

Abstract
Alpha-1 antitrypsin (AAT) deficiency (AATD) is an autosomal co-dominant condition that predisposes to the development of lung disease, primarily emphysema. Emphysema results from the breakdown of lung matrix elastin by proteases, including neutrophil elastase, a protease normally inhibited by AAT. AATD also predisposes to liver (cirrhosis) and skin (panniculitis) disease, and to vasculitis. The prevalence of AATD is estimated to be approximately 1 in 3,500 individuals in the United States. However, lack of awareness of AATD among some physicians, misperceptions regarding the absence of effective therapy, and the close overlap in symptoms with asthma and non-AATD chronic obstructive pulmonary disease are thought to contribute to under-recognition of the disease. In patients with AATD, treatment with intravenous AAT augmentation therapy is the only currently available treatment known to slow the progression of emphysema. Moreover, smoking cessation and other lifestyle interventions also help improve outcomes. Early diagnosis and intervention are of key importance due to the irreversible nature of the resultant emphysema. Liver disease is the second leading cause of death among patients with AATD and a minority of patients present with panniculitis or antineutrophil cytoplasmic antibody-associated vasculitis, thought to be directly related to AATD. Though no randomized trial has assessed the effectiveness of augmentation therapy for AATD-associated panniculitis, clinical experience and case series suggest there is a benefit. Other diseases putatively linked to AATD include aneurysmal disease and multiple neurological conditions, although these associations remain speculative in nature.
Keywords: Alpha-1 antitrypsin, Alpha-1 antitrypsin deficiency, antineutrophil cytoplasmic antibody-associated vasculitis, chronic obstructive pulmonary disease, emphysema, panniculitis
Introduction
Alpha-1 antitrypsin (AAT) deficiency (AATD) is an autosomal co-dominant condition that, in the most severe AATD genotypes, predisposes to disease in the lungs, liver, skin, and blood vessels. The primary pathogenic process in AATD-related lung disease is the breakdown of elastin in lung tissue as a result of excess uninhibited neutrophil elastase, a protease that is inhibited by AAT.1,2 Independent of elastase inhibition, AAT has a range of anti-inflammatory and immunomodulatory properties.3
AAT is a 52 kDa single-chain glycoprotein composed of 394 amino acid residues and three asparagine-linked complex carbohydrate side chains.1 Hepatocytes are the primary source of AAT, although macrophages and bronchial epithelial cells also synthesize the protein. AAT is known to inhibit a variety of serine proteinases, though its preferential target is the extracellular endopeptidase neutrophil elastase.1 AAT is encoded by the SERPINA1 gene, mutations in which can lead to the development of AATD.4,5 The M-type protease inhibitor (PI) is the most common and wild-type allele of AAT in the general population. PI*MM individuals have “normal” AAT plasma levels (>20 µmol/L) and the M-type AAT functions normally to bind and inactivate neutrophil elastase. Common deficient PI variants seen in patients with AATD include the Z and S variants; homozygotes for the Z allele, called PI*ZZ individuals, have plasma levels around 5–6 µmol/L, and PI*SS individuals have plasma levels approximately 60% of normal (14–20 µmol/L).1,6 As discussed below, the risk for lung disease and extrapulmonary manifestations of AATD are dependent upon the individual’s AATD genotype and AAT serum level.
The genotype most commonly associated with severe AATD is PI*ZZ, the prevalence of which in the United States has been estimated in various ways, all with concordant estimates of approximately 1 in 3,500. An early study in the 1970s by the Oregon State Public Health Laboratory reported a prevalence of 1 in 5,097;7 another study in 1989 conducted in St. Louis reported a prevalence as high as 1 in 2,857.8 Based on epidemiologic reports and by using mathematical modeling, the prevalence of the PI*ZZ genotype was recently estimated to be 1 in 4,126 among non-Hispanic whites in the United States.9 Notably, some geographic variation in prevalence rates was predicted, with higher values in the Eastern states that progressively decreased in the Western and South-Eastern areas of the country.9 However, as numerous other at-risk genotypes exist, the true prevalence and disease burden associated with AATD is difficult to estimate. In addition, low disease awareness has hampered detection efforts, and most affected individuals remain undiagnosed.10 Diagnostic delay in AATD is an ongoing challenge, with evidence that individuals may be symptomatic for 5–8 years and see many physicians before initial diagnosis of AATD.11 Furthermore, delay in diagnosis has been shown to be associated with worsened clinical status at the time of diagnosis.12
The current chapter of this review series addresses the clinical manifestations of AATD, primarily focusing on lung disease. The use of imaging technology in AATD diagnosis, management, and clinical research is described in a separate chap-ter by Huang et al.13 In addition, the current chapter discusses extrapulmonary consequences of AATD, including associations with panniculitis and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Liver disease associated with AATD is the consequence of a so-called intra-hepatic “toxic gain of function” and is also discussed in a later chapter of this series of reviews by Patel and Teckman.14 The current chapter closes with a brief discussion of other diseases in which AATD has been reportedly implicated, recognizing that clear evidence in support of such disease associations remains sparse.
Lung disease
AATD-related lung disease does not typically manifest until individuals are at least in their 30s, when respiratory symptoms such as dyspnea, cough, and wheezing may begin.15 However, these symptoms are non-specific and not pathognomonic of AATD.10 As a result, AATD is often overlooked and under-recognized. A number of key drivers for the under-recognition of AATD among healthcare providers have been identified and are summarized in Table 1.
Table 1.
Key drivers of the under-recognition of AATD.
| Driver | Additional information |
|---|---|
| Poor knowledge of AATD by physicians | • In a web-based survey comprising 30 multiple-choice questions aimed at testing the knowledge of physicians and respiratory therapists about AATD, respondents answered only approximately 50% of questions correctly.16 |
| • In another survey, only 14% of respondents reported “knowing very well” about AATD.17 | |
| Underappreciation of and non-compliance with guidelines by physicians | • A literature study identified a number of common potential barriers to guideline adherence, including lack of familiarity and awareness of guidelines, self-efficacy, outcome expectancy, and inability to overcome the inertia of previous practice.18 |
| • Another study found that it is not always clear to primary care physicians when to test for AATD, or when to refer patients to a specialist.19 | |
| “Therapeutic nihilism” | • The incorrect belief that therapy is not available has been reported.20 |
AATD, alpha-1 antitrypsin deficiency.
Patients with AATD report symptoms consistent with a range of conditions, including emphysema, chronic bronchitis, or asthma, often in combination (Figure 1).21 However, not all patients with AATD will develop symptoms in their lifetime and the impact of AATD is highly variable.2 There are a number of factors that may affect clinical disease severity in addition to AATD genotype and serum AAT level. Smoking is known to be a major risk factor, and patients who smoke are at a far higher risk of developing lung disease compared with non-smokers.22,23 Smoking is also associated with a significant increase in mortality. A study using data from the Swedish National AATD Registry estimated the standardized mortality ratio for smokers at 4.80 (95% confidence interval [CI]: 4.20–5.50) compared with 2.80 (95% CI: 2.30–3.40) for never-smokers.23 Some asymptomatic never-smokers with AATD (often detected through familial testing) have a normal lifespan.23 Occupational choice is another factor known to affect outcomes in patients with AATD, with occupational exposure to mineral dust, smoke, and fumes being related to poorer lung-related outcomes.24 Such exposures are commonly reported in people who work in construction and farming industries.24
Figure 1.
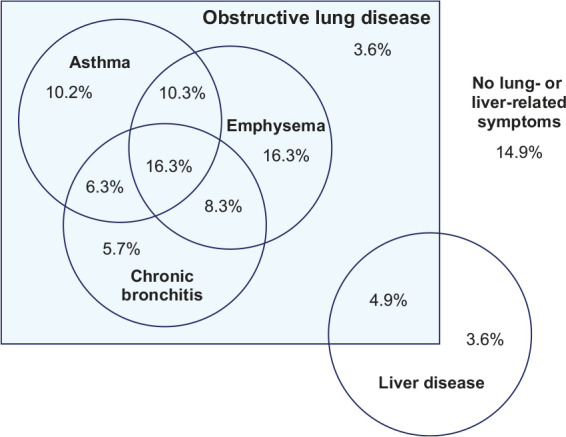
Overlap of patient-reported symptoms in AATD.21
Reproduced with permission from Strange et al.21
AATD, alpha-1 antitrypsin deficiency.
The major pulmonary complication of AATD is emphysema, which usually manifests earlier in life than non-AATD-related emphysema.1,2 The risk of emphysema in individuals with the PI*MM, PI*MS, and PI*SS genotypes appears to be no different than the general population.25 Similarly, never-smoking PI*MZ and PI*SZ individuals do not have higher than expected risk of emphysema.2 In individuals with the PI*MZ and PI*SZ genotypes, smoking appears to be the predominant risk factor for an increased risk of lung disease.25–28 Although potentially confounded by ascertainment bias, those with PI*Null-Null genotypes appear to have a 100% emphysema risk.25 Histologic assessment of tissue from 42 individuals enrolled in the Registry of Individuals with Severe Deficiency of AAT showed emphysema in 34 (81%) of the patients.29 In general, AATD is most commonly associated with a pattern of panacinar emphysema, which appears predominantly in the basal regions.1,30 However, contrary to traditional teaching, over one-third (36%) of patients with AATD have a pattern of emphysema that is not basilar predominant but instead is a primarily apical disease.30
Emphysema related to AATD is irreversible, and smoking is known to accelerate decline in lung function.2,31 The cause of death in patients with AATD is most often respiratory-related, with respiratory failure accounting for the majority of deaths (approximately 70%).23 However, respiratory-related death is less common in patients who have never smoked (39% versus 68%), who instead show a greater proportion of cirrhosis-related mortality compared with smokers (21% versus 8%).23
Other disease manifestations in AATD
In addition to emphysema, AATD is associated with bronchiectasis along with extrapulmonary manifestations (Figure 2).
Figure 2.
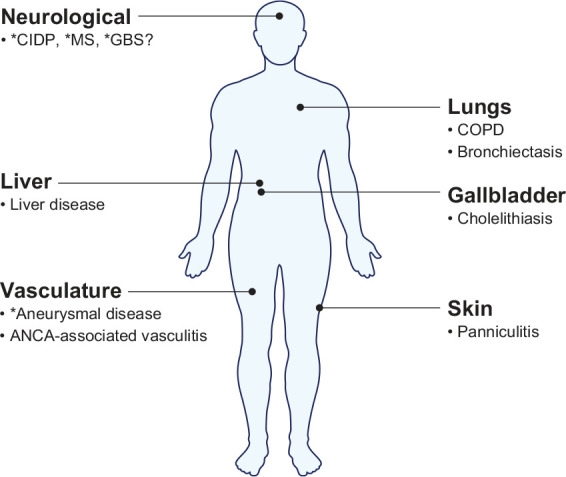
Clinical manifestations of AATD.
* Speculative association with AATD.
AATD, alpha-1 antitrypsin deficiency; ANCA, antineutrophil cytoplasmic antibody; CIDP, chronic inflammatory demyelinating polyneuropathy; COPD, chronic obstructive pulmonary disease; GBS, Guillain–Barré syndrome; MS, multiple sclerosis.
Pulmonary manifestations
Symptoms of chronic obstructive pulmonary disease (COPD) and asthma often overlap,32 and studies have shown that patients with AATD often exhibit a significant, albeit incomplete, bronchodilator response. In a study of the National Heart, Lung, and Blood Institute (NHLBI) Registry, 35% of subjects self-reported a history of asthma and more than 60% of subjects showed a post-bronchodilator response on three serial spirometry measurements.1 However, recent evidence suggests that AATD is no more prevalent in patients with asthma than in the general population, with no association found between AATD and specific asthma phenotypes.33 In addition, in a survey of individuals with AATD, approximately 40% also reported a diagnosis of chronic bronchitis.21 Furthermore, patients with AATD and abnormal lung function are significantly more likely to have a history of chronic bronchitis than those with normal lung function.15
As neutrophils are thought to have a key role in the development of bronchiectasis,34 it follows that an association between AATD and bronchiectasis is plausible. Indeed, a recent large study of high-resolution computed tomography scans by Carreto et al. found that 0.5% of 1,600 patients with bronchiectasis in the UK had AATD.35 Conversely, case series have reported clinical bronchiectasis among patients with AATD, with prevalence estimates ranging from 1% in the NHLBI Registry to 27% in a series by Parr et al.1,36 A recent targeted detection study found that bronchiectasis was particularly associated with the PI*ZZ genotype.35
Extrapulmonary manifestations
The systemic nature of AATD is underscored by the number of extrapulmonary conditions that have been reportedly linked to AATD. Pannicu-litis, defined as inflammation of the subcutaneous tissue, is an established but rare consequence of AATD (i.e., reported in only one of 1129 participants in the NHLBI Registry), and presents as migratory, painful red nodules on the skin.37 Common features of AATD-associated panniculitis include pathologic evidence of neutrophilic inflammation, primarily lobular involvement, and frequent ulceration, presenting with oily drainage (Figure 3).37
Figure 3.

Panniculitis in patients with AATD. (a) Violaceous nodule with overlying grouped yellowish pseudovesicles and telangiectasias on the right shoulder. (b) Erythematous, violaceous nodules on the left mons pubis with fat-like projections and ulcerations. (c) Erythematous, violaceous nodules and plaques on the right volar surface of the wrist with marked ulcerations. (d) Complete healing was documented within 3 weeks of initiating intravenous augmentation therapy.
Reproduced with permission from Elsensohn et al.38
AATD, alpha-1 antitrypsin deficiency.
Although not fully understood, panniculitis in AATD appears to be especially associated with the PI*ZZ genotype, where there is unopposed proteolytic damage in subcutaneous lipid tissue, analogous to the pathogenesis of pulmonary emphysema in individuals with severe deficiency of AAT.39,40 A systematic review of 32 case reports and four case series suggested that plasma-derived intravenous AAT therapy was effective in treating AATD-associated panniculitis.39 Although dapsone is considered by some to be the first line of therapy for this indication and achieves clinical resolution in 62% of cases, side effects can limit its use.39 When dapsone is either not effective or not tolerated, augmentation therapy with pooled human plasma-derived AAT is recommended.39 Other treatments, including antibiotics and immunosuppressants, have been associated with very low rates of clinical resolution, and non-steroidal anti-inflammatory drugs are ineffective.39
Another condition that has been shown to be associated with AATD and the Z allele is ANCA-associated vasculitis (AAV), an inflammatory state of the vasculature affecting multiple organ systems caused by the development of autoimmune antibodies to neutrophils (Figure 4).41,42 In a study of 142 patients with AAV (of whom 88 had granulomatosis with polyangiitis [GPA] and 54 had microscopic polyangiitis [MPA]), the frequencies of the Z and S alleles were 8% and 6%, respectively. There was no association between AATD and ANCA subtype or AAV phenotype, except for intra-alveolar hemorrhage, which was more frequent in patients with the Z or S alleles.43 The clinical significance of AAV in AATD is difficult to determine due to the low prevalence of AAV in patients with severe AATD genotypes; at least in one small study, the presence of AATD does not seem to expose patients to an increased risk of AAV.44 Further research is required to assess the need for augmentation therapy in individuals with AAV associated with AATD.
Figure 4.

Histological hallmarks for AAV: (a) only segmental necrosis (Masson trichrome stain); (b) segmental necrosis with small crescent (hematoxylin and eosin); (c) necrotizing extracapillary lesion in half of glomerulus (Masson trichrome stain); (d) massive necrosis with circumferential crescent (silver stain). All panels at ×20 magnification.
Reproduced with permission from Ferrario et al.57
AAV, ANCA-associated vasculitis.
In a recent genome-wide association study by Merkel et al.,45 an association between GPA and the Z allele was confirmed—an increased risk of AAV was found in patients with the single-nucleotide polymorphism variant rs141530233 at the SERPINA1 locus (Z allele). This association between GPA and the Z allele has also been confirmed by Mahr et al.,46 as well as an association between GPA risk and the S allele, strengthening the evidence of a link between AATD and GPA.
An increased risk of gallstone disease (cholelithiasis) has also been reportedly associated with AATD, the Z and S alleles in particular, and can result in patients requiring cholecystectomy.47 Liver dysfunction as a result of the toxic accumulation of AAT in the liver in individuals with the Z allele has been speculated to predispose patients to gallstone disease.47
Aneurysmal disease has also been associated with AATD.48 In one study of 100 patients with intracranial aneurysms, the rate of heterozygous AATD states (PI*MS and PI*MZ) was higher than in the general population (16% versus 7%, odds ratio: 2.56 [95% CI: 1.32–4.75; p = 0.005]).48 Another study reported that in a cohort of patients with emphysema, only patients with AATD were found to have age-related acceleration in aortic wall degeneration, suggesting a pathologic link between AATD and aortic dilatation.49 Links with neurological conditions such as chronic inflammatory demyelinating polyneuropathy, Guillain–Barré syndrome, and multiple sclerosis have also been suggested,48,50 though, at present, these associations remain speculative in nature.
Regarding other co-morbidities, recent data suggest that pulmonary hypertension, chronic kidney disease, and diabetes are more prevalent in patients with AATD than those with non-AATD asthma or emphysema.51 Conversely, cardiovascular diseases, specifically coronary artery disease and peripheral artery disease, appear to be less prevalent in patients with AATD than those with non-AATD COPD.52 However, the mechanisms underlying these proposed co-morbid associations are unclear.
Conclusions
AATD is a systemic condition that is associated with a broad spectrum of associated diseases, most commonly emphysema. The manifestations and presentation of lung disease in individuals with AATD is varied and generally similar to those with non-AATD COPD or asthma. As is discussed in the chapter in this series regarding rare and novel variants of SERPINA1 and the increasing complexity of testing for AATD, only AATD genotyping, AAT-phenotyping, or gene sequencing can identify the presence of AATD.53 Moreover, extrapulmonary manifestations of AATD, including panniculitis, AAV, and liver disease (which is discussed in more detail within the chapter by Patel and Teckman14) contribute to the disease burden of AATD.
The lack of awareness of AATD among many physicians poses a significant barrier to prompt diagnosis and proper treatment of individuals with AATD. Further efforts to increase awareness of the disease and management guidelines are warranted because early identification of patients with AATD can facilitate effective interventions (i.e., smoking cessation, occupational choice, and intravenous augmentation therapy where appropriate). Currently indicated non-pharmacological and pharmacological options for the management of lung disease in patients with AATD are discussed in detail within the chapter in this series on treatment, authored by Barjaktarevic and Campos,54 and emerging treatment strategies are discussed within the chapter by Rahaghi.55 When AATD progresses to end-stage lung disease and respiratory failure, lung transplantation may be the only option available, which is the focus of the chapter authored by Zamora and Ataya.56
Acknowledgments
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Medical writing assistance was provided by Ben McDermott and Steven Foster of Meridian HealthComms Ltd., Plumley, UK, in accordance with good publication practice (GPP3), funded by CSL Behring.
Footnotes
Author contributions: Both authors contributed to the writing of the manuscript, reviewed the manuscript, and approved the manuscript for submission.
Conflict of interest statement: JKS reports serving as a consultant to Grifols, Takeda, CSL Behring, 23andMe, Insmed, Vertex, InhibRx, Dicerna and Arrowhead Pharmaceuticals. VT reports no conflicts of interest.
ORCID iD: Vickram Tejwani  https://orcid.org/0000-0003-2100-6148
https://orcid.org/0000-0003-2100-6148
Contributor Information
Vickram Tejwani, Pulmonary and Critical Care, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
James K. Stoller, Education Institute, NA22, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195, USA.
References
- 1.American Thoracic Society and European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168: 818–900. [DOI] [PubMed] [Google Scholar]
- 2.Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in alpha1-antitrypsin deficiency. Eur Respir J 2017; 50: 1700610. [DOI] [PubMed] [Google Scholar]
- 3.Jonigk D, Al-Omari M, Maegel L, et al. Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci 2013; 110: 15007–15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoller JK, Aboussouan LS.A review of α1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185: 246–259. [DOI] [PubMed] [Google Scholar]
- 5.Hatipoğlu U, Stoller JK.α1-Antitrypsin deficiency. Clin Chest Med 2016; 37: 487–504. [DOI] [PubMed] [Google Scholar]
- 6.Ferrarotti I, Thun GA, Zorzetto M, et al. Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax 2012; 67: 669–674. [DOI] [PubMed] [Google Scholar]
- 7.O'Brien ML, Buist NRM, Murphey WH.Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92: 1006–1010. [DOI] [PubMed] [Google Scholar]
- 8.Silverman EK, Miletich JP, Pierce JA, et al. Alpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis 1989; 140: 961–966. [DOI] [PubMed] [Google Scholar]
- 9.Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis 2017; 12_suppl: 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogarth DK, Rachelefsky G.Screening and familial testing of patients for alpha 1-antitrypsin deficiency. Chest 2008; 133: 981–988. [DOI] [PubMed] [Google Scholar]
- 11.Horváth I, Canotilho M, Chlumský J, et al. Diagnosis and management of α1-antitrypsin deficiency in Europe: an expert survey. ERJ Open Res 2019; 5: 00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tejwani V, Nowacki AS, Fye E, et al. The impact of delayed diagnosis of alpha-1 antitrypsin deficiency: the association between diagnostic delay and worsened clinical status. Respir Care 2019; 64: 915–922. [DOI] [PubMed] [Google Scholar]
- 13.Huang YCT, Wencker M, Driehuys B.Imaging in alpha-1 antitrypsin deficiency: A window into the disease. Ther Adv Chronic Dis 2021; 12_suppl(2): 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel D, Teckman J.Liver disease with unknown etiology: have you ruled out alpha-1 antitrypsin deficiency? Ther Adv Chronic Dis 2021; 12_suppl(2): 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Chest 1997; 111: 394–403. [DOI] [PubMed] [Google Scholar]
- 16.Taliercio RM, Chatburn RL, Stoller JK.Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55: 322–327. [PubMed] [Google Scholar]
- 17.Esquinas C, Barrecheguren M, Sucena M, et al. Practice and knowledge about diagnosis and treatment of alpha-1 antitrypsin deficiency in Spain and Portugal. BMC Pulm Med 2016; 16: 64. [Google Scholar]
- 18.Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282: 1458–1465. [DOI] [PubMed] [Google Scholar]
- 19.Barrecheguren M, Monteagudo M, et al. Diagnosis of alpha-1 antitrypsin deficiency: a population-based study. Int J Chron Obstruct Pulmon Dis 2016; 11: 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greulich T, Vogelmeier CF.Alpha-1-antitrypsin deficiency: increasing awareness and improving diagnosis. Ther Adv Respir Dis 2016; 10: 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strange C, Stoller JK, Sandhaus RA, et al. Results of a survey of patients with alpha-1 antitrypsin deficiency. Respiration 2006; 73: 185–190. [DOI] [PubMed] [Google Scholar]
- 22.Seersholm N, Kok-Jensen A.Clinical features and prognosis of life time non-smokers with severe alpha 1-antitrypsin deficiency. Thorax 1998; 53: 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanash HA, Nilsson PM, Nilsson JÅ, et al. Survival in severe alpha-1-antitrypsin deficiency (PiZZ). Respir Res 2010; 11: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer AS, Bucher Stoller JK, et al. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162: 553–558. [DOI] [PubMed] [Google Scholar]
- 25.Stoller JK, Hupertz V, Aboussouan LS.Alpha-1 antitrypsin deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds) GeneReviews (Internet). https://www.ncbi.nlm.nih.gov/books/NBK1519/ (2020. [2006], accessed 11 February 2020).
- 26.Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189: 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turino GM, Barker AF, Brantly ML, et al. Clinical features of individuals with PI*SZ phenotype of alpha 1-antitrypsin deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med 1996; 154: 1718–1725. [DOI] [PubMed] [Google Scholar]
- 28.McElvaney GN, Sandhaus RA, Miravitlles M, et al. Clinical considerations in individuals with α1-antitrypsin PI*SZ genotype. Eur Respir J 2020; 55: 1902410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomashefski JF, Crystal RG, Wiedemann HP, et al. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the national registry for individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35: 1452–1461. [DOI] [PubMed] [Google Scholar]
- 30.Parr DG, Stoel BC, Stolk J, et al. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170: 1172–1178. [DOI] [PubMed] [Google Scholar]
- 31.Hutchison DC, Cooper D.Alpha-1-antitrypsin deficiency: smoking, decline in lung function and implications for therapeutic trials. Respir Med 2002; 96: 872–880. [DOI] [PubMed] [Google Scholar]
- 32.Leung JM, Sin DD.Asthma-COPD overlap syndrome: pathogenesis, clinical features, and therapeutic targets. BMJ 2017; 358: j3772. [DOI] [PubMed] [Google Scholar]
- 33.Suárez-Lorenzo I, de Castro FR, Cruz-Niesvaara D, et al. Alpha 1 antitrypsin distribution in an allergic asthmatic population sensitized to house dust mites. Clin Transl Allergy 2018; 8: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King PT.The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis 2009; 4: 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carreto L, Morrison M, Donovan J, et al. Utility of routine screening for alpha-1 antitrypsin deficiency in patients with bronchiectasis. Thorax 2020; 75: 592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parr DG, Guest PG, Reynolds JH, et al. Prevalence and impact of bronchiectasis in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2007; 176: 1215–1221. [DOI] [PubMed] [Google Scholar]
- 37.Stoller JK, Piliang M.Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med 2008; 15: 113–117. [Google Scholar]
- 38.Elsensohn AN, Curtis JA, Secrest AM, et al. Alpha-1-antitrypsin deficiency panniculitis presenting with severe anasarca, pulmonary embolus and hypogammaglobulinaemia. Br J Dermatol 2015; 173: 289–291. [DOI] [PubMed] [Google Scholar]
- 39.Sabbagh DK, Barmayehvar B, Nguyen T, et al. Managing panniculitis in alpha-1 antitrypsin deficiency: systematic review of evidence behind treatment. World J Dermatol 2018; 7: 1–8. [Google Scholar]
- 40.Gross B, Grebe M, Wencker M, et al. New findings in PiZZ α1-antitrypsin deficiency-related panniculitis. Dermatology 2009; 218: 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berden A, Goceroglu A, Jayne D, et al. Diagnosis and management of ANCA associated vasculitis. BMJ 2012; 344: e26. [DOI] [PubMed] [Google Scholar]
- 42.Morris H, Morgan MD, Wood AM, et al. ANCA-associated vasculitis is linked to carriage of the Z allele of α1 antitrypsin and its polymers. Ann Rheum Dis 2011; 70: 1851–1856. [DOI] [PubMed] [Google Scholar]
- 43.Deshayes S, Martin Silva N, Grandhomme F, et al. Clinical Effect of Alpha-1 Antitrypsin Deficiency in Antineutrophil Cytoplasmic Antibody–associated Vasculitis: Results from a French Retrospective Monocentric Cohort. J Rheumatol 2019; 46: 1502–1508. [DOI] [PubMed] [Google Scholar]
- 44.Deshayes S, Martin Silva N, Khoy K, et al. Prevalence of anti-neutrophil cytoplasmic antibodies and associated vasculitis in COPD associated with alpha-1 antitrypsin deficiency: an ancillary study to a prospective study on 180 French patients. Chest 2020; 158: 1919–1922. [DOI] [PubMed] [Google Scholar]
- 45.Merkel PA, Xie G, Monach PA, et al. Vasculitis clinical research consortium. Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol 2017; 69: 1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahr AD, Edberg JC, Stone JH, et al. Alpha1-antitrypsin deficiency-related alleles Z and S and the risk of Wegener's granulomatosis. Arthritis Rheum 2010; 62: 3760–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferkingstad E, Oddsson A, Gretarsdottir S, et al. Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat Commun 2018; 9: 5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schievink WI, Katzmann JA, Piepgras DG, et al. Alpha-1-antitrypsin phenotypes among patients with intracranial aneurysms. J Neurosurg 1996; 84: 781–784. [DOI] [PubMed] [Google Scholar]
- 49.Dako F, Zhao H, Mulvenna A, et al. Relationship between alpha-1-antitrypsin deficiency and ascending aortic distention. Eur Respir J 2019; 54: PA5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCombe PA, Clark P, Frith JA, et al. Alpha-1 antitrypsin phenotypes in demyelinating disease: an association between demyelinating disease and the allele PiM3. Ann Neurol 1985; 18: 514–516. [DOI] [PubMed] [Google Scholar]
- 51.Greulich T, Nell C, Hohmann D, et al. The prevalence of diagnosed α1-antitrypsin deficiency and its comorbidities: results from a large population-based database. Eur Respir J 2017; 49: 1600154. [DOI] [PubMed] [Google Scholar]
- 52.Fähndrich S, Biertz F, Karch A, et al. Cardiovascular risk in patients with alpha-1-antitrypsin deficiency. Respir Res 2017; 18: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foil KE.Rare and novel variants of SERPINA1 and the increasing complexity of testing for alpha-1 antitrypsin deficiency. Ther Adv Chronic Dis 2021; 12_suppl(2S): 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barjaktarevic I, Campos M.Management of lung disease in alpha-1 antitrypsin deficiency: what we do and what we don’t know. Ther Adv Chronic Dis 2021; 12(2S): 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahaghi FF.Alpha-1 antitrypsin deficiency research and emerging treatment strategies: what’s down the road? Ther Adv Chronic Dis 2021; 12(2S): 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zamora M, Ataya A.Lung and liver transplantation in patients with alpha-1 antitrypsin deficiency. Ther Adv Chronic Dis 2021; 12(2S): 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferrario F, Vanzati A, Pagni F.Pathology of ANCA-associated vasculitis. Clin Exp Nephrol 2013; 17: 652–658. [DOI] [PubMed] [Google Scholar]