

Graphical abstract

Keywords: COVID-19, Therapeutic targets, Drug targets, Docking, Drug suitability
Abstract
The severe acute respiratory syndrome coronavirus 2, famous as COVID-19, has recently emerged as a novel virus and imposed an unrecoverable loss to global health and the economy. At present, no effective drug against COVID-19 is available and currently available viral drugs targeting the viral key proteins of related RNA viruses have been found ineffective against COVID-19. This study evaluated the inhibitors of the viral proteases and other structural proteins, including Mpro (Main protease), RdRp (RNA-dependent RNA polymerase), and spike glycoprotein from synthetic and herbal sources. The molecular docking-based approach was used to identify and evaluate the putative inhibitors of key proteins involved in viral replication and survival. Furthermore, the pharmaceutical properties of these inhibitors were explored to predict the drug suitability as a therapeutic agent against COVID-19 by considering adsorption, distribution, metabolism, and excretion (ADME) using Lipinski’s rule or SwissADME. Trandolapril, Benazepril, and Moexipril were evaluated as the best non-carcinogenic and non-toxic potential inhibitors of spike glycoprotein, Mpro, and RdRp, respectively. The drugs showed significant binding affinities against the active sites of respective SARS_CoV-2 target proteins; hence, they can be used as potential therapeutic agents for the treatment of COVID-19.
1. Introduction
The coronavirus disease (COVID-19), caused by the severe acute respiratory syndrome coronavirus2 (SARS-CoV-2), is rapidly spread with > 4.1 million confirmed cases and > 310 thousand worldwide deaths, and the cases are still increasing [1] due to its high human-to-human transmission [2]. The SARS-CoV-2 is a positive-sense single RNA-containing enveloped virus belonging to the β-coronavirus family, composed of approximately 30,000 nucleotides [3]. Possible therapeutic targets of COVID-19 include main protease (Mpro), RNA-dependent RNA polymerase (RdRp), and spike glycoproteins, playing a role in the proteolytic maturation of the virus [4], replication [5], and survival against antibodies [6], respectively. Although various drugs have been proven effective against viruses belonging to the same group, none of them exhibits similar potential as a cure to COVID-19 [7,8]. The FDA approved malaria drugs, including hydroxychloroquine and chloroquine, but withdrew the authorization of these drugs against COVID-19 as found they were ineffective in later studies [9]. Similarly, Ivermectin and famotidine were also tested but found ineffective against COVID-19 [10]. Avigan, a potent inhibitor of many RNA viruses, was reported to shorten the recovery time but has several side effects such as anaphylactic shock and pneumonia [11]. The anti-inflammatory drug dexamethasone has been found to reduce mortality in patients with severe conditions but is not recommended for patients with mild symptoms [12]. Antiviral drugs, including remdesivir, favipiravir, and merimepodib, effectively reduce the mortality rate [13]. Still, we desperately need an effective and safe drug for the treatment of COVID-19. Therefore, to develop effective drugs against COVID-19, inhibitors of the drug targets need to be explored.
The computational docking approach provides the opportunity to identify and evaluate the binding affinities and efficiencies of various natural source and synthetic inhibitors using different molecular docking software such as AutoDock [14], MOE (Molecular Operating Environment) [15], MVD [16], and RosettaCommons [17]. After evaluating the efficient inhibitors, drug suitability could be determined by analyzing their pharmacological properties. Although validation of the putative therapeutic agents could only be done after testing experimentally, computational docking could provide a gateway towards developing effective drugs against diseases, including COVID-19.
Recently, several studies have proposed different compounds as potential drug candidates against COVID-19, for example, 11 compounds comprising macrolides antibiotics, antiarrhythmic agents, proton pump inhibitors, and CNS drugs [18]. Similarly, ten compounds with high coronavirus inhibition potential were screened using two different approaches, including the Docking Consensus Approach (DCA) and Common Hits Approach (CHA) [19]. Another study proposed the broad-spectrum anti-coronavirus drug remdesivir, as a potential drug against SARS-CoV-2 [20]. Simultaneously, several FDA-approved drugs and few in clinical trials have been screened as potential inhibitors of three viral proteins, i.e., Mpro, papain-like protease, and S-proteins [21]. In another study, three compounds, including bedaquiline, glibenclamide, and miconazole, have been computationally identified as promising inhibitors of Mpro [22]. Meanwhile, 3CL proteases of COVID-19 have also been chosen as a drug target, and several FDA-approved drugs, including oxytetracycline, doxorubicin, kanamycin, cefpiramide, teniposide, proanthocyanidin, and salvianolic acid B, have been predicted as a putative inhibitor of 3CL proteases [23].
In this study, we identified the binding affinities of 12 different drug candidates, including one herbal and animal source compound against the three COVID-19 key proteins (Mpro, RdRp, and spike glycoprotein) using two different tools (MOE and Discovery studio) and confirmation of drug suitability through pharmacological properties. The MOE is a reliable tool for determining the quantitative structure-activity relationship (QSAR) in micro molecules through high-throughput screening (HTS), docking, and interaction [24], which was used to evaluate the binding affinities of drug candidates with the three key proteins. Furthermore, to confirm the bioavailability of inhibitors, the ADMET analysis that includes absorption, distribution, metabolism, excretion, and toxicity of these inhibitors in the brain and gastrointestinal tract was performed using Lipinski’s Ro5 or Swiss adsorption, distribution, metabolism, and excretion (ADME) rules in this study [25]. Although approved drugs have favorable ADMET, their suggested therapeutic dosage depends on binding affinity with the molecular target and ADMET profile. Hence, the ADMET analysis was performed to provide fundamentals for drug discovery.
2. Material and methods
2.1. Protein preparation
The structures of the three key proteins were retrieved from the protein databank under PDB ID: 6XQB (RdRp), 6VSB (spike glycoprotein), and 6LU7 (Mpro). These structures were selected from the database because of their high-resolution power and the lowest R-value. R-value is the measure of how well the simulated diffraction pattern matches the experimentally observed diffraction pattern. The lower the R-value, the more reliable a structure is. The MOE software was used to balance the polar hydrogen and charges to increase protein susceptibility towards electronegative atoms (ligands). Energy minimization was performed to stabilize the target proteins to accomplish molecular docking at a stable protein phase. Dummies, i.e., dummy atoms comprised of lone pair (LP) atoms exhibiting no bonds, were created over the active sites of the target proteins to locate ligands. The locations of dummy atoms over the receptor protein binding sites were identified and defined in the MOE. The ligands remained restricted to residues in the binding sites where dummies were created. Dummies in the MOE were anonymous to a grid box in the Autodock vina, where a grid box covering 2–5 angstroms around binding sites was created to enhance interaction specificity.
2.2. Ligand preparation
For the molecular docking, ten synthetic compounds: Benazepril, Captopril, Cilazapril, Enalapril, Lisinopril, Moexipril, Perindopril, Quinapril, Ramipril, and Trandolapril; one animal source compound, i.e., Teprotide, and one herbal source compound, i.e., Allicin were screened (Table 1 ). All screened ligands have positive and greater than 0 ALogP values, indicating lipophilic screened compounds. AutoDockTools (ADT) was adopted to calculate the Gasteiger charges for selected ligands, and a torsion count widget was chosen to classify rotatable and non-rotatable ligand bonds. ADMET analysis revealed the activeness and inactiveness of compounds studied here (Fig. 1 ).
Table 1.
Information of Screened ligands used for molecular docking.
| Sr. # | ChEMBL ID | Compound | ALogP | Source |
|---|---|---|---|---|
| 1 | CHEMBL838 | BENAZEPRIL | 2.57 | Synthetic |
| 2 | CHEMBL1560 | CAPTOPRIL | 0.63 | Synthetic |
| 3 | CHEMBL515606 | CILAZAPRIL | 1.60 | Synthetic |
| 4 | CHEMBL578 | ENALAPRIL | 1.60 | Synthetic |
| 5 | CHEMBL1237 | LISINOPRIL | 1.21 | Synthetic |
| 6 | CHEMBL1165 | MOEXIPRIL | 2.58 | Synthetic |
| 7 | CHEMBL1581 | PERINDOPRIL | 1.94 | Synthetic |
| 8 | CHEMBL1592 | QUINAPRIL | 2.57 | Synthetic |
| 9 | CHEMBL1168 | RAMIPRIL | 2.38 | Synthetic |
| 10 | CHEMBL1519 | TRANDOLAPRIL | 2.77 | Synthetic |
| 11 | CHEMBL1519 | TEPROTIDE | N-A | Snake |
| 12 | CHEMBL359965 | ALLICIN | 1.76 | Garlic |
Fig. 1.

ADMET (absorption, distribution, metabolism and excretion, and toxicity) parameters disseminations of ligands based on the threshold of light blue ligands bars are actives and pink color bars representing in-actives in our benchmarking sets.
2.3. Molecular docking
The MOE was used for molecular docking in this study, while BIOVIA Discovery Studio was employed to visualize and validate the MOE results. MOE implements sophisticated optimization and forcefield algorithm for rescoring the rate of ligand atom’s appearance. The results were evaluated using an S-score (binding energy value) implemented by MOE, where the S-score is MM/GBVI binding free energy estimation. In addition, the top ten ligands exhibiting significant binding affinity with their target proteins were further analyzed to explore their pharmacokinetics and drug-likeness.
2.4. Binding sites chosen for molecular docking
Identification of binding hot spots in proteome is the fundamental step for performing molecular docking against putative inhibitors. Increase in the known repertoire of binding sites in proteome could provide an array of alternative targets for drug discovery. A considerable work has recently been performed to determine the potential druggable sites, for example, Cavasotto et al. [26] performed a computational analysis to explore the potential druggable sites in all the structural, non-structural, and accessory proteins of SARS-CoV-2. Binding hot spots identified in such studies serve to broad the therapeutic options for drug discovery. In addition to consider the already predicted binding sites in previous studies, we determined new binding sites and chosen sites in RdRp, Spike protein, and Mpro for docking against putative therapeutic agents are discussed below.
2.4.1. Binding sites in RdRp
Previous studies have reported that Tyr618, Cys622, Asn691, Asn695, Met755, Ile756, Leu757, Leu758, Ser759, Asp760, Asp761, Ala762, Val763, Glu811, Phe812, Cys813, and Ser814 are the main residues involved in the interaction with ligand molecues [[27], [28], [29]]. Asp618 is the most conserved residue in viral RdRp and the most common potential drug site observed in the studies. The binding potential of Moexipril (the ligand with the highest binding affinity) with RdRp is mostly regulated by H-bonds. Moexipril formed ten H-bonds with the active sites of Lys551 and Asp618 and with other important residues such as Trp617, Tyr619, Lys621, Ser682, Glu811, Lys621, Tyr619, Trp617, Ser682, and Glu811 with a docking score of −9.68 kcal/mol. The essential residues in the active site are neighboring aspartates, i.e., Lys551, Asp618, and Asp762, involved in the RdRp enzyme's real reaction.
2.4.2. Binding sites in spike glycoprotein (6VSB)
Chain A of the homotrimer spike protein was chosen to examine all domains. The binding sites of the target protein were predicted using Discovery studio's binding site module. The receptor-binding domain (Leu335 to Gly526) included the unique active site region or loop region, including the functional, active site (NAG of RCSB: 6VSB, chain A). The active site residues of Asn122 and Glu156 were selected as the grid center, with the following coordinates: the spacing was 0.753, the XYZ values were 76 126, 76, and the center coordinates were −36.336, 23.128, and 21.195.
The drug targets on the SARS-CoV-2 protein spike were also explored using PROCHECK, PDBsum, and DrugPort protein sequence searches, revealing the drug compounds and their target proteins. The medications targeted to the protein were discovered, and the docking server was used to perform molecular docking studies of those medications to the spike protein of SARS-CoV-2. The binding affinity in each case was identified using GlaxyWEb, global energy with PatchDock, followed by FireDock, or refinement and binding energy with MOE. Out of four proposed binding sites, the ligand in this study has shown potential inhibitory action (S-score = −6.9314) against Asn122 and Glu156 sites.
2.4.3. Binding sites in Mpro
Asn142, Gly143, Phe140, His164, Gln190, and His164 were chosen as potential drug binding sites in Mpro, and the same sites were selected in recent studies [30,31]. Among all ligands/drug candidates studied here, Benazepril has shown significant binding affinity (S-score = −8.884) with two of the potential binding sites, Asn142 and Gly143.
2.5. Pharmacokinetic properties and drug-likeness
The binding affinity of inhibitors with their target proteins does not ensure the suitability of inhibitors as potential drugs in a biological system. Therefore, the drug-likeness of inhibitors in a biological system needs to be explored using assessments such as the ADME analysis. We performed LRo5 and Egan’s BOILED-Egg methods to reveal the pharmacokinetics of ligands and their favorability in a biological system. Lipinski’s Ro5 interpret the drug-likeness based on the physicochemical properties such as the molecular weight (MW) should be less than 500 g/mol, hydrogen bond donors and acceptors should be less than 5 and 10, respectively, and lipophilicity should be less than or equal to 5 [32]. The evaluation of pharmacological activity and drug-likeness of compounds before performing in-vitro analysis to be used as an orally active drug is widely practiced these days.
Lipinski’s Ro5 [33] and Egan’s BOILED-Egg methods [34] were used to evaluating ligand suitability for biological systems. The ADME parameters were evaluated using SwissADME [35], a web-based accessible tool available to compute physiochemical and pharmacokinetic properties, drug-likeness, and medicinal chemistry. Furthermore, environmental evaluation of drug addicts and ADMET parameters were also performed to evaluate the drug suitability [36]. The ADMET database for the structure-activity link, abbreviated AdmetSAR, is an online service containing information on toxicity, carcinogenicity, and whether the medication complies with the Lipinski Regulations.
3. Results
3.1. Molecular docking
Drug candidates in this study demonstrated varying binding affinities with proteins via molecular docking of SARS-CoV-2 target proteins using MOE and Discovery. The results of Discovery were in line with those predicted through MOE, validating the docking results. The drug candidates were ranked based on their binding affinities; an inhibitor with a lower value of binding free energy is more likely to establish a strong interaction (Table 2 ). We docked 12 compounds, including 10 synthetics, an herbal source (Allicin), and an animal source (Teprotide) ligand against the active sites of 3 key therapeutic candidate proteins of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), including 2 spike glycoproteins, Mpro and RdRp. The MOE generated 200 possible poses with numerous S-scores (binding energy score). The ligands were ranked according to the complex (ligand + target protein) S-score, and ligands showing binding energy score > −6.0 kcal/mole were selected for further analysis. Table 2 lists the 200 candidates in the top 10 poses with their binding energy scores. Teprotide showed a maximum binding energy score (−6.9535) against spike glycoprotein, followed by Trandolapril (−6.9314). Similarly, Teprotide showed a maximum binding energy score (−10.9458) against Mpro, followed by Benazepril (−8.884). Meanwhile, Moexipril showed a maximum binding score (−10.2714) against the RdRp crystal structure (PDB ID: 6XQB).
Table 2.
Inhibitors showing binding affinities against active sites of target proteins.
| RdRp |
Mpro |
Spike glycoprotein |
||||||
|---|---|---|---|---|---|---|---|---|
| Sr. # | Ligand | PubChem ID | Energy score (kcal/mol) (MOE) Molecular Operating Environment | Binding Affinity (KD) Dissociation Constant (Discovery studio) | Energy score (kcal/mol) (MOE) | Binding Affinity (KD) (Discovery studio) | Energy score (kcal/mol) (MOE) | Binding Affinity (KD) (Discovery studio) |
| 1 | Teprotide | 443,376 | −7.25 | −9.7 | −10.95 | −12.4 | −6.95 | −10.9 |
| 2 | Moexipril | 91,270 | −10.27 | −13.5 | −7.55 | −7.1 | −5.93 | −7.3 |
| 3 | Cilazapril | 56,330 | −9.03 | −9.8 | −7.99 | −7.9 | −6.35 | −7.9 |
| 4 | Benazepril | 5,362,124 | −8.97 | −8.9 | −8.88 | −9.0 | −6.34 | −7.9 |
| 5 | Trandolapril | 5,484,727 | −6.76 | −8.7 | −8.04 | −8.4 | −6.93 | −8.0 |
| 6 | Quinapril | 54,892 | −7.74 | −8.5 | −8.45 | −8.7 | −6.32 | −7.7 |
| 7 | Lisinopril | 5,362,119 | −8.36 | −8.1 | −6.01 | −6.7 | −5.48 | −6.5 |
| 8 | Enalapril | 5,388,962 | −8.50 | −8.0 | −7.77 | −7.7 | −5.94 | −7.3 |
| 9 | Ramapril | 121,486,657 | −8.34 | −8.0 | −6.17 | −6.9 | −6.31 | −7.7 |
| 10 | Perindopril | 107,807 | N/A | −6.8 | −7.13 | −6.9 | −6.02 | −7.4 |
| 11 | Captopril | 44,093 | −5.40 | −6.5 | −6.00 | −6.2 | −5.34 | −6.5 |
| 12 | Allicin | 65,036 | No interaction | −4.9 | −5.90 | −4.7 | −5.21 | −4.9 |
3.2. Drug-likeness
The docking results revealed that although SARS-CoV-2 (PDB ID: 6VSB, spike glycoprotein) demonstrated the highest affinity to Teprotide, it does not follow Lipinski’s Ro5. In contrast, Trandolapril, which is ranked second, with a binding energy score of −6.9314, followed the Ro5. All six selected ligands fulfilled the parameters of Lipinski’s’ Ro5, demonstrating that these chemical compounds (ligands) are suitable for drug bioavailability (Table 3 ). Therefore, Trandolapril could be a better choice among all the ligands studied to be used as a potential therapeutic agent against SARS-CoV-2 spike glycoprotein to treat COVID-19.
Table 3.
Lipinski’s rule of five for ADME analysis of our inhibitors (ligands).
| No. | Name | Lipinski’s Rule of Five |
Drug-likeness | ||||
|---|---|---|---|---|---|---|---|
| Molecular weight (g/mol) | Lipophilicity (MLogP) |
Hydrogen bond donors | Hydrogen bond acceptors | No. of Rule Violations | |||
| Less than 500 Da | Less than 5 | Less than 5 | Less than 10 | Less than 2 violations | Lipinski’s rule follow | ||
| 13. | Allicin | 162 | 1.18 | 0 | 1 | 0 | Yes |
| 11. | Benzapril | 424 | 2.23 | 2 | 6 | 0 | Yes |
| 10. | Captopril | 217 | 0.45 | 1 | 3 | 0 | Yes |
| 6. | Cilizapril | 417 | 1.79 | 2 | 7 | 0 | Yes |
| 8. | Enalapril | 376 | 1.32 | 2 | 6 | 0 | Yes |
| 2. | Fosinopril | 563 | 3.74 | 1 | 7 | 0 | Yes |
| 5. | Lisinopril | 405 | −1.46 | 4 | 7 | 0 | Yes |
| 3. | Moexipril | 498 | 1.54 | 2 | 8 | 0 | Yes |
| 12. | Perindopril | 368 | 1.36 | 2 | 6 | 0 | Yes |
| 4. | Quinapril | 438 | 2.17 | 2 | 6 | 0 | Yes |
| 9. | Ramipril | 416 | 1.98 | 2 | 6 | 0 | Yes |
| 1. | Teprotide | 1101 | −3.11 | 10 | 13 | 3: MW > 500, NH or OH > 5, N or O > 10, 1: MW > 500 |
No |
| 7. | Trandolapril | 430 | 2.19 | 2 | 6 | 0 | Yes |
Similarly, Teprotide showed a maximum binding energy score (−10.9458), followed by Benazepril (−8.884) against Mpro. As Teprotide does not follow Lipinski’s Ro5, Benazepril is the best among the 12 candidates to inhibit Mpro activity. Allicin does not show any interaction against the active sites of SARS-CoV-2 RdRp, while Moexipril shows the highest binding energy score of −10.2714 and does not violate the Ro5 (Table 3). The pose confirmation of ligand-protein interactions of Trandolapril, Benazepril, and Moexipril against spike glycoproteins, Mpro, and RdRp demonstrates that the binding energy scores are due to ligand-protein interactions against the active sites of SARS-CoV-2 proteins (Table 4 ).
Table 4.
SARS-CoV-2 key proteins and best ligand interaction against active sites covering pockets and generating pose by making ligand-receptor complex.
| Spike Glycoprotein | Mpro | RdRp |
|---|---|---|
| Trandolapril (S-score -6.93) | Benazepril (S-score -8.88) | Moexipril(S-score -10.27) |
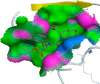 |
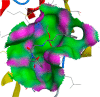 |
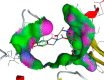 |
 |
 |
 |
SwissADME [35] was used to model the drug-like behavior of inhibitors, including the ingestion, delivery, metabolism, and excretion of these inhibitors within the body. Egan's BOILED-egg process, available in the SwissADME instrument, was used to determine the absorption of inhibitors in the gastrointestinal tract and brain. The BOILED-Egg (Brain Or IntestinaL EstimateD) permeation predictive model, also referred to as an Egan’s egg, was used with a threshold of WLOGP ≤ 5.88 and TPSA ≤ 131.6 and a simple graphical description of how far a molecular structure is for successful absorption from the optimal physiochemical area. The yolk area displays the molecules that can actively infiltrate through the blood-brain barrier (BBB) in this 2-D graphical representation.
The molecules found in the white region are supposed to be actively absorbed by the gastrointestinal (GI) tract. Results of the BOILED egg demonstrating the probability of absorption and penetration in the GI tract and brain using WLOGP and TPSA parameters are shown in Fig. 2 . All ligands in this study followed Lipinski's law except Teprotide, which violates the three criteria (MW > 500, number of donors of hydrogen bonds > 5, and number of acceptors of hydrogen bonds > 10); hence, violating the system of BOILED-egg. Although Teprotide has an optimum binding affinity to Human angiotensin-converting enzyme (ACE), it is not recommended as an orally active drug due to the violation of the Lipinski law. The Egan’s inhibitor egg graph produced with SwissADME revealed that brain absorption was only possible in the case of Allicin. However, the rest of the inhibitors demonstrated an appropriate range of GI absorption, excluding Teprotide and Lisinopril (WLOGP > 5.88 and TPSA > 131.6) in an acceptable range (Fig. 2).
Fig. 2.

Evaluation of ligands permeability through the gastrointestinal tract and brain by BOILED-Egg method: yolk region is for those molecules that permeate passively through the blood-brain barrier; white region represents those molecules that can passively be absorbed by the gastrointestinal tract.
Any chemical compound must be non-toxic to the body and do not show liability with different transporters and channels such as the hERG channel of hurt. The chemical compound toxicity assessment and ADMET study concluded that the three chemical compounds (Trandolapril, Benazepril, and Moexipril; showing high binding affinities against active sites of respective target proteins using ligands), follow Lipinski’s Ro5 (Table 3). These compounds do not exhibit liability towards the hERG channel and blood-brain barrier (Table 5 ). However, Trandolapril showed human intestinal absorption, Benazepril showed hepatotoxicity to some extent, and Moexipril has shown liability with estrogen receptor binding and androgen receptor binding transporters as well have hepatotoxicity. The dosage for the compounds is optimized based upon their toxicity to human.
Table 5.
Drug toxicity and body channels liability profiling of candidate’s compounds.
 |
Green color: Don’t show liability with transporters.
Light green color: Slightly show transporter liability.
Red Color: Potential liable with transporter/channel.
Light red color: Liable with transporter/channel.
The docking results, Lipinski’s Ro5, and drug toxicity assessment concluded that Trandolapril, Benazepril, and Moexipril are non-carcinogenic and non-toxic compounds. The compounds showed significant binding affinities against active sites of SARS-CoV-2 target proteins; hence, they might prove as potential therapeutic agents to treat COVID-19 after clinical trials.
Declaration of Competing Interest
The authors report no declarations of interest.
Acknowledgements
The authors are thankful to the Institute of Research and Consulting Studies at King Khalid University for supporting this research through grant number 9-49-S-2020. The authors also acknowledge the support from the Department of Computer Science, University of Agriculture Faisalabad, Pakistan for providing literature, computational and all other facilities.
References
- 1.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan J.F.-W., Yuan S., Kok K.-H., To K.K.-W., Chu H., Yang J., Xing F., Liu J., Yip C.C.-Y., Poon R.W.-S. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lillicrap D., Morrissey J.H. COVID‐19: 2020 a year in turmoil. J. Thromb. Haemost. 2020;18:993. doi: 10.1111/jth.14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Pawlik B., Wang X., Jochmans D., Neyts J., Mlynarski W., Hilgenfeld R., Drag M. SARS-CoV-2 M(pro) inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2021;17(2):222–228. doi: 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Y., Yin W., Xu H.E. RNA-dependent RNA polymerase: structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun. 2021;538:47–53. doi: 10.1016/j.bbrc.2020.08.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duan L., Zheng Q., Zhang H., Niu Y., Lou Y., Wang H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike-based vaccine immunogens. Front. Immunol. 2020;11:2593. doi: 10.3389/fimmu.2020.576622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan Q., Jin Y.J. Ostavimir is ineffective against COVID-19: in silico assessment, in vitro and retrospective study. medRxiv. 2020 doi: 10.1016/j.bioorg.2020.104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J., Xie B., Hashimoto K. Current status of potential therapeutic candidates for the COVID-19 crisis. Brain Behav. Immun. 2020;87:59–73. doi: 10.1016/j.bbi.2020.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yazdany J., Kim A.H. Use of hydroxychloroquine and chloroquine during the COVID-19 pandemic: what every clinician should know. Am. Coll. Physicians Obs. 2020 doi: 10.7326/M20-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panoutsopoulos A.A. Known drugs and small molecules in the battle for COVID-19 treatment. Genes Dis. 2020;7:528–534. doi: 10.1016/j.gendis.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pilkington V., Pepperrell T., Hill A. A review of the safety of favipiravir–a potential treatment in the COVID-19 pandemic. J. Virus Erad. 2020;6:45–51. doi: 10.1016/S2055-6640(20)30016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liou T.G., Adler F.R., Hatton N.D. The uncertain role of corticosteroids in the treatment of COVID-19. JAMA Intern. Med. 2021;181(1):139–140. doi: 10.1001/jamainternmed.2020.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eroglu E., Toprak C. Overview of favipiravir and remdesivir treatment for COVID-19. Int. J. Pharmceut. Sci. Res. 2021:1950–1957. [Google Scholar]
- 14.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vilar S., Cozza G., Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008;8:1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 16.Bitencourt-Ferreira G., de Azevedo W.F. Molegro virtual docker for docking. Methods Mol. Biol. 2019;2053:149–167. doi: 10.1007/978-1-4939-9752-7_10. [DOI] [PubMed] [Google Scholar]
- 17.Conchúir S.Ó., Barlow K.A., Pache R.A., Ollikainen N., Kundert K., O’Meara M.J., Smith C.A., Kortemme T.J.P. A web resource for standardized benchmark datasets, metrics, and Rosetta protocols for macromolecular modeling and design. PLoS One. 2015;10(9) doi: 10.1371/journal.pone.0130433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Touret F., Gilles M., Barral K., Nougairède A., Helden J., Decroly E., Lamballerie X., Coutard B.J. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci. Rep. 2020;10(1):1–8. doi: 10.1038/s41598-020-70143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Battisti V., Wieder O., Garon A., Seidel T., Urban E., Langer T.J.M. A computational approach to identify potential novel inhibitors against the coronavirus SARS‐CoV‐2. Mol. Infor. 2020;39(10) doi: 10.1002/minf.202000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shyr Z.A., Gorshkov K., Chen C.Z., Zheng W.J., Therapeutics E. Drug discovery strategies for SARS-CoV-2. J. Pharmacol. Exp. Ther. 2020;375(1):127–138. doi: 10.1124/jpet.120.000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavasotto C.N., Di Filippo J.I. In Silico drug repurposing for COVID‐19: targeting SARS‐CoV‐2 proteins through docking and consensus ranking. Mol. Infor. 2021;40 doi: 10.1002/minf.202000115. [DOI] [PubMed] [Google Scholar]
- 22.Ferraz W.R., Gomes R.A., Novaes A.L., Goulart G.H. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Med. Chem. 2020;12(20):1815–1828. doi: 10.4155/fmc-2020-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer-Almes F.J. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: virtual screening and structure based drug design. Comput. Biol. Chem. 2020;88 doi: 10.1016/j.compbiolchem.2020.107351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy U., Luck L.A. Molecular modeling of estrogen receptor using molecular operating environment. Biochem. Mol. Biol. Educ. 2007;35(4):238–243. doi: 10.1002/bmb.65. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M.-Q., Wilkinson B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007;18(6):478–488. doi: 10.1016/j.copbio.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Cavasotto C.N., Lamas M.S., Maggini J. Functional and druggability analysis of the SARS-CoV-2 proteome. Eur. J. Pharmacol. 2021;890 doi: 10.1016/j.ejphar.2020.173705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saha S., Nandi R., Vishwakarma P., Prakash A., Kumar D.J.F. Discovering potential RNA dependent polymerase inhibitors as prospective drugs against COVID-19: an in silico approach. Front. Pharmacol. 2021;12:267. doi: 10.3389/fphar.2021.634047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koulgi S., Jani V., Uppuladinne M., Sonavane U., Joshi R.J.P. Natural plant products as potential inhibitors of RNA dependent RNA polymerase of Severe Acute Respiratory Syndrome Coronavirus-2. PLoS One. 2021;16(5) doi: 10.1371/journal.pone.0251801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Zhou R.J.T. Structural basis of the potential binding mechanism of remdesivir to SARS-CoV-2 RNA-dependent RNA polymerase. J. Phys. Chem. B. 2020;124(32):6955–6962. doi: 10.1021/acs.jpcb.0c04198. [DOI] [PubMed] [Google Scholar]
- 30.Choudhary M.I., Shaikh M., Tul-Wahab A., Ur-Rahman A. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS One. 2020;15(7) doi: 10.1371/journal.pone.0235030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tachoua W., Kabrine M., Mushtaq M., Ul-Haq Z.J. Modelling, An in-silico evaluation of COVID-19 main protease with clinically approved drugs. J. Mol. Graph. Model. 2020;101 doi: 10.1016/j.jmgm.2020.107758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krüger A., Gonçalves Maltarollo V., Wrenger C., Kronenberger T.J., Advances D.N. ADME profiling in drug discovery and a new path paved on silica. Drug Discov. Develop. 2020;1:1–32. [Google Scholar]
- 33.Lipinski C.A. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004;1(4):337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Daina A., Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van De Waterbeemd H., Gifford E. ADMET in silico modelling: towards prediction paradise. Nat. Rev. Drug Discov. 2003;2(3):192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]