

INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm and a clonal disorder of hematopoietic stem or progenitor cells and is thought to arise from the granulocyte-macrophage progenitor.1 The median age of diagnosis is 67 years on the basis of SEER data. Most patients (85%-90%) with CML present in chronic phase, but a small percentage of patients present in either accelerated or blast phase. Fewer than 10% of patients with CML present in blast phase. After a median time of 3-5 years, untreated chronic phase CML will progress to either accelerated or blast phase CML. The median overall survival is 7-11 months even in the tyrosine kinase inhibitor (TKI) era.2,3 Blast phase CML is an aggressive malignancy that has a high rate of refractoriness and often requires multiagent chemotherapy in addition to high-potency TKI therapy. Therapeutic options are extremely limited in the relapsed and refractory setting.
The defining molecular abnormality that drives leukemogenesis in CML is BCR-ABL1 fusion gene, resulting from a reciprocal translocation between chromosomes 9 and 22.1 The breakpoint cluster region on chromosome 22 fuses with the Abelson-1 nonreceptor tyrosine kinase on chromosome 9 to produce a novel oncoprotein. This molecular aberration is also the therapeutic target of TKI therapy. TKI therapy forms the basis of treatment for chronic, accelerated, and blast phase CML. The goal of blast phase therapy is to convert the disease back into chronic phase and proceed with curative intent therapy in the form of allogeneic stem cell transplant for suitable patients. However, induction of remission for blast phase CML is a formidable task, and the treatment path is commonly interrupted by frequent relapses with TKI-resistant mutations.
CASE PRESENTATION
A 56-year-old man with no significant past medical history presented with severe fatigue, shortness of breath, and bifrontal headache for two months. Laboratory workup showed a WBC count of 122,900/μL with 15% band neutrophils, hemoglobin of 8 g/dL, and a platelet count of 6,000/μL. Flow cytometry of peripheral blood showed 11% lymphoblasts. Bone marrow aspirate or biopsy showed lymphoid blast phase CML with a cellularity of 90%-100% composed predominantly of lymphoblasts (Fig 1A). Flow cytometry of bone marrow showed positivity for c-TdT, CD34, CD19, CD22, CD71, CD10, c-CD79a, and human leukocyte antigen-DR, but negativity for CD20 (Fig 1B). Chromosome analysis showed 46,XY,t(9;22)(q34;q11.2)[21]/46,sl,−9,+der(22)t(9;22)[2] (Fig 1C). B-cell clonality studies showed clonal immunoglobulin kappa gene rearrangement (Fig 1E).
FIG 1.
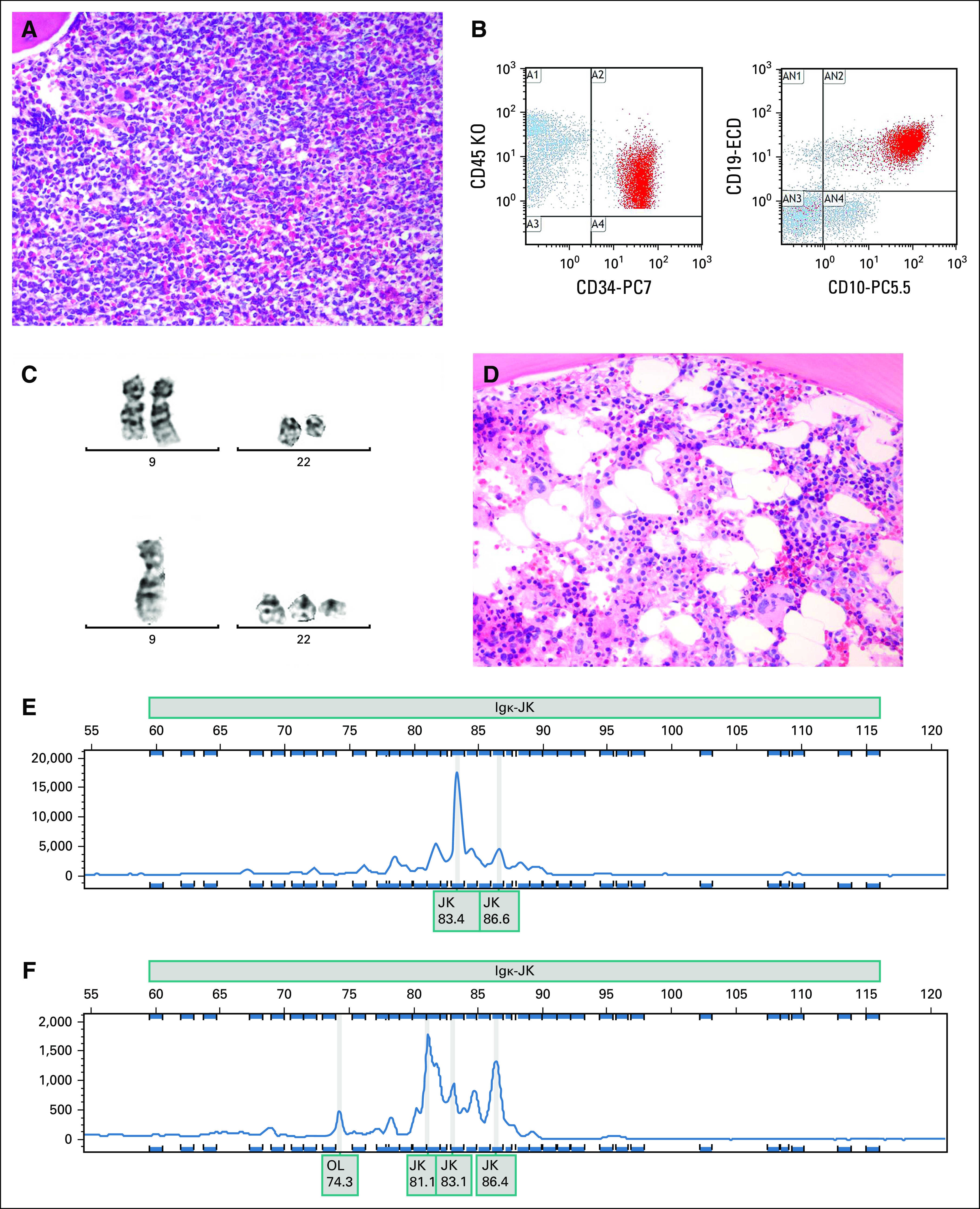
(A) Bone marrow biopsy at diagnosis with a diffuse infiltrate of medium-sized primitive lymphoid cells consistent with blasts. (B) Multiparameter flow cytometry shows a CD45(dim)CD34(+)CD19(+)CD10(+) immunophenotype. (C) Top: Chromosome analysis showed that 21 of 23 cells contained a reciprocal translocation between chromosomes 9 and 22, with breakpoints in bands 9q34 and 22q11.2, indicative of BCR-ABL1 rearrangement, which was confirmed by fluorescence in situ hybridization. Bottom: Two of 23 cells also demonstrated loss of chromosome 9 and an additional derivative chromosome 22. (D) Remission marrow after treatment with maturing trilineage hematopoiesis. (E) Multiplex polymerase chain reaction–based B-cell clonality studies identified a clonal Igκ gene rearrangement with a monoclonal peak with an electrophoretic mobility of 83.4. (F) B-cell clonality studies on the post-treatment specimen showed an oligoclonal pattern with no evidence of the previously characterized monoclonal Igκ rearrangement. Igκ, immunoglobulin kappa; JK, joining region of the immunoglobulin kappa gene; OL, immunoglobulin lambda light chain.
He subsequently received induction with hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone (HyperCVAD) part 1A plus dasatinib 140 mg once daily. BCR-ABL1 tyrosine kinase domain (TKD) mutation T315I was not detected at that time. Bone marrow aspirate or biopsy on day 26 showed a cellularity of 70%-80%, with 2% blasts. Chromosome analysis showed 46,XY,t(9;22) (q34;q11.2)[3]/46,XY[17], consistent with a partial cytogenetic response. BCR-ABL1 polymerase chain reaction (PCR) showed the p210 breakpoint with an International Scale (IS) of 28.2% in the bone marrow.
Three months after diagnosis, bone marrow aspirate or biopsy showed 90% cellularity and 90% blasts, consistent with relapse. BCR-ABL1 PCR showed an IS of 1,155% with the p210 breakpoint. He received HyperCVAD part 1B. The TKD mutation T315I was detected, and he was started on ponatinib 45 mg orally once daily. One month later, BCR-ABL1 PCR showed an IS of 1.18% with the p210 breakpoint. Two months after starting ponatinib, bone marrow biopsy showed relapse with 90% cellularity and 63% blasts. Cytogenetics showed t(9;22) in 70% of metaphases, consistent with loss of partial cytogenetic response. BCR-ABL1 PCR showed the IS of 522% from bone marrow and 397% from peripheral blood.
At the time of this second relapse, blasts were positive for CD19. He received cyclophosphamide 60 mg/kg (one dose only) for cytoreduction and then started blinatumomab 9 μg/d once daily induction 2 days later. Four days after the start of blinatumomab, he developed laboratory evidence of cytokine release syndrome with an elevated C-reactive protein of 157 mg/L but did not develop immune effector cell–associated neurotoxicity, tumor lysis syndrome, or infectious complications at that time. He did not have clinical deterioration from cytokine release syndrome, and his C-reactive protein decreased after day 4. He continued ponatinib. After a 28-day induction of blinatumomab, BCR-ABL1 PCR from peripheral blood showed an IS of 0.014%, consistent with major molecular response with 4-log reduction. Bone marrow biopsy showed trilineage hematopoiesis with a cellularity of 50% and no evidence of lymphoblasts by morphology, immunohistochemistry, or flow cytometry (Fig 1D). Chromosome analysis showed normal metaphases. BCR-ABL1 PCR from the bone marrow showed an IS of 0.1% (Fig 2). Complete blood count showed a WBC of 5,100/μL, hemoglobin of 9.8 g/dL, a platelet count of 159,000/μL, and a blast count of 0%. This was consistent with complete hematologic response, complete cytogenetic response, and major molecular response. B-cell clonality studies after blinatumomab treatment showed oligoclonality with no immunoglobulin kappa rearrangement (Fig 1F). After a 14-day treatment-free interval, he started cycle 2 blinatumomab and proceeded with haploidentical allogeneic stem-cell transplant from a related donor. He received myeloablative conditioning with fludarabine 30 mg/m2 once daily for 7 days and busulfan 3.2 mg/kg once daily for 3 days, followed by a cell dose of 7.51 × 106 CD34(+) cells/kg, which was infused 8 days after the completion of blinatumomab. Bone marrow biopsy on day +30 after transplant showed full donor chimerism and a BCR-ABL1 IS of 0% in both bone marrow and peripheral blood (Fig 2).
FIG 2.

Kinetics of molecular response of blast phase chronic myeloid leukemia with the addition of blinatumomab to ponatinib. Labels on the x-axis indicate the start time of therapy. CMR, complete molecular remission; HCT, hematopoietic cell transplant; HyperCVAD, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone; IS, International Scale; MMR, major molecular response.
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
DISCUSSION
CML is a myeloproliferative neoplasm and accounts for 15% of leukemias in adults. Chronic phase CML carries a risk of progression to advanced-phase disease in 3-5 years in the absence of treatment.1-3 Clinical manifestations include constitutional symptoms, rash, erythromelalgia, pruritus, hepatosplenomegaly, and bleeding. Initial treatment for chronic phase CML involves the use of a BCR-ABL1 TKI as monotherapy. The T315I mutation is a resistance mutation that occurs in 56% of patients with blast phase at diagnosis, compared with 30% of patients with chronic phase at diagnosis.4
Blast phase CML is defined variably by multiple organizations: blast phase is defined by the presence of 20% or more blasts in the bone marrow or peripheral blood per 2016 WHO criteria and by the presence of 30% or more blasts by the 2020 European LeukemiaNet criteria.5,6 Despite the absence of a unified diagnostic definition, blast phase CML is uncommon and very challenging to treat given the aggressive disease biology and limited treatment options. Lymphoid blast phase accounts for about 30% of blast phase CML and is defined by a B lineage immunophenotype, in contrast to myeloid blast phase that accounts for 70% of blast phase cases.7 The rates of complete hematologic response, complete cytogenetic remission, and complete molecular remission are 90%, 58%, and 25%, respectively, with established treatment involving HyperCVAD plus TKI.8 Although response rates have been reported to be high, the durability of response is limited (a median duration of remission of 14 months), and median overall survival in the post-TKI era remains poor (17 months).8 High-potency TKI therapy (dasatinib 140 mg daily), compared with imatinib, is preferred and carries a major hematologic response rate of 42% and a major cytogenetic response rate of 50%. However, long-term outcomes are still poor (21% overall survival at 24 months).9 There are very few long-term survivors of blast phase CML.
Relapsed and refractory B-cell acute lymphoblastic leukemia (ALL) can be treated with a variety of novel agents including blinatumomab, inotuzumab ozogamicin, or chimeric antigen receptor-T therapy, but options for relapsed blast phase CML are often limited to cytotoxic multiagent chemotherapy plus TKIs.10,11 Prognosis is dismal unless patients proceed with allogeneic transplant, which is reported to improve the median overall survival to 93 months.8 However, patients often do not make it to allogeneic transplant with curative intent. TKD resistance mutations can pose further barriers to effective definitive treatment for blast phase CML. Large-scale clinical studies are limited because of the rarity of this disease.
The distinction between Ph(+) B-cell ALL and lymphoid blast phase CML is often challenging. Both entities show similar morphologic and immunophenotypic features, and the distinction requires integration of molecular and clinical features, including whether the lymphoblast proliferation occurs in a patient with previously established chronic or accelerated phase CML (Table 1). The cell of origin in Ph(+) B-cell ALL is the pre-B-cell, whereas the cell of origin in blast phase CML is the granulocyte-macrophage progenitor.1 The BCR-ABL1 transcript is more commonly e1a2 in Ph(+) B-cell ALL, compared with e13a2 or e14a2 in CML. Therefore, the oncoprotein product in Ph(+) B-cell ALL is more commonly p190, whereas that in blast phase CML is more commonly p210.12 Furthermore, genomic differences also exist between the two entities: Ph(+) B-cell ALL is more commonly characterized by gains of chromosome 9p21-24 with losses of other genomic regions.13 Analysis of the nonlymphoid population for BCR-ABL1 fusion may help with differentiating de novo Ph(+) B-cell ALL from lymphoid blast phase CML. The prognosis can be quite favorable for younger patients with Ph(+) B-cell ALL, and the disease is curable with allogeneic transplant especially in the younger and fit population. The treatment of adolescents with Ph(+) B-cell ALL usually involves a pediatric-inspired regimen, and others may receive therapy with HyperCVAD, with the addition of rituximab for CD20-positive disease.14 Allogeneic transplant is commonly recommended for patients with high-risk disease who can tolerate transplant. Blinatumomab can also be used if measurable residual disease (MRD) persists at a level beyond 0.1%.15
TABLE 1.
Distinction Between Ph(+) B-Cell ALL and Lymphoid Blast Phase CML

To date, there are very few reports of blinatumomab in blast phase CML. A previous report of remission after blinatumomab plus TKI studied mostly patients with Ph(+) B-cell ALL and not blast phase CML.16 In the previous report, only one patient had overt blast phase CML.16 The time to response was 56 days, compared with our patient who responded in 29 days. The novelty of this case is that we describe a different cell of origin that has responded to a bispecific T-cell engager. Blinatumomab is already US Food and Drug Administration–approved for Ph(+) B-cell ALL but not for lymphoid blast phase CML, and we show a more rapid response in the setting of blast phase CML.
Since the incidence of blast phase has decreased significantly in the post-TKI era and since lymphoid blast crisis is even rarer, accrual of patients on a clinical trial is challenging. Currently, a phase II clinical trial (NCT03263572) is actively recruiting patients and is studying the combination of blinatumomab and ponatinib, plus intrathecal methotrexate and cytarabine, in patients with lymphoid blast phase CML or Ph(+) B-cell ALL (clinicaltrials.gov). The trial allows for patients in the treatment-naive or relapsed or refractory settings. If completed, this trial will likely provide the largest body of evidence for using blinatumomab for these patients.
Our approach to lymphoid blast phase CML is to perform allogeneic transplant in first remission if the depth of remission is sufficiently adequate. If MRD testing is positive at a low to moderate level, we favor proceeding with allogeneic transplant, as this will offer the best chance of sustained remission. If MRD testing is positive at a high level in the setting of morphologic remission, we consider treatment with blinatumomab first (as an attempt to achieve a deeper response), followed by allogeneic transplant. In B-cell ALL, blinatumomab therapy in patients harboring MRD > 0.1% has been shown to result in a high rate (78%) of MRD complete response, which was associated with a longer relapse-free survival and overall survival.15,17 However, the data for blinatumomab are very limited for blast phase CML. If MRD testing is negative, we still favor transplant although it is reasonable to consider other options and weigh the risks and benefits on the basis of transplant-related mortality. We ultimately favor incorporation of kinetics of molecular response and depth of remission into decision making about allogeneic transplant.
In summary, we show rapid and deep remission induced by blinatumomab in CD19(+) blast phase CML. Clinicians may consider the use of bispecific T-cell engager therapy as a bridge to transplant. Additional studies are needed before expanding the US Food and Drug Administration indication of blinatumomab to include lymphoid blast phase CML.
ACKNOWLEDGMENT
We thank the patient for providing consent to publish this report.
Shyam A. Patel
Consulting or Advisory Role: Dedham Group
Jonathan M. Gerber
Patents, Royalties, Other Intellectual Property: US Patent No. 9,012,215 and US Patent No. 10,222,376
No other potential conflicts of interest were reported.
AUTHOR CONTRIBUTIONS
Conception and design: Shyam A. Patel, Jonathan M. Gerber
Financial support: Jonathan M. Gerber
Administrative support: Jonathan M. Gerber
Provision of study materials or patients: Shyam A. Patel, Lloyd Hutchinson, Jonathan M. Gerber
Collection and assembly of data: Shyam A. Patel, Anne W. Higgins
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by the authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Shyam A. Patel
Consulting or Advisory Role: Dedham Group
Jonathan M. Gerber
Patents, Royalties, Other Intellectual Property: US Patent No. 9,012,215 and US Patent No. 10,222,376
No other potential conflicts of interest were reported.
REFERENCES
- 1.Jamieson CHM, Ailles LE, Dylla SJ, et al. :Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351:657-667, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, Guilhot F, O'Brien SG, et al. :Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 355:2408-2417, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Sokal JE:Evaluation of survival data for chronic myelocytic leukemia. Am J Hematol 1:493-500, 1976 [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Yang H, Xu X, et al. :Mutations in the BCR-ABL1 kinase domain in patients with chronic myeloid leukaemia treated with TKIs or at diagnosis. Oncol Lett 20:1071-1076, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arber DA, Orazi A, Hasserjian R, et al. :The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391-2405, 2016 [DOI] [PubMed] [Google Scholar]
- 6.Hochhaus A, Baccarani M, Silver RT, et al. :European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 34:966-984, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swerdlow SH, Campo E, Harris NL, et al. : WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Volume 2 (ed 4). Lyon, France, IARC, 2017 [Google Scholar]
- 8.Strati P, Kantarjian H, Thomas D, et al. :HCVAD plus imatinib or dasatinib in lymphoid blastic phase chronic myeloid leukemia. Cancer 120:373-380, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saglio G, Hochhaus A, Goh YT, et al. :Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: Efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer 116:3852-3861, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kantarjian HM, DeAngelo DJ, Stelljes M, et al. :Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med 375:740-753, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kantarjian H, Stein A, Gökbuget N, et al. :Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med 376:836-847, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arana-Trejo RM, Ruíz Sánchez E, Ignacio-Ibarra G, et al. :BCR/ABL p210, p190 and p230 fusion genes in 250 Mexican patients with chronic myeloid leukaemia (CML). Clin Lab Haematol 24:145-150, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Grace C, Nacheva EP:Significance analysis of microarrays (SAM) offers clues to differences between the genomes of adult Philadelphia positive ALL and the lymphoid blast transformation of CML. Cancer Inform 11:173-183, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maury S, Chevret S, Thomas X, et al. :Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med 375:1044-1053, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Curran E, Stock W:Taking a “BiTE out of ALL”: Blinatumomab approval for MRD-positive ALL. Blood 133:1715-1719, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Assi R, Kantarjian H, Short NJ, et al. :Safety and efficacy of blinatumomab in combination with a tyrosine kinase inhibitor for the treatment of relapsed Philadelphia chromosome-positive leukemia. Clin Lymphoma Myeloma Leuk 17:897-901, 2017 [DOI] [PubMed] [Google Scholar]
- 17.Gokbuget N, Dombret H, Bonifacio M, et al. :Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 131:1522-1531, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]