

Abstract
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2) is responsible for the global COVID-19 pandemic and millions of deaths worldwide. In December 2020, a new alpha strain of SARS-CoV2 was identified in the United Kingdom. It was referred to as VUI 202012/01 (Alpha strain modelled under investigation, 2020, month 12, number 01). The interaction between spike protein and ACE2 receptor is a prerequisite for entering virion into the host cell. The present study is focussed on the spike protein of the SARS-COV 2, involving the comparison of binding affinity of new alpha strain modelled spike with previous strain spike (PDB ID:7DDN) using in silico molecular docking, dynamics and simulation studies. The molecular docking studies of the alpha strain modelled spike protein confirmed its higher affinity for the ACE2 receptor than the spike protein of the dominant strain. Similar computational approaches have also been used to investigate the potency of FDA approved drugs from the ZINC Database against the spike protein of new alpha strain modelled and old ones. The drug molecules which showed strong affinity for both the spike proteins are then subjected to ADME analysis. The overall binding energy of Conivaptan (-107.503 kJ/mol) and Trosec (-94.029 kJ/mol) is indicative of their strong binding affinities, well supported by interactions with critical residues.
Keywords: Molecular docking, SARS-CoV2 spike protein, Drug repurposing, Alpha strain modelled, Molecular dynamics and simulation, COVID-19 pandemic
Highlights
-
•
We investigated the potential FDA drugs for repurposing against the spike protein of alpha strain modelled of SARS-CoV-2.
-
•
Spike protein of alpha strain modelled of SARS-CoV-2 has more affinity for the ACE2 receptor of host cell than previous strains and, therefore, is more contagious.
-
•
Conivaptan, Ecamsule and Trosec are common drugs that bind strongly with spike protein of both the strains .
-
•
Molecular Docking and simulation studies show that Conivaptan and Trosec emerged as potential inhibitors of the alpha strain modelled spike protein.
Molecular docking, SARS-CoV2 spike protein, Drug repurposing, Alpha strain modelled, Molecular dynamics and simulation, COVID-19 pandemic.
1. Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARSCoV2), responsible for the worldwide COVID-19 pandemic, emerged first in China (Wuhan) in 2019. In 2002, much before this COVID-19 pandemic, China had already faced an epidemic caused by Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), a deadly pneumonia virus [1]. Both the viruses are the members of β-coronavirus genera of the coronaviridae family. The electron microscopic images of these viruses confirm the crown-like morphology, so the term coronavirus was coined for them [2]. Out of seven known human coronavirus species, four of them (hCoV-29E, hCoV-OC43, hCoV-NL63, hCoV-HKU1) causes only mild flu-like symptoms in humans, while Middle East Respiratory Syndrome-CoV(MERS-CoV), SARS-CoV and SARS-CoV2 cause severe respiratory disorders and even death if not treated timely and appropriately [3, 4, 5]. SARS-CoV2 is highly homologous to SARS-CoV and shares 80% nucleotide identity and phylogenetic similarity with it. The SARS-CoV 2 virion diameter is 50–200nm with positively sensed single-strand RNA as genetic material [6]. The RNA of SARS-CoV2 is 29,811 nucleotides long and codes for ten open reading frames, producing 29 proteins required for viral pathogenesis, replication and survival [6, 7]. Polyprotein 1ab encoded by ORF1ab comprises proteins required for the replication and transcription process of the RNA genome. The 15 non-structural proteins are then released from polyprotein 1ab by the proteolytic activity of two proteases, namely PLpro and 3CLproat different sites [8]. The four structural proteins Spike(S), Envelope(E), Membrane(M) and Nucleocapsid(N) are encoded by S, E M and N gene, respectively. The S, E and M protein is required for the coat of the virus, while N protein facilitates the packing of the genome in the viral coat.
The transmembrane densely glycosylates the spike protein, which plays a vital role in helping entry of the virus into the host cell. The protein is more than 1200 amino acids long with a molecular weight ranging from 180-200 kDa. The basic structure of coronavirus spike protein comprises three domains: extracellular N-terminal domain, transmembrane domain (anchored in virus envelope) and a small C-terminal intracellular tail [9]. The S protein belonging to class1 viral fusion protein is a homotrimer that protrudes from the envelope of the virus. The S protein has two functional subunits S1 and S2, responsible for host recognition and entrance into host cells. The S1subunit directly interacts with the hACE2 receptor on the host cell. This binding induces a conformational change in the S2 subunit, facilitating the viral membrane fusion with the host cell membrane, a prerequisite for viral entry [10, 11]. The S2 subunit of SARS-CoV2 is highly conserved and show approximately 99% structural homology, whereas the S1 subunit of S protein evolved differently and shows only 70% structural similarity with SARS-CoV and bat SARS-CoV [12, 13]. The S1 subunit of SARS-CoV2 recognises angiotensin-converting enzyme 2 (ACE2) present on the epithelial cell linings of the heart, lungs, kidney, intestine of the host organism through its receptor-binding domain (RBD) [14, 15]. The binding affinity of RBD of SARS CoV-2 spike protein for ACE2 receptor is ten times that of SARS-CoV spike protein. Several substitutions in the RBD region of SARS CoV-2 contributes to its higher affinity for ACE2 receptors on host cells. Some of these substitutions from SARS-CoV to SARS CoV-2 are R426 to N439, Y484 to Q498, T487 to N501, Y442 to L445, L443 to F456 to F460 toY473, N479 toQ493 and V404 to K417 [12]. These substitutions increase the RBD domain's compactness and strengthen the interaction between the interfacial residues of ACE2 (specifically Lys31 and Lys 353) receptor and RBD of SARS-CoV2 [13]. Not only this, SARS CoV2 spike protein has multiple furin cleavage sites, which make it susceptible to cleavage by host cell protease into S1 and S2 subunits. This cleavage facilitates the fusion of the viral membrane with the host cell membrane and allows the viral genome's entry into the host cell [16]. All these features make the SARS CoV-2 more contagious than SARS-CoV. The sense RNA of CoVs is susceptible to frequent mutation and recombination and, therefore, is recognised as immensely evolving viruses.
In the fall of 2020, a new alpha strain of SARS-CoV2 was identified in the United Kingdom. It has been referred to as VUI 202012/01 variant under investigation, 2020, month 12, number 01). It is currently referred to as the alpha strain by World Health Organization. The alpha strain belongs to lineage B.1.1.7 [17,18]. Twenty-three mutations compared to the original Wuhan strain have been reported in the new alpha strain, out of which eight are in spike protein. The three deletions at positions 69, 70 and 144 remove histidine, valine and tyrosine amino acids and replace asparagine at 501 by tyrosine, which has been found in the S1 subunit of the spike protein. N501Y substitution occurs in the receptor-binding domain of spike protein of SARS CoV2, which comprises amino acids residues from 319 to 541. Five more point substitution mutations have been located in the S2 subunit of the spike protein of SARS-CoV2. These are alanine to aspartic acid, proline to histidine, threonine to isoleucine, serine to alanine and aspartic acid to histidine at positions 570, 681, 716, 982 and 1118, respectively.
All these mutations have made it more contagious and transmissible than earlier strain. To prevent the spread of the disease, UK had imposed a complete lockdown. Other countries were also on alert and taking necessary steps to contain the virus [19, 20]. The present study focuses on the spike protein of the SARS-COV-2, involving the binding affinity of potential drugs against the new alpha strain compared to the previous strain using in silico molecular docking. In the present study, the PDB ID 7DDN represents the spike protein of the previous strain as it was the most recent structure submitted to Protein Data Bank when the study was carried out. The spike protein of the alpha strain was modelled by including all the mutations in the previous strain. Similar computational approaches have also been used to investigate the potency of FDA-approved drugs from the ZINC Database or from other Databases against the spike protein, although mainly against the original Wuhan strain. These drugs are already FDA approved and have passed all the safety procedures for administration in humans, so the repurposing of drugs saves time and allows immediate use of them to treat the disease. This strategy is quite powerful to deal with the pandemic situation. The current study shows a few FDA-approved drugs’ binding affinities to the original and the alpha strain.
2. Materials and method
2.1. Protein molecular modelling of spike protein of alpha strain modelled (B.1.1.7 lineage)
We downloaded the available 3D structure of spike protein, PDB ID-7DDN, from the RCSB protein database [21]. At the time the study was carried out, 7DDN was the latest SARS CoV-2 structure available. The FASTA sequence was also retrieved, and all the eight reported mutations involving deletions (HV-69-70 del, Y-144del) and substitutions (N501Y, A 570D, P681H, T716I, S982A and D1118H) at respective positions were incorporated in the sequence before depositing it to SWISS-MODEL workspace. The SWISS-MODEL template library (SMTL version 2020-12-23, PDB release 2020-12-18) was searched with BLAST [22] and HHBlits [23] for related evolutionary structures matching the target sequence. Overall, we found 1917 templates for the target sequence. We chose the template with the highest quality to build the model based on template-target alignment using ProMod 3. PROMOD II was used to make an alternative model, and for the loop modelling, we used Promod 3fails [24]. We selected the best model among obtained models based on its GMQE (Global model Quality Estimation) score, QMEAN Score [25] and Ramachandran Plot. GMQE score estimates the quality of the model based on the target template alignment and template structure. The score values lie between 0 and 1, with higher values reflecting higher reliability of the model. QMEAN score measure different geometrical properties and provide information about both global (for entire structure) and local (per residue) quality estimate of the absolute model [25]. We used PROCHECK [26] and MolProbity [27] for generating the Ramachandran plots for the various models. The SWISS-MODEL structure assessment page runs MolProbity version 4.4.
2.2. Molecular docking
2.2.1. Protein preparation
We selected the most recently available electron microscopic structure of the spike protein of SARS-CoV2 PDB ID:7DDN [28]. We removed all the attached ligands and chains B and C. The protein was then energy minimised using Swiss-PDB viewer and converted into PDBQT format using auto dock tools for molecular docking purposes. We generated the Grid Parameter File (GPF) using the Autodock tools [29]. The modelled spike protein of the alpha strain was also prepared in the same way.
2.2.2. Ligand preparation
We chose FDA approved drugs from the ZINC database for our docking with the spike protein (PDB: 7DDN) of the SARS CoV-2, and the alpha strain modelled spike protein was prepared comprising all the mutations. We downloaded the available 3D-SDF structure of 1565 drug molecules from the ZINC database, an open repository of commercially available compounds for virtual screening [30]. We converted the 3D-SDF structures into Mol2 form using Open Babel version 2.4.1 [31]. We energy minimised the ligands, using the steepest descent method for 100 steps with a step size of 0.02, added hydrogens and assigned Gasteiger charges employing Assisted Model Building with Energy Refinement (AMBER) force field using UCSF Chimera v1.14 [32,33].
2.2.3. Docking parameters
We used the RBD (319aa-541aa) domain of the spike protein, PDB ID 7DDN and modelled spike of alpha strain as the template to prepare the grid for docking purposes. As the name itself implies, Receptor Binding Domain (RBD) interacts with ACE2 receptors on host cells and therefore play a crucial role in the entrance of viral in the host cell. Except for the N501Y mutation in the RBD region, the other mutations were in other spike protein parts. To see the effect of all these mutations on the RBD region, we prepared the grid accordingly. The parameter mentioned above file was generated through the ADT 4.2 tool. This tool follows the Lamarckian Genetic Algorithm (LGA), which uses the AMBER force field to run the docking between the receptor and the ligand. For preparing the GPF of spike protein (PDB ID: 7DDN), we defined 3-D grid centres with 225.56, 263.908 and 289.107 as X-, Y- and Z-coordinates, respectively. The alpha strain modelled spike's X-, Y- and Z-coordinates were 242.041, 222.301 and 171.022, respectively. Additionally, in the GPF, the number of grid points for spike protein (7DDN) was 126, 90, and 112, and for the alpha, strain modelled model was 84,126 and 100 in X-, Y-, and Z-coordinates, respectively. The spacing value was set as 0.375 Å. Further, in silico drug screening among the selected 1565 FDA-approved drugs were performed using AutoDock Vina with default configuration parameters with an exhaustiveness value of 8 to evaluate potentially effective drug candidates based on the favourable binding energy of the individual drug [34].
2.2.4. Molecular docking between spike proteins (7DDN/alpha strain modelled spike) and human ACE2 receptor
Human ACE2 receptor (PDB ID-1R42) was used as a receptor to see the interaction between spike protein (PDB ID-7DDN) and alpha strain modelled spike, using ClusPro 2.2 online webserver [35]. ClusPro uses the below equation to generate the model score and the lowest binding energy
| E = 0.40Erep+ −0.40Eatt + 600Eelec + 1.00EDARS |
The repulsive (re), attractive (att), electrostatic (elec) forces and interactions extracted from the decoys as the reference state (DARS) are measured using molecular docking study [36].
2.2.5. Post docking analysis
We chose the best conformation of the ligands based on lower binding energy and orientation of the ligand within our specified grid for further analysis using various softwares. We employed BIOVIA Discovery Studio Visualiser to generate 2D diagrams depicting the interactions between the ligand (Drug) and the protein models, both wild type and alpha strain models. All the settings used were set at default. We further used Protein-Ligand Interaction Profiler (PLIP) available online for analysing the hydrophobic contacts between the ligands and the protein [37, 38]. The 3D coordinates of the spike protein model of the alpha strain were superimposed with the spike protein (PDB ID: 7DDN) using the “super” command available in PyMol [39]. The docked structure from the ClusPro 2.2 webserver was also analysed using PyMol.
2.2.6. Assessment of drug likeness of the ligands (ADME analysis)
As a standard recommended protocol to check the candidature of any ligands as potential drug molecules, the ADME analysis is used to verify the various physicochemical parameters of the molecule. For this, the drug molecule has to satisfy Lipinski's rule of five. The rule defines a criterion according to which a newly discovered orally effective drug should comply with four out of five suggested criteria: molecular mass, lipophilicity, number of hydrogen donors and acceptors, and molar refractivity. The SMILES form of the ligands retrieved from the ZINC database were used as input structures in the Swiss ADME analysis server. This server depicts the ligand's suitability as a drug molecule based on Lipinski's rule and used to predict pharmacokinetics such as absorption, distribution, metabolism, and excretion (ADME) [40].
2.2.7. Molecular dynamics and simulation
Molecular Dynamics simulation and studies are routinely used along with molecular docking to gain advanced insights into mechanisms involving protein-ligand interactions, small molecules, peptides, or even nanoparticles [41, 42]. We performed molecular dynamics and simulation of the three protein-ligand complexes using the GROMACS v.2018 [42,43]. The complexes were chosen based on the specific high-affinity drugs that showed stable interactions with PDB ID:7DDN and alpha strain modelled spike proteins. The three common ligands Ecamsule, Trosec and Conivaptan, showed low binding energy and high affinity for both spike proteins in molecular docking. The complexes of these ligands with the modelled spike of the alpha strain are selected for Molecular Dynamics Simulation studies. We ran the MD simulation for 60 ns for all the systems using the GROMOS 54a7 forcefield. We used the previously reported and highly cited PRODRG Server to generate the ligand topology files for smooth analysis in the GROMACS environment [43, 44]. We solvated all the systems using the cubic box with an edge of 1.2nm using the SPC/E water model. The systems were neutralised using the Na+ ions appropriately. We employed the steepest descent method for energy minimisation of the systems, with maximum steps set at 50,000 and tolerance of 1000 kJ/mol/nm, system pressure was 1atm. Isotropic NPT equilibration was also carried using the widely accepted Parinello-Rahman coupling having a time constant of 2 fs and reference pressure of 1bar. We employed the standard Berendsen thermostat Velocity recoupling method at 310K with no pressure coupling in the NVT step, time constant being 2 fs [42, 45, 43]. The final MD production step was run using leapfrog integration using Nose-Hoover and Parinello-Rahman Couplings [46] and a time step of 2 fs [43]. LINCS algorithm was employed for constraining the bond lengths [45, 43, 47]. We used the Particle Mesh Ewald scheme (PME) for long-range electrostatic interactions using Fourier spacing of 0.16 nm and short-range electrostatic interactions cut off values set at 1.0 nm for NPT, NVT and MD production run.
2.2.8. MM/PBSA free energy analysis
We employed the widely used Poisson Boltzmann Surface Area (PBSA) free energy analysis method to check the complexes' post-simulation binding energy and affinity. This algorithm to compute protein-ligand energy complexes’ free energy is well accepted and highly used for such purposes [42, 43, 48, 49]. We used the g_mmpbsa utility of the GROMACS to calculate the free energy of the complex and the per residue-free energy analysis. The g_mmpbsa tool computes the energy and non-polar solvation energy using the standard equation:
| Gbind = Eelec + EVDW + Gpolar + GSASA [42, 43, 50] |
3. Results
We subjected a total of 1565 FDA drug molecules from the ZINC Database to molecular docking studies for both spike proteins (modelled spike of alpha strain and prevailing strain spike-PDB ID-7DDN) as receptors. The top molecules with the highest negative binding scores from both the docking studies were chosen for ADME analysis. The best drug molecules were then selected by combining docking and ADME analysis for molecular dynamics and simulation. The schematic representation of the overall methodology employed in the present study has shown in Figure 1.
Figure 1.

Schematic flowchart of critical steps in the accomplishment of the current study.
3.1. 3-D structure modelling and validation
7DDN (PDB ID) spike protein is chosen for study to represent a predominant strain of SARS CoV2 as it was the most recent structure deposited in PDB then. In this structure, the spike protein is in an open state and without any ligands or inhibitors bound to it. Only N501Y mutation of alpha strain falls in the RBD region, whereas all other mutations are reported around the RBD region like HV-69-70del, Y-144del or after it like A570D, P681H, T716I, S982A and D1118H. The point mutations in one part of the protein molecule significantly affect its three-dimensional structure and physiological role [51]. To see the effect of these mutations on the overall morphology and function of the modelled spike protein, including any preferential changes in its interaction with ACE2 receptors and with other ligand molecules (here FDA approved Drug molecules), we modelled the entire spike protein comprising all the mutations. We compared the structure and affinity of a modelled spike of alpha strain for ACE2 and drug molecules of ZINC database than spike protein of dominant strain (7DDN-PDB ID).
We used the 6Z97.1. A template for modelling the 3D structure of the spike protein (Figure 2A) of the alpha strain using the SWISS-MODEL server [24]. The spike protein has a sequence identity of 98.95% and coverage of 98% with the template 6Z97.1.A. The template has the description as Spike glycoprotein and covered the range 27–1144. The modelled structure obtained from the SWISS-MODEL server had a QMEAN value of -1.97. Negative QMEAN Z score values closer to zero implies a better agreement between the modelled and experimental structures in a similar size range. The GMQE score of the model is 0.67. Usually, the GMQE value lies between 0-1. Higher value is related to a more favourable model. The secondary structure of the alpha strain modelled spike protein was further validated by the Ramachandran plot using MolProbity version 4.4 [52]. The MolProbity score of the model is 1.4. The modelled structure had 92.8% residues in the favoured region, 1.49% in Ramachandran Outliers and 1.34% in Rotamer Outliers (Figure 2B). Ramachandran plot analysis from PROCHECK for the modelled protein shows that out of 2964 residues, 2573 (86.8%) residues are in the most favoured region, 361 (12.2%) residues in the additional allowed region, 22 (0.7%) residues in the generously allowed region and only 8 (0.3%) residues in the disallowed region [53]. We have provided the result of PROCHECK analysis in supplementary material in pdf form as (Fig.S1). As we have used the 6Z97 structure as a template for modelling the spike protein of alpha strain, whereas for the rest of comparative studies, we are using 7DDN structure, therefore to avoid any discrepancy, the single chain of 7DDN was superimposed on the 6Z97 structure (Fig.S2).
Figure 2.

A- Spike protein of alpha strain from SWISS-MODEL and B- Ramachandran plot from the MolProbity version 4.4.
We superimposed the modelled alpha strain spike with 7DDN spike for the trimeric and single-A chain structures (Figure 3A, B). The structural analysis revealed that sequences from 144Tyrosine-155Serine (Shown as green sticks on a cyan coloured single chain of 7DDN spike) are part of the loop connecting beta-sheets, but tyrosine Y-144del in the alpha strain modelled spike has made it more disorganised, as shown as orange sticks on a magenta-coloured single chain of alpha strain modelled spike protein in Figure 3A. Other mutations like A570D, P681H, T716I, S982A, and D1118H do not significantly affect the spike protein structure. They continued to be part of the same secondary element as in 7DDN (PDB ID) spike protein structure. The sequences from 828-843 amino acid residues (shown in orange) of the modelled alpha strain modelled spike do not align with the 7DDN spike protein structure as the corresponding structural elements are missing in the original PDB structure of the molecule. The multiple sequence alignment of the FASTA sequence of the modelled spike of alpha strain with 7DDN was also done, and the result is included in Supplementary Materials as Fig. S4. For docking studies single chain of 7DDN and modelled spike of the alpha chain is used. When we superimposed the structure of the RBD domain of spike with ACE2 receptor (PDB ID:6lzg) on the trimeric structure of 7DDN spike, we found that the RBD of 6lzg structure entirely coincides with the RBD of a single chain of the trimeric spike (Fig.S3, S10).
Figure 3.

A-Superimposed structure of the model of the single chain of the modelled spike protein of alpha strain variant on 7DDN (PDB ID). The Colour scheme is as follows: alpha strain spike chain - magenta, 7DDNspike chain -Cyan. Sequences 144Y–155S of 7DDN Spike-Green (Sticks) correspondingly sequence of the alpha strain model spike in orange (Sticks). Amino acid sequence from 828-843 of alpha strain modelled spike is in orange sticks. B. Superimposed 3D structure of alpha strain modelled spike (Magenta) and 7DDN spike (Cyan). All the figures are generated in PyMol software.
3.2. Molecular docking between human ACE2 receptor and modelled spike of alpha strain
Protein protein interaction between host cell receptor ACE2 (PDB ID-1R42) [54] and modelled spike protein is studied using an online web server ClusPro2.2. The cluster zero with 65 members and lowest binding energy value -968.8 kcal/mol was chosen to see the interactions between the different amino acids of ACE2 and alpha strain modelled spike protein. The corresponding value of spike (PDB ID: 7DDN) for cluster zero with 40 members is -729.3 kcal/mol. The energy value indicates that the receptor (ACE2) has a strong binding affinity for the alpha strain modelled spike protein. The essential interactions among amino acids of ACE2 and modelled spike protein are shown in Figure 4F. The 441K (lysine) of the alpha strain modelled spike is showing strong polar interaction with 150E (Glutamic acid) of ACE2, form two hydrogen bonds with distances value as measured by PyMol is 1.7 and 1.8, respectively (Figure 4A). Similarly, 418Y, 470Y and T467 of the alpha strain modelled spike interact with E56, Y50 and S128 of ACE2. The corresponding interactions are shown in Figure 4B, C and D, respectively. 348Y of alpha strain modelled spike protein interacts strongly with two amino acids T129 and K131 of ACE2 (Figure 4E). The higher negative value of binding energy (-1072.6 kcal/mol) and all these strong hydrophilic interactions may contribute to the spike protein's high affinity of the alpha strain modelled SARS CoV2 for ACE2 receptor.
Figure 4.

ACE2 receptor and modelled spike of alpha strain main chains are shown in magenta and yellow, respectively. Amino acids showing crucial interactions are in coloured sticks as their main chain. A- 150E of ACE2 and 441K of modelled spike B- 56E of ACE2 and 418Y of the modelled spike C– 50Y of ACE2 and Y470 of the modelled spike D- 128S of ACE2 and T467 of the modelled spike E- 129T and 131K of ACE2 and Y348 of modelled spike F- All the fundamental interactions at the interface of ACE2 and modelled spike of alpha strain. All the figures are generated in PyMol software.
3.3. Molecular docking results
Docking studies revealed that top-most binding drug molecules possess docking scores ranging between -8.7 to -7.8 kcal/mol. The top-nine drug molecules and the details of their 2-D and 3-D interactions are shown in Figure 5(A-F) and Fig. S5 and discussed in Table 1. Among all the ligands (FDA approved ZINC Database), Ecamsule and Trypan blue have higher binding energy for the alpha strain spike protein model modelled UK strain with energy values -8.7 and –8.4 Kcal/mole, respectively. Ecamsule forms three H-bonds, whereas Trypan blue forms two H-bonds with the alpha strain modelled spike protein residues. H bonds play a crucial role in binding affinity, selectivity and the stability of the receptor-ligand complex. The other attractive forces like non-covalent interactions, Van De Waal and hydrophobic interactions also stabilise the complex. Although these interactions are weak forces, their cumulative effect is sufficient to increase the docked complex's stability. The details of the top drug molecules from docking studies with the ZINC database have discussed in Table 2. For Ecamsule, Trypan blue and Digoxin, most of the interacting amino acid residues of the receptor fall within Receptor Binding Motif (438–506), a crucial element that interacts directly with ACE2 receptor on the host cell. Similarly, Naldemedine, Ponatinib, Conivaptan and Orap interact with amino acid residues, which comprise either RBM or RBD (319–541) part of the modelled spike protein. Trosec interacts with the amino acid residues of the RBM region of the alpha strain modelled spike protein, forming hydrogen bonds and exhibiting hydrophobic interaction with Tyr498, which is mutated Asn501 (N501Y). Because of three deletions at 69-70HV and 144Y, the 501 position of spike protein (PDB ID: 7DDN) corresponds to the 498th position in the alpha strain modelled spike protein. The binding of drug ligands will occupy the RBM and thus hinder or weaken the interaction between the viral spike protein and ACE2 receptor on the host cell. The recognition of the host cell receptor by viral spike protein is a prerequisite for virion entry in the host cell. The blocking of this interaction will inhibit the virion's entrance into the host cell and reduce the damage caused to the host organism.
Figure 5.

Visualisation of the docked spike (alpha Strain)-ligand complexes. 2D diagrams of Protein-Ligand Complexes generated by BIOVIA Discovery Studio Visualiser. Different colours dashed lines show different interactions—Green Dash-H-Bond; Dark purple-pi stacking; Light purple-pi-alkyl stacking; Orange Dash-pi-cation; Red Dash-unfavourable interactions. The interaction of the receptor with different drug molecules is shown as A. Ecamsule B. Conivaptan C. Trosec D. Trypan Blue E. Naldemedine. F. Ponatinib.
Table 1.
Binding interactions between the alpha strain modelled spike protein and the ligands. The interactions are calculated using PLIP online server and Discovery Studio Visualizer for the docked complex.
| Ligand | Binding Energy (kcal/mol) | Hydrogen Bonds | VDW | Non-Covalent Interactions | Hydrophobic interactions |
|---|---|---|---|---|---|
| Ecamsule | -8.7 | Arg405 Gln406 |
Asp402, Glu403, Thr412, Gly413, Lys414, Ser491, Tyr492, Tyr502, Gly493, Tyr498, | 400Arg(π-cation) 405Arg (Saltbridge) 502Tyr(πstacking) |
402Asp,414Lys,450Tyr,492Tyr |
| Trypan Blue | -8.4 | Tyr450 Ser491 Tyr502 |
Tyr348,446,498 Asn447, Leu452, Glu490, Gly493 |
Leu449(π-sigma) Lys414, Arg400 |
446Tyr,449Leu, 490Gln,502Tyr |
| Digoxin | -8.3 | Tyr450 | NA | NA | Tyr 446,492 Leu492 |
| Naldemedine | -8.1 | Arg343 | Glu337, Ser396,346 Phe344, Ala345, Tyr348, Arg463, Asn351,447 |
Val338, Ala341,349, Lys353, Ile465 | Ala341,349 Arg343, Asn351 Lys353, Ile465 |
| Ergotamine | -7.9 | NA | NA | Phe487 | Ala349 Phe487 |
| Ponatinib | -7.9 | Asn447 Ser346 |
NA | Phe487(πstacking) Ala349, Leu449 Asn351, Phe344 Tyr348(halogen bond) |
Leu449, Phe487 |
| Conivaptan | -7.9 | NA | NA | Arg343, Tyr448, Leu449 |
Arg343, Asn447 Tyr448, Leu449 Phe487 |
| Orap | -7.9 | Arg343 Tyr446 Ser491 |
NA | Tyr448((π-(π) Ala345,349(π-sigma) Leu449 |
Arg343, Asn447 Tyr448, Leu449 Phe487 |
| Trosec | -7.8 | Tyr498 | NA | Tyr502(πstacked) | Tyr446,492, Tyr498,502 Gln490 |
Table 2.
Top compounds from the docking studies of the ZINC database with their ZINC ID, Compound name, structure and medical use.
| ZINC ID | Compound Name | Structure | Function |
|---|---|---|---|
| ZINC000100370145 | Ecamsule | The active component of Sunscreen | |
| ZINC000169289767 | Trypan Blue |  |
As dye in cataract surgery |
| ZINC000242548690 | Digoxin |  |
Cardiac glycosides |
| ZINC000100378061 | Naldemedine |  |
Opioid antagonist |
| ZINC000052955754 | Ergotamine |  |
vasoconstrictor |
| ZINC000036701290 | Ponatinib |  |
Treat chronic myeloid leukaemia |
| ZINC000012503187 | Conivaptan | 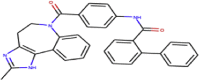 |
Treatment of hyponatremia |
| ZINC000004175630 | Orap | 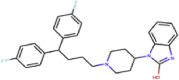 |
anti-psychotic |
| ZINC000100018598 | Trosec |  |
antispasmodic |
| ZINC0000142108876 | Eluxadoline | 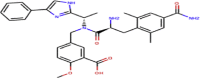 |
Treatment of Irritable Bowel Syndrome |
| ZINC000003927200 | Yaz | 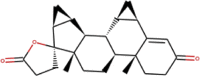 |
Oral contraceptive pill |
3.4. Molecular docking study of FDA approved drug molecules from ZINC database with the spike protein (PDB ID-7DDN)
Docking studies revealed that the docking scores of top-most binding drug molecules (4 ligands) ranges from -8.2 to -8.0 Kcal/mole. The 2-D details of the interaction between receptor spike protein (7DDN) and the drug molecules are shown in Figure 6 and discussed in Table 3. The 3-D interactions are given in Supplementary Materials as Fig. S6. Like alpha strain modelled spike protein, 7DDN spike protein also shows the highest affinity for Ecamsule. Similarly, Ergotamine, Eluxadoline and Yaz also have a strong affinity for receptor protein residues, which falls in the spike protein's RBM or RBD region. Besides Ecamsule, spike protein (7DDN) also exhibits a strong affinity for Trosec, Naldemedine, Conivaptan, Orap and Ponatinib with binding energy values from -7.2 to -6.5 Kcal/mole.
Figure 6.

Visualisation of the docked spike (7DDN wild type Strain)-ligand complexes. 2D diagrams of Protein-Ligand Complexes generated by BIOVIA Discovery Studio Visualiser. Different colours dashed lines show different interactions—Green Dash-H-Bond; Dark purple-pi stacking; Light purple-pi-alkyl stacking; Orange Dash-pi-cation; Fluorescent Dash-halogen bond; Red Dash-unfavourable interactions. The interaction of the receptor with different drug molecules is shown as A. Ecamsule B. Conivaptan C. Trosec D. Eluxadoline E. Naldemedine.F. Ponatinib.
Table 3.
Binding interactions between the spike protein (PDB ID: 7DDN) and the ligands. The interactions are calculated using PLIP online server and Discovery Studio Visualizer for the docked complex.
| Ligand | Binding Energy (kcal/mol) | Hydrogen Bonds | VDW | Non-Covalent Interactions | Hydrophobic interactions |
|---|---|---|---|---|---|
| Ecamsule | -8.2 | Phe347 | Ala348 | Ala352 | Tyr351, Ala352, Asn450, Ile468, Thr470 |
| Eluxadoline | -8.0 | Tyr451, Ser349, Phe347, Ala352(2), Trp353 | Arg346, Asn450, Ile468 | Tyr351(π- πT shaped), Arg346, Ile468 | Ala352, Asn450, Ile468, Leu492 |
| Ergotamione | -8.0 | Ser349 Asn450(2) |
Ala352, Leu492 | Arg346 | Arg346, Ala348, Ala352, Leu452, Ile468, Thr470, Leu492 |
| Yaz | -8.0 | Tyr351 | NA | Ala348, Tyr351, Ala352, Ile468 | Ala348,352, Asn450, Ile468, Leu492 |
| Trosec | -7.2 | Ser349 | NA | Ala348,352 | Ala348,352, Asn450, Tyr451, Ile468, Leu492 |
| Naldemedine | -7.1 | Phe347, Ser349, Asn450 |
Asn450, Leu452,492 | Ala348,352 Leu452,492, Ile468 |
Leu452,492 Ile468, Phe490 |
| Conivaptan | -7.1 | Asn450 | Ala352, Ile468 | Ala,348,352(2), Arg346, Ile468 | Arg346, Ala348,352, Asn450, Ile468, Leu492 |
| Orap | -6.5 | Asn354 | Ala352 | Ala348, Ile468(halogen bond) |
Tyr351, Asn450, Leu452, Ile468 |
| Ponatinib | -6.5 | Ser349 | Ala348,352 | Ala348,352, (π-sigma) Tyr351(π-Tshaped) |
Ala348,352, Tyr351,451 Asn450, Thr470, Leu492 |
3.5. Common ligands with high affinity for both alpha strains modelled spike and spike (PDB ID-7DDN)
The comparative molecular docking studies of the FDA approved drug ligand from the ZINC Database with both the spike proteins suggested some common drug molecules that are quite effective against both of them. These common ligands engage most of the spike proteins' important positions in binding with the ACE2 receptor. Comparing the ligands’ docking analysis results with two proteins, alpha strain modelled spike protein; prevailing strain spike 7DDN- the first energy value corresponds to modelled spike, and the second value corresponds to 7DDN spike. We report that Ecamsule has the highest negative binding energy for both the proteins (-8.7; -8.1 kcal/mol), followed by Ergotamine (-7.9; -8.0 kcal/mol), Naldemedine (-8.1; -7.1 kcal/mol), Conivaptan (-7.9; -7.1 kcal/mol), Ponatinib (-7.9;-6.5 kcal/mol), Orap (-7.9;-6.8 kcal/mol) and Trosec (-7.8;-7.2 kcal/mol). Ponatinib and Orap have less binding affinity for spike protein (PDB ID: 7DDN) than the other five ligands. Ecamsule and Trosec interact with the hydrophilic pocket (Fig.S9A, S8A), whereas Conivaptan extends its interaction from hydrophilic to hydrophobic pockets (Fig.S7A) amino acid residues of the alpha strain modelled spike protein. For spike (PDB ID-7DDN), Conivaptan, Trosec and Ecamsule all occupying both hydrophilic and hydrophobic surfaces of the receptor protein (Fig. S7B, S8B, S9B). Common drugs are discussed in the supplementary paper. The references mentioned are cited in the references section.
3.6. ADME analysis of common drug molecules with high affinity for both spike proteins
After sorting the common drug ligands for both spike proteins based on their highest negative binding energies value, the above mentioned seven drug ligands were subjected to test their drug likeliness using online analysis server Swiss-ADME [55]. The most popular and authentic rule for confirming the drug-likeness of the Ligand is Lipinski's rule. According to the rule, the molecular weight of drug-like molecule should be less than 500 g/mol, the lipophilicity denoted by Log-P value should be lower than 5 (MLogP<4.15), the number of H-bond donors (NH or OH) and acceptors (N or O) should be less than or equal to 5 and 10, respectively [56]. Conivaptan and Trosec passed Lipinski's rule without any violation among the top compounds in the present study. However, many drugs that do not pass through the Lipinski's filter but have immense pharmacological properties have been approved by the FDA as potential drugs for clinical purposes [57]. Ecamsule, Naldemedine and Ergotamine also qualified Lipinski's rule of five with one violation concerning molecular weight. Their molecular weights are slightly more than the recommended values but within the permissible limit. Orap also qualifies Lipinski's rule with one violation of MLogP value. Except for Orap, all other drug molecules showed the value of MLogP less than 4.15, which indicates that the molecules are more likely to be in the hydrophilic environment and are favourable for their drug-likeness. The detailed comparative analysis of all other parameters of Lipinski's filter and ADME properties for all the eight drugs' ligands are given in Table 4 and Table 5, respectively. Overall, the drugs used in this study successfully qualified the filtering criteria based on Lipinski's rule of five and follow the ADME properties for being a potent orally active drug.
Table 4.
Detailed analyses of parameters of Lipinski's filter for the drug-likeness. The elaborated comparative analysis of all the seven drugs concerning parameters of Lipinski's filter.
| Compound | Molecular weight g/mole | H-Bond donors | H-bond acceptors | MLogP | Drug-likeliness |
|---|---|---|---|---|---|
| Ecamsule | 562.69 | 2 | 8 | 3.05 | Yes,1Violation |
| Naldemedine | 570.64 | 4 | 9 | 1.49 | Yes,1Violation |
| Ergotamine | 581.66 | 3 | 6 | 1.84 | Yes,1Violation |
| Ponatinib | 532.56 | 1 | 8 | 3.90 | Yes,1 Violation |
| Conivaptan | 498.57 | 2 | 3 | 4.07 | No Violation |
| Orap | 461.55 | 1 | 5 | 5.38 | Yes,1Violation (MLogP>4.15 |
| Trosec | 392.51 | 1 | 3 | -0.14 | No Violation |
Table 5.
Detailed list of parameters to predict the ADME properties. Comparative analysis of all the seven compounds concerning parameters of predicting the ADME properties are done. TPSA represents the topological polar surface area, and LogS indicates the water solubility, PAINS is the acronym used for Pans assay interference structures, and BS is the bioavailability score.
| Compound | TPSA (Å2) | LogS | PAINS | BS |
|---|---|---|---|---|
| Ecamsule | 159.64 | -4.98 | No alerts | 0.11 |
| Naldemedine | 141.18 | -5.24 | No alerts | 0.56 |
| Ergotamine | 118.21 | -5.3 | No alerts | 0.55 |
| Ponatinib | 65.77 | -5.73 | No alerts | 0.55 |
| Conivaptan | 78.09 | -6.67 | No alerts | 0.55 |
| Orap | 41.29 | -6.63 | No alerts | 0.55 |
| Trosec | 46.53 | -5.0 | No alerts | 0.55 |
3.7. Molecular dynamics and simulation analysis
We performed molecular dynamics of the modelled spike apoprotein of alpha strain and the three complexes with Ecamsule, Trosec and Conivaptan for 60ns. Conivaptan and Trosec complexes were chosen as they fully satisfied all the parameters of Lipinski's rule, whereas Ponatinib, Naldemedine [58], Ergotamine and Orap showed one violation of Lipinski's rule of five. Ecamsule also showed one violation of the rule, but still, its complex with the alpha strain modelled spike proceeded for molecular dynamics simulation as it has the highest binding affinity for both the receptors in docking studies. To comprehend the molecular dynamics results, we performed several analyses, including RMSD (gmx_rms), RMSF (gmx_rmsf), Radius of Gyration (gmx_gyrate), hydrogen bond analyses (gmx_hbond) and the free energy analysis using various inbuilt tools in GROMACS. The RMSD analysis of the apoprotein showed high fluctuations in the first 30ns of the simulation, beyond which it gained considerable stability. Conivaptan (MSC) complex stabilised the fluctuations seen in the apoprotein RMSD, indicating its binding within the receptor-binding motif and the receptor-binding domain of the alpha strain modelled spike significantly stabilises the protein. The maximum RMSD of the apoprotein is around 1.6nm, whereas, in the complex with Conivaptan, it is around 1nm. The other two complexes with Ecamsule (MSE) and Trosec (MST) show different behaviour.
The alpha strain modelled spike protein in the presence of Ecamsule has a highly smooth trajectory without any significant fluctuations in RMSD. The maximum RMSD in the presence of Ecamsule reaches above 1.5 nm. The system also gains stability within the first 10ns of the simulation. The alpha strain modelled spike protein complex with Trosec as the ligand is highly disrupted, as seen in the RMSD trajectory. There are significant fluctuations seen in the overall trajectory. Significant fluctuations in the presence of Trosec is indicative of structural instability of the mutant spike protein. Any ligand can act on the target spike protein in two ways. The ligand's binding could stabilise the protein's overall structure, and the system's overall stability would be very high, which shows tight binding of the ligands within the receptor-binding domain. Another way ligand binding could affect the spike protein is by causing significant disruptions in the protein's structure, affecting various vital contacts within the protein's RBD. This study shows Trosec showing the latter mechanism as evident from the RMSD, RMSF and the Radius of gyration trajectories (Figure 7A, B).
Figure 7.

Molecular Dynamic and Simulation graphs (A) RMSD (B) Radius of Gyration and (C) RMSF trajectories of Apoprotein (Black), Conivaptan complex (MSC) in Red, Ecamsule complex in Blue (MSE) and Trosec complex (MST) in green. Here MS stands for the modelled spike of the alpha strain of SARS CoV2.
The Radius of gyration trajectory of all three complexes agrees with the RMSD trajectories. The apoprotein Radius of gyration shows high fluctuation, indicating a lack in compactness of the protein. The maximum Rg is 4.6 nm at the beginning of the simulation, finally converging around 4.1nm. Even in the ligands' presence, the Rg of the complexes at the beginning of the simulation is around 4.6 nm although, the final convergence is least for Ecamsule at 3.8 nm, followed by Conivaptan at 4.0 nm and Trosec at 4.1 nm. The maximum Rg is 4.6 nm at the beginning of the simulation, finally converging around 4.1nm. Even in the ligands’ presence, the Rg of the complexes at the beginning of the simulation is around 4.6 nm although, the final convergence is least for Ecamsule at 3.8 nm, followed by Conivaptan at 4.0 nm and Trosec at 4.1 nm.
As anticipated, the root means square fluctuation of both the apoprotein and the complexes aligns with the RMSD and the Rg trajectory (Figure 7C). The maximum side-chain fluctuations are reported in the complex, with Trosec with fluctuations reaching above 2nm. Both Ecamsule and Conivaptan complexes have much lower average RMSF in the total protein compared to the apoprotein. We further highlighted the RMSF of the most critical residues for ligand interaction in the alpha strain modelled spike protein's receptor-binding motif (Figure 8 A-D). The effect of Trosec is visible as the presence of Trosec makes the protein lose its overall structural integrity in the binding motif. This makes Trosec an essential potential inhibitor. On the other hand, Conivaptan and Ecamsule complexes show the reverse effect. In the presence of Conivaptan, the amino acid residues in the receptor-binding domain have lower side-chain flexibility indicating tight binding of the ligand with the amino acid residues. Ecamsule also showed a similar trend in RMSF as Conivaptan.
Figure 8.

Bar graph representation of the RMSF of the apoprotein, Conivaptan (MSC) and Trosec complex (MST) in specific regions of the receptor-binding motif (A) 343–352 (B) 445–450 (C) 480–489 and (D) 490-502.
3.7.1. Hydrogen bond analyses
We performed hydrogen bond analyses calculating the average number of intraprotein hydrogen bonds and the average number of hydrogen bonds between protein-ligand for all the systems. The average number of intraprotein hydrogen bonds in the apoprotein is 4.8/ns throughout the simulation. The Conivaptan complex had 4.9 intraprotein hydrogen bonds/ns and approximately one hydrogen bond/ns between protein and ligand. Arg343, Ser346, Tyr348,446, Asn445, 447, Leu449, Thr487, Tyr486, Gln490, Ser491, Leu480,489, Glu481 all played a role in hydrogen bonds. The alpha strain modelled spike protein complex with Ecamsule also had approximately 4.8 intraprotein hydrogen bonds/ns and no hydrogen bonds formed between protein-ligand. Even in the presence of Trosec, the average intraprotein hydrogen bonds are 4.9, and on average, approximately one hydrogen bond is formed between Trosec and the protein. Arg343, Ser346, Tyr348,446, Asn445, 447, Leu449, Thr467, 487, Gly479, Tyr486, Gln490, Ser491, Leu489, Glu481 were the residues involved in hydrogen bonds during the simulation. As seen here, there are not many significant differences in the number of intraprotein hydrogen bonds between the apoprotein and the protein-ligand complexes, indicating that it has little role in the protein's overall stability. We would also like to add that most hydrogen bond interactions occur in the loop regions containing the residues 442–447 and 471–489 [59]. These interactions between the protein and the ligand are deemed substantial. Loop regions in any protein structure are highly variable. Conivaptan and Ecamsule both reduce these fluctuations by forming a stable interaction with the receptor protein. In contrast, Trosec destabilises the loop region and increases the side-chain flexibility and thus affects the overall structural integrity of the alpha strain modelled spike protein. The details of the amino acid residues participating as hydrogen bond donors and acceptors between the ligands and alpha strain modelled spike protein are given in Supplementary Materials as Table S1.
3.7.2. MM/PBSA free energy analyses
We performed MM/PBSA free energy analyses for two of our complexes: Conivaptan and Trosec. We performed the energy analyses in the respective complexes once the systems were stabilised. Conivaptan free energy analysis was performed between 30-50 ns, and Trosec free energy analysis was performed between 50-60ns. In both cases, energy was calculated at every 0.1ns time step. The overall binding energy of the Conivpatan complex was -107.503 ± 13.14, and for the Trosec complex -94.029 ± 17.31. In both the complexes, maximum contribution in the overall free energy was due to Van der Waal's interactions, which indicates that hydrophobic amino acids play an essential role in the protein-ligand complex's overall stability and interaction. Electrostatic interactions in the Conivaptan complex and SASA energy in the Trosec complex contributed significantly to the overall binding energy. We have represented the exact values of the overall binding energy in Table 6. Also, overall positive values in polar solvation energies show that the polar contribution to the solvation energy does not favour the ligand interactions, and the non-polar contribution is higher.
Table 6.
Details of MM/PBSA free energy analyses of the Conivaptan (MSC) and Trosec (MST) complex with the alpha strain modelled spike protein.
| Protein Model | VDW (kcal/mol) | Electrostatic Interaction (kcal/mol) | Polar Solvation (kcal/mol) | SASA (kcal/mol) | Binding energy (kcal/mol) |
|---|---|---|---|---|---|
| MSC | -132.067 ± 13.24 | -21.81 ± 7.35 | 60.35 ± 9.76 | -13.97 ± 1.44 | -107.50 ± 13.14 |
| MST | -110.92 ± 11.22 | -1.45 ± 3.26 | 29.52 ± 14.49 | -11.17 ± 1.55 | -94.02 ± 17.31 |
4. Discussion
Drug discovery and development requires critical information about potential drug targets in an organism [44]. Repurposing drugs has been a valuable strategy for emerging diseases and situations like a pandemic outbreak. Although extensive computational studies are available in the repurposing of FDA approved drugs with various targets in the SARS CoV-2 virus, we must keep this search on for alpha strain modelled spike protein that is now well-established in various countries. It is from this perspective that the current manuscript adds to the growing source of literature. The primary purpose of this manuscript is to disseminate some knowledge about necessary drugs which could be used subject to trial results. Previous studies mainly report terpenes [44], phytomedicine [60], extracts from spices [61], marine products [62], plants secondary metabolites and various other forms of drugs as potential candidates as SARS CoV-2 inhibitors [63].
The main strategies for drug development or designing against SARS CoV-2 include targeting viral growth progress within the living cell. The envelope of the SARS CoV-2 virus contains the spike protein, which interacts primarily with the ACE2 receptor cells inside the host through the RBD domain of the spike protein. Preventing the interaction of the spike protein's RBD domain with the ACE2 receptor is a crucial strategy for drug development in this case [44]. An essential objective of the present study is to inhibit or decrease the interaction frequency between the receptor (ACE2) and the spike protein. For this, the 1565 molecules from ZINC Database (FDA approved) have docked with the modelled spike protein (alpha Strain) and the spike protein (PDB ID: 7DDN) of the dominant strain. The top molecules of the docking studies interacted with most of the residues comprising either the RBM or RBD part of the protein. By engaging these crucial residues, the interaction between the ACE2 receptor and spike can be hampered [64]. ADME analysis of the common ligands, which show a high binding affinity for both the spike proteins, revealed that Conivaptan and Trosec are the most suitable drug ligands, followed by Ecamsule, Naldemedine, Ergotamine and Ponatinib. The binding affinity values indicate that Orap and Ponatinib have low binding affinity for spike protein (PDB ID: 7DDN). Combining both the analyses results with giving more weightage to ADME analysis and without much compromising on the binding affinity values for both the spike proteins, Conivaptan, Trosec and Ecamsule emerged as the most suitable drug ligands followed by Ergotamine and Naldemedine. Comparative studies like this are currently limited. Further, we selected those compounds which showed similar results with both the dominant strain and alpha strain spike protein. This manuscript helped us identify drugs that could be used in both cases.
Several other notable studies also report FDA approved drug repurposing as SARS CoV-2 inhibitors. A study that carried out drug repurposing specifically of three classes of drugs: antivirals, antimalarial, and peptides report Velpatasvir and Glecaprevir as potential drugs, which shows excellent promise [65]. Another study that carried out in vitro studies with more than 1400 approved drugs reports that drugs such as Clomiphene, Vortioxetine and Asenapine showed low IC50 values [66]. There are also drug repurposing studies against the SARS CoV-2 main protease [67, 68, 69], reporting a wide range of drugs that could potentially inhibit the SARS CoV 2 main protease. The current manuscript highlights that this is an effort to study both predominant strain and the alpha strain modelled spike protein's activity simultaneously in the presence of several drugs which looks like potential inhibitors.
Further, our results show that different drugs have different mechanisms for interacting with the RBD domain of the alpha strain modelled spike protein. Conivaptan and Ecamsule binding stabilises the RBD domain, preventing it from interacting with the ACE2 receptor. Trosec destabilises this region (RBD) domain which creates a loss in overall protein structural integrity. This activity will also have a similar impact if the RBD domain loses important contacts helpful in interacting with the ACE2 receptor. The manuscript emphasises this hypothesis. Significant interactions between various critical amino acids of the RBD domain and the ligands also show their potential role in engaging critical residues and preventing them from interacting with the ACE2 receptor.
5. Concluding remarks
The present work's first objective focuses on the modelled spike protein of the new alpha strain of SARSCoV2 reported first in the United Kingdom. The alpha strain of the Covid-19 virus has 23 mutations. Out of which, eight are in the spike protein. The spike protein helps in the recognition of the host cell through ACE2 receptors. The host cell's recognition by spike protein of virus is a prerequisite for its entry into the host system. The new alpha strain of SARS CoV2 is found to be more infectious than the present strain. Our study's computational analysis of the interaction between the modelled spike (alpha strain) and ACE2 receptor confirms that the new alpha strain modelled spike has more affinity for the ACE2 receptor than the previous strain (spike PDBID:7DDN), and therefore it is highly transmissible than the previous strains. The primary threat that prevails in the current situation is the high rate of the mutations continuously reported in the SARS CoV2 virus, resulting in the variant structural (Spike protein) and functional proteins (Protease). Although the extensive vaccination program has been started in many countries, it will still take a long time to complete. Therefore, the search for effective drugs against this constantly mutating virus will be essential to control to avert any potential outbreak again. Repurposing drugs will be a valuable strategy to combat such a high rate of transmission and mortality associated with some strains of SARS CoV2 and will be of high priority. Drug development against viral infections remain one of the most elusive challenges of medical biotechnology, and we must strive forward towards the ever-growing challenge with a sound literature background.
Declarations
Author contribution statement
Monu Pande: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Debanjan Kundu: Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Ragini Srivastava: Analyzed and interpreted the data; Wrote the paper.
Funding statement
Monu Pande was supported by DST, Government of India in the form of Woman Scientist A (WoS-A) research grant [Project no: SR/WOS-A/LS-478-2017]. Debanjan Kundu was supported by the research fellowship provided by IIT-BHU.
Data availability statement
Data included in article/supplementary material/referenced in article.
Declaration of interests statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
The support and the resources provided by the PARAM Shivay Facility under the National Supercomputing Mission, Government of India at the Indian Institute of Technology, Varanasi are gratefully acknowledged.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.R. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Gui M., Song W., Zhou H., Xu J., Chen S., Xiang Y., Wang X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017;27:119–129. doi: 10.1038/cr.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication and pathogenesis 418. J. Med. Virol. 2020;92(4):418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Wit E., Van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14(8):523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ul Qamar M.T., Saleem S., Ashfaq U.A., Bari A., Anwar F., Alqahtani S. Epitope based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: an immune informatics study. J. Transl. Med. 2019;17:362. doi: 10.1186/s12967-019-2116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. PMID: 31978945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. PMID: 32015508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sardar R., Satish D., Birla S., Gupta D. Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. bioRxiv. 2020 doi: 10.1016/j.heliyon.2020.e04658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterisation of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 12.Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition in SARS CoV 2. Nature. 2020;581:221. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choudhary S., Malik Y.S., Tomar S. 2020. Identification of SARS CoV 2 Cell Entry Inhibitors by Drug Repurposing Using in Silico Structure-Based Virtual Screening Approach Chemrxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrarlo C.M. ACE2: more of ang-(1–7) or less ang II? Curr. Opin. Nephrol. Hypertens. 2011 doi: 10.1097/MNH.0b013e3283406f57. PMID: 21045683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004 doi: 10.1074/jbc.M311191200. PMID: 14754895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N.G., Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.SARS-CoV-2 Lineages. 2020. https://cov-lineages.org/ [Internet] Available from: [Google Scholar]
- 18.Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5(11):1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rapid Increase of a SARS-CoV-2 Alpha Strain Modelled with Multiple Spike Protein Mutations Observed in the United Kingdom. Threat Assessment Brief; Dec 20 2020. [Google Scholar]
- 20.Alpha Strain Modelled Coronavirus in the United Kingdom Sets off Alarms, but its Importance Remains Unclear. AAAS, Science; Dec 20, 2020. By Kai Kupferschmidt. [Google Scholar]
- 21.Zhang C., Wang Y., Zhu Y. Development and structural basis of a two-MAb cocktail for treating SARS-CoV-2 infections. Nat. Commun. 2021;12(1):264. doi: 10.1038/s41467-020-20465-w. PMID: 33431876; PMCID: PMC7801428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden TL BLAST+: architecture and applications. BMC Bioinf. 2009;10:421–430. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger S.J., Söding J. HH-suite 3 for fast remote homology detection and deep protein annotation. BMC Bioinf. 2019;20:473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guex N., Peitsch M.C., Schwede T. Automated comparative protein structure modelling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30:S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 25.Studer G. QMEAN DisCo—distance constraints applied on model quality estimation. Bioinformatics. 2020;36:1765–1771. doi: 10.1093/bioinformatics/btz828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laskowski R.A., MacArthur M.W., Thornton J.M. In: Crystallography of Biological Macromolecules. Rossmann M.G., Arnold E.D., editors. Kluwer Academic Publishers; The Netherlands: 2001. PROCHECK: validation of protein structure coordinates, in international tables of crystallography, volume F; pp. 722–725. [Google Scholar]
- 27.Chen V.B. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66(1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cong Y., Liu C.X. 2020. 7DDN SARS-Cov 2 S Protein at Open State. [Google Scholar]
- 29.Morris G.M., Olson A.J. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recogn. 1996;9(1):1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 30.Irwin J.J., Sterling T., Mysinger M.M., Bolstad E.S., Coleman R.G. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52(7):1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: an open chemical toolbox. J. Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.EF P., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualisation system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Pettersen E.F., Goddard T.D., Huang C.C. UCSF Chimera--a visualisation system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 34.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimisation, and multithreading. J. Comput. Chem. 2009;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kozakov D. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozakov D. How good is automated protein docking? Prot. Struct. Funct. Bioinf. 2013;81(12):2159–2166. doi: 10.1002/prot.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dassault Systèmes BIOVIA . Dassault Systèmes; San Diego: 2020. Discovery Studio Visualiser, v.20.1.0.19295. [Google Scholar]
- 38.Salentin S. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ordog R. PyDeT, a PyMOL plug-in for visualising geometric concepts around proteins. Bioinformation. 2008;2:346–347. doi: 10.6026/97320630002346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwede T., Kopp J., Guex N., Peitsch M.C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31(13):3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jimenez Alberto A., Ribas Aparicio R.M., Aparicio-Ozores G. Virtual screening of approved drugs as potential SARS CoV2 main protease inhibitors. Biol. Chem. 2020;88(2020):107325. doi: 10.1016/j.compbiolchem.2020.107325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borkotoky S., Banerjee M., Modi G.P., Dubey V.K. Identification of high affinity and low molecular alternatives of boceprevir against SARS CoV2 main protease: a virtual screening approach. Chem. Phys. Lett. 2021;770:138446. doi: 10.1016/j.cplett.2021.138446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pande M., Kundu D., Srivastava R. Vitamin C and Vitamin D3 show strong binding with the amyloidogenic region of G555F alpha strain modelled of Fibrinogen A alpha-chain associated with renal amyloidosis: proposed possible therapeutic intervention. Mol. Divers. 2021 doi: 10.1007/s11030-021-10205-7. [DOI] [PubMed] [Google Scholar]
- 44.Muhseen T.Z., Hameed R.A., Al-Hasani M.H.H. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (RBD) attachment inhibitors to the human ACE2 receptor: integrated computational approach. J. Mol. Liq. 2020;320(2020):114493. doi: 10.1016/j.molliq.2020.114493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasidharan S., Selvaraj C., Singh S.K., Dubey V.K. Bacterial protein azurin and derived peptides as potential anti-SARS-CoV-2 agents: insights from molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1787864. [DOI] [PubMed] [Google Scholar]
- 46.Martonak R., Laio A., Parrinello M. Predicting crystal structures: the Parrinello- Rahman method revisited. Phys. Rev. Lett. 2003;90(7) doi: 10.1103/PhysRevLett.90.075503. [DOI] [PubMed] [Google Scholar]
- 47.Hess B., Bekker H., Berendsen H.J., Fraaije J.G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18(12):1463–1472. [Google Scholar]
- 48.Chen J-g., Wu S-f., Zhang Q-f., Yin Z-f., Zhang L. α-Glucosidase inhibitory effect of anthocyanins from Cinnamomum camphora fruit: inhibition kinetics and mechanistic insights through in vitro and in silico studies. Int. J. Biol. Macromol. 2020;143:696–703. doi: 10.1016/j.ijbiomac.2019.09.091. [DOI] [PubMed] [Google Scholar]
- 49.Ma R., Wong S.W., Ge L., Shaw C., Siu S.W., Kwok H.W. In vitro and MD simulation study to explore physicochemical parameters for antibacterial peptide to become potent anticancer peptide. Mol. Therap. Oncolytics. 2020;16:7–19. doi: 10.1016/j.omto.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumari R., Kumar R., Lynn A. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 51.Pande M., Srivastava R. Molecular and clinical insights into protein misfolding and associated amyloidosis. Eur. J. Med. Chem. 2019 Dec 15;184:111753. doi: 10.1016/j.ejmech.2019.111753. Epub 2019 Oct 7. PMID: 31622853. [DOI] [PubMed] [Google Scholar]
- 52.Lovell S.C. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Protein Struct. Funct. Genet. 2002;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 53.Laskowski R.A., MacArthur M.W., Thornton J.M.( In: International Tables of Crystallography, Volume F. Crystallography of Biological Macromolecules. Rossmann M.G., Arnold E., editors. Dordrecht, Kluwer Academic Publishers; The Netherlands: 2001. PROCHECK: validation of protein structure coordinates; pp. 722–725. [Google Scholar]
- 54.Deflandre A., Lang G. Photostability assessment of sunscreens. Benzylidene camphor and dibenzoylmethane derivatives. Int. J. Cosmet. Sci. 1988;10(2):53–62. doi: 10.1111/j.1467-2494.1988.tb00002.x. PMID 19456910. [DOI] [PubMed] [Google Scholar]
- 55.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J.( Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 56.Degoey D.A., Chen H.J., Cox P.B., Wendt M.D. Beyond the rule of 5: lessons learned from AbbVie’s drugs and compound collection. J. Med. Chem. 2018;61(7):2636–2651. doi: 10.1021/acs.jmedchem.7b00717. [DOI] [PubMed] [Google Scholar]
- 57.Drugbank database site. https://go.drugbank.com/
- 58.National Center for Biotechnology Information PubChem compound summary for CID 54732242,Naldemedine. 2021. https://pubchem.ncbi.nlm.nih.gov/compound/Naldemedine
- 59.Villoutreix B.O., Calvez V., Marcelin A.-G., Khatib A.-M. In silico investigation of the new UK (B.1.1.7) and South African (501Y.V2) SARS-CoV-2 alpha strain modelleds with a focus at the ACE2–spike RBD interface. Int. J. Mol. Sci. 2021;22:1695. doi: 10.3390/ijms22041695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mandal A., Jha A.K., Hazra B. Plant products as inhibitors of coronavirus 3CL protease. Front. Pharmacol. 2021;12:167. doi: 10.3389/fphar.2021.583387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Umesh Kundu D., Selvaraj C., Singh S.K., Dubey V.K. Identification of new anti- nCoV drug chemical compounds from Indian spices exploiting SARS- CoV-2 main protease as target. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1763202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar V., Parate S., Yoon S. Computational simulations identified marine-derived natural bioactive compounds as replication inhibitors of SARS-CoV-2. Front. Microbiol. 2021;12:583. doi: 10.3389/fmicb.2021.647295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Unni S., Aouti S., Thiyagarajan S. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020;45(2020):130. doi: 10.1007/s12038-020-00102-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Towler P. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279(17):17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ray A.K., Gupta P.S.S., Panda S.K., Biswal S., Rana M.K. Cambridge Open Engage; Cambridge: 2020. Repurposing of FDA Approved Drugs for the Identification of Potential Inhibitors of SARS-CoV-2 Main Protease. ChemRxiv. [Google Scholar]
- 66.Xong L.H., Cao L.J., Shen G.C. Several FDA-approved drugs effectively inhibit SARS-CoV-2 infection in vitro. Front. Pharmacol. 2021 doi: 10.3389/fphar.2020.609592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kouznetsova V.L., Huang D.Z., Tsigelny I.F. Potential SARS-CoV-2 protease Mpro inhibitors: repurposing FDA-approved drugs. Phys. Biol. 2021;18(2) doi: 10.1088/1478-3975/abcb66. [DOI] [PubMed] [Google Scholar]
- 68.Tejera E., Munteanu R.C., Cortez L.A. Drugs repurposing using QSAR, docking and molecular dynamics for possible inhibitors of the SARS-CoV-2 Mpro protease. Molecules. 2020;25:5172. doi: 10.3390/molecules25215172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sensanski M., Perovic V., Pajovic B.S. Drug repurposing for candidate SARS-CoV-2 main protease inhibitors by a novel in silico method. Molecules. 2020;25:3830. doi: 10.3390/molecules25173830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data included in article/supplementary material/referenced in article.