

Abstract
Inhibition of human ornithine δ-aminotransferase (hOAT) is a potential therapeutic approach to treat hepatocellular carcinoma. In this work, (S)-3-amino-4,4-difluorocyclopent-1-enecarboxylic acid (SS-1–148, 6) was identified as a potent mechanism-based inactivator of hOAT while showing excellent selectivity over other related aminotransferases (e.g., GABA-AT). An integrated mechanistic study was performed to investigate the turnover and inactivation mechanisms of 6. A monofluorinated ketone (M10) was identified as the primary metabolite of 6 in hOAT. By soaking hOAT holoenzyme crystals with 6, a precursor to M10 was successfully captured. This gem-diamine intermediate, covalently bound to Lys292, observed for the first time in hOAT/ligand crystals, validates the turnover mechanism proposed for 6. Co-crystallization yielded hOAT in complex with 6 and revealed a novel noncovalent inactivation mechanism in hOAT. Native protein mass spectrometry was utilized for the first time in a study of an aminotransferase inactivator to validate the noncovalent interactions between the ligand and the enzyme; a covalently-bonded complex was also identified as a minor form observed in the denaturing intact protein mass spectrum. Spectral and stopped-flow kinetic experiments supported a lysine-assisted E2 fluoride ion elimination, which has never been observed experimentally in other studies of related aminotransferase inactivators. This elimination generated the second external aldimine directly from the initial external aldimine, rather than the typical E1cB elimination mechanism, forming a quinonoid transient state between the two external aldimines. The use of native protein mass spectrometry, X-ray crystallography employing both soaking and co-crystallization methods, and stopped-flow kinetics allowed detailed elucidation of unusual turnover and inactivation pathways.
Keywords: cyclopentene, amino acid, fluoride ion elimination, noncovalent, inactivation, hepatocellular carcinoma, structure-based drug design
Graphical Abstract
INTRODUCTION
Human ornithine δ-aminotransferase (hOAT; EC2.6.1.13) is a pyridoxal-5’-phosphate (PLP)-dependent enzyme that catalyzes two coupled half-reactions, converting L-ornithine (L-Orn) in the first half-reaction to L-glutamate-γ-semialdehyde (L-GSA) and generating L-glutamate (L-Glu) from α-ketoglutarate (α-KG) in the second half-reaction (Figure 1A).1 The product, L-GSA, is in equilibrium with Δ1-pyrroline-5-carboxylate (P5C), which is converted to L-proline by P5C reductases (PYCRs).2 The generated L-Glu can also be converted to P5C by pyrroline-5-carboxylate synthase (P5CS), and therefore also participating in proline metabolism (Figure 1A).2 Proline biosynthesis was identified as the most substantially altered amino acid metabolism in human tumor tissues of hepatocellular carcinoma (HCC), featured by accelerated proline consumption, hydroxyproline accumulation, and increased α-fetoprotein (AFP) levels, which are correlated with poor prognosis in HCC.3 In addition, glutamine synthetase (GS) catalyzes L-Glu’s conversion to L-glutamine (L-Gln).4 L-Gln is required by cancer cells to support the abnormally elevated anabolic processes, thus promoting cellular proliferation.
Figure 1.
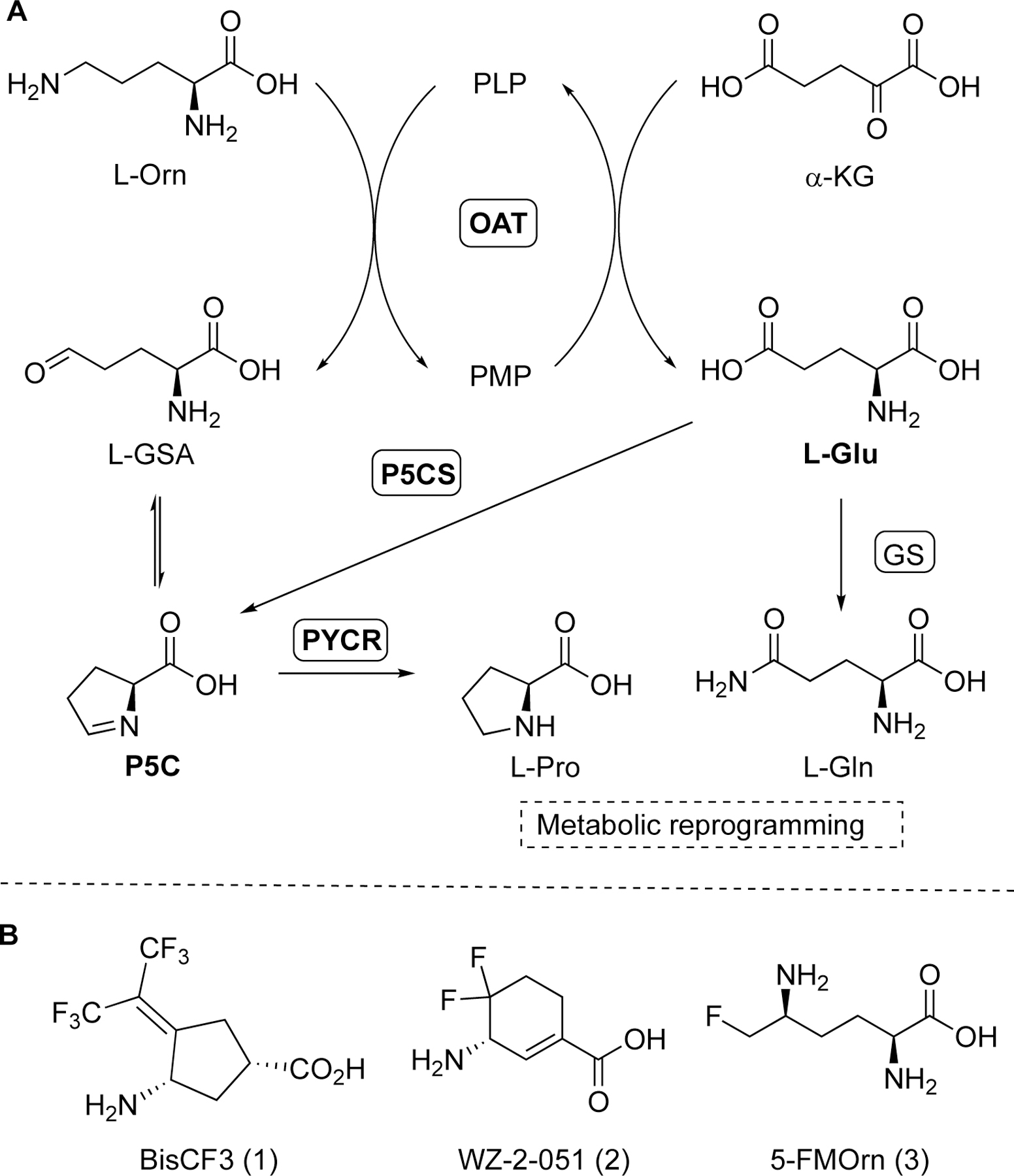
Transamination reactions of hOAT and related metabolic pathways (A); Structures of hOAT mechanism-based inactivators 1–3 (B).
HCC is the predominant liver malignancy and ranks among the most common causes of cancer-associated mortality worldwide.5 Our prior DNA microarray analyses identified the OAT gene as one of seven overexpressed genes in the spontaneous HCC-developing livers from Psammomys obesus (sand rat).6 Moreover, the treatment (at 0.1 and 1.0 mg/kg; po) of selective hOAT mechanism-based inactivator (MBI) BisCF3 (1) remarkably decreased the serum AFP levels and inhibited tumor growth in a human-derived HCC mouse model,6 underscoring the antitumor effects of selective hOAT inhibition. A MBI is a molecule that initially acts as an alternative substrate for the target enzyme and is converted by this enzyme to a species that inactivates that enzyme.7–9 MBIs are typically unreactive before the initial binding with the active site of the target enzyme and usually exhibit significant target specificity and selectivity.10 Overall, hOAT is a potential therapeutic target for HCC, and selectively inactivating hOAT may provide a novel opportunity to discover an effective HCC treatment.
Optimization of reversible inhibitors mainly focuses on improving the binding affinity (Ki) or IC50 values. In contrast, kinact and KI values are two critical kinetic parameters for the development of MBIs.11 The kinact value is the maximal rate constant for enzyme inactivation. On the other hand, the KI value, the concentration of inactivator that gives half-maximal inactivation rate, is the inhibition constant, which represents the ability of an MBI to initially bind to the active site of the target enzyme and compete with the substrate. The ratio kinact/KI is used to evaluate the inactivation efficiency of MBIs.11
A major challenge for discovering a selective MBI of hOAT is to overcome the irreversible inhibition of other aminotransferases,7 especially γ-aminobutyric acid aminotransferase (GABA-AT), which has a high structural similarity with hOAT.1 There are only two significant differences in the active site pocket of their homodimer structures: Tyr85 and Tyr55 in hOAT are replaced by Ile72 and Phe351* (asterisk denotes arising from the adjacent subunit) in GABA-AT, respectively (Figure 2).1 Moreover, Ile72 and Phe351* are responsible for the slightly narrower and more hydrophobic active site of GABA-AT relative to hOAT. In contrast, the hydroxyl group of Tyr55 serves as a hydrogen bond acceptor to interact with the charged C-2 amino group of substrates, while Tyr85 is a significant determinant of substrate specificity and conformational flexibility to adopt bulky substrates.1
Figure 2.
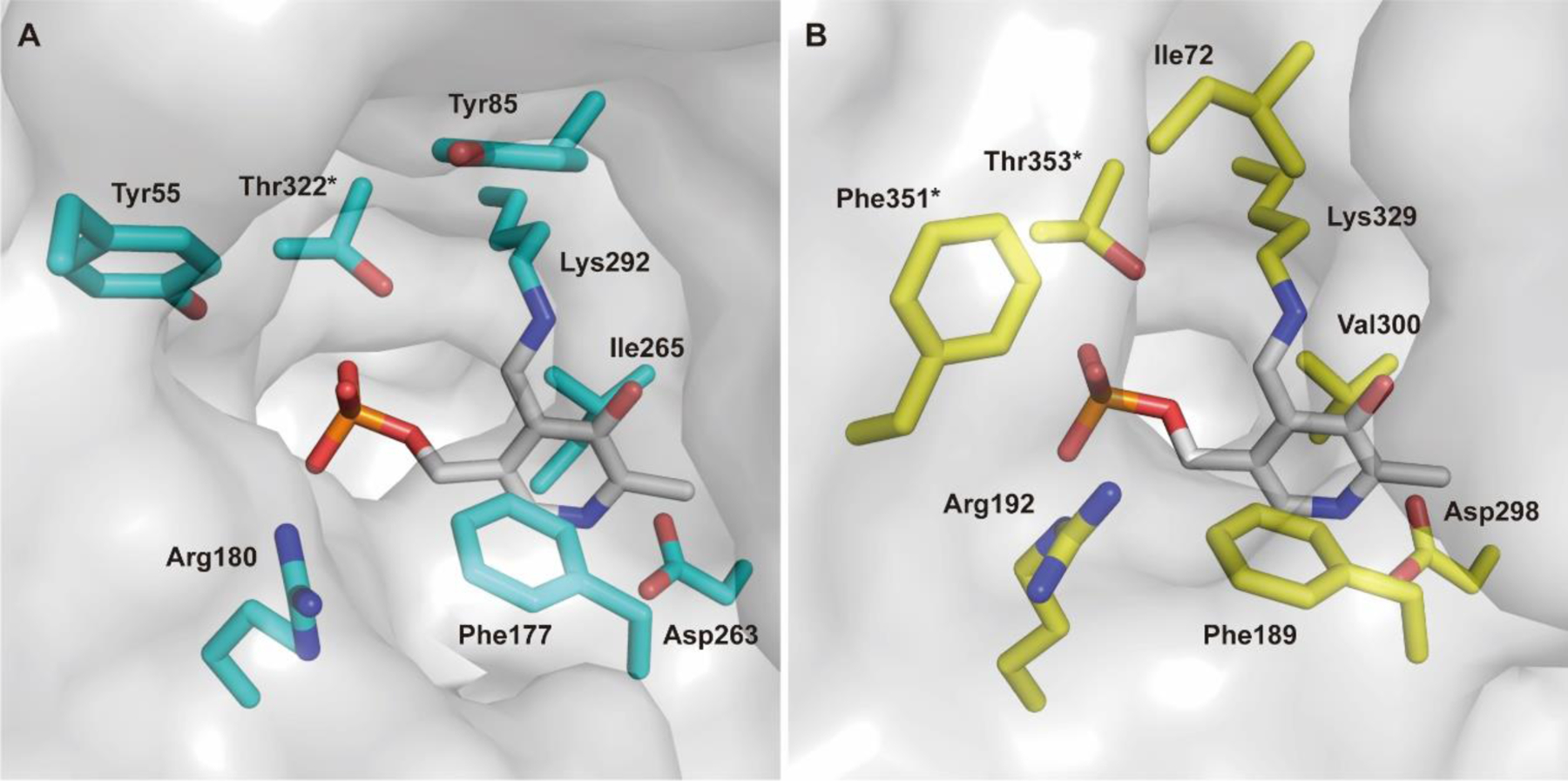
Active site comparison of hOAT (A; PDB entry 1OAT) and GABA-AT (B; PDB entry 1OHV)
Because of the high similarity between these two aminotransferases, a preliminary screening against hOAT was carried out previously using our stock GABA-AT inhibitors.6 A cyclopentane-based analog, termed BisCF3 (1, Figure 1B), bearing a bis(trifluoromethyl) group as its warhead, was identified to be a selective MBI of hOAT while only showing millimolar reversible inhibition of GABA-AT. Recent mechanistic studies have revealed that one of its trifluoromethyl groups undergoes fluoride ion elimination, leading the ligand to covalently modify the catalytic Lys292 residue by conjugate addition (Scheme 1A).9, 12 It is considered that the sterically bulky bis(trifluoromethyl) group may not access the more narrow pocket of GABA-AT as readily, influencing the initial binding pose between the ligand and the enzyme, which may be responsible for its reversible inhibition of this enzyme. Compound 1 has been demonstrated to be effective in vivo6 and is being investigated further in HCC patient-derived xenograft (PDX) models. Based on a similar strategy, we enlarged the ring system and further developed a cyclohexene-based analog, WZ-2–051 (2, Figure 1B), bearing a difluoro group.13 Compound 2 exhibited a 23-fold improvement in inactivation efficiency (defined by the kinact/KI ratio) against hOAT compared to 1 while showing 13.3-fold selectivity over GABA-AT. The subsequent mechanistic studies disclosed that 2 undergoes a two-step fluoride ion elimination and finally inactivates hOAT through an addition-aromatization mechanism (Scheme 1B).13 An additional example of a selective hOAT inactivator is 5-fluoromethylornithine (5-FMOrn, 3) inspired by the structure of hOAT substrate L-Orn and related to the structure of non-selective GABA-AT inactivator (S)-4-amino-5-fluoropentanoic acid (AFPA).14 This molecule inactivates hOAT via an enamine pathway by forming a ternary adduct (Scheme 1C).15
Scheme 1.
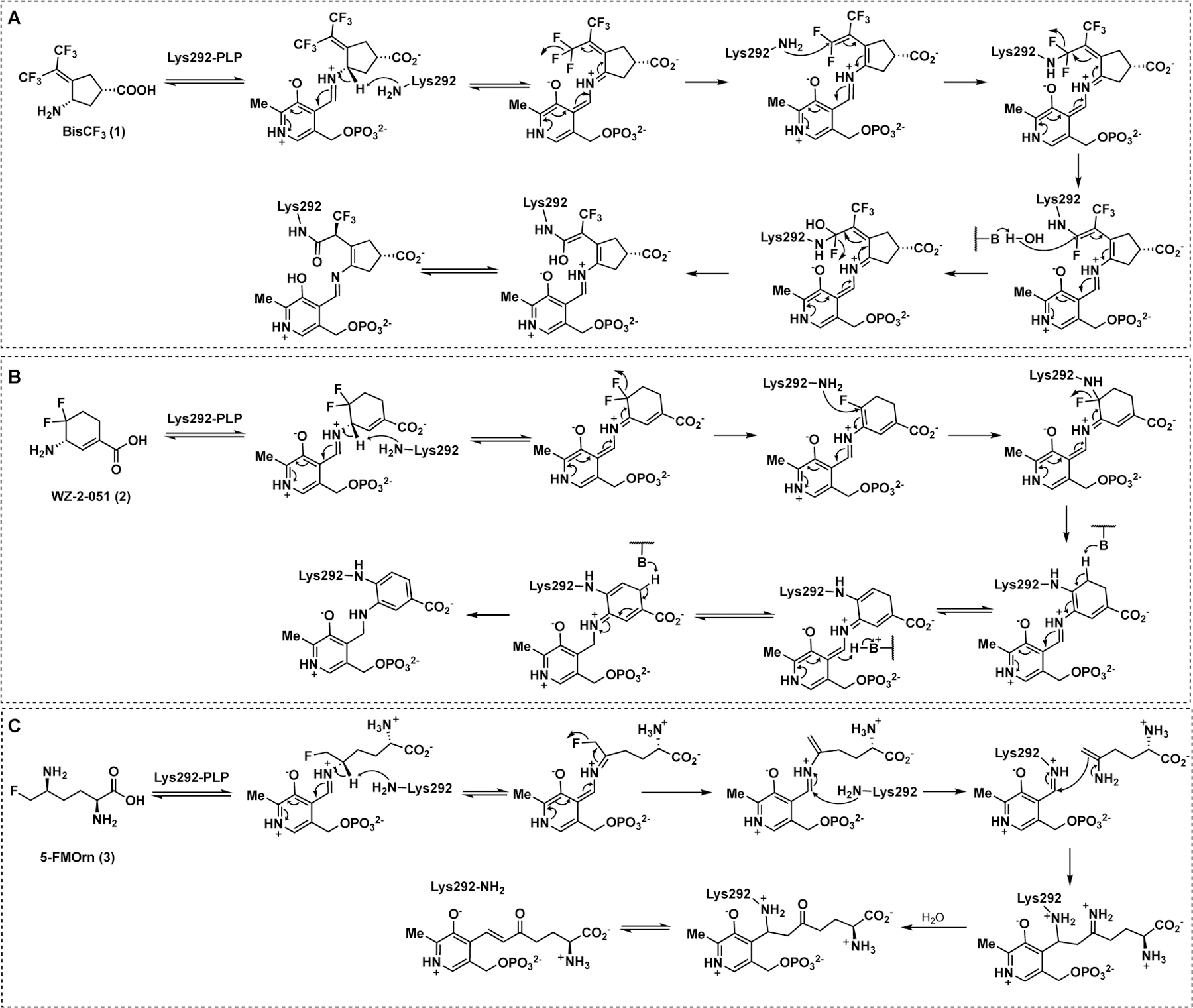
Inactivation mechanisms of hOAT by 1–3
It should be noted that the α-amino group of 5-FMOrn forms a strong hydrogen bond with the phenol group of Tyr55 in the hOAT crystal complex (PDB entry 2OAT).16 Moreover, we also observed hydrogen bonds between the carboxylate groups of 1 (PDB entry 6OIA) and 2 (PDB entry 6V8C) and Tyr55 in hOAT crystal complexes.12–13 Among the published hOAT inactivators, only 1 demonstrates (a) promising hOAT selectivity through potent irreversible inhibition of hOAT, (b) weak, reversible inhibition of GABA-AT (Ki = 4.2 mM), and (c) no inhibition of either aspartate aminotransferase (Asp-AT) or alanine aminotransferase (Ala-AT) (up to 4 mM).6
In 2000, (1R,4S)-4-amino-3,3-difluorocyclopentanecarboxylic acid (4, Figure 3) was found to be a reversible inhibitor against GABA-AT (Ki = 0.19 mM).17 Fifteen years later, it was further demonstrated to be an hOAT inactivator.6 However, 4 exhibits poor binding affinity (KI = 7.8 mM) and has a low maximum rate of inactivation (kinact = 0.02 min−1) against hOAT, only yielding a modest inactivation efficiency (kinact/KI = 0.003 min−1mM−1).
Figure 3.
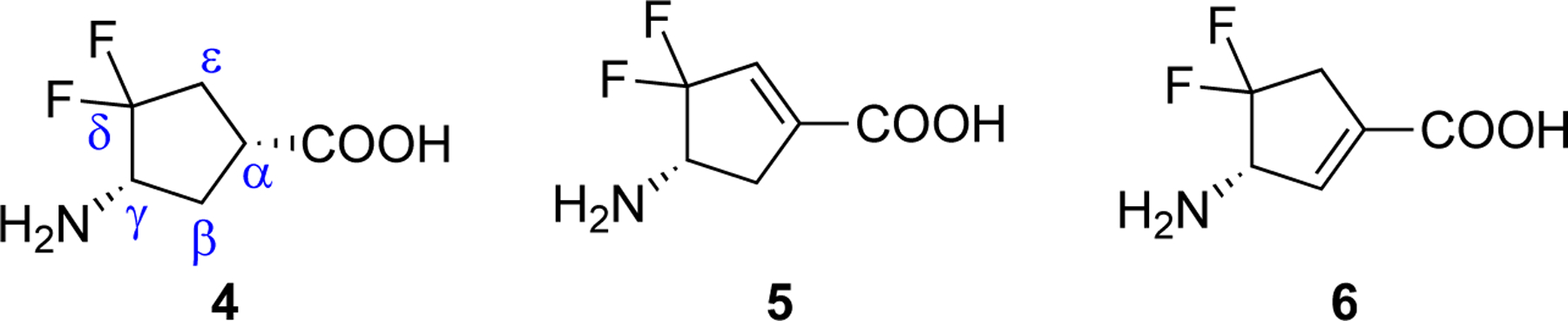
Structures of hOAT inactivators 4–6
In this work, we developed a novel cyclopentene-based analog 6 by incorporating an additional double bond into the cyclopentane ring system of 4, which was demonstrated to be a potent and selective hOAT inactivator. Furthermore, we performed mechanistic studies utilizing protein crystallography, multiple modes of mass spectrometry, transient-state spectrophotometric measurements, and computational simulations to reveal a novel noncovalent inactivation mechanism for 6.
RESULTS AND DISCUSSION
Synthesis of Cyclopentene Analogs 5 and 6 Bearing a gem-Difluoro Group.
On the basis of our previous experience with the discovery of GABA-AT inactivators, incorporation of a double bond into a cyclopentane ring has been demonstrated to be an effective strategy for improving inactivation efficiency, which influences the configuration of the initial external aldimine and the adjunct proton’s acidity resulting from the α,β-unsaturated carboxylate.18–19 Therefore, we designed cyclopentene-based analogs 5 and 6 bearing a gem-difluoro group based on the structure of parent compound 4 (Figure 3) with the intent to develop more potent hOAT inactivators.
The synthetic route to prepare compound 5 initiated from the enantiopure Vince lactam 20 (7; (1R)-(−)-2-azabicyclo[2.2.1hept-5-en-3-one; CAS#: 79200–56-9) to afford the key bicyclic intermediate 9 according to the procedure developed previously (Scheme 2).17 The acetyl group of 9 was then hydrolyzed under acid conditions followed by Swern oxidation, yielding ketone intermediate 11.
Scheme 2. The synthetic route to 4 and 5a.
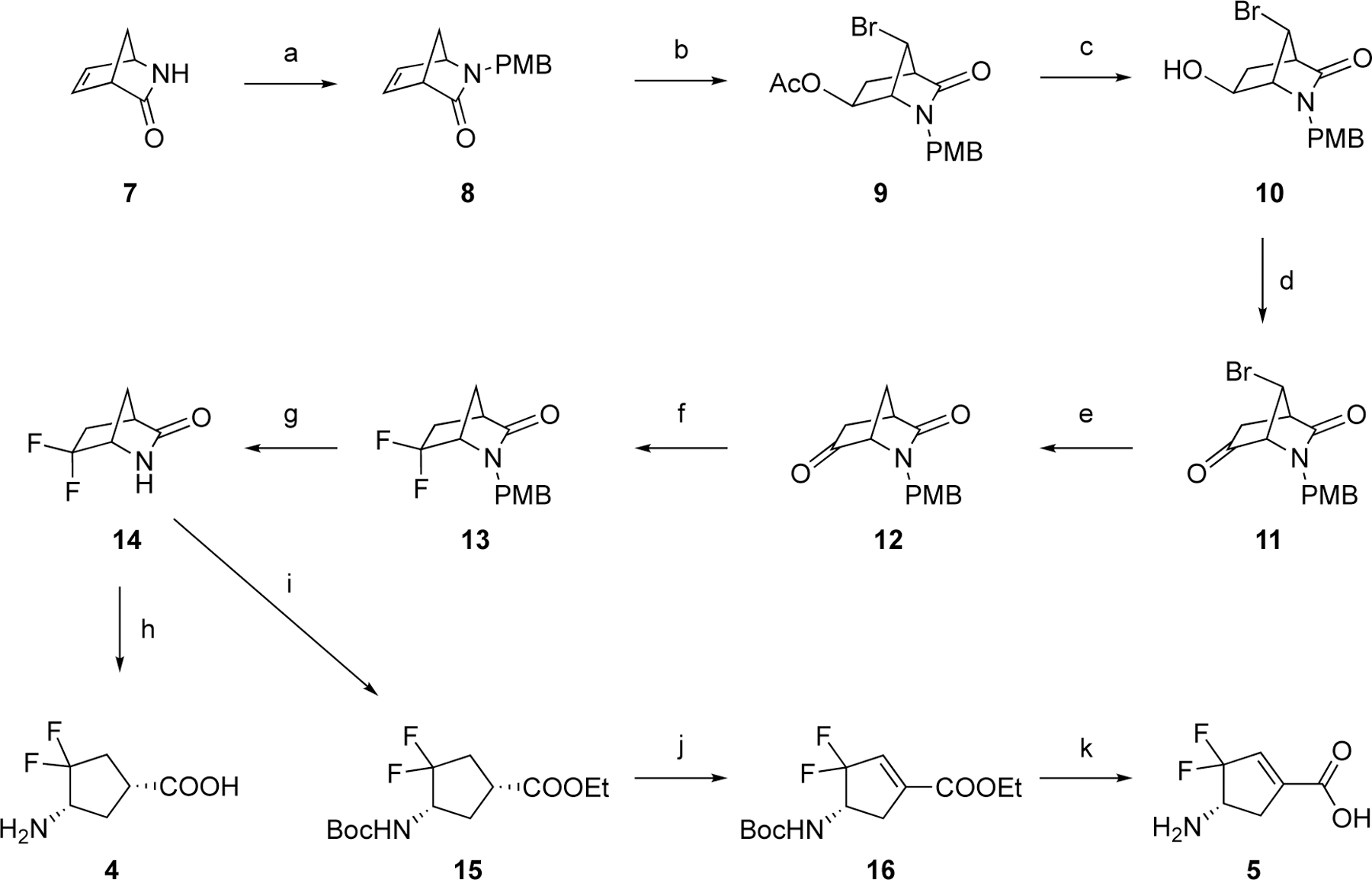
aReagents and conditions. (a) i) p-anisyl alcohol, conc. HCl, rt; ii) NaH, tetra-n-butylammonium iodide (TBAI), THF/DMF (10:1), 0 °C-rt; (b) 1,3-Dibromo-5,5-dimethylhydantoin (DBDMH), Ac2O, rt; (c) K2CO3, MeOH/H2O, rt; (d) (COCl)2, DMSO, TEA, THF, –78 °C-rt; (e) Bu3SnH, azobisisobutyronitrile (AIBN), benzene, reflux; (f) Deoxo-Fluor (2.7 M in toluene), THF, 120 °C (MW); (g) ceric ammonium nitrate, CH3CN/H2O, rt; (h) 4N HCl, AcOH, 70 °C; (i) i) HCl in EtOH (1.2 M), 70 °C; ii) Boc2O, TEA, DCM, rt; (j) PhSeCl, KHMDS (3.0 equiv., 0.5 M in toluene), –78 °C-rt; (k) 4N HCl, AcOH, 70 °C.
Subsequently, reduction of the bromo group on the bridgehead of 11 to form 12 was carried out with Bu3SnH/AIBN by a published procedure.17 Intermediate 12 was treated with Deoxo-Fluor reagent under microwave conditions to afford difluoro intermediate 13. The PMB protecting group of 13 was removed using ceric ammonium nitrate (CAM) to produce lactam 14. Treatment of 14 with HCl/EtOH under reflux conditions followed by Boc protection yielded 15. Unexpectantly, when we attempted to selenate 15 at the α-position of the ethyl ester with KHMDS (3.0 equiv) and PhSeCl for the follow-up α-elimination reaction,18 intermediate 16, bearing an α,ε-conjugated carboxylate group, was produced directly as the sole product. Because of the strong electron-withdrawing effect of the gem-difluorines, the theoretical pKa values of the hydrogens at the Cε position are considerably decreased, which are further decreased by the incorporation of the phenylselenyl group (Scheme S1). This should facilitate deprotonation in the presence of excess KHMDS, thereby causing elimination of the phenylselenyl group. 1D and 2D NMR spectra were obtained to validate the structure of 16. Cyclopentene-based analog 5 was generated after deprotection under acidic conditions. The parent cyclopentane-based analog (4) was also prepared from intermediate 14 by acid hydrolysis and was evaluated together with new analogs and 1 in subsequent kinetic studies.
The synthetic route to 6 started from the preparation of the critical bicyclic intermediate 17 from PMB-protected Vince lactam 8 following our recently published procedure (Scheme 3).19 The hydroxyl group of 17 was converted to ketone 18 by Swern oxidation. Difluoro intermediate 19 was obtained through the same fluorination conditions described in Scheme 2. The PMB group of 19 was removed by ceric ammonium nitrate followed by lactam hydrolysis and Boc protection, yielding cyclopentane intermediate 21. The hydroxyl group of 21 was dehydrated using Burgess reagent21–22 under reflux, forming cyclopentene intermediate 22. The final product (6) was afforded using the deprotection conditions in Scheme 2.
Scheme 3. Synthetic route to 6a.
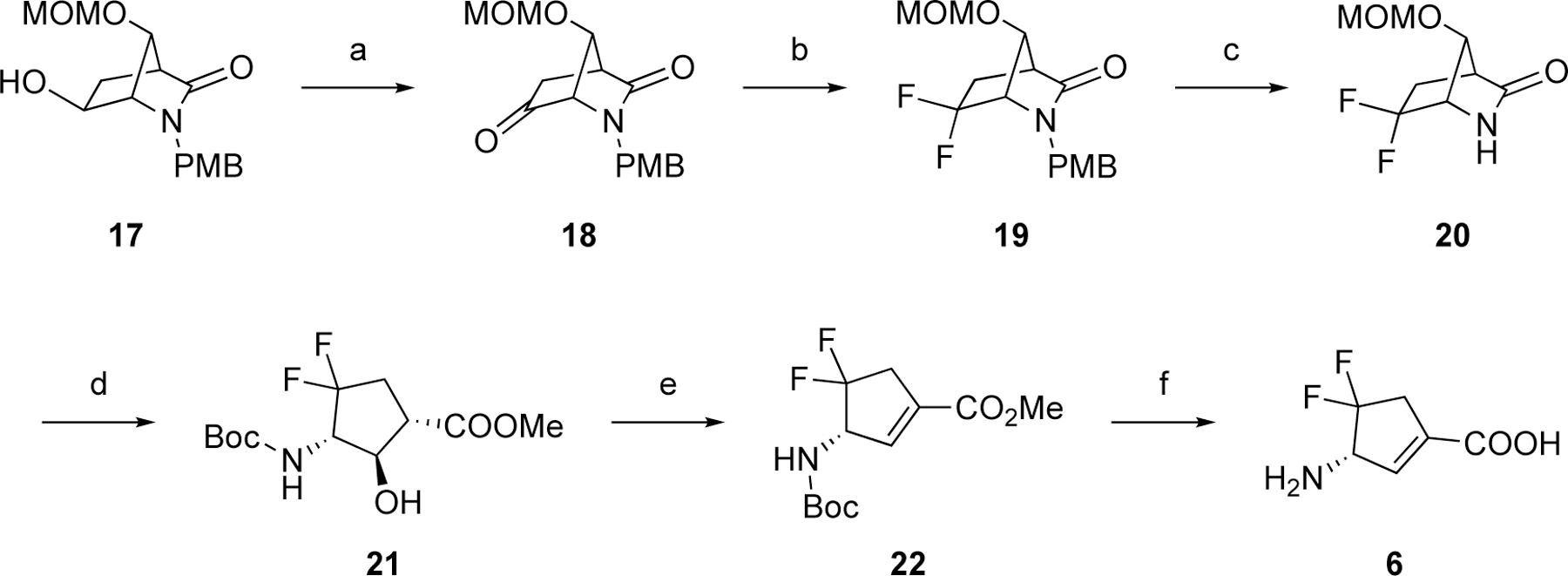
aReagents and conditions. (a) (COCl)2, DMSO, TEA, THF, –78 °C-rt; (b) Deoxo-Fluor (2.7 M in toluene), THF, 120 °C (MW); (c) ceric ammonium nitrate, CH3CN/H2O, rt; (d) i) HCl (1.2 M in MeOH), 85 °C, seal; ii) Boc2O, MeOH, rt; (e) Burgess reagent, THF, 70 °C; (f) 4N HCl, AcOH, 70 °C.
Kinetic Studies of Analogs 4–6.
The kinetic results in Table 1 indicate that all three difluoro-based compounds 4–6 are irreversible inhibitors of hOAT but reversible inhibitors of GABA-AT. Whereas inactivators 4 and 5 are weak-binding inactivators (KI = 4.0 and 2.0 mM, respectively) of hOAT, 6 exhibited significantly higher affinity (KI = 0.06 mM). The partition ratio is defined as the ratio of the number of equivalents of inactivator consumed as a substrate per active site compared to each equivalent of inactivator leading to inactivation.18, 23 The fluoride ion release result is determined as the equivalents of fluoride ions released per active site for each inactivation event, which can be measured with a fluoride ion-selective electrode.23 The partition ratio determination and fluoride ion release results in Table 1 revealed that the majority of reactions of 4 and 5 involves an alternative turnover pathway, resulting in the release of a large fraction of fluoride ions. Compound 6 showed the highest maximal rate constant of inactivation (kinact) relative to 4 and 5, thereby leading to superior inactivation efficiency (kinact /KI) (4 vs. 6, 0.003 vs. 1.300 min−1mM−1, a 400-fold improvement). Furthermore, the inhibitory activities of cyclopentene-based 5 (Ki = 1.40 mM) and 6 (Ki = 1.10 mM) toward GABA-AT are about 10-times weaker than that of cyclopentane-based 4 (Ki = 0.09 mM). It should be noted that, compared to parent cyclopentane 4 (OAT, KI = 4 mM; GABA-AT, Ki = 0.09 mM), newly developed cyclopentene 6 displayed significantly higher binding affinity for OAT but much lower binding affinity for GABA-AT (OAT, KI = 0.06 mM; GABA-AT, Ki = 1.10 mM), indicating 6 has an improved ability to more selectively bind with OAT relative to GABA-AT. Compound 6, also called SS-1–148, exhibited comparable inactivation efficiency against hOAT while retaining reversible inhibition of GABA-AT similar to that of the preclinical compound 1. It also did not show noticeable inhibition of Asp-AT or Ala-AT at concentrations up to 10 mM, motivating interest in the elucidation of the inactivation and turnover mechanisms of 6 with hOAT.
Table 1.
Kinetic Constants for the Inactivation of hOAT and Reversible Inhibition of GABA-AT by 4–6a
| Cmpd | Structure | hOAT | GABA-AT | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| kinact (min−1) | KI (mM) | kinact /KI (min−1mM−1) | Partition ratioc | Fluorideion release (eq.)d | Ki (mM) | ||
| 1 |
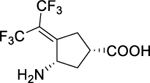
|
0.09 ± 0.01 | 0.09 ± 0.03 | 1.0 ± 0.4 | 12 ± 1b | 79 ± 2b | 5.2 ± 0.6 |
| 4 |
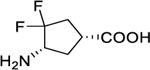
|
0.01 ± 0.00 | 4.00 ± 1.00 | 0.003 ± 0.001 | 2200 ± 70 | 3400 ± 30 | 0.1 ± 0.0 |
| 5 |
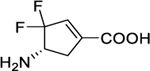
|
0.03 ± 0.01 | 2.00 ± 0.90 | 0.015 ± 0.006 | 790 ± 35 | 750 ± 60 | 1.4 ± 0.1 |
| 6 |
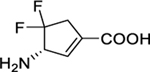
|
0.08 ± 0.01 | 0.06 ± 0.03 | 1.3 ± 0.7 | 34 ± 0 | 34 ± 1 | 1.1 ± 0.1 |
kinact and KI values were determined by the equation: kobs = kinact×[I]/(KI + [I]) and presented as means and standard errors. Ki values were calculated by the Cheng-Prusoff equation: Ki = IC50/(1+[S]/Km) and shown as means and standard errors. IC50 values were obtained using nonlinear regression analysis in GraphPad Prism 8 of a 9-point enzymatic assay with a 2-fold serial dilution against GABA-AT. The partition ratios were determined under conditions in the presence of α-KG, while fluoride ion release results were determined in the absence of α-KG.
Data were extracted from Reference 12.
Enzyme activity remaining was measured as a function of the number of equivalents of 4–6 relative to enzyme concentration. Linear regression analysis was used on the linear portion of the curves to obtain the x-intercept, which was the turnover number (partition ratio = turnover number −1). Data are shown as the means with standard errors.
The fluoride ion release experiments were performed in triplicates. Data are presented as the means with standard deviations.
Proposed Inactivation Mechanism Pathways of 6.
Based on our previous mechanistic studies of other related GABA-AT/hOAT inactivators,8, 13 we propose three potential pathways (Scheme 4). Initially, transimination with 6 would form the external aldimine M1. M1 would then undergo deprotonation to form the quinonoid species M2 followed by fluoride ion elimination, affording the intermediate (M3) that can branch into three different pathways.
Scheme 4.
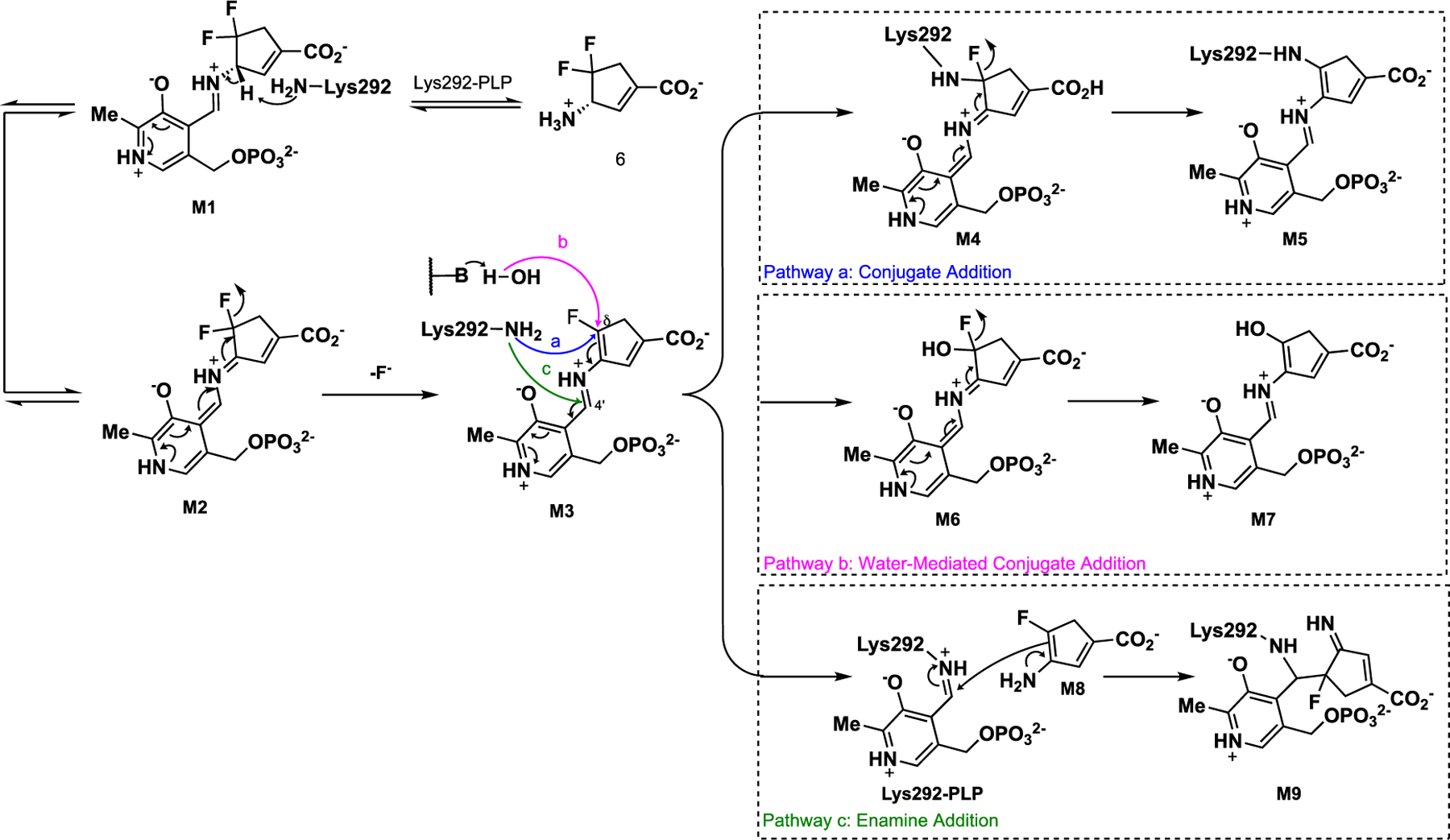
Possible Inactivation Mechanisms for 6
Pathway a is proposed based on our recent findings with the cyclohexene-based analog 2.13 The electrophilic Cδ position of intermediate M3 could undergo conjugate addition by Lys292, forming a covalent bond (M4). Quinonoid species M4 is also subject to a second fluoride ion elimination to give final adduct M5. Pathway b is inspired by the inactivation mechanisms of CPP-11523 and OV32918 with GABA-AT, which are achieved through a water-mediated mechanism resulting in tight electrostatic interactions between their carboxylates and arginine residues in the active site. Lys292 would then activate a water molecule that attacks the electrophilic Cδ position of intermediate M3 followed by a further fluoride ion elimination that would yield enol/carbonyl species M7 rather than establishing a covalent bond with the catalytic lysine residue in hOAT. Pathway c is proposed according to a typical enamine mechanism.19 Lys292 attacks the C4’ position of the aldimine instead of the Cδ position while releasing enamine intermediate M8, which attacks the imine linkage of the internal aldimine (PLP-Lys292) and produces covalent adduct M9.
Plausible Turnover Mechanisms of 6 with hOAT and GABA-AT.
Compound 6 exhibited a relatively high partition ratio (34-fold, Table 1 and Figure S1), indicating that 35 equivalents of 6 are turned over per active site for each equivalent of compound leading to inactivation. Moreover, 34 ± 1 equivalents of fluoride ion (Table 1) are released per inactivation event, indicating that the primary turnover pathway only involves a single fluoride ion elimination step. Previously, we carried out electrostatic potential (ESP) charge calculations to demonstrate that the fluorine atom decreases the nucleophilicity of the enamine intermediate (similar to the structure of M8),13 which may prevent enamine addition. A mass spectrometry (MS)-based analysis of 6 with hOAT (Figure 4A) showed that the molecular weight and fragmentations of the primary metabolite match the structure of M10 in Scheme 5, the hydrolyzed product of enamine intermediate M8. Conversely, we also identified the primary metabolite of 6 in GABA-AT (Figure 4B). The molecular weight and fragmentations suggest that 6 acts as a substrate with this enzyme to yield ketone M11, bearing a difluoro group (Scheme 5).
Figure 4.
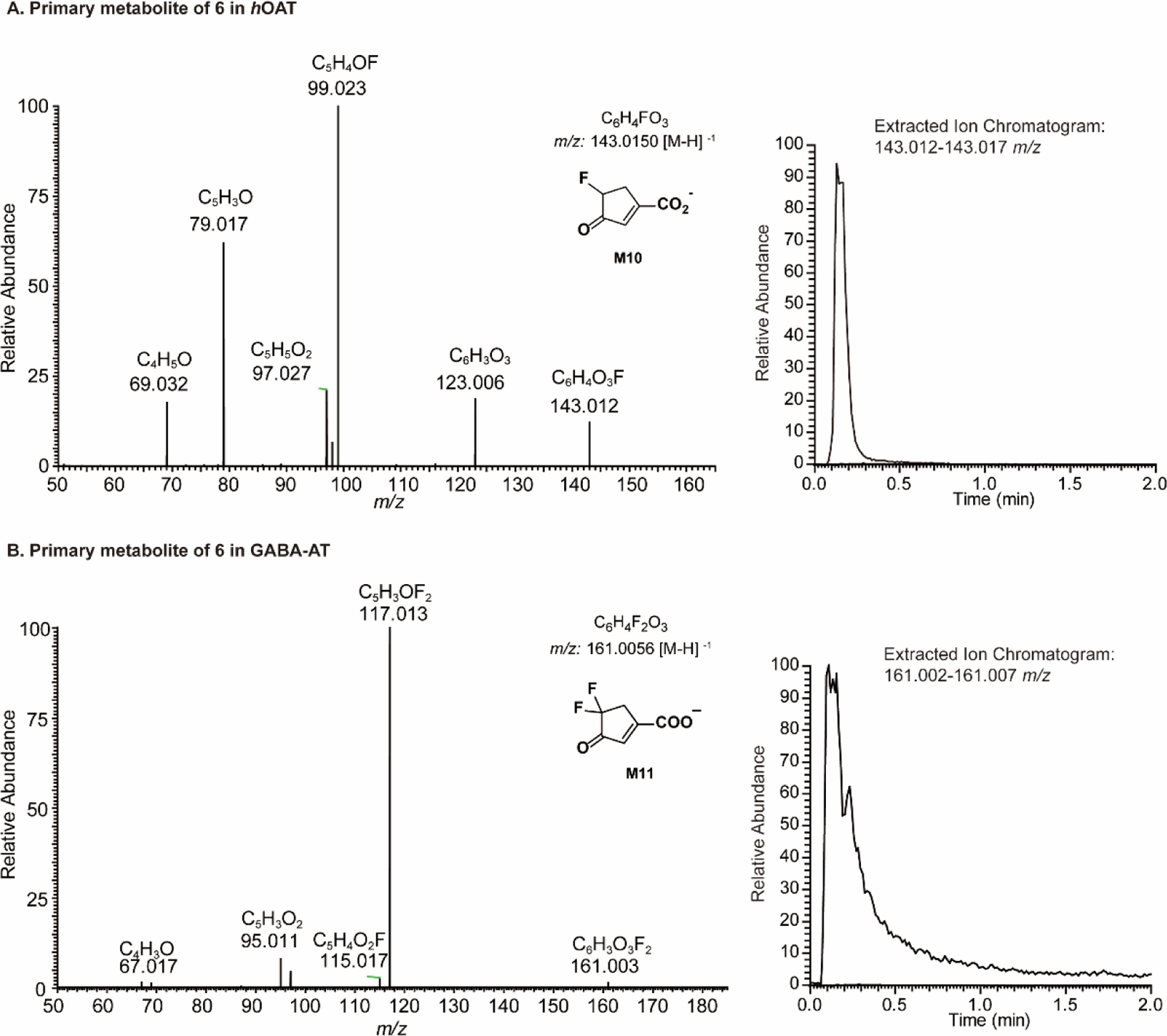
Primary metabolites of 6 in hOAT (A) and GABA-AT (B)
Scheme 5.
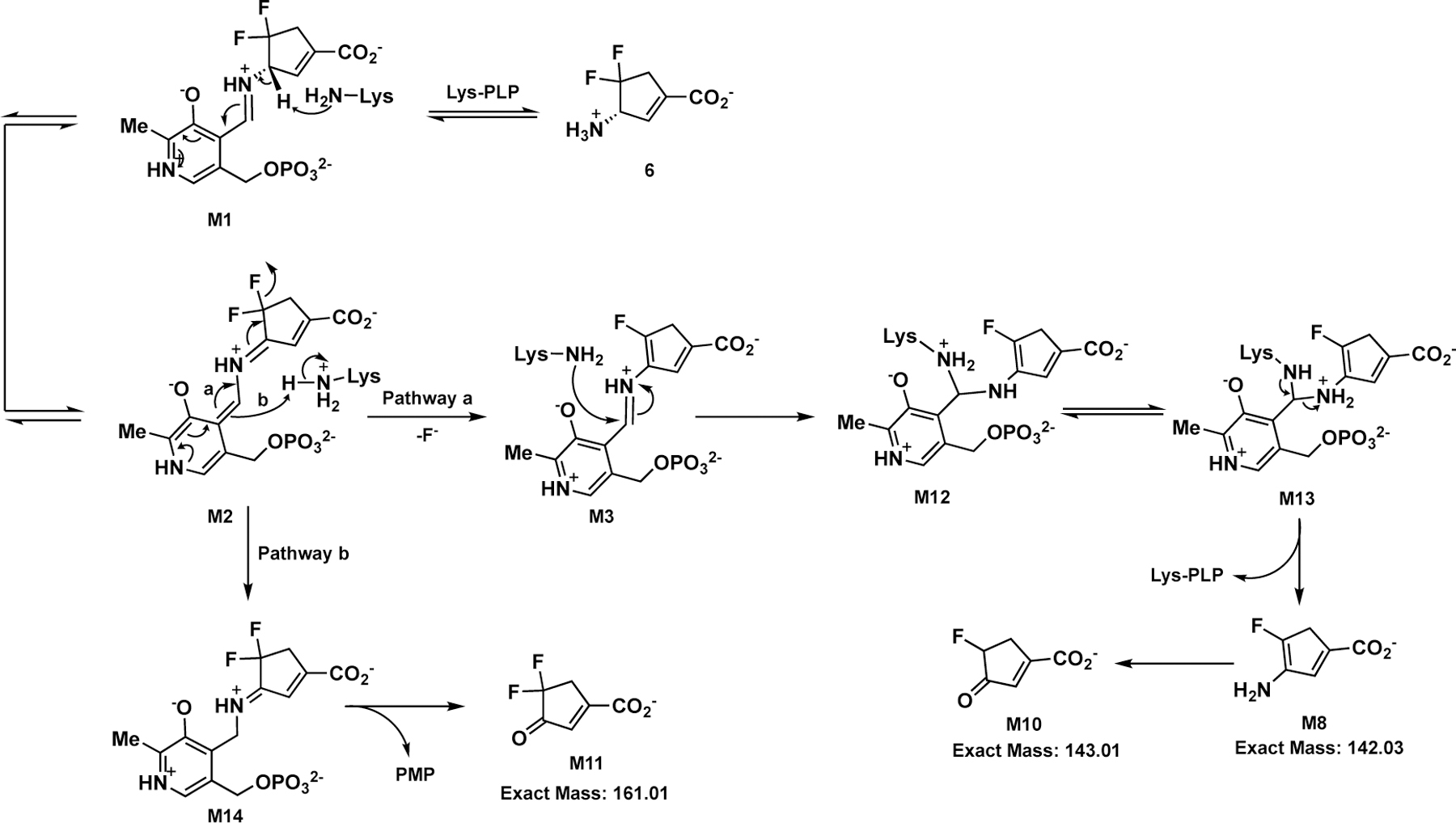
Plausible Turnover Mechanisms of 6 with hOAT and GABA-AT
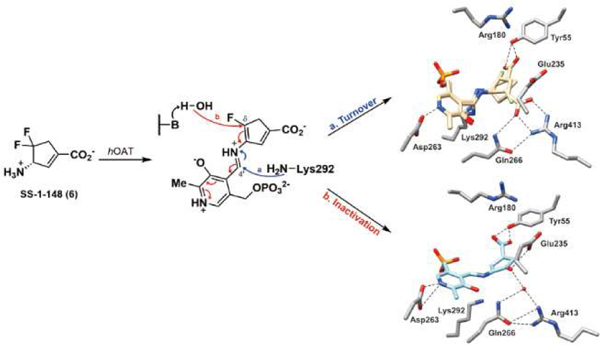
In an attempt to capture the primary intermediate of the non-inactivation pathway, hOAT holoenzyme crystals were utilized to perform one-hour soaking experiments with 6. The hOAT structure was solved by molecular replacement from a previously reported structure (PDB entry 1OAT). The space group for the 6 soaking structure was found to be P3221, and the structure contains three copies of the protein monomer in one asymmetric unit. The crystal structure (PDB entry 7LK1) shown in Figure 5A and Figure S3 indicates that PLP is covalently linked to 6, and a covalent bond between Lys292 and 6 tethering the compound to the enzyme. The covalent bond between Lys292 and 6 represents a stable gem-diamine species that has not been observed in any previous hOAT/ligand crystals.9, 12–13, 24 This observation further validated the gem-diamine precursors (M12 or M13, Scheme 5) of enamine intermediate M8. Moreover, two alternate conformations of this intermediate were observed, which differ in the position of the carboxylate group derived from 6. The first conformation forms a hydrogen bond between Tyr55 and the carboxylate of 6, while the second forms a salt bridge with Arg413. The interpretation that there are two alternate conformations for the intermediate structure was based on the positive density in proximity to Arg413 and Tyr55 as well as the relatively high B-factors for any single conformation. An alternative explanation could include two different, yet structurally similar, intermediate species that could interact with the protein active site in different ways.
Figure 5.
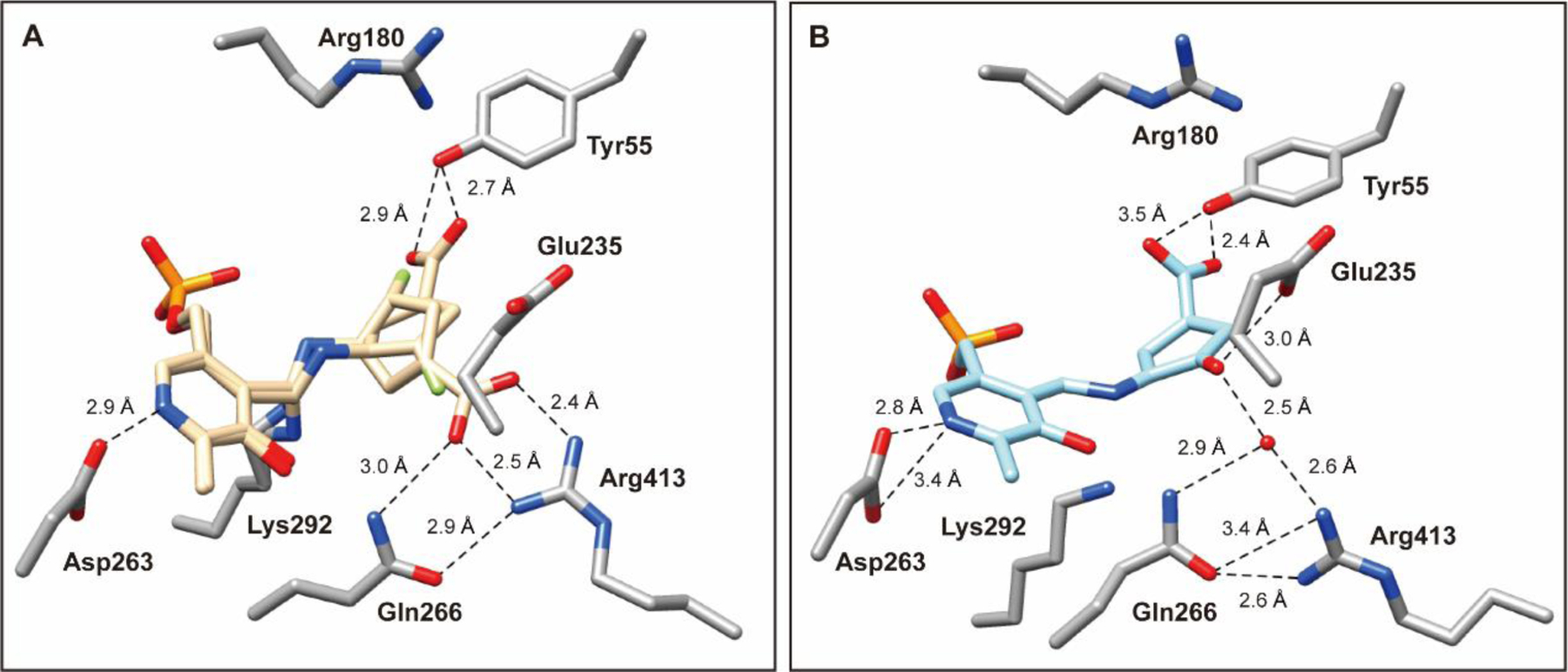
Crystal structures of hOAT resulting from soaking experiment (A; PDB entry 7LK1) and co-crystallization (B; PDB entry 7LK0) with 6. The 6 soaking structure is shown in two alternate conformations (beige): one in which the carboxylate group interacts with Tyr55 (conformation A) and the other in which the carboxylate forms a salt bridge with Arg413 (conformation B). For this specific chain, the refined occupancies of conformers are 0.51 (conformation A) and 0.49 (conformation B). hOAT residues are in stick representation with carbon atoms in the residues colored gray, nitrogen in blue, and oxygen in red; the water molecule is shown as a red sphere. Hydrogen bonding distances between atoms are in Ångstroms (Å) and are shown as black dashed lines.
The major turnover mechanisms for 6 in hOAT and GABA-AT are proposed in Scheme 5. After capturing the PLP coenzyme from Lys292, M1 undergoes deprotonation to give quinonoid M2 that causes elimination of a fluoride ion (Pathway a; Scheme 5) in hOAT to produce aldimine M3 bearing a single fluorine atom. The majority of M3 is attacked by Lys292 at its C4’ position, forming the first gem-diamine (M12), followed by proton transfer,25–26 which results in the second gem-diamine (M13), which is further converted to enamine metabolite M8 and the internal aldimine. M8 undergoes hydrolysis to ketone M10 as the primary metabolite of 6 in hOAT. In contrast, quinonoid intermediate M2 only undergoes electron transfer to yield ketimine M14, which hydrolyzes to release PMP and M11 as the primary metabolites in the GABA-AT reaction. The behavior of 6 in GABA-AT mimics a canonical transamination reaction without eliminating fluoride ions, consistent with its reversible inhibition of GABA-AT. Given the structural differences between hOAT and GABA-AT, the known selective hOAT inactivators (1–3) described above contain a bulky moiety or α-amino group to improve their selectivity. However, the selectivity of 6 for hOAT over GABA-AT occurs because of its competition with substrate GABA.
Plausible Inactivation Mechanisms of 6 with hOAT.
The irreversibility of the inhibition of hOAT by 6 was evaluated with a 48-hour time-dependent dialysis experiment against a buffer containing excess PLP and α-KG (Figure S2). The residual activity of hOAT was unchanged, demonstrating that 6 is an irreversible inhibitor of this enzyme. To reveal the structure of the final product of the inactivation reaction, we co-crystallized hOAT in the presence of excess 6. The crystal structure (PDB entry 7LK0) was solved using the same procedure described above. The space group for the 6 cocrystal (Figure 5B and Figure S4) was found to be P3112 and contained three subunits per asymmetric unit. Similar to the soaking crystal structure shown in Figure 5A and Figure S3, PLP is covalently linked to the 6 moiety in the cocrystal structure. However, no covalent bond is observed between Lys292 and the final product. Moreover, in the published crystal structure of the native enzyme (holo-hOAT; PDB entry 1OAT), Arg413 typically forms a salt bridge with Glu235, which was observed for several cocrystals of hOAT with different inactivators.16, 24, 27 In the hOAT/6 soaking structure (Figure 5A), the salt bridge of Arg413-Glu235 is broken because of the formation of an alternative salt bridge between the carboxylate of one intermediate and Arg413. Interestingly, the Arg413-Glu235 salt bridge is also found to be broken in the hOAT/6 cocrystal structure, while no direct interaction is observed between Arg413 and the ligand. Instead, Arg413, Gln266, and the final product are hydrogen-bonded to the same water molecule. Additionally, the carbonyl oxygen of the ligand shows a hydrogen bond with Glu235 (3.0 Å) (Figure 5B), which may contribute to stabilizing the ligand in the hOAT pocket. The ligand structure in the hOAT/6 cocrystal structure indicates that the inactivation may be attributed to Pathway b in Scheme 4, generating a final product that resembles M7 (theoretical mass: 370.06 Da).
We previously have applied denaturing intact protein mass spectrometry (denaturing MS) to probe covalent inactivation mechanisms of hOAT inactivators.12–13 Here, through denaturing MS using hOAT fully inactivated by 6, only ~16% of hOAT was found to be covalently modified, leading to a mass increase of 370.07 Da (Figure 6A, right). The majority of hOAT remained unmodified (Figure 6A, right) like the untreated hOAT used as a control (Figure 6A, left). The observed mass addition (370.07 ± 0.82 Da) corresponds to the theoretical mass of gem-diamine M15 (370.06 Da; Scheme 6) that could be in equilibrium with the noncovalent form M7. However, we would expect a higher abundance of the covalently bound enzyme under these conditions where the enzyme is fully inactivated. This finding supports a noncovalent inactivation mechanism between hOAT and 6.
Figure 6.
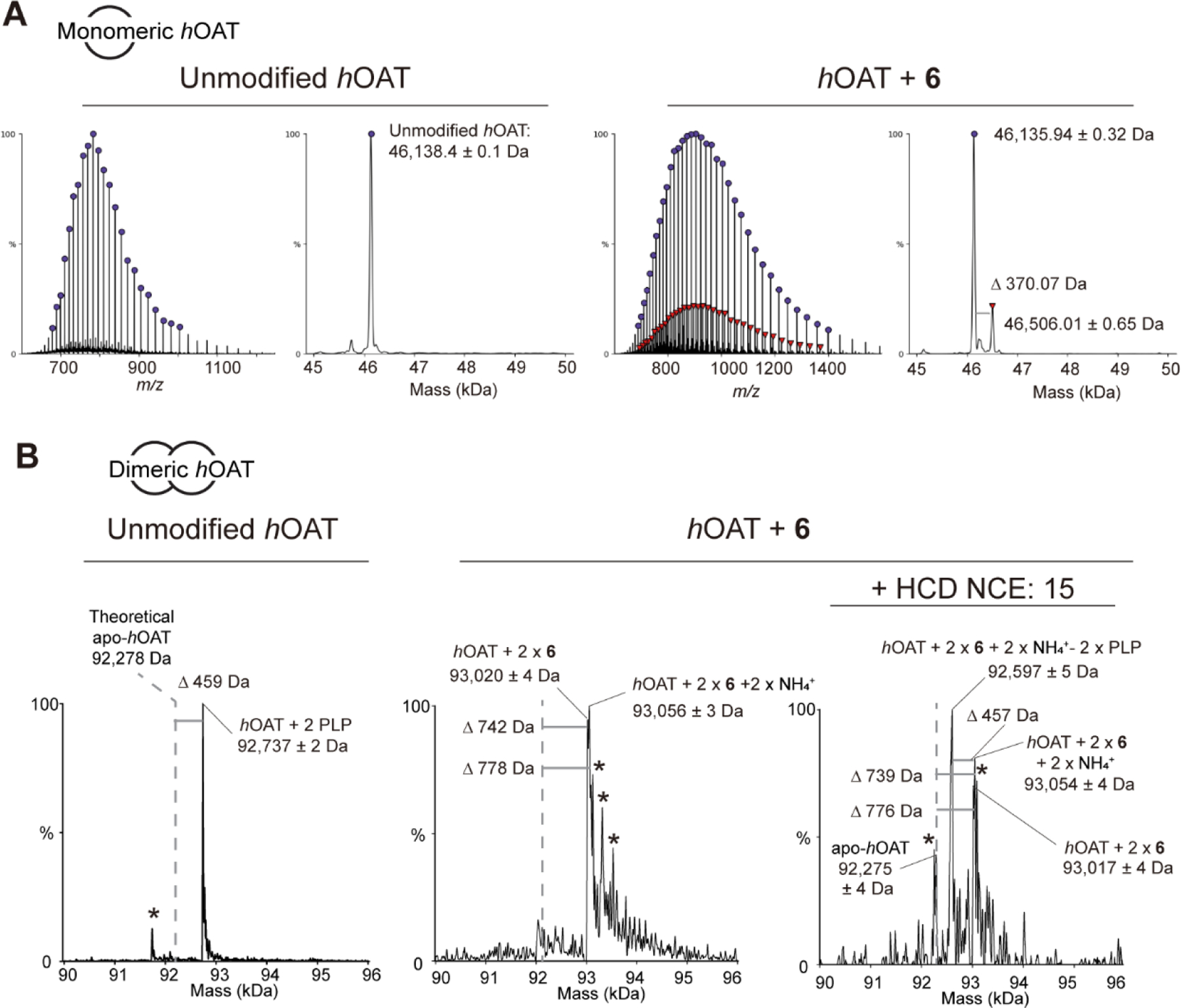
Denaturing (A) and native (B) intact protein mass spectrometry of hOAT untreated and inactivated by 6
Scheme 6.
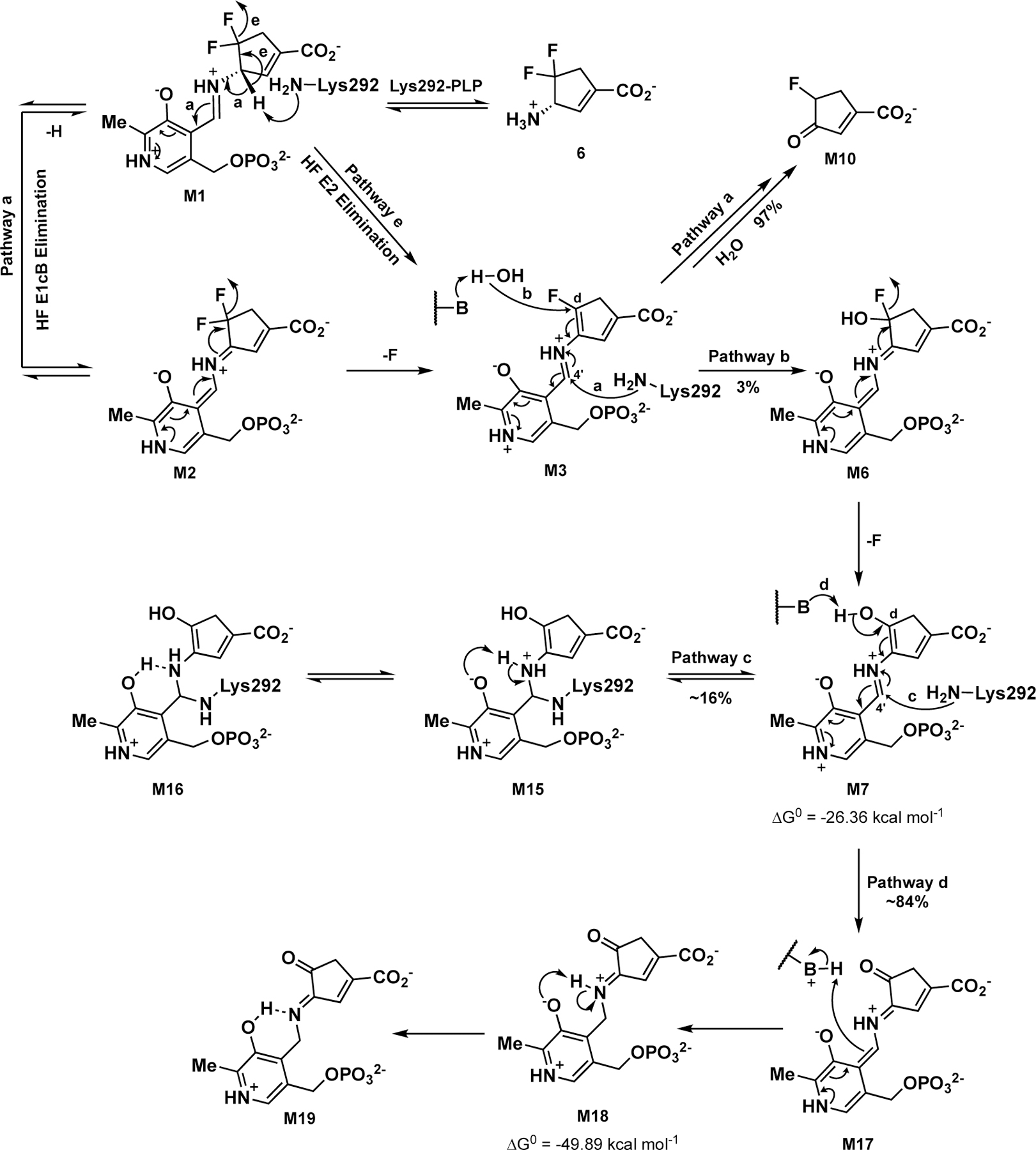
Possible Inactivation Mechanisms of 6 with hOAT
We further employed native mass spectrometry (native MS) to identify solution-state, noncovalent protein interactions that are preserved in the gas phase and characterized the binding of the 6 product in hOAT. The results shown in Figure 6B indicate that untreated hOAT appears as a dimer (92,737 ± 2 Da) that is 459 Da more than the mass of the apo-hOAT dimer. This mass shift is consistent with two PLP-bound internal aldimines that reside in the active sites of the dimer (459 Da shift observed; 460 Da theoretical). A proteolytic proteoform of hOAT was also observed, but only under untreated conditions (Figure 6B, left (*)). In contrast, native, dimeric hOAT fully inactivated by 6 was observed with two high-abundance masses of 93,020 ± 4 Da and 93,056 ± 3 Da, corresponding to a mass addition of 742 Da and 778 Da, respectively (Figure 6B, middle). Several additional masses were observed and can be attributed to salt adducts (Figure 6B, middle (*)). In addition, neither unmodified apo-hOAT nor PLP-bound hOAT was observed. The observed 93,020 ± 4 Da mass is consistent with the predicted mass of M7 (370.06 Da) bound in both active sites (theoretical: 93,018 Da). As the hOAT samples were desalted by dialyzing against 100 mM NH4OAc solution for one week, we think that the 93,056 ± 3 Da observed mass is the predicted mass of M7 complexed with one ammonium cation ([M+NH4]+ = 388.09 Da) in both protein chains (theoretical: 93,054 Da).
To further probe the lability of the 6-hOAT interaction by mass spectrometry, higher-energy collisional dissociation (HCD) was applied to untreated and treated hOAT to dissociate the protein-ligand interactions and eject ligands from the enzyme complex. To eject adducts from protein dimers, HCD was applied with normalized collisional energy (NCE) ranging from 5–15%. No mass shift was observed for untreated hOAT (Figure S5). Under these same conditions, two additional masses were produced for 6-inactivated hOAT (Figure 6B, right). One mass, consistent with apo-hOAT, was observed at 45% relative abundance (observed: 92,275 ± 4 Da; theoretical: 92,278 Da), and a second mass of 92,597 ± 5 Da was observed at 100% relative abundance. Compared to the previously observed dimer mass, which is consistent with each active site bound by M7 and one ammonium cation, this species has a 457 Da mass shift. Interestingly, the high abundance mass shift cannot be explained by the loss of a single M7 ± NH4+ adduct from the protein dimer (theoretical mass for M7-hOAT: 92,648 Da; theoretical mass for M7-hOAT + NH4+: 92,666 Da). The mass shift observed through HCD activation, however, can be explained by the loss of PLP from both active sites, while the 6 moiety with one ammonium cation was apparently retained (observed: 457 Da; theoretical: 460 Da). This finding is surprising, given that the same collisional energy does not eject PLP from the untreated protein dimer (Figure S5).
As no apo-enzyme is observed by native MS and approximately 84% of hOAT was in an apo-enzyme state under denaturing MS conditions at pH 2.5, these results indicate that noncovalent M7 is the primary form after inactivation, while covalently-bound M15 is a minor form that is in equilibrium with M7. Therefore, M7 generated from the water-mediated Pathway b (Scheme 4) seems to be the final product of 6 after inactivation. However, given its highly conjugated structure, M7 readily tautomerizes. A Gibb’s free energy calculation28 was performed on MOPAC to assess the stabilities of M7 and the related tautomeric forms in the hOAT’s active site. The results shown in Figure S6 suggest that, compared with enol form M7 (Tautomer 1; ΔGo = −26.36 kcal mol−1), the corresponding ketone (Tautomer 5, ΔGo = −48.07 kcal mol−1) and two potential ketimines (Tautomer 8, ΔGo = −49.89 kcal mol−1 and Tautomer 9, ΔGo = −49.07 kcal mol−1) are relatively more stable. Moreover, a species that shows absorbance at ~275 nm was determined as the final product in the subsequent transient-state spectrophotometric measurements (Figure 8C), thereby ruling out all external aldimines (e.g., M7 and Tautomer 5) that would display absorbance maxima at ~420 nm. The results above indicate that the final state is more likely a gem-diamine or ketimine that typically have absorbance maxima ~330–340 nm.25, 29 According to the literature, in comparison with the ketoenamine moiety, containing a protonated aldimine and deprotonated hydroxyl, the neutral enolimine of the hydroxyl and aldimine groups exhibit shifted absorption maximum (410 nm vs. 330 nm).30 Taken together, we believe that gem-diamine M15 (Scheme 6) is in equilibrium with M7, and ketimine M18 is tautomerized from M7; both M15 and M18 may then undergo additional electron transfer to form M16 and M19, respectively. Because of the neutral states of gem-diamine M16 and ketimine M19, their absorption transitions shift from 330 nm to 275 nm, consistent with the absorbance of the final species observed in the transient-state measurements.
Figure 8.
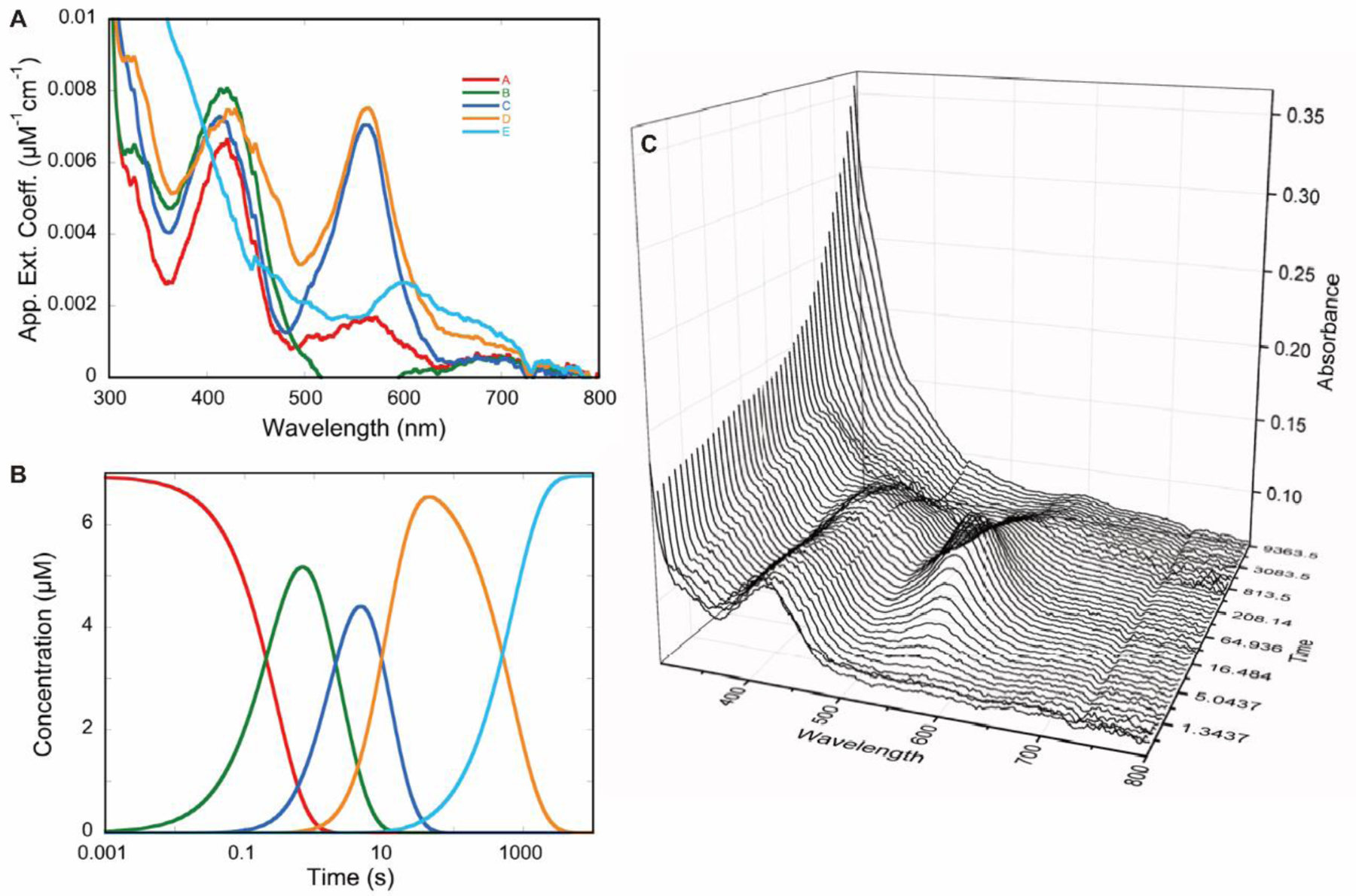
Partial deconvolution by singular value decomposition (SVD) of transient state absorption changes observed for hOAT reacting with 6. hOAT (6.94 µM; final concentration) was allowed to react in a stopped-flow spectrophotometer with 6 (1040 µM; final concentration) at 10 °C. To obtain time resolution sufficient to analyze kinetic rates spanning four orders of magnitude, a composite CCD absorbance dataset was prepared spanning 250–800 nm and 0.0137–9843 seconds by splicing together averaged short- and long time-frame datasets. These data were fit to a linear irreversible four-step model in which the rate constants were constrained to those determined from single-wavelength analyses (Figure 7). Deconvoluted composite spectra were derived from SVD analysis (A). The species concentration profile based on the rate constants were used to fit the dataset (B). A three-dimensional depiction of a subset of spectra from the dataset were analyzed (C).
Proposed inactivation pathways for 6 with hOAT are summarized in Scheme 6. The initial external aldimine M1 undergoes deprotonation, catalyzed by Lys292, and forms the first quinonoid (M2). Elimination of a single fluoride ion follows to yield monofluoro aldimine M3 from M2. The C4’ position of the majority of M3 (~97%; determined by its partition ratio) is attacked by Lys292, which releases an enamine metabolite that hydrolyzes to ketone M10 as the primary metabolite (Pathway a; Schemes 5 and 6).
The Cδ position of a small portion of M3 (~3%) goes through a water-mediated nucleophilic attack (Pathway b; Scheme 6), generating the second quinonoid (M6). Intermediate M6 undergoes another fluoride ion elimination to form M7. Subsequently, a small fraction of M7 (~16%) may covalently bond to Lys292 at its C4’ position via a gem-diamine form (M15) (Pathway c; Scheme 6), which is in equilibrium with M7 and also facilitate further proton transfer to generate the neutral gem-diamine (M16), which should be a more stable form. However, the majority of M7 (~84%) tautomerizes to a more favorable ketimine (M18, the most stable tautomeric form in Figure S6), which is followed by proton transfer to afford ketimine M19 as the primary final product (Pathway d; Scheme 6).
Transient-State Measurements of hOAT Inhibited by 6.
As a variety of transient states are involved in the proposed mechanisms, we performed rapid-mixing spectrophotometric measurements in an attempt to capture the kinetics of the inhibition of hOAT by 6. This approach takes advantage of the conjugated species that accumulate sequentially in PLP-dependent aminotransferase reactions.9 The inhibition reaction that occurs with 6 was interpreted in combination with crystal structures (mentioned above) acquired for different stages of the reaction progression. The experimental data shown in Figures 6 and 7 indicate that the reaction of 6 with hOAT is complex. Four discernable phases were observed with evidence of at least two parallel reaction paths (Figure 8), indicating that the mechanistic conclusions drawn are necessarily from undetermined models. Within the deadtime of the stopped-flow instrument, the reaction of 6 with hOAT formed a spectrum signal (~420 nm) consistent with an external aldimine (red spectrum, Figures 7A–B). Titration of hOAT with 6 modulated the rate and the extent of accumulation of a second external aldimine that is presumably additive with the aldimine formed in the deadtime (green spectrum, Figures 7A–B). The second external aldimine forms with a rate dependence that indicates a bimolecular reaction (6.2 ×103 M−1s−1) and suggests that 6 interacts with hOAT in the least two ways, resulting in parallel reaction paths (Figures 6A–B). The intensity of the combined external aldimine spectra decays partially with the accumulation of a spectral transition characteristic for a quinonoid species (~560 nm) at 0.4 s−1 (Figures 6A–B and 7A–B). The apparent quinonoid species is formed with concomitant and partial decay of the external aldimine transitions that are approximately equal in amplitude to that gained with the second external aldimine accumulation. This suggests that these species reside on the same reaction pathway.
Figure 7.
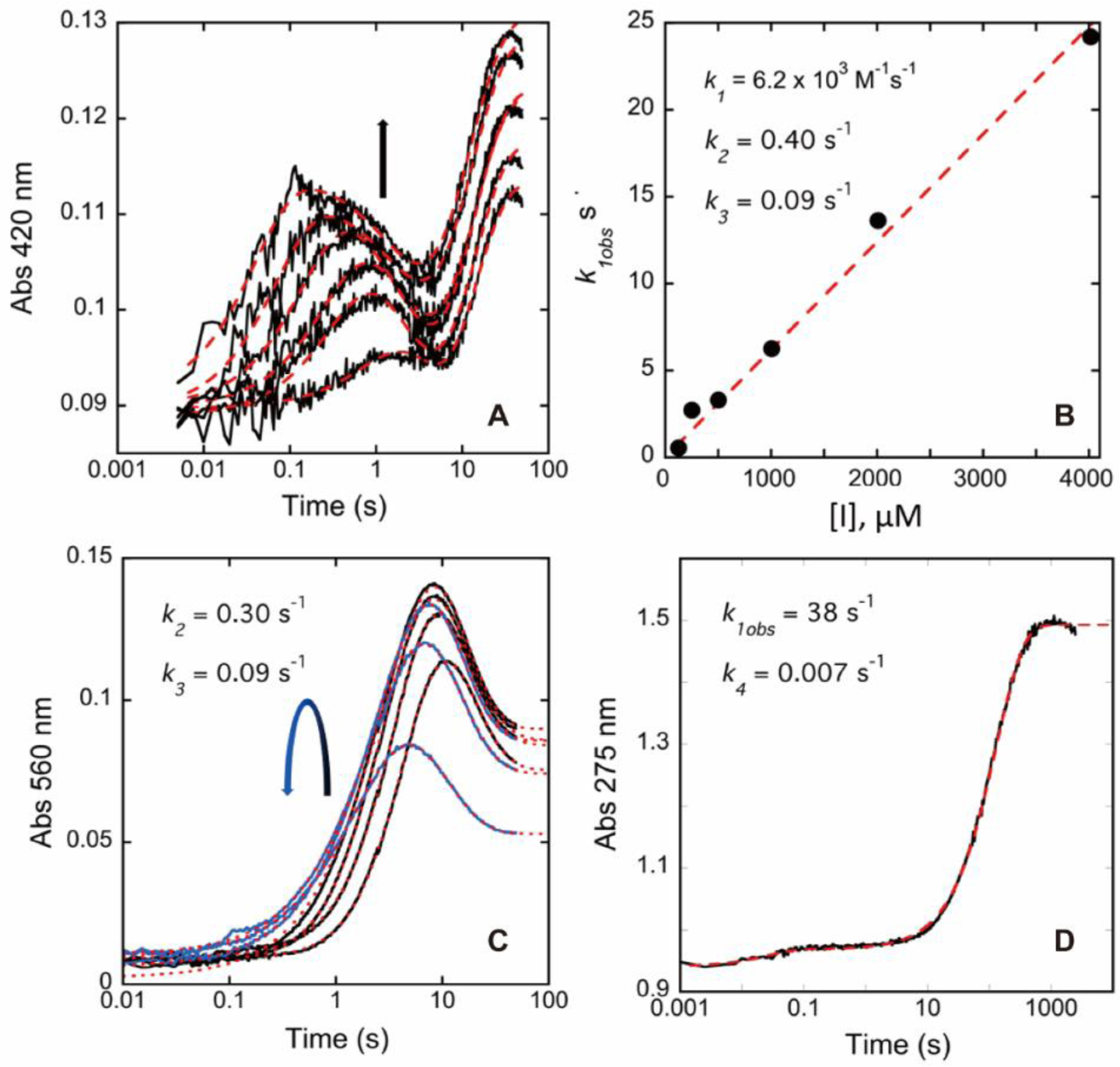
Transient state absorption changes observed at 275, 420, and 560 nm for hOAT reacting with 6. OAT (12.7 µM final) was mixed with 6 (126, 251, 502, 1004, 2008, 4016 µM), and CCD spectra were collected for the timeframe 0.009–49.2 seconds. (A) The data observed at 420 nm fit to a linear combination of three exponential terms according to equation (3) described in the Supporting Information. The arrow indicates the trend observed in amplitude for increasing inhibitor concentration. (B) The observed rate constant dependence of the first phase observed at 420 nm fits equation (5) described in the Supporting Information. The values for k2 and k3 indicated are the average values obtained from the fit in Figure 7A. The fit is shown by red dashes. (C) The data observed at 560 nm. The curved arrow indicates the trend observed in amplitude for increasing inhibitor concentration. These data were fit to a linear combination of two (1004, 2008, and 4016 µM, blue traces) or three (126, 251, and 502 µM, black traces) exponential terms according to equation (4) described in the Supporting Information. The fit is shown by red dashes. (D) The data observed at 275 nm over 2000 seconds obtained in the presence of 8032 µM 6 fit a linear combination of two exponential terms according to equation (4) in the Supporting Information. The fit is shown by red dashes.
It has been reported that the deprotonation of initial external aldimine M1 and stepwise fluoride elimination steps proposed in Scheme 6 are typically considered as an E1cB elimination mechanism.31–32 The electron-withdrawing effect of fluorine and the protonated nitrogen of the aldimine may stabilize the formed carbanion state during the elimination reaction.32–33 Quinonoid transient state M2 is expected to form between the first and second external aldimines (M1 and M3). However, the stopped-flow experimental results suggest that the antiperiplanar hydrogen and fluorine atoms in M1 undergo a Lys292-assisted E2 mechanism,31 i.e., concerted loss of a proton fluoride ion, with the formation of the alkene as a more favorable fluoride ion elimination pathway, affording the second external aldimine (M3) directly as the single transient state species (Pathway e; Scheme 6). This E2 elimination pathway has never been observed experimentally in previous mechanistic studies of other related PLP-dependent aminotransferase inactivators.1, 8
Assuming a quinonoid extinction coefficient of ~30 mM−1cm−1,9 the fractional accumulation of the quinonoid observed is ~20% of the total reacting species at 1 mM of 6. The quinonoid then decays at a rate of 0.09 s−1, while the residual transitions assigned to the external aldimine species broaden and persist (orange spectrum, Figures 7A–B). Quinonoid M6 seems to be a rare case of quinonoid species (~560 nm) that can be observed based on the turnover (Pathway a; Scheme 5) and inactivation (Pathway b; Scheme 6) mechanisms. The final phase observed occurs with a rate constant of 0.007 s−1; in this phase, the features of the external aldimine decay with a pronounced increase in absorption intensity at ~275 nm, indicative of a loss of conjugation (blue spectrum, Figures 7A–B), consistent with gem-diamine M16 and ketimine M19, which are proposed as the final products in Scheme 6.
Collectively, these data support a dominant pathway comprised of multiple distinct external aldimine species that ultimately decay to a less conjugated product (turnover mechanism; Pathways e then a to M10; Scheme 6) and a second minor pathway that forms an initial external aldimine more slowly but then proceeds through a quinonoid intermediate and decays to also form a nonconjugated product (inactivation mechanism; Pathways e-b-c and d to M16 and M19, Scheme 6). The data shown in Figure 8C indicate that the proportion of each pathway is dependent on the concentration of 6. Higher concentrations of 6 diminish the accumulation of the quinonoid species but do not alter the observed rates of accumulation and decay, suggesting that the more rapid and dominant pathway sequesters a larger fraction of enzyme at higher 6 concentrations.
The Significance of the Conjugated Alkene of 6.
After better understanding the inactivation and turnover mechanisms of 6, we carried out computational calculations to compare analogs 4–6. Our previous studies have revealed that incorporating an extra double bond into the cyclopentane ring system establishes an α,β-conjugated carboxylate and facilitates the deprotonation step as a result of the increased acidity of the adjunct proton, leading to the enhanced inactivation rate constant.18–19 In this work, we performed theoretical pKa calculations using the DFT/B3LYP method34 at 298K to predict the proton’s acidity at the Cγ position of difluoro analogs 4–6 (Figure 3). The results shown in Figure 9A suggest that the hydrogen (highlighted with red) of PLP-bound 6 (M1) with an α, β-conjugated carboxylate system displays the lowest pKa value among the three analogs, while the pKa value of the corresponding proton in PLP-bound 5 (M1”) is not noticeably affected by the introduced double bond at the Cα and Cε positions relative to the parent cyclopentane 4 (M1’). Taken together with their distinct kinact values (Table 1), our findings suggest that the more acidic hydrogen at the Cγ position facilitates the first external aldimine of 6 (M1), which initiates an E2 fluoride ion elimination step rather than the typical E1cB elimination reaction, thus contributing to its enhanced rate constant.
Figure 9.
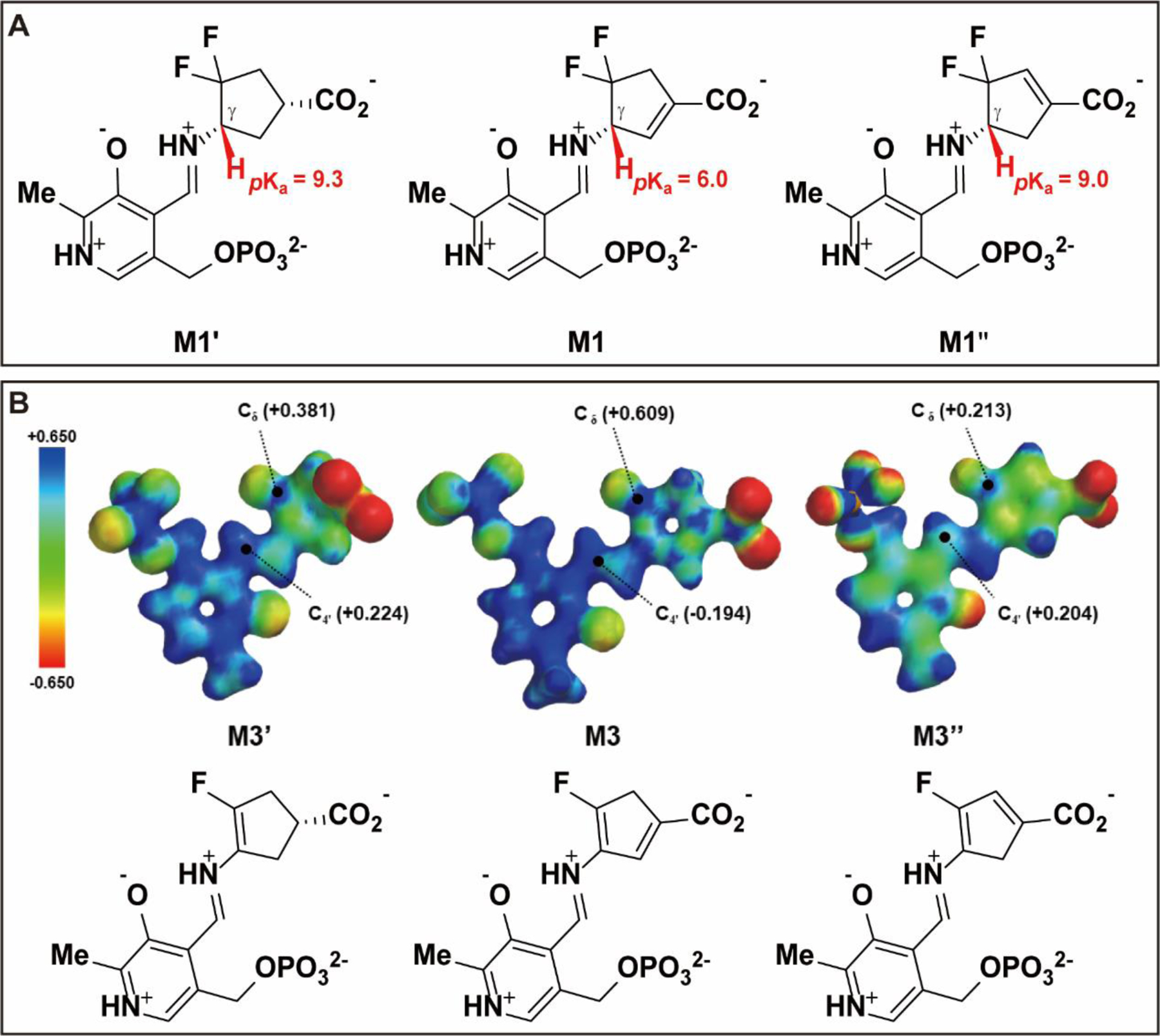
Theoretical pKa calculations of the hydrogen at the Cγ position using the DFT/B3LYP method (A) and electron density maps colored coded to the electrostatic potential of intermediates and ESP charges of Cδ and C4’ positions (B).
The partition ratio determination and fluoride ion release results (Table 1) elucidated that 4 and 5 predominantly participate in the alternative turnover pathway rather than the inactivation pathway, simultaneously releasing large amounts of fluoride ions. The electron density maps and ESP charge calculations13 (Figure 9B) indicate that the electrophilicity of Cδ in the transient state M3 of 6 is much higher than that in the corresponding transient states of 4 and 5 (M3’ for 4 and M3” for 5), suggesting that the Cδ position of M3 is more reactive than in the other two intermediates. The C4’ positions in the aldimine linkage of M3’ and M3” display comparable electrophilicity that is much greater than that of M3, which demonstrates that M3’ and M3” are easier to be attacked by catalytic Lys292 to trigger their turnover pathways, eventually resulting in significant partition ratios for 4 and 5. Furthermore, additional MS-base analyses in hOAT revealed that, similar to 6 in hOAT, cyclopentene analog 5 generated a ketone bearing a single fluorine atom as its primary metabolite (M5–1, Figure S7B). In contrast, cyclopentane 4 not only formed a monofluorinated ketone (M4–1, Figure S7A) but also generated a ketone metabolite bearing a hydroxyl group (M4–2, Figure S7A), which was not detected in the MS-base analyses of cyclopentene analogs. According to the proposed mechanisms for 6 in Scheme 6, it is likely that the incorporated double bond also plays a critical role in stabilizing the conjugated system.
CONCLUSION
Previously, we discovered (S)-3-amino-4,4-difluorocyclohex-1-enecarboxylic acid (WZ-2–051, 2), which inactivates hOAT through a covalent addition-aromatization mechanism (Scheme 1B). However, it also exhibited apparent inhibition of GABA-AT. In this work, (S)-3-amino-4,4-difluorocyclopent-1-enecarboxylic acid (SS-1–148, 6) exhibited comparable inactivation efficiency to that of preclinical stage selective hOAT inactivator BisCF3 (1) but was demonstrated to be a reversible inhibitor of GABA-AT. The kinetic studies and computational calculations provide evidence to support the notion that the conjugated alkene of 6 in its cyclopentene ring is essential for retaining high hOAT inactivation efficiency. A soaking experiment was performed to obtain a quasi-stable gem-diamine intermediate covalently bound to Lys292 in the soaked crystal, an intermediate that has never been captured in other studies of related aminotransferase inactivators. Co-crystallization of hOAT and 6 captured a stable noncovalent final product in the hOAT cocrystal complex. The critical salt bridge of Arg413-Glu235 in hOAT was found to be broken in both crystal complexes. In addition, we have applied native MS, for the first time, in studies of aminotransferase inactivators to further support the noncovalent pathway as the primary inactivation mechanism of 6. We also performed intact MS to support a covalent modification observed as a minor form, which appears to be a gem-diamine structure in equilibrium with the noncovalent form. Using rapid-mixing experiments, we observed, for the first time, that the first external aldimine of 6 undergoes a lysine-assisted E2 fluoride ion elimination instead of the typical E1cB elimination mechanism, forming the second external aldimine as the single transient state. Overall, we have carried out comprehensive mechanistic studies to demonstrate that 6 mainly inactivates hOAT through a noncovalent water-mediated mechanism. However, it is still unclear why there are distinct inactivation mechanisms for cyclopentene 6 and the corresponding cyclohexene 2.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (Grant R01 DA030604 to R.B.S. and Grant P30 DA018310 to N.L.K.) and National Science Foundation (Grant 2015210477 to P.F.D) for financial support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Comet Bridges Stampede2 through allocation TG-CHE190070, which is supported by National Science Foundation grant number ACI-1548562. This work made use of the IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN). X-ray diffraction data collection used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of financial support. Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DEAC02–06CH11357. The use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817). G.M.F. is a recipient of a fellowship from FAPESP, Brazil. We also thank Drs. Wei Zhu and Pathum M. Weerawarna for constructive comments and revision.
ABBREVIATIONS
- AFPA
(S)-4-amino-5-fluoropentanoic acid
- Boc2O
di-tert-butyl dicarbonate
- Deoxo-Fluor
bis(2-methoxyethyl)aminosulfur trifluoride
- DMPK
drug metabolism and pharmacokinetics
- DIPEA
N,N-diisopropylethylamine
- DBDMH
1,3-dibromo-5,5-dimethylhydantoin
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DCM
dichloromethane
- KHMDS
potassium bis(trimethylsilyl)amide
- PO
Per os administration
- THF
tetrahydrofuran
- TEA
trimethylamine
- TBAI
tetra-n-butylammonium iodide
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Supplementary figures, scheme, table, experimental methods, syntheses, spectra, crystallographic methods and data
Accession Codes
Atomic coordinates and corresponding structure factors for the soaking result and cocrystal complex have been deposited at the Protein Data Bank (PDB) as the 7LK1 and 7LK0 entries, respectively. Authors will release the atomic coordinates upon article publication.
The authors declare no competing financial interest.
REFERENCES
- 1.Lee H; Juncosa JI; Silverman RB, Ornithine aminotransferase versus GABA aminotransferase: implications for the design of new anticancer drugs. Med. Res. Rev 2015, 35, 286–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanner JJ; Fendt SM; Becker DF, The proline cycle as a potential cancer therapy target. Biochemistry 2018, 57, 3433–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang L; Zeng J; Geng P; Fang C; Wang Y; Sun M; Wang C; Wang J; Yin P; Hu C; Guo L; Yu J; Gao P; Li E; Zhuang Z; Xu G; Liu Y, Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin. Cancer Res 2018, 24, 474–485. [DOI] [PubMed] [Google Scholar]
- 4.Cadoret A; Ovejero C; Terris B; Souil E; Levy L; Lamers WH; Kitajewski J; Kahn A; Perret C, New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM; Zucman-Rossi J; Pikarsky E; Sangro B; Schwartz M; Sherman M; Gores G, Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [DOI] [PubMed] [Google Scholar]
- 6.Zigmond E; Ben Ya’acov A; Lee H; Lichtenstein Y; Shalev Z; Smith Y; Zolotarov L; Ziv E; Kalman R; Le HV; Lu H; Silverman RB; Ilan Y, Suppression of hepatocellular carcinoma by inhibition of overexpressed ornithine aminotransferase. ACS Med. Chem. Lett 2015, 6, 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eliot AC; Kirsch JF, Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem 2004, 73, 383–415. [DOI] [PubMed] [Google Scholar]
- 8.Silverman RB, Design and mechanism of GABA aminotransferase inactivators. Treatments for epilepsies and addictions. Chem. Rev 2018, 118, 4037–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butrin A; Beaupre BA; Kadamandla N; Zhao P; Shen S; Silverman RB; Moran GR; Liu D, Structural and kinetic analyses reveal the dual inhibition modes of ornithine aminotransferase by (1S,3S)-3-amino-4-(hexafluoropropan-2-ylidenyl)-cyclopentane-1-carboxylic acid (BCF3). ACS Chem. Biol 2021, 16, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan Y; Gerasimov MR; Kvist T; Wellendorph P; Madsen KK; Pera E; Lee H; Schousboe A; Chebib M; Brauner-Osborne H; Craft CM; Brodie JD; Schiffer WK; Dewey SL; Miller SR; Silverman RB, (1S, 3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115), a potent gamma-aminobutyric acid aminotransferase inactivator for the treatment of cocaine addiction. J. Med. Chem 2012, 55, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copeland RA, Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal 2005, 46, 1–265. [PubMed] [Google Scholar]
- 12.Moschitto MJ; Doubleday PF; Catlin DS; Kelleher NL; Liu D; Silverman RB, Mechanism of inactivation of ornithine aminotransferase by (1S,3S)-3-amino-4-(hexafluoropropan-2-ylidenyl)cyclopentane-1-carboxylic acid. J. Am. Chem. Soc 2019, 141, 10711–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu W; Doubleday PF; Catlin DS; Weerawarna PM; Butrin A; Shen S; Wawrzak Z; Kelleher NL; Liu D; Silverman RB, A remarkable difference that one fluorine atom confers on the mechanisms of inactivation of human ornithine aminotransferase by two cyclohexene analogues of gamma-aminobutyric acid. J. Am. Chem. Soc 2020, 142, 4892–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daune G; Gerhart F; Seiler N, 5-Fluoromethylornithine, an irreversible and specific inhibitor of L-ornithine:2-oxo-acid aminotransferase. Biochem. J 1988, 253, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolkenius FN; Knodgen B; Seiler N, DL-canaline and 5-fluoromethylornithine. Comparison of two inactivators of ornithine aminotransferase. Biochem. J 1990, 268, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Storici P; Capitani G; Muller R; Schirmer T; Jansonius JN, Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J. Mol. Biol 1999, 285, 297–309. [DOI] [PubMed] [Google Scholar]
- 17.Qiu J; Silverman RB, A new class of conformationally rigid analogues of 4-amino-5-halopentanoic acids, potent inactivators of gamma-aminobutyric acid aminotransferase. J. Med. Chem 2000, 43, 706–720. [DOI] [PubMed] [Google Scholar]
- 18.Juncosa JI; Takaya K; Le HV; Moschitto MJ; Weerawarna PM; Mascarenhas R; Liu D; Dewey SL; Silverman RB, Design and mechanism of (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid, a highly potent gamma-aminobutyric acid aminotransferase inactivator for the treatment of addiction. J. Am. Chem. Soc 2018, 140, 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen S; Doubleday PF; Weerawarna PM; Zhu W; Kelleher NL; Silverman RB, Mechanism-based design of 3-amino-4-halocyclopentenecarboxylic acids as inactivators of GABA aminotransferase. ACS Med. Chem. Lett 2020, 11, 1949–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh R; Vince R, 2-Azabicyclo[2.2.1]hept-5-en-3-one: chemical profile of a versatile synthetic building block and its impact on the development of therapeutics. Chem. Rev 2012, 112, 4642–4686. [DOI] [PubMed] [Google Scholar]
- 21.Wang BL; Gao HT; Li WD, Total synthesis of (+)-iresin. J. Org. Chem 2015, 80, 5296–5301. [DOI] [PubMed] [Google Scholar]
- 22.Gross LJ; Stark CBW, Regioselective dehydration of alpha-hydroxymethyl tetrahydrofurans using Burgess’ reagent under microwave irradiation. Org. Biomol. Chem 2017, 15, 4282–4285. [DOI] [PubMed] [Google Scholar]
- 23.Lee H; Doud EH; Wu R; Sanishvili R; Juncosa JI; Liu D; Kelleher NL; Silverman RB, Mechanism of inactivation of gamma-aminobutyric acid aminotransferase by (1S,3S)-3-amino-4-difluoromethylene-1-cyclopentanoic acid (CPP-115). J. Am. Chem. Soc 2015, 137, 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mascarenhas R; Le HV; Clevenger KD; Lehrer HJ; Ringe D; Kelleher NL; Silverman RB; Liu D, Selective targeting by a mechanism-based inactivator against pyridoxal 5’-phosphate-dependent enzymes: mechanisms of inactivation and alternative turnover. Biochemistry 2017, 56, 4951–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Salvo ML; Scarsdale JN; Kazanina G; Contestabile R; Schirch V; Wright HT, Structure-based mechanism for early PLP-mediated steps of rabbit cytosolic serine hydroxymethyltransferase reaction. Biomed. Res. Int 2013, 2013, 458571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soniya K; Awasthi S; Nair NN; Chandra A, Transimination reaction at the active site of aspartate aminotransferase: a proton hopping mechanism through pyridoxal 5’-phosphate. ACS Catalysis 2019, 9, 6276–6283. [Google Scholar]
- 27.Shah SA; Shen BW; Brunger AT, Human ornithine aminotransferase complexed with L-canaline and gabaculine: structural basis for substrate recognition. Structure 1997, 5, 1067–1075. [DOI] [PubMed] [Google Scholar]
- 28.Stewart JJ, MOPAC: a semiempirical molecular orbital program. J. Comput. Aided Mol. Des 1990, 4, 1–105. [DOI] [PubMed] [Google Scholar]
- 29.Karsten WE; Ohshiro T; Izumi Y; Cook PF, Reaction of serine-glyoxylate aminotransferase with the alternative substrate ketomalonate indicates rate-limiting protonation of a quinonoid intermediate. Biochemistry 2005, 44, 15930–15936. [DOI] [PubMed] [Google Scholar]
- 30.Thibodeaux CJ; Liu HW, Mechanistic studies of 1-aminocyclopropane-1-carboxylate deaminase: characterization of an unusual pyridoxal 5’-phosphate-dependent reaction. Biochemistry 2011, 50, 1950–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clift MD; Ji H; Deniau GP; O’Hagan D; Silverman RB, Enantiomers of 4-amino-3-fluorobutanoic acid as substrates for gamma-aminobutyric acid aminotransferase. Conformational probes for GABA binding. Biochemistry 2007, 46, 13819–13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gokcan H; Konuklar FA, Theoretical study on HF elimination and aromatization mechanisms: a case of pyridoxal 5’ phosphate-dependent enzyme. J. Org. Chem 2012, 77, 5533–5543. [DOI] [PubMed] [Google Scholar]
- 33.Alunni S; De Angelis F; Ottavi L; Papavasileiou M; Tarantelli F, Evidence of a borderline region between E1cb and E2 elimination reaction mechanisms: a combined experimental and theoretical study of systems activated by the pyridine ring. J. Am. Chem. Soc 2005, 127, 15151–15160. [DOI] [PubMed] [Google Scholar]
- 34.Ghalami-Choobar B; Dezhampanah H; Nikparsa P; Ghiami-Shomami A, Theoretical calculation of the pKa values of some drugs in aqueous solution. Int. J. Quantum Chem 2012, 112, 2275–2280. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.