; Yoshioka S; Takahashi S; Ishimori K; Hori H; Morishima I Biophys. J 2003, 84, 1998–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Barends TRM; Foucar L; Ardevol A; Nass K; Aquila A; Botha S; Doak RB; Falahati K; Hartmann E; Hilpert M; Heinz M; Hoffmann MC; Köfinger J; Koglin JE; Kovacsova G; Liang M; Milathianaki D; Lemke HT; Reinstein J; Roome CM; Shoeman RL; Williams GJ; Burghardt I; Hummer G Science 2015, 350, 445–450. [DOI] [PubMed] [Google Scholar]
- (29).Zhang W; Wong KK; Magliozzo RS; Kozarich JW Biochemistry 2001, 40, 4123–4130. [DOI] [PubMed] [Google Scholar]
- (30).Garrity JD; Bennett B; Crowder MW Biochemistry 2005, 44, 1078–1087. [DOI] [PubMed] [Google Scholar]
- (31).Yu MA; Egawa T; Shinzawa-Itoh K; Yoshikawa S; Yeh S-R; Rousseau DL; Gerfen GJ Biochim. Biophys. Acta 2011, 36, 1295–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bollinger JM; Tong WH; Ravi N; Huynh BH; Edmondson DE; Stubbe JA J. Am. Chem. Soc 1994, 116, 8015–8023. [Google Scholar]
- (33).George SJ; Seravalli J; Ragsdale SW J. Am. Chem. Soc 2005, 127, 13500–13501. [DOI] [PubMed] [Google Scholar]
- (34).Mehrabi P; Schulz EC; Agthe M; Horrell S; Bourenkov G; von Stetten D; Leimkohl JP; Schikora H; Schneider TR; Pearson AR; Tellkamp F; Miller RJD Nat. Methods 2019, 16, 979–982. [DOI] [PubMed] [Google Scholar]
- (35).Monteiro DCF; Von Stetten D; Stohrer C; Sans M; Pearson AR; Santoni G; Van Der Linden P; Trebbin M IUCrJ. 2020, 7, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Beyerlein KR; Dierksmeyer D; Mariani V; Kuhn M; Sarrou I; Ottaviano A; Awel S; Knoska J; Fuglerud S; Jönsson O; Stern S; Wiedorn MO; Yefanov O; Adriano L; Bean R; Burkhardt A; Fischer P; Heymann M; Horke DA; Jungnickel KEJ; Kovaleva E; Lorbeer O; Metz M; Meyer J; Morgan A; Pande K; Panneerselvam S; Seuring C; Tolstikova A; Lieske J; Aplin S; Roessle M; White TA; Chapman HN; Meents A; Oberthuer D IUCrJ. 2017, 4, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.