

Abstract
BACKGROUND AND PURPOSE:
Ganglionic eminence abnormalities on fetal MR imaging are associated with cerebral malformations. Their presumed genetic basis and associated postnatal outcomes remain largely unknown. We aimed to elucidate these through a multicenter study.
MATERIALS AND METHODS:
Between January 2010 and June 2020, seven hospitals in 2 countries performing fetal MR imaging examinations identified fetal MR imaging studies demonstrating ganglionic eminence enlargement, cavitation, or both. Cases with no genetic diagnosis, no whole exome sequencing, or no outcome of a liveborn child were excluded. Head size was classified as large (fronto-occipital diameter > 95th centile), small (fronto-occipital diameter <5th centile), or normal.
RESULTS:
Twenty-two fetuses with ganglionic eminence abnormalities were identified. Of 8 with large heads, 2 were diagnosed with MTOR mutations; 1 with PIK3CA mutation–producing megalencephaly, polymicrogyria, polydactyly, hydrocephalus (MPPH) syndrome; 3 with TSC mutations; 1 with megalencephaly capillary malformation syndrome; and 1 with hemimegalencephaly. Cardiac rhabdomyoma was present prenatally in all cases of TSC; mutation postaxial polydactyly accompanied megalencephaly capillary malformation and MPPH. Of 12 fetuses with small heads, 7 had TUBA1A mutations, 1 had a TUBB3 mutation, 2 had cobblestone lissencephaly postnatally with no genetic diagnosis, 1 had a PDHA1 mutation, and 1 had a fetal akinesia dyskinesia sequence with no pathogenic mutation on trio whole exome sequencing. One of the fetuses with a normal head size had an OPHN1 mutation with postnatal febrile seizures, and the other had peri-Sylvian polymicrogyria, seizures, and severe developmental delay but no explanatory mutation on whole exome sequencing.
CONCLUSIONS:
Fetal head size and extracranial prenatal sonographic findings can refine the phenotype and facilitate genetic diagnosis when ganglionic eminence abnormality is diagnosed with MR imaging.
The human ganglionic eminence (GE) is a focal thickening in the proliferative neuroepithelium lining the ventricles of the telencephalic lobes, situated inferolateral to the frontal horns of the lateral ventricles, and persists throughout nearly all of fetal life, longer than other proliferative areas.1,2 It is thickest at 20 weeks, decreases in size by approximately 50% between 26 and 28 weeks' gestation, and involutes by 34–36 weeks' gestation; this period of rapid involution of the GE through fibrinolysis has been theorized as a contributing factor to the selective vulnerability of preterm neonates to hemorrhage in this region.2 Most cortical GABAergic interneurons are generated in the GE, and they migrate to the cortex by first tangential and then radial migration. The GE also contributes to a population of thalamic neurons and is responsible for forming most of the basal ganglia. Furthermore, the GE represents an intermediate target for thalamocortical and corticothalamic axons and is a source of oligodendrocyte precursors.1-3
Abnormal persistence, enlargement, and cavitation of GEs on fetal MR imaging have been associated with markedly reduced fetal head size and underdevelopment of sulcation (microlissencephaly),4 as well as a range of other brain malformations, including mild partial callosal agenesis, vermis hypoplasia and rotation, cerebellar hypoplasia, ventriculomegaly, enlarged subarachnoid spaces, and molar tooth malformation.5
Although the normal GE is demonstrable with sonography using an endovaginal 3D technique between 9 and 13 weeks' gestation,6 the pathologic GE has been identified at or after the midtrimester only recently in isolated case reports,7,8 without postnatal outcome or genetic diagnosis. Coexistence of GE abnormalities with other brain malformations of the corpus callosum, cerebellum, or microcephaly may be the impetus for fetal MR imaging in which the GE abnormality is first recognized.
A very recent report9 on a large cohort of fetuses with GE alterations at MR imaging highlighted the more frequently associated brain anomalies. However, the authors themselves stressed that a scarcity of knowledge on the genetic substrate of such GE anomalies is the major drawback for the correct characterization and understanding of the pathophysiology. We have also noted an association of fetal GE abnormalities with extremely large and small head sizes.
Better understanding of the intracranial and extracranial associations of this abnormality, its genetic causes, and postnatal outcomes for liveborn children is pivotal for the following reasons:
Genetic diagnosis requires accurate fetal phenotyping; lack thereof is one of the greatest barriers to the use of prenatal whole genome and exome sequencing.10,11
Given the current lack of wide availability of fetal exome and genome sequencing during pregnancy, an understanding of the causes of GE abnormalities would facilitate prenatal counseling and decision-making within a clinically realistic timeframe.
We hypothesized that a multi-institutional study of prenatal sonographic findings, genetic analyses, and postnatal outcomes of fetuses with prenatal MR imaging showing an abnormality of the GE would enable us to do the following:
Establish the genetic abnormalities most often associated with GE abnormalities
Narrow the range of differential diagnoses for a given fetus by categorization based on cranial biometry and specific extracranial structural abnormalities
Develop a less biased picture of the relative likelihood of various genetic causes for the GE abnormalities by involving multiple subspecialist fetal MRI centers in case identification.
MATERIALS AND METHODS
Study Setting
Hospitals in all states of Australia and New Zealand involved in providing fetal MR imaging clinical services and the Vittore Buzzi Children's Hospital in Milan, Italy, were invited by e-mail from the principal investigator to participate in the study by contributing cases from their institutional fetal MR imaging databases. Participating sites provided deidentified sample JPEG images of candidate cases to the principal investigator to determine whether the images demonstrated a GE abnormality before applying other inclusion criteria and final enrollment of the subject.
Study Period
January 2010 to June 2020 was chosen due to the lesser availability of genomic testing before January 2010 at the participating sites.
Inclusion
We included all women older than 18 years of age with a singleton or multiple pregnancies who had fetal MR imaging for a suspected fetal cranial abnormality as a result of a referral for abnormal findings on a prior tertiary sonography during the study period (January 2010 to June 2020), on which the fetus had a GE abnormality reported in the initial clinical report or discovered on review of the case. Postnatal MR imaging with clinical outcome data or composite clinicoradiologic diagnosis (based on extracranial prenatal sonography and MR imaging findings specific for a disorder) or genomic testing (gene panel, singleton whole exome sequencing [WES], trio exome sequencing, or any combination of these) was a further requirement for inclusion. All fetuses had tertiary prenatal ultrasound imaging that resulted in their referral for fetal MR imaging. Before submitting cases for consideration, participating centers were provided with 4 sample cases of fetuses with abnormal ganglionic eminence enlargement or cavitation, with the gestational ages of the fetuses as well as references4,5 with cases of GE abnormalities on fetal MR images.
Case Ascertainment
Searches of radiology reports (using the radiology information system search functions) or local fetal MR imaging case databases (maintained by radiologists at their institutions) were conducted using the following search terms: “ganglionic eminence,” “lissencephaly,” “Walker Warburg,” “dystroglycanopathy,” “cobblestone,” “germinolytic,” “tuberous sclerosis,” “TSC,” “megalencephaly,” “microcephaly,” “hemimegalencephaly,” “SEGA (subependymal giant cell astrocytoma)", and “polymicrogyria.” The rationale for these terms is that these conditions and abnormalities have been previously shown to be associated with abnormal GEs. This search would potentially help identify cases with abnormal GEs, not appreciated or reported in the original clinical report, even if the correct diagnosis had been provided on the basis of other criteria and abnormal findings.
Exclusion
Submitted cases that, in the opinion of the lead authors, did not demonstrate GE abnormalities were excluded. The criterion for determining that the GEs were abnormally enlarged for gestational age or cavitated was a qualitative, visual assessment of size and assessment for any cavitation; currently there are no published data for fetal GE measurements by gestational age. Cases that had no genomic testing and also no postnatal imaging or clinical follow up that provided a genetic diagnosis were excluded.
For example, an infant with typical tuberous sclerosis complex (TSC) features on postnatal MR imaging would be included even if no genetic test results were available or genetic testing had not been performed. Similarly, a fetus of a terminated pregnancy, in which a cardiac rhabdomyoma was identified along with abnormal fetal cranial MR imaging findings consistent with tuberous sclerosis complex, would also be eligible for inclusion in the absence of genetic confirmation.
Finally, in all cases diffusion-weighted and T2* images were reviewed, and if the GE enlargement was attributable to hemorrhage based on this evaluation, the case was excluded.
Data Collection
Demographics.
The following data were collected: Gestational age at sonography before MR imaging; prenatal sonographic findings; gestation at the first fetal MR imaging examination demonstrating GE abnormality; gestation at any subsequent MRI; the nature of GE abnormality (uni- or bilateral, enlargement, cavitation); other abnormal cranial findings on MR imaging; fronto-occipital dimension of the fetal head on the first MR imaging demonstrating GE abnormality; brain stem and cerebellar biometry; and clinical outcomes and postnatal MR imaging findings for liveborn children. The radiologist collected identified patient and imaging data at the site where the study was performed, and all patient identification was removed before the subject was matched with a unique study identification number. An Excel (Microsoft) spreadsheet case report form containing this deidentified data was submitted to the coordinating site.
Genomic Data Collection.
The results of all genomic testing (panel and single-gene testing, singleton WES, trio WES) performed for study subjects were provided by genetics departments affiliated with participating sites. Variants were classified according to the American College of Medical Genetics and Genomics criteria.12
Fetal MR Image Acquisition Technique and Analysis.
Deidentified JPEG images were sent from the participating sites, including full axial, coronal, and sagittal single-shot T2-weighted HASTE or fast spin-echo as well as single-plane echo-planar diffusion-weighted sequences. Imaging was performed on both 1.5T and 3T MR imaging units with a variety of scanning platforms. A torso-array coil with or without a spinal array was used to acquire image data, and section thickness was typically 3–4 mm with in-plane resolution ranging from 0.5 to 1.2 mm. Other imaging sequences may have been acquired at the participating sites, as per local protocol, but these were not provided for this study. However, hemorrhage as the sole cause of GE enlargement was excluded by the participating sites by reviewing the acquired T2* gradient-echo sequences.
The fronto-occipital diameter of the fetal brain was measured at the participating sites as were transverse cerebellar diameter, vermis height, anteroposterior diameter of the pons, and maximum ventricular atrial diameters; centile was calculated at the coordinating site on the basis of gestational age and standard tables of biometric data for normal fetuses.13
Ethics
A site-specific waiver of formal ethics application was granted at all participating sites on the basis of audit methodology to permit sending deidentified clinical and imaging data to the coordinating site.
Key Outcome Measures
The key outcome measures were the following:
Final clinical outcome and imaging diagnoses and/or
Results of genomic testing
Fetal cranial biometry
Associated non-GE findings intra- and extracranial.
RESULTS
Individual institutional data bases with a total of 4135 fetal MR imaging studies were searched by the participating sites for cases meeting all the inclusion criteria, and these cases were submitted for consideration. No submitted case was excluded following review of the provided images by the coordinating site, resulting in 22 study subjects. Their gestational ages at the time of MR imaging ranged from 21 to 33 weeks. There were 9 pregnancy terminations and 13 live births. Genomic testing was performed in 17 of the 22. In 13/22, this was WES; in 2, it was trio exome sequencing; in 1, a prenatal microarray that identified a pathogenic mutation causing tuberous sclerosis; and in the final subject, neuromuscular gene panel testing was performed (More detailed information regarding associated malformations and clinical outcomes is found in the Online Supplemental Data).
Head Size and Genetic/Clinicoradiologic Diagnoses
Subjects 1–8 had fronto-occipital head dimension measurements of ≥2 SDs greater than the mean. Of 5 cases with genetic testing, an MTOR or PI3KCA mutation was identified in 3, with megalencephaly, polymicrogyria, polydactyly, and hydrocephalus syndrome diagnosed prenatally in case 2 because of sonographically demonstrated postaxial polydactyly (Figs 1 and 2). Tuberous sclerosis was confirmed on genetic testing in 2 cases with cardiac rhabdomyoma, and a putative diagnosis was made in a third case with fetal hemimegalencephaly, a markedly enlarged ipsilateral GE, and a cardiac rhabdomyoma (Fig 3). In 2 subjects with large head sizes and no genetic testing, megalencephaly and hemimegalencephaly were present on postnatal imaging.
FIG 1.

PIK3CA pathogenic variant. Case 2 at 31 weeks 3 days' gestation. Megalencephaly, polymicrogyria, polydactyly hydrocephalus syndrome due to a PIK3CA heterozygous pathogenic variant. The fronto-occipital diameter is >6 SDs above the mean for gestation. Enlarged bilateral ganglionic eminences (arrows, A) are seen on T2-weighted single-shot FSE and DWI (A and B). Diffusion-weighted b = 0 image (C) and T2*-weighted EPI (D) confirm the absence of hemorrhage as the cause for ganglionic eminence enlargement. Abnormal opercularization is present with Sylvian fissures lined by peri-Sylvian polymicrogyria (E). Postaxial polydactyly is seen on sonography with arrow indicating a rudimentary sixth digit medial to the fifth digit of the hand (F).
FIG 2.
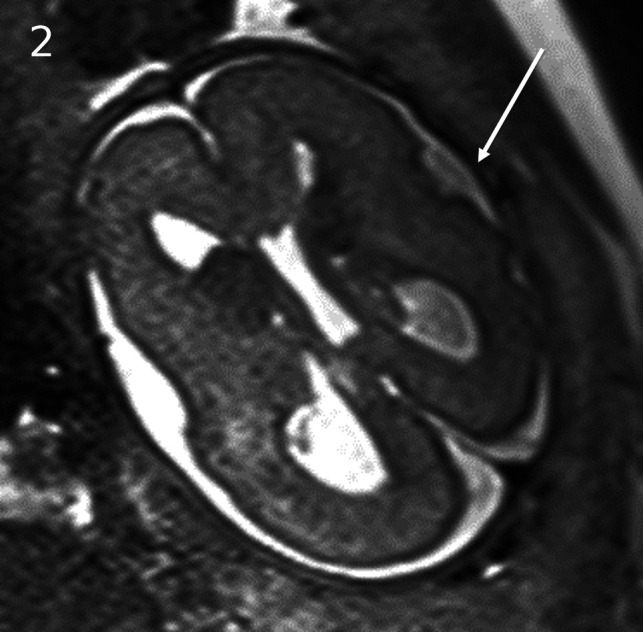
MTOR/PROS pathway mutations. Smith-Kingsmore syndrome (case 5) at 25 weeks' gestation demonstrates enlarged GEs on DWI and T2-weighted single-shot FSE. There is bilateral underopercularization and left peri-Sylvian polymicrogyria (arrow). The right hemisphere is mildly overgrown.
FIG 3.

Tuberous sclerosis complex (A and B). Case 1. Dichorionic diamniotic twin at 22 weeks 5 days' gestation. T2-weighted single-shot FSE image demonstrates hemimegalencephaly and marked enlargement of the ipsilateral GE, which merges with a hypointense masslike lesion in the enlarged right cerebral hemisphere. The fetus had left ventricular cardiac rhabdomyoma on prenatal sonography (arrow, B); the findings likely represent tuberous sclerosis complex with an associated hemispheric malformation or, less likely, coexistent subependymal giant cell astrocytoma. No postmortem data or confirmatory genetic testing was available. Case 8. T2 single-shot FSE at 25 weeks gestation (C) and T2 FSE at 1-week postnatal imaging (D) demonstrate mild enlargement of the right GE, ipsilateral and mild ventriculomegaly on fetal MR imaging, and typical changes of tuberous sclerosis complex postnatally. The fetus' father also had tuberous sclerosis complex.
Of 12 fetuses with small head sizes, 8 had tubulin mutations and 7 of these were mutations involving the TUBA1A gene; 2 had clinical and imaging findings consistent with Walker-Warburg syndrome and cobblestone lissencephaly postnatally; 1 had a PDHA1 mutation; and 1 had a fetal akinesia dyskinesia sequence (Fig 4).
FIG 4.

TUBA1A mutations (A–C). Case 18. T2-weighted single-shot FSE at 24 weeks 4 days' gestation. TUBA1A pathogenic heterozygous variant. The bilateral GEs are enlarged. The fronto-occipital diameter is 2.5, standard deviations below the mean. The corpus callosum and cerebellum are severely hypogenetic/hypoplastic. Underopercularization, severe ventriculomegaly, a thin kinked brainstem (A), a ventral pontine cleft suggestive of a Walker-Warburg phenotype, and apparent diencephalic-mesencephalic fusion or dysplasia (C) are also noted. D, Case 19 at 33 weeks. TUBA1A mutation with enlarged, cavitated GEs and abnormal persistence of hemispheric lamination.
Two fetuses had normal head size, one with an OPHN1 mutation and the other with no genetic diagnosis following WES, but severe developmental delay and seizures.
In no case were the GE abnormalities identified on sonography. Subjects were referred due to a wide variety of abnormalities on sonography, including ventriculomegaly, a small cerebellum, and agenesis of the corpus callosum.
DISCUSSION
Our work has gathered international data from centers performing fetal MR imaging to provide information about the genetic causes and postnatal outcomes of fetuses with GE abnormalities on prenatal MR imaging. We have identified a strong association between MTOR-PIK3CA (PROS) AKT pathway mutations, including tuberous sclerosis, and fetal head size greater than the 97th centile. TUBA1A mutations were the dominant cause of GE abnormalities when frontal-occipital dimensions were less than the third centile, with frequent absence or severe hypoplasia of the corpus callosum and Walker-Warburg imaging phenotype in some cases. Postaxial polydactyly and cardiac rhabdomyoma are useful findings in the fetus with megalencephaly (Frontoocccipital diameter = +2 SDs or more) in separating fetuses with tuberous sclerosis from those with MTOR/PROS spectrum disorders. Also, extreme increases in head size were most often due to MTOR-PIK3CA mutations rather than TSC mutations in our cohort.
Caution needs to be exercised with regard to the major differential diagnosis of pathologic GE enlargement, germinal matrix hemorrhage, which, when isolated in the fetus, does not have the severe neurodevelopmental implications that many of the conditions associated with GE enlargement have. T2*-weighted imaging and DWI should be a routine part of fetal brain imaging, and the b = 0 DWI and T2*-weighted images must be evaluated for hemorrhage before concluding that the GE is abnormally prominent due to another cause. Notably, in no patient in our series was a GE abnormality seen in the absence of other major structural malformations involving one or more of the corpus callosum, cerebellum, brainstem, microcephaly, or megalencephaly, or a malformation of cortical development. No case of GE abnormality in fetuses with tuberous sclerosis complex was seen in the absence of cardiac rhabdomyoma.
Several findings that lay outside our intended aims are worthy of mention:
In 1 fetus with tuberous sclerosis complex, the only cranial abnormalities were mild ventriculomegaly and subtle asymmetric enlargement of 1 GE (Fig 3). The fetus also had a father with tuberous sclerosis complex and a cardiac rhabdomyoma on prenatal sonography, and it would have been possible to dismiss the subtle GE abnormality or perhaps attribute it to germinal matrix hemorrhage without this clinical and sonographic information that increases the pretest probability of such an abnormality reflecting tuberous sclerosis complex. This finding underlines the importance of the acquisition of T2*-weighted sequences to exclude hemorrhage as the cause of asymmetric GE enlargement and the importance of knowledge of the prenatal sonographic findings and family history at the time of MR imaging interpretation.
The imaging of our single case of pyruvate dehydrogenase deficiency (Fig 5) manifested as bilateral cavitated GE and agenesis of the corpus callosum with microcephaly, superficially resembling findings in some fetuses with tubulinopathy. The combination of ACC with germinolytic cysts is well-recognized as the postnatal imaging phenotype of PDHA1 mutations causing pyruvate dehydrogenase deficiency.14 It may be that the GE cavitations “collapse” with physiologic resolution of the cavitated GE, resulting in germinolytic cysts postnatally.
We have confirmed that the Walker-Warburg imaging phenotype with cobblestone lissencephaly may be due to tubulinopathy. To our knowledge, there is only a single case report of this in the literature15 in which a TUBB3 mutation was found to be the causative pathogenic variant, but no images were provided. Cobblestone lissencephaly is considered pathognomonic of a group of autosomal recessive disorders characterized by ocular and muscular deficits, including Walker–Warburg syndrome, muscle-eye-brain disease, and Fukuyama muscular dystrophy. All these conditions grouped under the term “α-dystroglycanopathies” result in neuroglial overmigration and the typical cobblestone appearance of the cortex seen on postnatal but often not on fetal MR imaging examinations. Two-thirds of the cases of cobblestone lissencephaly can be attributed to mutations in 1 of 6 genes, particularly POMT1, POMT2, and FKPR, which are involved in the most severe form of α-dystroglycanopathy, Walker-Warburg syndrome.16 However, if a dystroglycanopathy or muscular dystrophy panel is used to identify the causative genetic variant in cobblestone lissencephaly, the diagnosis may be missed.15 Indeed, one of our subjects with postnatal cobblestone lissencephaly had muscular dystrophy gene panel testing, not including all of the tubulin genes, which was negative for an explanatory mutation; this case is now the subject of re-evaluation with WES. Another (case 16) had initially negative genomic testing findings following pregnancy termination, with cobblestone lissencephaly diagnosed at postmortem examination; a diagnosis of a TUBA1A pathogenic variant in this fetus was only made later, following the neonatal death of a younger sibling diagnosed with this pathogenic variant (case 17). In this case, the presumption is that one of the parents has germline mosaicism, but this has not yet been investigated with genomic testing. Genetic re-evaluation of fetuses with structural brain abnormalities is important.
FIG 5.

PDHA1 and OPHN1 mutations. A and B, PDHA1 mutation. Case 20 at 23 weeks 3 days' gestation. PDHA1 mutation. Agenesis of the corpus callosum with the anterior commissure present (short arrow), cavitated enlarged GEs, and anterior temporal pole subependymal pseudocysts (germinolytic cysts) (long arrows). C and D, Case 21 at 29 weeks. OPHN1 mutation with mild ventriculomegaly (11 mm) and bilaterally enlarged GEs.
Our study has limitations. The retrospective methodology of case identification and search terms used may have biased our results by finding what we were suspecting, and there was the risk that other less common causes of GE abnormality have been overlooked. However, it was not feasible for participating sites to review their entire fetal MR imaging database in search of potential cases for this exploratory study. We sought to maximize the yield of database review by directing our search. In addition, fetuses with GE abnormalities but no genetic testing or postnatal outcome data were, a priori, not included in the study, introducing possible selection bias.
CONCLUSIONS
Our work will raise awareness of the radiologic appearance of the abnormal fetal GE and the associated malformations. It will thus facilitate prenatal counseling and identification of further genetic causes for this disorder. Increasing access to genomic testing for prenatal cases will increase genetic diagnoses in fetuses presenting with features suggestive of a GE disorder, and improvements in turnaround time of results of such testing will allow genomic testing results in this setting to guide decision-making in pregnancy. Information on the genotype-phenotype correlation in this group of patients will continue to improve and inform future work in this area.
Supplementary Material
ABBREVIATIONS:
- GE
ganglionic eminence
- WES
whole exome sequencing
- TSC
tuberous sclerosis complex
Footnotes
This work was supported by the Research Grant for Clinical Radiology awarded by The Royal Australian and New Zealand College of Radiologists (2020/RANZCR/004).
Paper previously presented in part at: Virtual World Congress of the International Society of Ultrasound in Obstetrics and Gynaecology, October 16–18, 2020; Virtual
Disclosures: Stacy K. Goergen—RELATED: Grant: Royal Australian and New Zealand College of Radiologists, Comments: We received a grant of A$5000 (3860.50 US dollars) from the 2020 Royal Australian and New Zealand College of Radiologists Research Grant funding round to support the conduct and publication of this study. The grant disbursement will be managed by the Monash University School of Clinical Sciences at Monash Medical Center, Clayton, Victoria, Australia.* Michael Fahey—UNRELATED: Board Membership: Australian New Zealand Child Neurology Society, Comments: voluntary board membership; Expert Testimony: multiple expert testimonies with no influence over the work; Grants/Grants Pending: NHMRC, CPA, Comments: no influence over the work; Stock/Stock Options: Sigma Pharmaceuticals, Comments: of no influence over the work. Laura Fender—UNRELATED: Employment: hospital consultant radiologist, Comments: full-time employment at Public Hospital; Sir Charles Gairdner Hospital, Perth, Western Australia, Australia; King Edward Memorial Hospital, Perth, Western Australia, Australia. Jason R. Pinner—UNRELATED: Employment: Sydney Children's Hospital. Andrea Righini—UNRELATED: Expert Testimony: 2396.39 US dollars paid to the author's institution, Comments: Expert testimony in medicolegal cases. *Money paid to the institution.
References
- 1.Ulfig N. Ganglionic eminence of the human fetal brain: new vistas. Anat Rec 2002;267:191–95 10.1002/ar.10104 [DOI] [PubMed] [Google Scholar]
- 2.Del Bigio MR. Cell proliferation in human ganglionic eminence and suppression after prematurity-associated haemorrhage. Brain 2011;134:1344–61 10.1093/brain/awr052 [DOI] [PubMed] [Google Scholar]
- 3.Vasung L, Lepage C, Rados M, et al. Quantitative and qualitative analysis of transient fetal compartments during prenatal human brain development. Front Neuroanat 2016;10:11. 10.3389/fnana.2016.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Righini A, Frassoni C, Inverardi F, et al. Bilateral cavitations of ganglionic eminence: a fetal MR imaging sign of halted brain development. AJNR Am J Neuroradiol 2013;34:1841–45 10.3174/ajnr.A3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Righini A, Cesaretti C, Conte G, et al. Expanding the spectrum of human ganglionic eminence region anomalies on fetal magnetic resonance imaging. Neuroradiology 2016;58:293–300 10.1007/s00234-015-1622-5 [DOI] [PubMed] [Google Scholar]
- 6.Boitor-Borza D, Kovacs T, Stamatian F. Ganglionic eminence within the early developing brain visualised by 3D transvaginal ultrasound. Med Ultrason 2015;17:289–94 10.11152/mu.2013.2066.173.rbb [DOI] [PubMed] [Google Scholar]
- 7.Prefumo F, Petrilli G, Palumbo G, et al. Prenatal ultrasound diagnosis of cavitation of the ganglionic eminence. Ultrasound Obstet Gynecol 2019;54:558–60 10.1002/uog.20236 [DOI] [PubMed] [Google Scholar]
- 8.Pooh RK. Normal ganglionic eminence and blighted ganglionic eminence in neuronal migration disorder cases detected by transvaginal neuroscan. In: Proceedings of 27th World Congress on Ultrasound in Obstetrics and Gynecology, Vienna, Austria; September 16–19, 2017 [Google Scholar]
- 9.Scarabello M, Righini A, Severino M, et al. Ganglionic eminence anomalies and coexisting cerebral developmental anomalies on fetal MR imaging: Multicenter-based review of 60 cases. AJNR Am J Neuroradiol 2021;42:1151–56 10.3174/ajnr.A7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Best S, Wou K, Vora N, et al. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn 2018;38:10–19 10.1002/pd.5102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pratt M, Garritty C, Thuku M, et al. Application of exome sequencing for prenatal diagnosis: a rapid scoping review. Genet Med 2020;22:1925–34 10.1038/s41436-020-0918-y [DOI] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kline-Fath B, Bulas D, Bahado-Singh R. Fundamental and Advanced Fetal Imaging Ultrasound and MRI. Wolters Kluwer Health; 2015 [Google Scholar]
- 14.Poretti A, Blaser SI, Lequin MH, et al. Neonatal neuroimaging findings in inborn errors of metabolism. J Magn Reson Imaging 2013;37:294–312 10.1002/jmri.23693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powis Z, Chamberlin AC, Alamillo CL, et al. Postmortem diagnostic exome sequencing identifies a de novo TUBB3 alteration in a newborn with prenatally diagnosed hydrocephalus and suspected Walker-Warburg syndrome. Pediatr Dev Pathol 2018;21:319–23 10.1177/1093526617698611 [DOI] [PubMed] [Google Scholar]
- 16.Devisme L, Bouchet C, Gonzalès M, et al. Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain 2012;135:469–82 10.1093/brain/awr357 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.