

Abstract
Alpha-1 antitrypsin deficiency (AATD) is caused by mutations in the SERPINA1 gene, which encodes the alpha-1 antitrypsin (AAT) protein. Currently, over 200 SERPINA1 variants have been identified, many of which cause the quantitative and/or qualitative changes in AAT responsible for AATD-associated lung and liver disease. The types of these pathogenic mutations are varied, often resulting in misfolding, or truncating of the AAT amino acid sequence, and improvements in sequencing technology are helping to identify known and novel genetic variants. However, due to the diversity and novelty of rare variants, the clinical significance of many is largely unknown. There is, therefore, a lack of guidance on how patients should be monitored and treated when the clinical significance of their variant combination is unclear or variable. Nevertheless, it is important that physicians understand the advantages and disadvantages of the different testing methodologies available to diagnose AATD. Owing to the autosomal inheritance of the genetic mutations responsible for AATD, genetic testing should be offered not only to patients at increased AATD risk (e.g. patients with chronic obstructive pulmonary disease), but also to relatives of those with an abnormal result. Genetic counseling may help patients and family members understand the possible outcomes of testing and the implications for the family. While stress/anxiety can arise from genetic diagnosis or confirmation of carrier status, there can be positive consequences to genetic testing, including improved lifestyle choices, directed medical care, and empowered family planning. As genetic testing technology grows and becomes more popular, testing without physician referral is becoming more prevalent, irrespective of the availability of genetic counseling. Therefore, the Alpha-1 Foundation offers genetic counseling, as well as other support and educational material, for patients with AATD, as well as their families and physicians, to help improve the understanding of potential benefits and consequences of genetic testing.
Keywords: alpha-1 antitrypsin, alpha-1 antitrypsin deficiency, genetic counseling, rare variants, SERPINA1, testing
Introduction
The SERPINA1 gene that encodes alpha-1 antitrypsin (AAT) is a highly polymorphic 12.2kb gene clustered with other serpin genes on the long arm of chromosome 14 at position 32.13 (14q32.13), and consists of three noncoding regions (1a–c) and four protein-coding regions numbered 2–5 (Figure 1).1 AAT deficiency (AATD) is caused by variations/mutations in the SERPINA1 gene sequence, the different types of which lead to different changes in the AAT protein (conformational and concentration), and consequently, have different pathological implications. The normal AAT protease inhibitor (PI) is designated with the letter M and is encoded by the wild-type SERPINA1 sequences, also termed the M alleles; individuals with biallelic M alleles are therefore designated as PI*MM individuals. Serum AAT levels in PI*MM individuals are approximately 100–220 mg/dL, or 20–53 µM.2 The M allele encompasses several common benign subvariants, such as M1 (Ala213), M1 (Val213), M2, M3 and M4, which can be differentiated by some but not all testing methodologies.2 Other benign allelic variants also exist, such as E, G, and Zpratt, in which the respective sequence variation encodes a variant AAT protein that does not diminish AAT levels or function.3–5
Figure 1.
SERPINA1 gene.
Located on the long arm of chromosome 14 at position 14q32.13, the SERPINA1 gene consists of three transcriptional promoters (1a–c) upstream of the transcriptional start codon and four protein-coding regions (2–5). The S and F mutations occur in the third protein-coding region, and the I and Z mutations occur in coding regions 2 and 5, respectively. The green bar in coding region 2 represents the AUG transcriptional start codon and the red bar in coding region 5 represents the TAA stop codon.

The allele most commonly associated with severe AATD is the Z allele. Individuals that are homozygous for the Z allele (PI*ZZ) are at high risk of lung and liver disease, and have serum AAT levels that are approximately 10–20% of normal (20–45 mg/dL, or 2.5–7 µM).2,6 Other alleles associated with severe AATD include MMalton, MHeerlen, select other rare variants, and the family of null alleles, which produce no AAT protein product. Some other alleles, such as the S variant, are associated with less severe deficiency and intermediate forms of AATD, resulting in serum AAT levels that are reduced compared to PI*MM levels but higher than PI*ZZ levels. Such alleles may contribute to AATD when combined with a pathogenic variant on the other allele. A serum AAT level of <57 mg/dL, or <11 µM, conventionally defines AATD,2 but attention to moderate AAT levels and corresponding genotypes will continue to elucidate the full spectrum of AATD-associated risks and phenotypes. Yet other SERPINA1 variants have unique molecular mechanisms, or significance, that is not yet greatly understood.
Nomenclature of SERPINA1 alleles is unconventional. AAT protein variants were originally identified and named based on their electrophoretic properties long before the encoding SERPINA1 gene was identified. The normal AAT protein (M) received its name based on its migration to the middle of the isoelectric focusing (IEF) gel. Other alleles were designated with a letter A–L or N–Z depending on their proximal or distal location, respectively, to the M protein band. When challenged with no unused letter for a new allele, numerical figures (for polymorphisms with >0.01 allele frequency) or place of origin names (for rare alleles) were used with the letter of the closest anodal allele, giving rise to allele names such as M1 and MProcida.7 Upon identification of the SERPINA1 gene, the sequence variant corresponding to each PI allele received the same name for fidelity of allele and protein product (e.g. Z allele encodes PI*Z AAT) in AATD testing, literature and clinical practice. Null alleles, which produce no AAT protein, were originally collectively named ‘null’ (or −), and later, when corresponding sequence variations were elucidated, were individually named with the prefix ‘Q0’ and place of origin suffix (i.e. the birthplace of the proband). Genomic nomenclature has evolved with expanding use of genetic testing technologies, and the Human Genome Variation Society (HGVS) has published guidelines for the consistent and unambiguous description of DNA variants.8 Many more recently identified SERPINA1 variants are named in accordance with newer guidelines, but conventional AAT nomenclature still widely persists in the literature and in AAT specialty laboratories, and previously named alleles now have multiple aliases (e.g. PI*Z, Z, and c.1096G>A refer to the same variant).
In the United States, the most common abnormal alleles linked to AATD in order of prevalence are S, Z, F and I;9 beyond these, numerous other SERPINA1 variants exist. Over 200 variants of SERPINA1 are now described in the ClinVar database of the National Center for Biotechnology Information,10 and with next-generation sequencing (NGS) technology, more novel variants are likely to be found.
As previously discussed in the first part of this review series by Tejwani and Stoller,11 AATD cannot be diagnosed in the clinic based on symptomology alone; a range of quantitative and qualitative methods are required to identify the presence of AATD. However, each of these techniques have advantages and disadvantages, which will be discussed within this part. In addition, an overview of the diversity of the genetic changes that can lead to AATD will also be discussed, along with the implications for clinical practice, as well as the role that genetic counseling can play in the care of patients with AATD and their families.
The spectrum of variants in AATD
AATD is most commonly associated with single-nucleotide polymorphisms (SNPs); for example, the Z and S allelic variants result from the base pair substitutions c.1096G>A and c.863A>T leading to the amino acid changes p.Glu366Lys and p.Glu288Val, respectively, in the AAT protein (ClinVar accession VCV000017967.13 and VCV000017969.9; Table 1).10 Small changes to the AAT amino acid sequence can affect the structure/folding of the protein and reduce its secretion from hepatocytes, and/or reduce the binding affinity of AAT for neutrophil elastase (NE), with or without protein polymerization. However, AATD can also be caused by null variants (or nonexpressing SERPINA1 alleles), which can arise from missense mutations, nonsense mutations, frameshifts, large deletions, and complete absence of the SERPINA1 gene.12
Table 1.
Examples of common and rare SERPINA1 variants and their clinical significance.
| SERPINA1 variants | Type of mutation10 (nucleotide/amino acid change) | Clinical significance | Approximate serum AAT level | Reference | ClinVar accession number |
|---|---|---|---|---|---|
| Common | |||||
| I | Point mutation (c.187C>T; p.Arg63Cys) | Uncertain | <120 mg/dL, (22 µM)24 | Graham et al.25 | VCV000017974.6 |
| F | Point mutation (c.739C>T; p.Arg247Cys) | Uncertain | 160 mg/dL, (30 µM)26 | Okayama et al.26 | VCV000017961.5 |
| S | Point mutation (c.863A>T; p.Glu288Val) | Uncertain | 82 mg/dL, (15 µM)27 | Tan et al.28 | VCV000017969.9 |
| Z | Point mutation (c.1096G>A; p.Glu366Lys) | Pathogenic: linked to development of emphysema and liver disease | 20–45 mg/dL, (2.5–7 µM)6 | Laurell and Eriksson29 | VCV000017967.13 |
| Rare | |||||
| MHeerlen | Point mutation (c.1178C>T; p.Pro393Leu) | Pathogenic: linked to development of lung disease | 5 mg/dL, (~1 µM)30 | Hofker et al.30 | VCV000017965.2 |
| MMalton | Point mutation (c.226_228del; p.Phe76_del) | Pathogenic: linked to development of emphysema and liver disease | 64–87 mg/dL, (11–16 µM)31 | Curiel et al.15 | RCV000201853.1 |
| MProcida | Point mutation (c.194T>C; p.Leu65Pro) | Pathogenic: intermediate pathogenicity | <10 mg/dL, (<2 µM)19 | Takahashi et al.19 | RCV000019571.3 |
| MWürzburg | Point mutation (c.1177C>T; p.Pro393Ser) | Pathogenic: linked to development of lung disease | 94 mg/dL, (17 µM)17 | Poller et al.17 | RCV000336993.8 |
| NNagato | Point mutation (c.899T>C; p.Leu300Arg) | Uncertain significance: likely benign | ~180 mg/dL, (~33 µM)32 | Yuasa et al.32 | VCV000594462.3 |
| PBrescia | Point mutation (c.745G>C; p.Gly249Arg) | Pathogenic: linked to lung and liver disease | 61 mg/dL, (11 µM)16 | Medicina et al.16 | VCV000189018.1 |
| PLowell | Point mutation (c.839A>T; p.Asp280Val) | Pathogenic: linked to lung disease | 77 mg/dL, (14 µM)33 | Cook et al.13 | VCV000017975.4 |
| Q0Amersfoort | Null mutation – stop codon insertion (c.552C>G; p.Tyr184Ter) *Occurs in the same codon as Q0Granite Falls (c.552delC; p.Tyr184Terfs) | Pathogenic: emphysema | No AAT expression | Prins et al.20 | VCV000017976.1 |
| Q0Mattawa | Null mutation – frameshift resulting in a premature stop codon (c.1131A>T; p.Leu377Phe) | Pathogenic: emphysema | No AAT expression | Cox and Levison34 | VCV000017978.1 |
| Q0Bellingham | Null mutation – stop codon insertion (c.721A>T; p.Lys241Ter) | Pathogenic: emphysema | No AAT expression | Satoh et al.21 | VCV000017977.2 |
| SIiyama | Point mutation (c.230C>T; p.Ser77Phe) | Pathogenic: emphysema | 33 mg/dL, (6 µM)35 | Yuasa et al.36 | VCV000017992.1 |
| VMunich | Point mutation (c.77A>C; p.Asp26Ala) | None | 150–250 mg/dL, (29–46 µM)37 | Holmes et al.37 | VCV000017983.1 |
| WBethesda | Point mutation (c.1078G>A; p.Ala360Thr) | Uncertain | ~170 mg/dL, (~32 µM)38 | Holmes et al.39 | VCV000017985.1 |
| X | Point mutation (c.682G>A; p.Glu228Lys) | None | Serum AAT levels likely within the normal range40 | Crystal et al.40 | VCV000017963.1 |
| XChristchurch | Point mutation (c.1159G>A; p.Glu387Lys) | Uncertain: likely benign | Serum AAT levels likely within the normal range40 | Brennan and Carrell41 | VCV000017964.4 |
| ZAugsburg | Point mutation (c.1096G>A; p.Glu366Lys) | Pathogenic: linked to development of emphysema and liver disease | 83 mg/dL (15 µM)4 | Faber et al.42 | VCV000017967.13 |
| ZWrexham | Point mutation (c.17C>T; p.Ser6Leu) | Uncertain | Serum AAT levels likely as for PI*Z43 | Graham et al.43 | VCV000017970.1 |
Amino acid numbers described in protein mutations have been updated according to recommendations outlined by the Human Genome Variation Society and therefore may differ slightly in comparison to corresponding mutations reported in the literature.8
AAT, alpha-1 antitrypsin.
Many rare AAT variants, such as the more common/well-characterized MHeerlen, MMalton, MProcida, MWürzburg, PBrescia, and PLowell variants (not a comprehensive list) have been linked to the development of lung disease and/or liver disease as a result of changes in AAT quantity, polymerization, and/or a reduction in ability to inhibit NE (Table 1).13–19 In particular, the MMalton single amino acid deletion at position 76, is strongly linked to emphysema and liver disease, and is the highest frequency rare variant reported in a recent Portuguese study.18 Null variants are designated with the prefix ‘Q0’ and the examples of these given in Table 1, Q0Amersfoort, Q0Mattawa and Q0Bellingham (not a comprehensive list), all result in premature termination of the AAT amino acid sequence, and consequently, undetectable levels of serum AAT. In addition, null variants are strongly linked to the development of emphysema,12,14,20,21 but in line with all null variants, do not confer a risk of liver disease as they do not induce the polymerization of AAT responsible for liver damage.12 There is, therefore, a wide spectrum of SERPINA1 genetic variants that cause AATD (Table 1), but currently there is only clear clinical guidance for the management and treatment of the most common (e.g. PI*ZZ) or severe (PI*Znull) forms of the disease.22 Detailed information on the management and treatment of AATD can be found in the next part of this review series authored by Barjaktarevic and Campos.23
Establishing clinical relevance
Guidelines are clear for the most common or severe AATD-causing genotypes with regard to how patients should be monitored and treated, but they are unclear for variants with variable or uncertain clinical consequence, and even for combinations of moderate frequency alleles associated with moderate deficiency (e.g. PI*SF). The first indicator of whether a rare variant is pathogenic is often the associated serum AAT level. In general, patients who are heterozygous for a pathogenic SERPINA1 variant and the normal allele (e.g. PI*MZ) are unlikely to have severely low serum AAT levels, but may have mild-to-moderate reduction compared with normal due to the codominant allelic expression of AAT. When pathogenic rare variants are present in combination with other pathogenic variants (e.g. the Z allele), serum AAT levels may be well below normal; such patients may benefit from, or should be considered for, intravenous AAT therapy when clinical criteria for augmentation therapy are met. For example, an individual with the PI*ZMProcida genotype was recently reported as having a serum AAT level of 24 mg/dL (~4.4 µM).44 Likewise, when rare pathogenic variants are present in combination with null variants, AAT levels may be very low or undetectable (although this is extremely rare).12 Case reports have reported an individual with the PI*ZQ0Ourém genotype as having a serum AAT level of 14.5 mg/dL (~2.7 µM),45 and individuals homozygous for the Q0Cairo allele with serum AAT levels below 10 mg/dL (~1.8 µM).46
Although AAT level is often useful in interpretation of results and inferring clinical risk, several pathogenic variants result in near-normal levels of serum AAT but are functionally deficient in their ability to inhibit NE. This is thought to be the case with the F variant;47 individuals with AATD involving the F allele may have serum AAT levels that appear adequate, despite their genetic risk. Testing assays measuring AAT function are available, but not widely used in testing setups. Other factors can also affect serum AAT levels, such as inflammation, cigarette smoke and estrogen.48,49 As AAT is an acute phase reactive protein, serum AAT levels increase during stages of acute inflammation. Therefore, the presence of systemic inflammation at the time of sample collection for AATD testing may mask usually low baseline AAT levels.48 Furthermore, different AAT protein phenotypes react differently in response to inflammation, with M-type AAT proteins showing a strong response and Z-type AAT proteins exhibiting a reduced reaction.48 Serum AAT levels have also been shown to increase with tobacco smoke exposure in a dose-dependent manner.49 In a population-based cohort of 5187 adults, those who were exposed to environmental tobacco smoke had higher serum AAT levels than nonexposed never-smokers, and serum AAT levels were highest in active smokers who consumed at least 15 cigarettes per day.49 In women within the same cohort, menopausal status was significantly associated with a decrease in serum AAT levels (p = 0.003), and intake of female hormones, such as oral contraceptives and hormone replacement therapy, was significantly associated with an increase in serum AAT (p < 0.001 for both).49 Therefore, serum AAT levels should be interpreted with mindfulness of these factors and should not be used alone for establishing the clinical relevance of rare and novel SERPINA1 variants.
Determining the clinical significance of novel variants where there are no previous reports of pathogenicity can be challenging; however, computational approaches are being utilized to identify variants with severe pathogenic potential, which can be tested for secretory deficiencies in cell culture models.50,51 For example, PolyPhen-2 is an easy-to-use and freely accessible software that can predict the possible impact of amino acid substitutions on protein structure and function, and is one of the interpretation tools used in several studies of SERPINA1 variants.18,51–53 However, many different programs are available and being used to apply pathogenicity predictions on SERPINA1 mutations, developing algorithms to identify variants of interest for in vitro studies.50 Although it is increasingly possible to predict the pathogenicity of rare and novel SERPINA1 variants, much investigation is still required to fully understand their clinical consequences and what the appropriate management of patients with these variants should entail.
Testing
Based on the latest evidence, clinical practice guidelines recommend testing for AATD in any patient with chronic obstructive pulmonary disease (COPD), emphysema and/or chronic bronchitis, regardless of age or ethnicity, in patients with incompletely reversible asthma, those with unexplained chronic liver disease (see the part of this review series by Patel and Teckman),54 necrotizing panniculitis, granulomatosis with polyangiitis, and unexplained bronchiectasis.22 Furthermore, parents, siblings, children, and extended family of patients identified with an abnormal AAT gene should be provided with genetic counseling and offered AATD testing. For family testing, testing AAT level alone is not recommended as it does not fully characterize AATD disease risk;22 further testing is required. For diagnostic testing of symptomatic patients, the 2016 US Clinical Practice Guidelines recommend genotyping for at least the S and Z alleles because >95% of all individuals with severe AATD have PI*ZZ or PI*SZ genotypes, with confirmatory testing considered using PI phenotyping, AAT level testing and/or expanded genotyping.22 The 2017 European Respiratory Society (ERS) statement identifies AAT level testing as a crucial first test to identify AATD, but notes that it must be supported by qualitative tests to identify the causative mutations.55 In practice, AATD testing consists of at least one of four fundamental steps: determination of AAT serum levels, IEF phenotyping, allele-specific genotyping, and direct sequencing.56 Measurement of AAT serum levels can detect some, but not all severe AAT deficiencies. When serum levels are severely low, additional testing is required for comprehensive diagnosis and risk assessment. A clinician may order a specific test(s) based on clinical circumstance (e.g. presence of a null allele in the family, patient history of liver transplant, whether a patient is receiving augmentation therapy).57 Different laboratories employ different testing algorithms, with NGS increasingly incorporated for select samples at AATD specialty labs, and as a standalone AATD test at select genetics labs.
Quantitative testing
As AATD is most commonly characterized by low levels of antigenic AAT, measuring serum levels of AAT provides an initial indication of AAT deficiency irrespective of a patient’s genetic variant status, and is a widely available diagnostic test. Nephelometry is the most widely used method in the literature;6 however, the most modern technique is immunoturbidimetry. Both techniques measure the turbidity of a sample to determine levels of an analyte; however, whereas nephelometry is a measure of light scattered by a sample, immunoturbidimetry is a measure of light absorbed by a sample. Immunoturbidimetry is a higher-throughput method with lower associated costs but provides similar results to nephelometry.27,58 In the past, quantitative techniques for identifying AATD were performed on whole blood samples. However, more recently, quantitative techniques using dried blood samples are more widely available, facilitating testing.55,59 Whole or dried blood spots allows application of any of the major testing methods discussed within this part. Use of saliva and buccal samples is gaining popularity for genetic testing; for AATD, DNA-based testing (i.e. genotyping and sequencing) may be done on a buccal or saliva sample, but blood is required for quantitating AAT serum level and PI phenotyping.
Serum AAT reference ranges reported in the 2003 American Thoracic Society (ATS)/ERS statement are still widely used today; by nephelometry, the normal range is given as 83–220 mg/dL, and the range given for patients homozygous for the Z variant (PI*ZZ), is 20–45 mg/dL.6 However, these reference ranges were largely based on older studies and may contain data obtained from patients with inflammation, which as mentioned earlier, can affect serum AAT levels.48 Serum AAT levels can increase by 3–4-fold in response to inflammation and infection in AAT replete individuals,60 and therefore reduce the accuracy of the reference ranges. Although the acute phase response is negligible in terms of AAT elevation in severe AATD, patients with genotypes such as PI*MZ, PI*MS and PI*SS still retain a substantial acute phase response.48 Therefore, C-reactive protein (CRP) may be tested simultaneously as an indicator of inflammation at the time of AAT testing. Alternative AAT reference ranges from a cohort of individuals with normal (<5 mg/L) CRP levels are shown in Table 2.
Table 2.
Alternative AAT reference ranges by genotype based on data from a large US screening program,48 with reference ranges from the 2003 ATS/ERS statement6 for comparison.
| Normal range | ||||
| Genotype | PI*MM | |||
| AAT levels: mg/dL (µM) | Sanders et al.48† | 126.9–159.2 (23–29) | ||
| ATS/ERS statement | 150–350 (27–64) | |||
| Intermediate deficiency genotypes | ||||
| Genotype | PI*MS | PI*SS | PI*MZ | |
| AAT levels: mg/dL (µM) | Sanders et al.48† | 105.0–131.4 (19–24) | 88.2–107.5 (16–20) | 75.8–96.6 (14–18) |
| ATS/ERS statement | Not provided | 100–200 (18–37) | 90–210 (17–39) | |
| Severe deficiency genotypes | ||||
| Genotype | PI*ZF | PI*SZ | PI*ZZ | |
| AAT levels: mg/dL (µM) | Sanders et al.48† | 64.5–79.2 (12–15) | 46.4–61.0 (8–11) | 0–28.2‡ (0–5) |
| ATS/ERS statement | Not provided | 75–120 (13–22) | 20–45 (3–8) | |
Data are interquartile ranges of AAT levels (measured by immunoturbidimetry) from individuals with normal CRP levels (CRP < 5 mg/dL).
lower quartile was below the assay detection limit of 20 mg/dL.
AAT, alpha-1 antitrypsin; ATS, American Thoracic Society; CRP, C-reactive protein; ERS, European Respiratory Society; PI, protease inhibitor.
Measuring serum AAT levels alone is useful for identifying ‘classic’ severe AATD but is inadequate for detecting all types of severe AATD and for identifying genotypes. For example, PI*ZZ and PI*ZMHeerlen would have comparable severe AATD, but differentiating the genotypes is important for accurate family testing and estimating the risk for liver disease to the patient. Measuring serum AAT levels alone is also inadequate for detection of alleles that are associated with variable, or near-normal AAT levels with reduced AAT function. The F variant is an example of this, as it is associated with near-normal circulating levels of AAT, but abnormal AAT function, which contributes to risk of lung disease.47 Measuring serum levels alone also limits the identification of individuals harboring an abnormal genotype that have AAT levels overlapping the normal range.48 Examples of this include individuals with the PI*MZ genotype, where individuals have enhanced susceptibility and familial risk,61 in addition to the PI*SS genotype, which is of variable consequence.27 Furthermore, AAT distribution is not uniform and so serum AAT levels do not reflect levels of AAT in the lungs, which is where AAT primarily functions to inhibit NE.60 Given these limitations of basing diagnosis on serum AAT levels alone, additional qualitative assessment of AATD genotype/phenotype is essential.
Qualitative testing
Qualitative analysis is performed to identify AAT protein variants and pathogenic alleles. IEF, also called Pi-typing, or phenotyping, is one method used to identify AAT protein variants. This method is a high-resolution electrophoretic technique that separates proteins based upon their charge, which can be used to identify the AAT protein types, both normal and abnormal, present in patient sera. IEF requires expert interpretation of results due to the large number of AAT variants and technical aspects of such assays.62 IEF is unable to identify heterozygosity for null AAT variants and is not appropriate for patients receiving AAT therapy. PI*ZZ patients receiving AAT therapy will be identified as PI*MZ through IEF due to the introduction of exogenous M-type AAT. For patients on AAT therapy for whom genotype is unknown, genetic testing must be performed.
Targeted polymerase chain reaction (PCR) testing provides rapid and accurate identification of common/well-known genetic variants (e.g. Z, S, and increasingly a subset of other well-characterized pathogenic variants), but given the diversity of SERPINA1 variations, it would be too costly to assemble a comprehensive panel of probes to detect all known deleterious SERPINA1 variants. Furthermore, targeted PCR will not detect novel SERPINA1 variants.62 Multiplex PCR, the simultaneous detection of multiple genotypes in a single reaction, is possible for a limited number of AATD genotypes, including rare alleles with a known genetic sequence, and allows for more variants to be tested from a single sample.63 However, multiplex genotyping is also unable to detect novel SERPINA1 variants. Furthermore, as the prevalence of AATD genotypes varies worldwide,64 and the prevalence of rare AATD genotypes is largely unknown, it is likely that the panels of probes in multiplex PCR tests will be better suited to some countries than others. Only complete genetic sequencing can identify all SERPINA1 variants.
Complete genetic sequencing of SERPINA1 using the Sanger method can provide full details of all mutations present, including rare/novel SNPs and null variants.47,65 However, NGS has a higher throughput and lower cost than Sanger sequencing and is therefore becoming the gold standard sequencing methodology for AATD. NGS can also identify novel variants and should allow the discovery of further rare SERPINA1 variants.49,50 Nevertheless, irrespective of the sequencing method used, it is essential that defined laboratory protocols are followed in order to ensure error rates are minimized.55
Screening for AATD
At present, the majority of epidemiological data on AATD comes from the United States and Western Europe. While AATD has been identified at the highest frequencies in these populations, there is a considerable lack of information available for ~50% of the world’s United Nations, hindering our global understanding of the disease.66,67 Many studies of AATD detection have targeted or drawn populations with enhanced suspicion for AATD, such as relatives of a proband or those with clinical features (e.g. early-onset COPD).55 Population-wide screening studies for AATD can be challenging to perform on a large scale, but several have been successfully conducted and they can provide a less-biased estimate of disease prevalence and natural history.68 Due to the differing methodologies used, the reported prevalence of AATD and associated variants may not be directly comparable across studies.24 It has also been suggested that some rare alleles may not be as rare as previously assumed.24 Determining the frequency and clinical significance of both rare and common variants is key to understanding the true burden of AATD.
Targeted screening of enriched patient populations with respiratory symptoms results in a far higher detection rate than population-wide screening and so has been employed to great effect in several countries worldwide.27,69,70 A potential disadvantage of targeted screening programs is missing individuals with severe AATD who have not been diagnosed with emphysema or COPD.69,71,72 This cohort is likely composed of (1) symptomatic individuals who have not sought care/received diagnosis, (2) pre-symptomatic individuals, and (3) those who will remain asymptomatic, with largely unknown distribution among these groups. Some targeted screening programs also test relatives of diagnosed patients (familial screening), which again has advantages and disadvantages, which will be discussed in the ‘Genetic counseling’ section.70
The largest population-based screening study conducted in AATD was performed in 200,000 Swedish infants in 1976, which identified 127 PI*Z and 48 PI*SZ individuals who have been followed longitudinally.73 Studies such as this are informative but can be prohibitively expensive and therefore, population-wide screening has seen limited application.33 Benefits of population-based screening leading to early and widespread identification of affected individuals include lifestyle recommendations (e.g. smoking avoidance) to reduce disease risk, earlier symptom recognition and treatment, and the opportunity for genetic counseling. Limitations include potential psychological effects, potential genetic discrimination, and medical resource utilization in (the often many) pre-symptomatic years, and these have remained barriers to adoption of standardized newborn screening for AATD, so additional data are needed.74 Current testing recommendations from the Alpha-1 Foundation and the ATS/ERS are shown in Table 3.6,22
Table 3.
| Pulmonary | Extra-pulmonary | Familial testing |
|---|---|---|
| Testing should occur in those with: • COPD • Incompletely reversible asthma • Unexplained bronchiectasis |
Testing should occur for those with: • Unexplained chronic liver disease • Necrotizing panniculitis • Granulomatosis with polyangiitis |
Testing should be offered to: • Parents, children, siblings, and extended family of an individual with an abnormal gene |
AATD, alpha-1 antitrypsin deficiency; COPD, chronic obstructive pulmonary disease.
Several US commercial screening programs currently provide cost-free AATD testing for use in clinical settings (i.e. the test must be ordered by and resulted to a healthcare provider).65 Clinicians may also order AATD testing through a variety of institutional and for-fee commercial laboratories. AATD testing is also increasingly available direct to consumers without physician referral, and, therefore, without pre-test genetic counseling to fully inform patients of the implications the results may bring. The Alpha-1 Foundation has offered free and confidential testing for AATD for over 15 years through a research program,75 though with associated online educational materials and access to genetic counseling. More recently, AATD is among the conditions included on genomic health screens offered direct-to-consumer by companies such as 23andMe, and others. Additionally, in recent years, AATD has been added to expanded prenatal/preconception genetic carrier screens. While such screens are aimed at detecting couples with reproductive risk, individuals genetically affected with conditions such as AATD are occasionally identified.76,77
The hopeful dissemination of deidentified data from genomic tests that include AATD, but are not performed due to concern for AATD specifically, will contribute to elucidating the true prevalence of AATD variants, and facilitate the continued understanding of AATD’s disease burden and natural history through projects such as patient registries. The American College of Medical Genetics and Genomics (ACMG) recommends that all labs performing whole-exome and genome sequencing report secondary findings in 59 genes associated with select highly penetrant genetic disorders, for which established interventions are available to reduce morbidity and mortality.78 Of note, despite increasing consumer access to AATD results via inclusion on genomic health and expanded carrier screens, the SERPINA1 gene is not currently on the ACMG list of genes in which to report secondary findings; therefore, individuals receiving clinical exome or genome testing generally do not learn their AATD genotype even though early recognition may improve outcomes. Such tests, however, are usually ordered in an attempt to find the etiology of an unexplained clinical phenotype, and the reporting paradigm will likely change if exomes/genomes are pursued for broader predictive purposes in the future.
Inheritance of AATD variants and genetic counseling
As AATD follows the autosomal pattern of inheritance, one of each parents’ two SERPINA1 alleles is passed to each child. In addition, the SERPINA1 alleles that encode AAT are co-dominantly expressed,1 meaning that in AATD heterozygotes, half of AAT produced is normal, while half is abnormal in accordance with the genetic variant. The risks to a patient’s offspring therefore depend upon the genetic status of both parents (Figure 2).79 If only one parent is heterozygous for a pathogenic allele and the other is an unaffected noncarrier, there will be a 50% chance that each offspring will be heterozygous for the pathogenic allele and a 50% chance that they will be an unaffected noncarrier [Figure 2(a)]. If one parent is homozygous (e.g. PI*ZZ) or compound heterozygous (e.g. PI*SZ) for pathogenic alleles and the other parent is an unaffected noncarrier, each offspring will have a 100% chance of carrying one pathogenic allele [Figure 2(b)]. If both parents are heterozygous for a pathogenic allele, there will be a 25% chance of each offspring being an unaffected noncarrier, a 50% chance of being heterozygous for a pathogenic allele, and a 25% chance of being affected with biallelic pathogenic variants [homozygous or compound heterozygous, based on parental genotypes; Figure 2(c)]. More significantly, if one parent themselves is homozygous or compound heterozygous and the other parent is also heterozygous, there is a 50% chance of the offspring being severely deficient in AAT [Figure 2(d)]. In the rare event that both parents have biallelic pathogenic alleles, then each offspring will also be affected. Furthermore, the likelihood of inheriting two different pathogenic alleles is becoming increasingly apparent with the growing number of recognized deficiency genotypes, as well as the commonality of heterozygotes in the general population.
Figure 2.
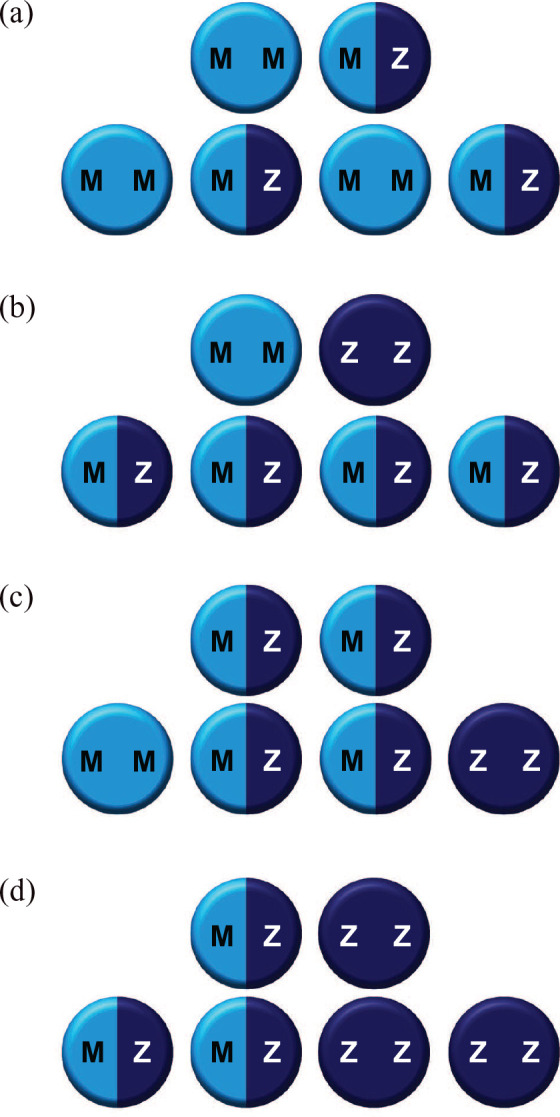
Autosomal inheritance of SERPINA1 alleles.
Autosomal inheritance of SERPINA1 alleles if parents are: ‘normal’ and heterozygous (a), ‘normal’ and homozygous (b), both heterozygous (c), and heterozygous and homozygous (d). Only eventualities c and d confer risk of the offspring being severely deficient in AAT (25% and 50% chance, respectively).
Estimated worldwide prevalence:80
MZ, 42,564,136; ZZ, 181,894.
It is important to note that genetic testing only informs patients whether or not they harbor any pathogenic SERPINA1 alleles and does not predict the age of AATD disease onset, the type of symptoms patients will present with, the severity of these symptoms, or the rate of AATD disease progression.22 However, knowledge of AAT pathogenic variant status allows for risk modification (e.g. smoking avoidance) and allows proper monitoring for disease onset or progression.22 Owing to the autosomal inheritance described above, testing for AATD should be offered to parents, siblings, children, and extended family members of individuals found to have AATD (whether homozygous or heterozygous), with siblings of severely deficient individuals at highest priority due to their greater genetic risk.22 However, genetic testing has implications for the whole family and the possible outcomes of the results should be discussed before testing.6 Patients and family members should be offered genetic counseling for this purpose. For children, ethical considerations are more complicated; predictive testing in adolescents should be undertaken only if both themselves and their parents give informed consent, and if they are mature enough to understand the implications of testing and the possible outcomes. For younger children, parental/guardian consent is required, but it has been shown that parents value genetic testing for their children at risk for AATD when it can be done in a confidential setting.81 A discussion of potential risks and benefits should occur surrounding pre-dispositional diagnostic testing for a patient of any age, weighing medical utility with potential anxiety, impact on insurability (e.g. life, disability), and, for children, autonomy. Testing children for AATD is only clearly indicated when clinically directed (e.g. in the presence of liver disease), where testing follows the previously discussed guidelines.
Where genetic counseling is unavailable, withholding testing based on the absence of a genetic counselor referral is not recommended and a knowledgeable provider can advise patients on the possible outcomes of testing. However, it is helpful for providers ordering AATD testing to be familiar with key areas of pre- and post-test genetic counseling, including the natural history, pattern of inheritance, reduced penetrance and clinical variability, and genetic risk to family members based on the results. The information patients receive from genetic testing can be personally unsettling and create stresses and anxiety within the family. In accordance with the 2008 Genetic Information Non-discrimination Act (GINA) in the United States, genetic testing results cannot affect a patient’s ability to obtain health insurance or employment; however, GINA does not address life, disability or long-term care insurance, or apply to employers with fewer than 15 employees or the military. Potential effect of results on eligibility or premiums for life, disability, or long-term care insurance may further exacerbate stress and anxiety. Despite the potential for negative psychological impacts from AATD testing, there is evidence to suggest that knowledge of AATD status can enable patients to feel peace of mind and in control of their lives, as well as improve medical outcomes.75 Specific information about genetic counseling for AATD is available through the Alpha-1 Foundation website (www.alpha1.org).
Conclusions
There are many known SERPINA1 variants associated with AATD and the increasing availability of higher throughput sequencing methodologies, particularly NGS, is contributing to continued detection of rare and novel variants responsible for AATD. Several studies suggest that some variants are not as rare as previously thought, indicating that additional data from standardized studies and/or population-level testing are required to refine prevalence data and determine AATD’s true extent and disease burden. Deciding the best course of treatment in individuals with rare/novel variants can be challenging. Computational modeling, quantitative AAT testing and clinical assessment can assist in proper diagnosis and management of patients. To better inform patients and healthcare professionals about the genetic variants related to AATD, a dedicated online database now exists, which is being updated as further data become available (Alpha-1 Alleles; www.alpha1research.org/allele_search). Irrespective of which genetic variant(s) of SERPINA1 a patient carries, laboratory testing is the cornerstone of AATD diagnosis, and it is important that clinicians have an understanding of the advantages/disadvantages of the different diagnostic tests available. Expert analysis and utilization of multiple methods in inconclusive cases can help provide comprehensive analysis and accurate diagnosis. Where possible, genetic counseling should be offered with genetic testing and to those diagnosed with AATD so that patients can better understand the broader implications of AATD variants to themselves and their families.
Acknowledgments
Medical writing assistance was provided by Ben McDermott and Steven Foster of Meridian HealthComms Ltd., Plumley, United Kingdom, in accordance with good publication practice (GPP3), funded by CSL Behring.
Footnotes
Author contributions: KF contributed to the writing of the manuscript, reviewed the manuscript, and approved the manuscript for submission.
Conflict of interest statement: The author declares that there is no conflict of interest.
References
- 1.Stoller JK, Aboussouan LS.A review of α1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185: 246–259. [DOI] [PubMed] [Google Scholar]
- 2.Stoller JK, Hupertz V, Aboussouan LS.Alpha-1 antitrypsin deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds) GeneReviews® [Internet]. Seattle, WA: University of Washington, https://www.ncbi.nlm.nih.gov/books/NBK1519/ (2006, accessed 11 February 2020). [PubMed] [Google Scholar]
- 3.Hug G, Chuck G, Slemmer TM, et al. Pi Ecincinnati: a new alpha 1-antitrypsin allele in three Negro families. Hum Genet 1980; 54: 361–364. [DOI] [PubMed] [Google Scholar]
- 4.Renoux C, Odou M-F, Tosato G, et al. Description of 22 new alpha-1 antitrypsin genetic variants. Orphanet J Rare Dis 2018; 13: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hug G, Chuck G, Bowles B, et al. PiZpratt: a new alpha 1-antitrypsin allele in an American Negro family. Hum Genet 1980; 56: 107–110. [DOI] [PubMed] [Google Scholar]
- 6.American Thoracic Society and European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168: 818–900. [DOI] [PubMed] [Google Scholar]
- 7.Cox DW, Johnson AM, Fagerhol MK.Report of nomenclature meeting for alpha 1-antitrypsin, INSERM, Rouen/Bois-Guillaume-1978. Hum Genet 1980; 53: 429–433. [DOI] [PubMed] [Google Scholar]
- 8.den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 2016; 37: 564–569. [DOI] [PubMed] [Google Scholar]
- 9.Rouhani F, Oshins R, Charleston C, et al. Results of a large scale gene-based screening program for alpha-1-antitrypsin deficiency in the USA. Am J Respir Crit Care Med 2017; 195: A7402. [Google Scholar]
- 10.ClinVar. SERPINA1[gene]. Bethesda, MD: The National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/clinvar/?term=SERPINA1[gene] (accessed 5 February 2020). [Google Scholar]
- 11.Tejwani V, Stoller JK.The spectrum of clinical sequalae associated with alpha-1 antitrypsin deficiency. Ther Adv Chronic Dis 2021; 12_suppl(2): 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrarotti I, Carroll TP, Ottaviani S, et al. Identification and characterisation of eight novel SERPINA1 null mutations. Orphanet J Rare Dis 2014; 9: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook L, Burdon J, Brenton S, et al. Alpha-1-antitrypsin PLowell: a normally functioning variant present in low concentration. Aust N Z J Med 1995; 25: 695–697. [DOI] [PubMed] [Google Scholar]
- 14.Curiel D, Brantly M, Curiel E, et al. Alpha 1-antitrypsin deficiency caused by the alpha 1-antitrypsin Nullmattawa gene. An insertion mutation rendering the alpha 1-antitrypsin gene incapable of producing alpha 1-antitrypsin. J Clin Invest 1989; 83: 1144–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curiel DT, Holmes MD, Okayama H, et al. Molecular basis of the liver and lung disease associated with the alpha 1-antitrypsin deficiency allele Mmalton. J Biol Chem 1989; 264: 13938–13945. [PubMed] [Google Scholar]
- 16.Medicina D, Montani N, Fra AM, et al. Molecular characterization of the new defective P(brescia) alpha1-antitrypsin allele. Hum Mutat 2009; 30: E771–E781. [DOI] [PubMed] [Google Scholar]
- 17.Poller W, Merklein F, Schneider-Rasp S, et al. Molecular characterisation of the defective alpha 1-antitrypsin alleles PI Mwurzburg (Pro369Ser), Mheerlen (Pro369Leu), and Q0lisbon (Thr68Ile). Eur J Hum Genet 1999; 7: 321–331. [DOI] [PubMed] [Google Scholar]
- 18.Silva D, Oliveira MJ, Guimaraes M, et al. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir Med 2016; 116: 8–18. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi H, Nukiwa T, Satoh K, et al. Characterization of the gene and protein of the alpha 1-antitrypsin “deficiency” allele Mprocida. J Biol Chem 1988; 263: 15528–15534. [PubMed] [Google Scholar]
- 20.Prins J, van der Meijden BB, Kraaijenhagen RJ, et al. Inherited chronic obstructive pulmonary disease: new selective-sequencing workup for alpha1 antitrypsin deficiency identifies 2 previously unidentified null alleles. Clin Chem 2008; 54: 101–107. [DOI] [PubMed] [Google Scholar]
- 21.Satoh K, Nukiwa T, Brantly M, et al. Emphysema associated with complete absence of alpha 1-antitrypsin in serum and the homozygous inheritance [corrected] of a stop codon in an alpha 1-antitrypsin-coding exon. Am J Hum Genet 1988; 42: 77–83. [PMC free article] [PubMed] [Google Scholar]
- 22.Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis 2016; 3: 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barjaktarevic I, Campos M.Management of lung disease in alpha-1 antitrypsin deficiency: what we do and what we don’t know. Ther Adv Chronic Dis 2021; 12_suppl(2): 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez-Frias F, Miravitlles M, Vidal R, et al. Rare alpha-1-antitrypsin variants: are they really so rare? Ther Adv Respir Dis 2012; 6: 79–85. [DOI] [PubMed] [Google Scholar]
- 25.Graham A, Kalsheker NA, Newton CR, et al. Molecular characterisation of three alpha-1-antitrypsin deficiency variants: proteinase inhibitor (Pi) nullcardiff (Asp256––Val); PiMmalton (Phe51––deletion) and PiI (Arg39––Cys). Hum Genet 1989; 84: 55–58. [DOI] [PubMed] [Google Scholar]
- 26.Okayama H, Brantly M, Holmes M, et al. Characterization of the molecular basis of the alpha 1-antitrypsin F allele. Am J Hum Genet 1991; 48: 1154–1158. [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrarotti I, Thun GA, Zorzetto M, et al. Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax 2012; 67: 669–674. [DOI] [PubMed] [Google Scholar]
- 28.Alpha-1-antitrypsin: molecular abnormality of S variant. Br Med J 1976; 1: 130–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laurell CB, Eriksson S.The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15: 132–140. [Google Scholar]
- 30.Hofker MH, Nukiwa T, van Paassen HM, et al. A Pro––Leu substitution in codon 369 of the alpha-1-antitrypsin deficiency variant PI MHeerlen. Hum Genet 1989; 81: 264–268. [DOI] [PubMed] [Google Scholar]
- 31.Aiello M, Fantin A, Longo C, et al. Clinical manifestations in patients with PI*MMMalton genotypes. A matter still unsolved in alpha-1 antitrypsin deficiency. Respirol Case Rep 2020; 8: e00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuasa I, Suenaga K, Gotoh Y, et al. PI(alpha 1-antitrypsin) polymorphism in the Japanese: confirmation of PI*M4 and description of new PI variants. Hum Genet 1984; 67: 209–212. [DOI] [PubMed] [Google Scholar]
- 33.Corda L, Medicina D, La Piana GE, et al. Population genetic screening for alpha1-antitrypsin deficiency in a high-prevalence area. Respiration 2011; 82: 418–425. [DOI] [PubMed] [Google Scholar]
- 34.Cox DW, Levison H.Emphysema of early onset associated with a complete deficiency of alpha-1-antitrypsin (null homozygotes). Am Rev Respir Dis 1988; 137: 371–375. [DOI] [PubMed] [Google Scholar]
- 35.Ko D-H, Chang HE, Song SH, et al. Identification of compound heterozygous mutation in a Korean patient with alpha 1-antitrypsin deficiency. Korean J Lab Med 2011; 31: 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuasa I, Sugimoto Y, Ichinose M, et al. PI*S(iiyama), a deficiency gene of alpha 1-antitrypsin: evidence for the occurrence in Western Japan. Jpn J Hum Genet 1993; 38: 185–191. [DOI] [PubMed] [Google Scholar]
- 37.Holmes MD, Brantly ML, Curiel DT, et al. Characterization of the normal alpha 1-antitrypsin allele Vmunich: a variant associated with a unique protein isoelectric focusing pattern. Am J Hum Genet 1990; 46: 810–816. [PMC free article] [PubMed] [Google Scholar]
- 38.Holmes M.Molecular analysis of alpha-1 antitrypsin variation and deficiency. Unpublished Doctor of Medicine Thesis, The University of Adelaide, South Australia, https://digital.library.adelaide.edu.au/dspace/bitstream/2440/38329/2/02whole.pdf (1992, accessed 26 January 2021). [Google Scholar]
- 39.Holmes MD, Brantly ML, Fells GA, et al. Alpha 1-antitrypsin Wbethesda: molecular basis of an unusual alpha 1-antitrypsin deficiency variant. Biochem Biophys Res Commun 1990; 170: 1013–1020. [DOI] [PubMed] [Google Scholar]
- 40.Crystal RG.Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest 1990; 85: 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brennan SO, Carrell RW.Alpha 1-antitrypsin Christchurch, 363 Glu––Lys: mutation at the P’5 position does not affect inhibitory activity. Biochim Biophys Acta 1986; 873: 13–19. [DOI] [PubMed] [Google Scholar]
- 42.Faber JP, Weidinger S, Olek K.Sequence data of the rare deficient alpha 1-antitrypsin variant PI Zaugsburg. Am J Hum Genet 1990; 46: 1158–1162. [PMC free article] [PubMed] [Google Scholar]
- 43.Graham A, Kalsheker NA, Bamforth FJ, et al. Molecular characterisation of two alpha-1-antitrypsin deficiency variants: proteinase inhibitor (Pi) Null(Newport) (Gly115––Ser) and (Pi) Z Wrexham (Ser-19––Leu). Hum Genet 1990; 85: 537–540. [DOI] [PubMed] [Google Scholar]
- 44.Maddaloni V, Pepe N, Morano F, et al. Alpha 1 antitrypsin deficiency: a rare allele in patients from South of Italy. J Lung Pulm Respir Res 2019; 6: 32–35. [Google Scholar]
- 45.Hernández Pérez JM, Ramos Díaz R, Fumero García S, et al. Description of alpha-1-antitrypsin deficiency associated with PI*Q0ourém allele in La Palma Island (Spain) and a genotyping assay for its detection. Arch Bronconeumol 2015; 51: e1–e3. [DOI] [PubMed] [Google Scholar]
- 46.Jouhadi Z, Odou MF, Zerimech F, et al. Alpha1 antitrypsin deficiency due to an homozygous PI*Null Q0Cairo mutation: early onset of pulmonary manifestations and variability of clinical expression. Respir Med Case Rep 2018; 24: 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinden NJ, Koura F, Stockley RA.The significance of the F variant of alpha-1-antitrypsin and unique case report of a PiFF homozygote. BMC Pulm Med 2014; 14: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanders CL, Ponte A, Kueppers F.The effects of inflammation on alpha 1 antitrypsin levels in a national screening cohort. COPD 2018; 15: 10–16. [DOI] [PubMed] [Google Scholar]
- 49.Senn O, Russi EW, Schindler C, et al. Circulating alpha1-antitrypsin in the general population: determinants and association with lung function. Respir Res 2008; 9: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giacopuzzi E, Laffranchi M, Berardelli R, et al. Real-world clinical applicability of pathogenicity predictors assessed on SERPINA1 mutations in alpha-1-antitrypsin deficiency. Hum Mutat 2018; 39: 1203–1213. [DOI] [PubMed] [Google Scholar]
- 51.Matamala N, Lara B, Gomez-Mariano G, et al. Characterization of novel missense variants of SERPINA1 gene causing alpha-1 antitrypsin deficiency. Am J Respir Cell Mol Biol 2018; 58: 706–716. [DOI] [PubMed] [Google Scholar]
- 52.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kueppers F, Andrake MD, Xu Q, et al. Protein modeling to assess the pathogenicity of rare variants of SERPINA1 in patients suspected of having alpha 1 antitrypsin deficiency. BMC Med Genet 2019; 20: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel D, Teckman J.Liver disease with unknown etiology: have you ruled out alpha-1 antitrypsin deficiency? Ther Adv Chronic Dis 2021; 12_suppl(2): 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in alpha1-antitrypsin deficiency. Eur Respir J 2017; 50: 1700610. [DOI] [PubMed] [Google Scholar]
- 56.Belmonte I, Barrecheguren M, Esquinas C, et al. Genetic diagnosis of α1-antitrypsin deficiency using DNA from buccal swab and serum samples. Clin Chem Lab Med 2017; 55: 1276–1283. [DOI] [PubMed] [Google Scholar]
- 57.Foil KE, Blanton MG, Sanders C, et al. Sequencing alpha-1 MZ individuals shows frequent biallelic mutations. Pulm Med 2018; 2018: 2836389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ledue TB, Collins MF.Development and validation of 14 human serum protein assays on the Roche cobas® c 501. J Clin Lab Anal 2011; 25: 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Costa X, Jardi R, Rodriguez F, et al. Simple method for alpha1-antitrypsin deficiency screening by use of dried blood spot specimens. Eur Respir J 2000; 15: 1111–1115. [DOI] [PubMed] [Google Scholar]
- 60.Janciauskiene SM, Bals R, Koczulla R, et al. The discovery of α1-antitrypsin and its role in health and disease. Respir Med 2011; 105: 1129–1139. [DOI] [PubMed] [Google Scholar]
- 61.Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189: 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rachelefsky G, Hogarth DK.Issues in the diagnosis of alpha 1-antitrypsin deficiency. J Allergy Clin Immunol 2008; 121: 833–838. [DOI] [PubMed] [Google Scholar]
- 63.Veith M, Klemmer A, Anton I, et al. Diagnosing alpha-1-antitrypsin deficiency using a PCR/luminescence-based technology. Int J Chron Obstruct Pulmon Dis 2019; 14: 2535–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luisetti M, Seersholm N.α1-antitrypsin deficiency. 1: Epidemiology of α1-antitrypsin deficiency. Thorax 2004; 59: 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kueppers F, Sanders C.State-of-the-art testing for alpha-1 antitrypsin deficiency. Allergy Asthma Proc 2017; 38: 108–114. [DOI] [PubMed] [Google Scholar]
- 66.Chorostowska-Wynimko J.Targeted screening programmes in COPD: how to identify individuals with α1-antitrypsin deficiency. Eur Respir Rev 2015; 24: 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blanco I, de Serres FJ, Cárcaba V, et al. Alpha-1 antitrypsin deficiency PI*Z and PI*S gene frequency distribution using on maps of the world by an Inverse Distance Weighting (IDW) multivariate interpolation method. Hepat Mon 2012; 12_suppl: e7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stoller JK.Detecting alpha-1 antitrypsin deficiency. Ann Am Thorac Soc 2016; 13: S317–S325. [DOI] [PubMed] [Google Scholar]
- 69.Greulich T, Nell C, Herr C, et al. Results from a large targeted screening program for alpha-1-antitrypsin deficiency: 2003 - 2015. Orphanet J Rare Dis 2016; 11: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carroll TP, O’Connor CA, Floyd O, et al. The prevalence of alpha-1 antitrypsin deficiency in Ireland. Respir Res 2011; 12_suppl: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eriksson S.A 30-year perspective on alpha 1-antitrypsin deficiency. Chest 1996; 110(Suppl. 6): 237S–242S. [DOI] [PubMed] [Google Scholar]
- 72.Parr DG, Stoel BC, Stolk J, et al. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170: 1172–1178. [DOI] [PubMed] [Google Scholar]
- 73.Sveger T.Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294: 1316–1321. [DOI] [PubMed] [Google Scholar]
- 74.Teckman J, Pardee E, Howell RR, et al. Appropriateness of newborn screening for α1-antitrypsin deficiency. J Pediatr Gastroenterol Nutr 2014; 58: 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strange C, Dickson R, Carter C, et al. Genetic testing for alpha1-antitrypsin deficiency. Genet Med 2004; 6: 204–210. [DOI] [PubMed] [Google Scholar]
- 76.Lazarin GA, Haque IS, Nazareth S, et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med 2013; 15: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wienke S, Brown K, Farmer M, et al. Expanded carrier screening panels—does bigger mean better? J Community Genet 2014; 5: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017; 19: 249–255. [DOI] [PubMed] [Google Scholar]
- 79.Stoller JK, Aboussouan LS.Alpha1-antitrypsin deficiency. Lancet 2005; 365: 2225–2236. [DOI] [PubMed] [Google Scholar]
- 80.De Serres FJ, Blanco I. Prevalence of α1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review. Ther Adv Respir Dis 2012; 6: 277–295. [DOI] [PubMed] [Google Scholar]
- 81.Strange C, Moseley MA, Jones Y, et al. Genetic testing of minors for alpha1-antitrypsin deficiency. Arch Pediatr Adolesc Med 2006; 160: 531–534. [DOI] [PubMed] [Google Scholar]