

Abstract
Background
p53 plays a key role in the DNA repair process and response to ionising radiation. We sought to determine the clinical phenotype of TP53 mutations and p53 pathway alterations in patients with rhabdomyosarcoma (RMS) and Ewing sarcoma (ES) treated with radiation.
Methods
Of patients with available genomic sequencing, we identified 109 patients with RMS and ES treated to a total of 286 radiation sites. We compared irradiated tumour control among tumours with TP53 mutations (n = 40) to those that were TP53 wild-type (n = 246). We additionally compared irradiated tumour control among tumours with any p53 pathway alteration (defined as tumours with TP53 mutations or TP53 wild-type tumours identified to have MDM2/4 amplification and/or CDKN2A/B deletion, n = 78) to those without such alterations (n = 208).
Results
The median follow-up was 26 months from radiation. TP53 mutations were associated with worse irradiated tumour control among the entire cohort (hazard ratio, HR = 2.8, P < 0.0001). Tumours with any p53 pathway alteration also had inferior irradiated tumour control (HR = 2.0, P = 0.003). On multivariable analysis, after controlling for tumour histology, intent of radiation, presence of gross disease, and biologically effective dose, TP53 mutations continued to be associated with a radioresistant phenotype (HR = 7.1, P < 0.0001).
Conclusions
Our results show that TP53 mutations are associated with increased radioresistance in RMS and ES. Novel strategies to overcome this radioresistance are important for improved outcomes in p53 disruptive RMS and ES.
Subject terms: Paediatric cancer, Cancer genomics, Cancer genomics, Tumour-suppressor proteins
Background
For patients with paediatric sarcomas including rhabdomyosarcoma (RMS) and Ewing sarcoma (ES), the prognosis for patients who relapse remains very poor, with few long-term survivors.1,2 Failure after radiation therapy (RT) is a common cause of relapse, with clinical factors such as tumour size and tumour location often associated with local tumour failure.3,4 Current strategies to improve irradiated tumour control in paediatric sarcomas such as RT dose escalation are currently applied relatively homogenously without regard for underlying molecular characteristics. Although there is a clear heterogeneity of tumour response after RT in paediatric sarcomas, it is unknown on a molecular level what drives radioresistance in some tumours versus radiosensitivity in others. Given radiotherapy’s critical role in the treatment of both the primary site and sites of distant metastases in RMS and ES, it is important to further understand the genomic determinants of radiation response.
TP53 is the most commonly altered gene in cancer, and mutations confer cell growth and survival advantages through a combination of loss of tumour suppressor functions and gain of oncogenic activity. Even in the absence of genetic alteration, the p53 protein is often dysregulated. For example, p53 is targeted for degradation by the E3-ubiquitin ligases, MDM2 and MDM4, and these genes are frequently amplified in TP53 wild-type tumours as a means of suppressing p53 levels and its associated activity.5 Similarly, the tumour suppressor, ARF, which is encoded by the CDKN2A/B locus, inhibits the MDM2–p53 interaction, and genetic deletion of the CDKN2A/B locus is a common means of inactivating the p53 pathway.6
The p53 pathway plays an important role in radiation-induced DNA repair and the cell cycle. Defective p53 has been associated in vitro with radioresistance in various paediatric cell lines, including medulloblastoma,7 diffuse intrinsic pontine gliomas (DIPG)8 and neuroblastoma.9 In addition, TP53 mutations have been associated with clinical radioresistance in patients with DIPG,8 endometrial cancer10 and head and neck cancer.11 We hypothesised that TP53 mutations and other p53 pathway alterations (specifically MDM2/4 amplification and CDKN2A/B deletion) might also confer radioresistance in RMS and ES. Accordingly, the goal of this study was to determine the clinical phenotype of TP53 mutations and p53 pathway alterations in RMS and ES.
Materials and methods
Study design
All patients with RMS and ES who underwent prospective genomic profiling utilising our institutional 468-gene oncopanel, MSK-IMPACT, from 11/2014-3/2020 and received RT were analysed. Matched peripheral blood samples were collected to distinguish germline from somatic mutations. Alterations of interest included TP53 mutations and other p53 pathway alterations, defined as MDM2/4 amplification and/or CDKN2A/B deletion. Baseline patient, tumour and treatment characteristics were collected, including age, histology, primary tumour site, stage, biologically effective dose (BED) of radiation, radiation site, the presence of gross disease at the time of radiation and radiation intent (definitive versus palliative). This analysis was approved by our Institutional Review Board.
Statistical analysis
The primary outcome was irradiated tumour progression among TP53 mutant versus TP53 wild-type tumours. Irradiated tumour progression was defined as progression or relapse within the radiation treatment field plus a margin of 1 cm or less, and was determined based on either (1) pathologic confirmation of viable tumour, (2) radiographic growth of disease on at least two progressive scans and/or (3) growth resulting in a change in systemic therapy. Imaging was reviewed by two independent clinicians, with a third clinician available to resolve any discrepancies. In addition, irradiated tumour progression was compared among tumours with or without any p53 pathway alteration (defined as above as any TP53 mutation, MDM2/4 amplification and/or CDKN2A/B deletion).
Secondary outcomes included initial radiographic tumour response after irradiation in TP53 mutant versus TP53 wild-type tumours, with response defined as the percent change in maximal dimension of the tumour from pre-radiation imaging (CT and/or MRI) to post-radiation imaging (first CT and/or MRI ≥ 4 weeks after radiation). Other secondary outcomes included distant progression outside of the irradiated field and overall survival in TP53 mutant versus TP53 wild-type tumours. Baseline patient, tumour and treatment characteristics between TP53 mutant and TP53 wild-type groups were compared with Chi-square and unpaired two-tailed t tests. Cumulative incidence functions, the Kaplan–Meier method with log-rank test, and Cox proportional hazard regression analyses were utilised to compare tumour control outcomes by TP53 status. A P value <0.05 was considered significant.
Results
Study population
One hundred and forty patients with RMS and 113 with ES underwent targeted sequencing utilising MSK-IMPACT from 11/2014-3/2020. Among this cohort, 59 with RMS and 50 with ES underwent RT to a total of 286 sites (126 RMS, 160 ES) and were included in our analysis (Fig. 1). The site of radiation treatment included most commonly the primary site (n = 119) and sites of bony metastases (n = 89). Forty-eight sites of metastases were treated with stereotactic body radiation therapy and 4 brain metastases with stereotactic radiosurgery. In total, 19 patients were treated with whole-lung irradiation. The median follow-up was 26 months from radiation.
Fig. 1. CONSORT.
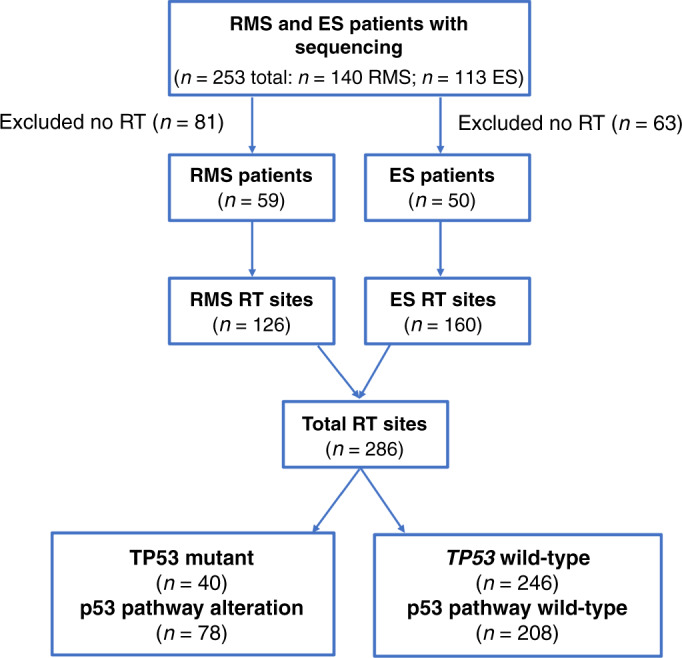
CONSORT diagram of the patient population and TP53 status (RMS rhabdomyosarcoma, ES Ewing sarcoma, RT radiation therapy).
The median age was slightly higher in those with TP53 mutations compared to wild-type patients (26 years versus 17 years, P = 0.09, Supplementary Fig. 1). Other characteristics such as tumour histology (RMS versus ES), stage at the time of first RT course, intent of RT (definitive versus palliative), gross disease at the time of RT, and BED were similar along with TP53 mutant versus TP53 wild-type tumours (Table 1).
Table 1.
Baseline patient and radiation characteristics of TP53 mutant versus TP53 wild-type patients.
| Patient cohort (n = 109) |
TP53 mutant (n = 17) |
TP53 wild-type (n = 92) |
P | |
|---|---|---|---|---|
| Age (median, range) | 18 (<1–74) | 26 (5–55) | 17 (<1–74) | 0.09 |
| Diagnosis | ||||
| ES | 50 (46%) | 9 (53%) | 41 (45%) | 0.52 |
| RMS | 59 (54%) | 8 (47%) | 51 (55%) | |
| Stage at time of first RT course | ||||
| Localised | 63 (58%) | 9 (53%) | 54 (59%) | 0.66 |
| Metastatic | 46 (42%) | 8 (47%) | 38 (41%) | |
| Radiated sites (n = 286) |
TP53 mutant (n = 40) |
TP53 wild-type (n = 246) |
P | |
|---|---|---|---|---|
| Intent of RT (by course) | ||||
| Definitive | 140 (49%) | 15 (38%) | 125 (51%) | 0.12 |
| Palliative | 146 (51%) | 25 (63%) | 121 (49%) | |
| Gross disease at time of RT (by course) | ||||
| No | 50 (17%) | 7 (17%) | 43 (14%) | 0.99 |
| Yes | 236 (83%) | 33 (83%) | 203 (86%) | |
| BED (Gy, median, range, by course) | 48 (14–121) | 43 (14–72) | 48 (14–121) | 0.97 |
RT radiation therapy, ES Ewing sarcoma, RMS rhabdomyosarcoma.
Genomics
Sequencing was performed from the primary tumour in 69%; from a regional node in 6%; and from a site of metastasis in 25%. Genomic profiling was performed from tissue taken before initiation of any treatment (including chemotherapy) in 72% of patients. Eight of 59 (14%) patients with RMS harboured TP53 mutations, while 9 of 50 patients (18%) with ES did. See Fig. 2 for an oncoprint demonstrating the distribution of p53 pathway alterations. Among the 286 sites treated, 40 (14%: 10% RMS, 18% ES) were TP53 mutated. Among the 246 TP53 wild-type sites treated, we identified 38 sites with other p53 pathway alterations, including 19 with MDM2/4 amplification, 18 with CDKN2A/B deletion, and one site that harboured both an MDM2/4 amplification and CDKN2A/B deletion. TP53 mutations were predominately missense mutations (77%), with the remaining mutations resulting in truncated proteins due to nonsense or frameshift mutations (Supplementary Fig. 2). There were no cases of TP53 deletion or inactivating translocation events. All alterations were somatic except for one patient who harboured a TP53 germline mutation. As expected, MDM2/4 amplifications and CDKN2A/B deletions were found predominantly in TP53 wild-type tumours and were mutually exclusive with TP53 mutations (P = 0.03).
Fig. 2. Oncoprint.

Oncoprint demonstrating the distribution of p53 pathway mutations (defined as TP53 mutation, MDM2/4 amplification and/or CDKN2A/B deletion).
The median number of total genomic alterations (somatic mutations, germline mutations, copy number alterations or gene fusions) identified by MSK-IMPACT testing was 3, and was not different between TP53 wild-type and mutant tumours (mean of 3.9 and 5.8, respectively, P = 0.09). Comparing the TP53 mutant tumours to wild-type tumours did not identify any additional statistically significant genomic alterations that were enriched in either group.
Radiation response and TP53 mutations
On univariate analysis (UVA), TP53 mutations were associated with an increased risk of irradiated tumour progression (hazard ratio (HR) = 2.8, 95% confidence interval (CI) 1.6–5.0, P < 0.0001, Fig. 3a). To avoid any potentially confounding effects based on the inclusion of multiple irradiated sites per patient, an additional per-patient analysis limited to the first radiated site for each patient was performed. On this per-patient analysis, TP53 mutations remained associated with increased irradiated tumour progression (HR = 2.7, 95% CI 1.2–6.0, P = 0.01), consistent with the site-level analysis. Including MDM2/4 amplification and CDKN2A/B deletion, any p53 pathway alteration was also associated with worse irradiated tumour control (HR = 2.0, 95% CI 1.3–2.2, P = 0.003), although alterations in MDM2/4 or CDKN2A/B were not individually associated with irradiated tumour progression (HR = 1.1, P = 0.90 and HR = 1.3, P = 0.52, respectively).
Fig. 3. Irradiated tumour progression and survival analyses by TP53 status.

Irradiated tumour progression by TP53 status in the (a) entire cohort, (b) subset of rhabdomyosarcoma and (c) subset of Ewing sarcoma; (d) distant tumour progression by TP53 status (outside of the irradiated field); and (e) overall survival by TP53 status.
Among the subgroup of patients with RMS, TP53 mutations were again associated with significantly increased irradiated tumour progression (HR = 3.8, 95% CI 1.8–8.4, P = 0.001, Fig. 3b), although this did not reach statistical significance in the ES subgroup (HR = 1.8, 95% CI 0.6–5.2, P = 0.27, Fig. 3c). The radioresistant phenotype of TP53 mutations was also observed whether patients were treated with palliative intent (HR = 4.0, 95% CI 1.4–10.9, P = 0.008) or definitive intent (HR = 3.0, 95% CI 1.4–6.1, P = 0.004). See Table 2 for UVA demonstrating the effect of the different p53 pathway mutations on irradiated tumour progression among various subgroups. On multivariable analysis (MVA) including all factors associated with irradiated tumour progression on UVA (TP53 status, intent of radiation (palliative versus definitive), presence of gross disease at time of RT, histology (RMS versus ES) and BED), TP53 mutations continued to be associated with worse irradiated tumour control (HR = 7.1, 95% CI 3.7–13.7, P < 0.0001, Supplementary Table 1). In addition, the radiographic response of irradiated tumours as determined by pre- and post-treatment imaging was worse in TP53 mutant tumours, with a median decrease in tumour size of 2% compared to 18% in TP53 wild-type tumours (P = 0.03, Supplementary Fig. 3).
Table 2.
Effect of p53 pathway mutations on irradiated tumour control in the entire cohort and subsets of patients with rhabdomyosarcoma (RMS), Ewing sarcoma (ES), patients treated with palliative intent and patients treated with definitive intent (univariate analysis).
| Gene | Entire cohort n = 286 (HR, 95% CI) |
P | RMS n = 126 (HR, 95% CI) |
P | ES n = 160 (HR, 95% CI) |
P | Palliative n = 146 (HR, 95% CI) |
P | Definitive n = 140 (HR, 95% CI) |
P |
|---|---|---|---|---|---|---|---|---|---|---|
| TP53 | 2.8 (1.6–5.0) | <0.0001 | 3.8 (1.8–8.4) | 0.001 | 1.8 (0.6–5.2) | 0.27 | 4.0 (1.4–10.9) | 0.008 | 3.0 (1.4–6.1) | 0.004 |
| MDM2/4 | 1.1 (0.4–2.6) | 0.90 | 0.83 (0.3–2.1) | 0.69 | n/a | n/a | 2.6 (0.6–11.2) | 0.20 | 0.9 (0.3–3.1) | 0.93 |
| CDKN2A/B | 1.3 (0.6–2.8) | 0.52 | 0.89 (0.3–2.5) | 0.81 | 1.1 (0.1–8.0) | 0.94 | 0.6 (0.08–4.4) | 0.61 | 1.9 (0.8–4.5) | 0.15 |
| Any alteration | 2.0 (1.3–3.2) | 0.003 | 1.7 (0.9–3.0) | 0.10 | 1.6 (0.6–4.2) | 0.32 | 2.4 (1.1–5.5) | 0.04 | 2.4 (1.3–4.3) | 0.003 |
To further evaluate whether the TP53-mutant phenotype was specific to radiation, local progression was evaluated among a cohort of ES patients with available genomic sequencing who were treated with surgery alone for local tumour control. In this surgical cohort (n = 43), TP53 mutations alone or alterations in the p53 pathway were not associated with increased risk of local progression (P = 0.99 and P = 0.79, respectively).
Distant tumour progression and overall survival
Distant tumour progression outside of the irradiated field was also increased in TP53 mutant tumours compared to TP53 wild-type tumours, although not to the same extent as tumour progression within the irradiated field (HR = 1.6, 95% CI 1.1–2.4, P = 0.03, Fig. 3d). In addition, overall survival (OS) was worse in patients with TP53 mutations (HR = 3.4, 95% CI 1.8–6.7, P < 0.0001, Fig. 3e). This remained true in the subgroups of patients with RMS (HR = 4.3, 95% CI 1.4–12.4, P = 0.008) and ES (HR = 4.4, 95% CI 1.7–11.3, P = 0.002). CDKN2A/B deletion was also individually associated with worse OS in patients with ES (HR = 4.3, 95% CI 1.1–16.5, P = 0.03), but not RMS. MDM2/4 amplification was not associated with OS in patients with RMS or ES. On MVA, including the other factors associated with OS on UVA (histology and stage), the association of TP53 with worse OS persisted (HR = 7.4, 95% CI 3.5–16.0, P < 0.0001).
Discussion
Our data support the hypothesis that TP53 mutations are associated with a radioresistant phenotype and poor survival in patients with RMS and ES. The association with both worse irradiated tumour control and worse OS suggests not only a radioresistant phenotype but also an overall unfavourable biologic behaviour of TP53 mutant tumours. Collectively, the poor outcomes for TP53 mutant tumours are likely due to a multitude of factors, including radioresistance, possible chemoresistance, and a highly aggressive underlying biology. Interestingly, among patients with ES treated with surgical resection alone, local tumour progression was not different in TP53 mutant versus TP53 wild-type tumours. This suggests that the increased risk of irradiated tumour progression observed in TP53 mutant tumours may be due to intrinsic radioresistance. However, given the small number of patients who underwent surgery alone, validation in a larger cohort is necessary to fully assess the impact of TP53 mutations on surgical local control.
Although findings from the COG did not show TP53 and/or CDKN2A to be reliable prognostic biomarkers in ES,12 our data are consistent with other studies demonstrating a markedly poor survival among RMS patients with TP53 mutations13 and ES patients with either TP53 mutations and/or p16/p14ARF deletion.14,15 Despite the established prognostic value of TP53 mutations, it has been previously unknown whether TP53 mutant tumours are specifically resistant to irradiation in RMS and ES, and/or whether this radioresistance contributes to the dismal survival observed in patients with TP53 mutations. This is the first study, to our knowledge, to demonstrate the radioresistant phenotype of TP53 mutations in patients with RMS and ES. These results support the design of prospective studies to further assess whether TP53 mutations are predictive of radioresistance and whether strategies that improve local control can also improve the prognosis for these patients. Whether other p53 pathway alterations are also associated with a radioresistant phenotype remains possible, but given the relatively small number of MDM2/4 and CDKN2A/B alterations in our cohort, we were unable to fully ascertain a similar phenotype to that of TP53 mutations. Of note, in ES, a particularly poor survival has been observed among those harbouring both STAG2 and TP53 mutations.16 Among our cohort of ES patients, only 3 were STAG2- and TP53-co-mutated, limiting our ability to corroborate the poor prognosis previously observed with the co-occurrence of these mutations.
With an incidence of ~15% in RMS and ES, TP53 mutations are not as common as they are in other tumours such as osteosarcoma,17 HPV-negative head and neck cancer,18 and triple-negative breast cancer.19 However, the strength of the effect of TP53 mutations on radioresponse and prognosis in our cohort reinforces the clinical relevance of these mutations. In addition, RT is a prevalent treatment modality for ES and RMS, where it is used in the upfront, relapsed and metastatic settings, further highlighting the potential significance of these mutations for clinical practice. This contrasts with osteosarcoma, in which >90% of tumours harbour a TP53 mutation,17 but where RT is rarely used secondary to the known intrinsic radioresistance; this radioresistance may be due in large part to the prevalence of TP53 mutations. Consistent with our findings in RMS and ES, TP53 mutations have been shown to impart a radioresistant phenotype in other paediatric tumours, such as neuroblastoma in the preclinical setting9 and DIPG in both the preclinical and clinical setting.8
p53, known as the “guardian of the genome,” plays a critical role in the DNA damage response, the cell cycle (specifically the G1 checkpoint), apoptosis, and cellular senescence. One mechanism behind the radioresistance of p53 mutant tumours is the absence of G1 arrest in response to radiation20,21 and subsequent inhibition of radiation-induced cell death.22 In addition, dysfunctional p53 has been demonstrated within head and neck cancers to result in inhibition of radiation-induced senescence.11 With refined knowledge of the pathways involved in p53-mediated radioresistance, methods to exploit TP53 dysfunction and reverse this radioresistance are possible. For example, with a defective G1 checkpoint, p53-deficient tumours must rely on ATR and CHK1 to respond to DNA damage and arrest in the S and G2 phases.23 Accordingly, inhibition of CHK1 has been shown to selectively overcome the radioresistance of TP53 mutated cells in response to radiation and other DNA damaging agents;24–26 and ATR inhibitors have also been shown to induce synthetic lethality in TP53 disruptive cells.27,28 Other strategies to mitigate radioresistance may include MET inhibition,29 DNA PK inhibition,30–32 or novel fractionation regimens that exploit the vulnerability and dependence of TP53 defective tumours on the G2-M and intra-S checkpoints.
Limitations of our analysis include the heterogeneity of patients in our cohort with ES and RMS, with various stages at the time of RT and various timing of genomic sequencing. In addition, local treatment paradigms for RMS and ES differ, in that almost all patients with initially unresected RMS will receive RT, whereas over half of patients with ES will have surgery for local control, often without the need for postoperative RT. Of note, the tumour control and overall survival outcomes in this cohort are less favourable than has been described on prospective randomised ES and RMS trials. This may reflect a bias in patient selection and the nature in which patients are identified for tumour sequencing with MSK-IMPACT.
However, even among this heterogeneous cohort, after controlling for clinical factors, the strength of the association of TP53 mutations with radioresistance strongly persisted. Further studies examining the generalisability of these observations are warranted. In addition, although sequencing was performed from metastatic samples rather than the primary tumour in 25% of patients, the source and timing of sequencing do not change the conclusion that a TP53 mutation, when discovered, can serve as a marker of poor response to RT and an overall unfavourable prognosis.
Our results call into question the application of uniform treatment strategies to genotypes that are likely to be treatment-resistant to conventional radiation methods in ES and RMS. Novel strategies to individualise therapies by exploiting the p53 pathway are critical for improvement in outcomes in patients with TP53 mutated ES and RMS.
Supplementary information
Author contributions
D.L.C., K.L.P., L.H.W., E.K.S., G.P.G. and S.L.W. designed the study. D.L.C., K.L.P., L.H.W. and S.L.W. collected the data. D.L.C., K.L.P., L.H.W., E.K.S., G.P.G. and S.L.W. contributed to the formal analysis and methodology. D.L.C., K.L.P. and S.L.W. wrote the original draft. D.L.C., K.L.P., L.H.W., E.K.S., G.P.G. and S.L.W. reviewed and edited.
Ethics approval and consent to participate
Consent for genomic testing was obtained under the prospective protocol (#12–245) through the Memorial Sloan Kettering Cancer Center Institutional Review Board. Consent to participate in this retrospective analysis was waived by the Memorial Sloan Kettering Cancer Center Institutional Review Board approved protocol (#16–1125). This study was performed in accordance with the Declaration of Helsinki.
Consent to publish
No personal data or identifying information are being submitted.
Data availability
Data are relevant to MSKCC patients and not publicly available. We will make de-identified data available upon request after institutional approval.
Competing interests
D.L.C., K.L.P., L.H.W., E.K.S., G.P.G. and S.L.W. declare no competing interests. L.H.W. held a consultant position at EUSA Pharma in 2019, not relevant to this work.
Funding information
National Institutes of Health/National Cancer Institute Cancer Center Support Grant (P30CA008748).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01438-2.
References
- 1.Grunewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Alava E, Kovar H, et al. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018;4:5. doi: 10.1038/s41572-018-0003-x. [DOI] [PubMed] [Google Scholar]
- 2.Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019;5:1. doi: 10.1038/s41572-018-0051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casey DL, Chi YY, Donaldson SS, Hawkins DS, Tian J, Arndt CA, et al. Increased local failure for patients with intermediate-risk rhabdomyosarcoma on ARST0531: a report from the Children’s Oncology Group. Cancer. 2019;125:3242–3248. doi: 10.1002/cncr.32204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed SK, Randall RL, DuBois SG, Harmsen WS, Krailo M, Marcus KJ, et al. Identification of patients with localized Ewing sarcoma at higher risk for local failure: a report from the Children’s Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2017;99:1286–1294. doi: 10.1016/j.ijrobp.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 6.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhukova N, Ramaswamy V, Remke M, Martin DC, Castelo-Branco P, Zhang CH, et al. WNT activation by lithium abrogates TP53 mutation associated radiation resistance in medulloblastoma. Acta Neuropathol. Commun. 2014;2:174. doi: 10.1186/s40478-014-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Werbrouck C, Evangelista CCS, Lobon-Iglesias MJ, Barret E, Le Teuff G, Merlevede J, et al. TP53 pathway alterations drive radioresistance in diffuse intrinsic pontine gliomas (DIPG) Clin. Cancer Res. 2019;25:6788–6800. doi: 10.1158/1078-0432.CCR-19-0126. [DOI] [PubMed] [Google Scholar]
- 9.Yogev O, Barker K, Sikka A, Almeida GS, Hallsworth A, Smith LM, et al. p53 loss in MYC-driven neuroblastoma leads to metabolic adaptations supporting radioresistance. Cancer Res. 2016;76:3025–3035. doi: 10.1158/0008-5472.CAN-15-1939. [DOI] [PubMed] [Google Scholar]
- 10.Akiyama A, Minaguchi T, Fujieda K, Hosokawa Y, Nishida K, Shikama A, et al. Abnormal accumulation of p53 predicts radioresistance leading to poor survival in patients with endometrial carcinoma. Oncol. Lett. 2019;18:5952–5958. doi: 10.3892/ol.2019.10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin. Cancer Res. 2012;18:290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lerman DM, Monument MJ, McIlvaine E, Liu XQ, Huang D, Monovich L, et al. Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children’s Oncology Group. Pediatr. Blood Cancer. 2015;62:759–765. doi: 10.1002/pbc.25340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casey DL, Wexler LH, Pitter KL, Samstein RM, Slotkin EK, Wolden SL. Genomic determinants of clinical outcomes in rhabdomyosarcoma. Clin. Cancer Res. 2020;26:1135–1140. doi: 10.1158/1078-0432.CCR-19-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Alava E, Antonescu CR, Panizo A, Leung D, Meyers PA, Huvos AG, et al. Prognostic impact of P53 status in Ewing sarcoma. Cancer. 2000;89:783–792. doi: 10.1002/1097-0142(20000815)89:4<783::AID-CNCR10>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 15.Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH, et al. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J. Clin. Oncol. 2005;23:548–558. doi: 10.1200/JCO.2005.02.081. [DOI] [PubMed] [Google Scholar]
- 16.Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4:1342–1353. doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2007;357:2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McIlwrath AJ, Vasey PA, Ross GM, Brown R. Cell cycle arrests and radiosensitivity of human tumor cell lines: dependence on wild-type p53 for radiosensitivity. Cancer Res. 1994;54:3718–3722. [PubMed] [Google Scholar]
- 21.Siles E, Villalobos M, Valenzuela MT, Nunez MI, Gordon A, McMillan TJ, et al. Relationship between p53 status and radiosensitivity in human tumour cell lines. Br. J. Cancer. 1996;73:581–588. doi: 10.1038/bjc.1996.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shu HK, Kim MM, Chen P, Furman F, Julin CM, Israel MA. The intrinsic radioresistance of glioblastoma-derived cell lines is associated with a failure of p53 to induce p21(BAX) expression. Proc. Natl Acad. Sci. USA. 1998;95:14453–14458. doi: 10.1073/pnas.95.24.14453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 24.Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J. Clin. Investig. 2012;122:1541–1552. doi: 10.1172/JCI58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vance S, Liu E, Zhao L, Parsels JD, Parsels LA, Brown JL, et al. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle. 2011;10:4321–4329. doi: 10.4161/cc.10.24.18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Lai J, Du Z, Gao J, Yang S, Gorityala S, et al. Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress. Oncotarget. 2016;7:34688–34702. doi: 10.18632/oncotarget.9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127:582–595. doi: 10.1182/blood-2015-05-644872. [DOI] [PubMed] [Google Scholar]
- 28.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011;7:428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 29.Mikami K, Medova M, Nisa L, Francica P, Gluck AA, Tschan MP, et al. Impact of p53 status on radiosensitization of tumor cells by MET inhibition-associated checkpoint abrogation. Mol. Cancer Res. 2015;13:1544–1553. doi: 10.1158/1541-7786.MCR-15-0022. [DOI] [PubMed] [Google Scholar]
- 30.Hafsi H, Dillon MT, Barker HE, Kyula JN, Schick U, Paget JT, et al. Combined ATR and DNA-PK inhibition radiosensitizes tumor cells independently of their p53 status. Front. Oncol. 2018;8:245. doi: 10.3389/fonc.2018.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ortiz T, Burguillos MA, Lopez-Lluch G, Navas P, Herrador M, Gonzalez I, et al. Enhanced induction of apoptosis in a radio-resistant bladder tumor cell line by combined treatments with X-rays and wortmannin. Radiat. Environ. Biophys. 2008;47:445–452. doi: 10.1007/s00411-008-0188-6. [DOI] [PubMed] [Google Scholar]
- 32.Kumar, R. J., Chao, H. X., Simpson, D. A., Feng, W., Cho, M. G., Roberts, V. R. et al. Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency. NAR Cancer. 2, zcaa038 (2020). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are relevant to MSKCC patients and not publicly available. We will make de-identified data available upon request after institutional approval.