

Graphical abstract
Keywords: Deep learning, Metastasis, Multi-omics, Pan-cancer, Autoencoder, Cancer, Machine learning, Clinical decision support
Abstract
Predicting metastasis in the early stages means that clinicians have more time to adjust a treatment regimen to target the primary and metastasized cancer. In this regard, several computational approaches are being developed to identify metastasis early. However, most of the approaches focus on changes on one genomic level only, and they are not being developed from a pan-cancer perspective. Thus, we here present a deep learning (DL)–based model, MetaCancer, that differentiates pan-cancer metastasis status based on three heterogeneous data layers. In particular, we built the DL-based model using 400 patients’ data that includes RNA sequencing (RNA-Seq), microRNA sequencing (microRNA-Seq), and DNA methylation data from The Cancer Genome Atlas (TCGA). We quantitatively assess the proposed convolutional variational autoencoder (CVAE) and alternative feature extraction methods. We further show that integrating mRNA, microRNA, and DNA methylation data as features improves our model's performance compared to when we used mRNA data only. In addition, we show that the mRNA-related features make a more significant contribution when attempting to distinguish the primary tumors from metastatic ones computationally. Lastly, we show that our DL model significantly outperformed a machine learning (ML) ensemble method based on various metrics.
1. Introduction
In silico metastasis-related research has generally been directed towards predicting if cancer has metastasized, classifying sites as primary or secondary, and identifying potential therapeutic approaches [1], [2]. The reason being, it is believed that administering drugs that also target the metastases can curb cancer-related deaths, but only if we find a way to identify metastasis with more precision than currently used blood tests and imaging technology [2], [3], [4], [5]. In this regard, different omics data types have been used to build models that predict metastasis mainly using two approaches, i.e., network-based and ranked-based.
Network-based approaches include one developed by He and colleagues [6]. They used microarray-based gene expression data to classify metastatic and non-metastatic osteosarcoma (OS) patient samples, using a support vector machine (SVM) based classifier. Using statistical methods, at first, they identified the differentially expressed genes (DEGs), which they used to construct a protein–protein interaction (PPI) network. They then ranked the genes based on the network property, betweenness centrality (BC), and used the SVM classifier's top-ranking genes to predict metastasis. But during the same timeframe, Metri and colleagues [7] also built multigene models to differentiate metastatic melanoma from primary melanoma using adaptive boosting (Adaboost) and random forest (RF) classifiers. Later, Wei and colleagues [8], and Tuo and colleagues [9] built SVM models to classify cutaneous melanoma and breast cancer samples, respectively, as metastatic and non-metastatic. Both SVM models achieved significantly improved accuracy compared to the Adaboost and RF models. More recently, though, Chereda and colleagues [10] developed a deep learning (DL) model that applies the graph convolutional neural networks (CNN) technique by exploiting the PPI graph as prior knowledge for predicting breast cancer metastasis. They showed the graph CNN model’s strength by comparing it with different machine learning (ML) models, and it showed superior accuracy.
On the other hand, the rank-based approaches use an iterative gene selection approach that determines the subset of genes better suited to serve as features. Wu and colleagues [11] used this approach with DNA methylation data to build an RF classifier to predict lymph node (LN) metastasis status in stomach cancer. They performed three preprocessing steps. First, they carried out differential methylation analysis to extract significantly differentiating probes between metastatic and non-metastatic samples. Next, the feature selection technique, minimum redundancy maximum relevance (mRMR), was applied to remove redundant features. The final step implemented a genetic algorithm-based method to extract the most relevant probes fed to the RF model. Several of the probes were known to be associated with LN metastasis-related genes such as HOXD1, NMT1, and SEMA3E. Ahsen and colleagues [12] employed microarray-based genome-wide microRNA expression profiling to identify robust molecular signatures to predict LN metastasis risk in endometrial cancer. They proposed the lone star algorithm specifically developed to identify a small number of discriminative features when the number of features is less than the number of samples. They fed the top discriminative microRNAs that target 23 cancer-associated genes to a weighted SVM classifier. Similar to the above study, Zhao and colleagues [13] used microarray-based microRNA expression data to identify brain metastasis-related (BM) microRNAs in the lung adenocarcinoma (LUAD) samples. They used RF to select the most correlated microRNAs (with the BM classification according to the important permutation score) and classify samples. Karabulut and colleagues [14] proposed a discriminative deep belief network (DDBN) to demonstrate the DL approach's ability to produce a powerful decision support model using gene expression data. They used their proposed DL model to distinguish between metastatic and non-metastatic colorectal cancer. They implemented preprocessing steps such as 1) selecting essential features using the information gain technique and 2) oversampling the minority class using synthetic minority over-sampling technique (SMOTE). The proposed DDBN outperformed other ML models such as SVM, RF, and k-nearest neighbor (KNN).
Most methods predict metastasis using only a single-omics data type such as mRNA [7], [8], [9], [10], microRNA [12], [13], or DNA methylation [11], [15]. However, multiple omics layers contribute to an observed biological phenotype; therefore, several studies are now integrating multi-omics data [16], [17], [18], [19], [20], [21], [22]. In this regard, Bhalla and colleagues [23] used multi-omics data (mRNA expression, microRNA expression, and DNA methylation data) with ML to classify metastatic and primary skin cutaneous melanoma tumors. They developed an ensemble learning model that takes the prediction scores from three models (one model for each omics data type) as input features for an SVM to predict the metastasis status. This study revealed the genes CASP7, S100A7, C7, KRT14, MMP3, LOC642587, and microRNAs hsa-mir-203b and hsa-mir-205 as potential key genomic features that contribute to the oncogenesis of melanoma and further suggested CDK14, ESM1, NFATC3, ZNF827, C7orf4, and ZSWIM7 as novel putative markers for SKCM metastasis. Despite this work and others achieving good prediction accuracy and precision, none of the models are generic. In addition, to the best of our knowledge, researchers do not know if a DL method’s performance with multi-omics data yields a better result or which combination of multi-omics data will produce the best result.
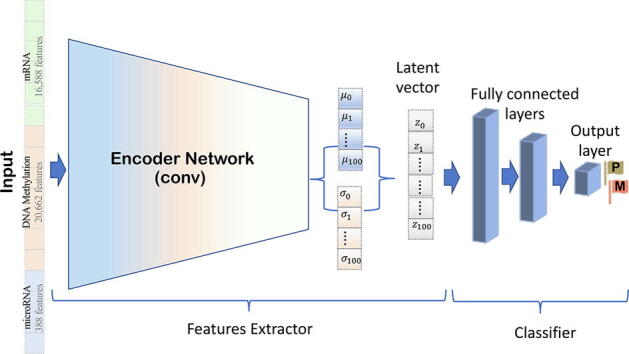
This work represents the first attempt to use DL with multi-omics pan-cancer data to predict metastasis. We used 420 samples from the TCGA multi-omics cohort, which have mRNA expression, microRNA expression, DNA methylation, and clinical information. We used a convolutional variational autoencoder (CVAE) to automatically extract relevant features that we fed to a deep neural network (DNN) model to predict which tumors have metastasized (M) and which ones are primary (P).
2. Materials and method
2.1. Data collection and preprocessing
We sifted through 33 TCGA projects in search of primary tumor samples (with/without distant metastasis) that have mRNA, microRNA, and DNA methylation data available. We found samples from 11 types of cancer with the three types of omics data available, and at least ten of the samples are metastasized based on American Joint Committee on Cancer (AJCC) categorization [24]. AJCC distant metastasis (M) categories assign M0 to samples that show no evidence of distant metastasis and M1 to others that show evidence of distant metastasis. Specifically, for M1 samples, we had:
-
•
10 cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) samples.
-
•
10 thyroid carcinoma (THCA) samples.
-
•
10 kidney renal papillary cell carcinoma (KIRP) samples.
-
•
10 urothelial bladder urothelial carcinoma (UBC) samples.
-
•
10 esophageal carcinoma (ESCA).
-
•
11 rectum adenocarcinoma (READ) samples.
-
•
20 stomach adenocarcinoma (STAD) samples.
-
•
17 invasive breast carcinoma (BRCA) samples.
-
•
19 lung adenocarcinoma (LUAD) samples.
-
•
41 colon adenocarcinoma (COAD) samples.
-
•
52 kidney renal clear cell carcinoma samples.
We collated the same number of samples from the respective associated primary cancer samples (M0) to generate a balanced dataset. We used a total of 420 samples (210 metastasized and 210 primary) in this study. We obtained the data using the TCGAbiolinks R package [25], [26] that provides programmatic access to the Genomic Data Commons (GDC) Data Portal, a platform that includes TCGA, among other resources. We used the TCGAbiolinks package to preprocess the harmonized TCGA dataset as well. In the preprocessing phase, we used the Enhancer Linking by Methylation/Expression Relationship (ELMER) R package to map CpG islands within 1500 bp ahead of the transcription start sites (TSS) of genes and averaged their methylation values. Moreover, we performed three preprocessing steps to deal with missing values as described by Chaudhary and colleagues [27]. Briefly, we first removed the biological features (e.g., mRNA/microRNAs) showing zeros in more than 25% of the patients. Second, we removed samples with less than 75% features remaining after the first step. Finally, we used the R impute function [28] which is a function to impute missing expression data, using nearest neighbor averaging to fill the remaining missing values. Preprocessing steps have been done independently on each test set. After preprocessing, there are 200 samples with all three data types, and the number of features for mRNA is 16,588, DNA methylation is 20,662, and microRNA is 388.
2.2. Deep learning framework
2.2.1. Convolutional variational autoencoder for feature extraction
We used an autoencoder (AE)-based architecture [29] to automatically extract features and reduce the input data’s dimensionality. AE is an unsupervised DL technique in which we leverage neural networks for the task of representation learning. A typical AE consists of 1) an encoder, which maps the high dimensional input data into a latent variable embedding having lower dimensionality than the input, and 2) a decoder, which attempts to reconstruct the input data from the embedding. The training phase aims to retain as much information as possible when encoding to minimize the reconstruction error when decoding. In particular, we used the so-called variational autoencoders (VAE) [30] for their ability to better generalize to different input data.
The VAE's goal is to learn the probability distribution parameters modeling the data or calculating the posterior . It builds a variational inference model q that approximates the true posterior [31]. Given a data-point x, it produces a distribution over the latent values z from where it could have been drawn, and this is called a probabilistic encoder (recognition model). As a result, each latent variable is related to a corresponding observation in the data-point x through the likelihood , called a probabilistic decoder. Given a latent values z, VAE decodes it into a distribution over the observation × (generative model) [31].
Two loss functions are used to train the VAE model. First, the reconstruction loss function is computed as the cross-entropy loss to force the decoded samples to match the initial inputs. Second, the regularizing constraint is computed as the Kullback-Leibler (KL) divergence to force the latent embeddings z to conform to a normal distribution with zero mean and one standard deviation (Eq. (1)) [31].
| (1) |
where and denote the parameters of the encoder (recognition model) and decoder (generative model), respectively.
Rather than using regular feedforward layers in our VAE, we applied convolutional VAE (CVAE) because they utilize sliding filters to recognize better local patterns independent of their position in the data. Fig. 1 illustrates the CVAE architecture. It includes the two symmetrical and reversed encoder and decoder structures; each comprise two convolutional layers and one dense layer. We used the mean and the standard deviation layers (i.e., μ and σ) to generate the latent 100-dimensional vector (refer to Table 1 for details). We implemented CVAE using Python Keras library (https://github.com/fchollet/keras) [32]. The CVAE input was a stack of three omics data (mRNA, microRNA, and DNA methylation) matrices, and we employed the Stochastic Gradient Descent (SGD) algorithm with the default parameters as the optimizer. The number of epochs = 100 and batch size = 8, and we used the early stopping technique [33] to avoid overfitting.
Fig. 1.
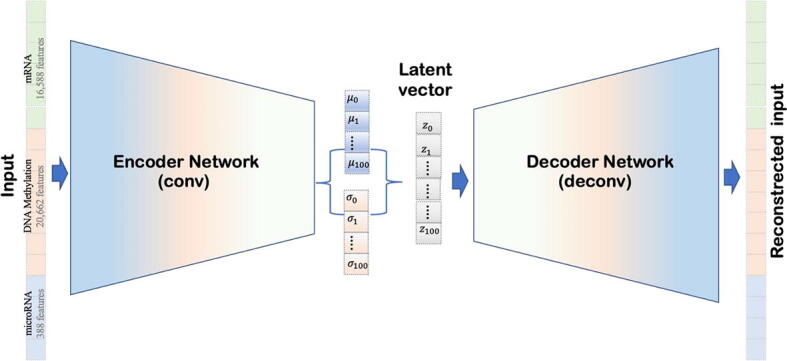
An overview of the proposed convolutional variational autoencoder (CVAE) model. This architecture includes two parts, an encoder and a decoder that are two symmetrical and reversed structures. Each one is composed of two convolutional layers and one dense layer. The latent 100-dimensional vector is the sampling layer (Z) generated using the mean and the standard deviation layers (i.e., μ and σ). We trained this architecture to take the input data, a stack of three integrated omics data (mRNA, microRNA, and DNA methylation), and automatically learn the latent vector’s distinguishing features.
Table1.
The main layers in the proposed CVAE architectures.
| Main layer | Parameters |
|---|---|
| Input layer | Stack of integrated omics data |
| Convolutional layer | filters = 32, kernel_size = 3, strides = 1, padding = ‘same’, activation = ‘tanh’. |
| Convolutional layer | filters = 64, kernel_size = 3, strides = 1, padding = ‘same’, activation = ‘tanh’. |
| Dense layer | units = 256, activation = ‘tanh’, kernel_regularizer = ‘l2’, bias_regularizer = ‘l2’. |
| Mean layer | units = 100 |
| Standard deviation layer | units = 100 |
| Sampling layer (latent vector) | units = 100 |
| Dense layer | units = 256, activation = ‘tanh’, kernel_regularizer = ‘l2’, bias_regularizer = ‘l2’. |
| Deconvolutional layer | filters = 32, kernel_size = 3, strides = 1, padding = ‘same’, activation = ‘tanh’. |
| Deconvolutional layer | filters = 32, kernel_size = 3, strides = 1, padding = ‘same’, activation = ‘tanh’. |
| Output layer (reconstructed input) | Stack of reconstructed integrated omics data |
2.2.2. Deep neural network for classification
The model we are building requires the highest capacity to discriminate between metastasis and primary cases. As a consequence, even a slight gain in accuracy is essential. For this reason, we choose deep neural network (DNN), the common approach used to solve similar problems, which is capable of pattern recognition in complex data [34] to build the classifier.
Thus, after completing the CVAE training process, the encoder part is fed to a DNN classifier, as illustrated in Fig. 2. We used the latent vector as input to train the classifier to predict whether the input data is from a metastasized (M) tumor or a primary tumor (P). The encoder parameters obtained by the previous step are frozen during the classifier training step (Fig. 2). We implemented a DNN using the Python Keras library (https://github.com/fchollet/keras). We employed the SGD algorithm with the default parameters as the optimizer and used cross-entropy to compute the loss between actual and predicted labels. The number of epochs was set to 100, and the batch size to 8. We used the early stopping technique [33] and the dropout technique [35] with a drop rate of 0.3 to avoid overfitting. Table 2 provides the details of the DNN model parameters.
Fig. 2.

The latent vector used as input to train the DNN to classify metastasized (M) or primary tumor (P) samples correctly.
Table 2.
Main layers in the proposed DNN architectures.
| Main layer | Parameters |
|---|---|
| Input layer | 100 D-vector (latent vector) |
| Dense layer | units = 60, activation = ‘tanh’, kernel_regularizer = ‘l2’, bias_regularizer = ‘l2’. |
| Dense layer | units = 30, activation = ‘tanh’, kernel_regularizer = ‘l2’, bias_regularizer = ‘l2’. |
| Output layer | units = 1, activation = ‘sigmoid’. |
2.2.3. Data partitioning and robustness assessment
To assess the model’s robustness, we used a cross-validation (CV)–like procedure to partition the TCGA dataset. We split the TCGA data 80%, 20% for training and test sets, respectively, to have sufficient test samples to generate evaluation metrics. That is, we randomly split the 420 TCGA samples into five folds in the stratified mode where each fold should have the same percentage of positive and negative samples, then used one-fold as the test set and the remaining four folds as the training set. For each split, we constructed a model that extracts features (latent vector) from 80% of samples (training set) using CVAE, and then we used DNN to learn the predictions. Then, we tested the model to predict the labels of the 20% of samples held out (test set). Note we used each of the 5-folds as test sets in 5 different models and extracted the latent vector during the building of each model, and the final metrics were averaged over the folds.
2.2.4. Alternative feature extraction processes
We compared the DL framework's performance with two commonly used alternatives, i.e., protein–protein interaction (PPI) network construction (network-based) and recursive feature elimination (rank-based), in metastasis-related work.
For the network-based approach, we downloaded the PPI information from Database of Protein, Chemical, and Genetic Interactions (BioGRID) [36], Human Protein Reference Database (HPRD) [37], and Database of Interacting Proteins (DIP) [38]. We merged this information to construct a huge network using Cytoscape [39] and then mapped the DEGs onto the network to obtain a smaller, focused DEGs’ PPI network [40]. We calculated betweenness centrality (BC) for each node to reflect the node’s hubness in the PPI network. BC score ranges between 0 and 1, with the larger indicating a higher degree of hubness. Thus, essential proteins involved in the densely interconnected biological processes are more likely to be represented by hub nodes. The top 100 BCE nodes (genes) are selected to be fed to the classifier.
In the rank-based approach, the goal is to select the top genes that are more relevant in predicting the metastatic status (P or M) by recursively considering smaller and smaller sets of genes. Recursive feature elimination (RFE) is an efficient approach for eliminating genes and for feature selection. The process starts by training an estimator on all genes’ in the initial set and then ranks the genes from the most important to the least. Then, the least important genes are discarded, and the model is re-trained on the remaining set of genes. This process is recursive until a specified number of top genes remains. The top 100 genes are then fed to a classifier to predict whether the input data indicate a metastasized (M) or primary tumor (P). The estimator used in this work is SVM and applied using Scikit-learn [41].
3. Results and discussion
3.1. Comparing feature extraction methods
Several factors affect a model’s prediction performance, such as the effectiveness of the feature extraction method implemented. Thus, we quantitatively assessed the proposed DL approach’s performance when using CVAE preprocessing and alternative feature extraction preprocessing methods. We computed the statistical measures such as accuracy, sensitivity (a.k.a. recall), specificity, precision, and F1-score. These metrics were calculated based on the prediction of positive (Metastasized) or negative (Primary) samples. All reported results are the average performance obtained from 5 models on the hold-out test sets.
As the two described alternative feature extraction approaches were proposed based on using only mRNA data, we similarly trained and tested our DL approach using mRNA only to make a fair comparison. In Table 3, we provide the averaged results with the standard deviation used in the comparison. The results show superior performance for CVAE preprocessing across all the metrics. The model’s performance using the rank-based feature extraction method is lower than that achieved by the network-based feature extraction method in all metrics except sensitivity. However, the rank-based method identifies the metastasized samples better than the network-based method, while the latter better identifies primary samples.
Table 3.
Comparing performance using alternative feature extraction processes (using mRNA data only).
| Approach (mRNA only) | Accuracy | Precision | Sensitivity (Recall) | F1 score | Specificity |
|---|---|---|---|---|---|
| Rank-based | 74.16 (±2.23) | 67.33 (±3.61) | 80.15 (±2.13) | 75.92 (±2.18) | 67.66 (±2.09) |
| Network-based | 78.46 (±2.48) | 77.50 (±1.71) | 76.66 (±2.24) | 79.65 (±1.99) | 80.90 (±2.89) |
| CVAE-based | 83.83 (±0.44) | 85.98 (±0.54) | 81.33 (±0.16) | 83.59 (±0.60) | 86.33 (±0.35) |
Since the features produced by the CVAE-based method better distinguish the primary tumors from the metastatic ones, compared to the commonly used feature extraction methods, it would be interesting to know which features the CVAE-based method is extracting or omitting that benefit this classification process. However, the CVAE-based method’s power comes with a price such as the lack of interpretable and exploitable features in the latent vector [42], which prevents us from seeing these results. Therefore, efforts are ongoing to make DL models more interpretable, more accessible, and more useful to biologists [42].
3.2. Evaluating the contribution of different omics data types
After assessing alternative feature selection methods’ performance, we sought to investigate how using multiple omics data layers improves performance. The results of this analysis are shown in Fig. 3, which reports the performance scores obtained using MetaCancer with the mRNA data layer only or using the full (mRNA, microRNA, and DNA methylation) dataset. The results show that MetaCancer achieves superior performance when integrating multiple omics data layers compared to using mRNA data.
Fig. 3.

MetaCancer’s performance when using the integrated omics (mRNA, microRNA, DNA methylation) dataset or mRNA data alone.
We then sought to analyze the contribution of each data layer to the observed performance. Fig. 4 illustrates the area under the curve (AUC) evaluation metric when using each or a combination of omics types. When using a single-omics type, mRNA performed the best with AUC = 88.28, and microRNA had the lowest performance with AUC = 76.93. The DNA methylation ranked second with AUC = 86.17. However, using integrated multi-omics data improved the performance and achieved AUC = 91.19 (see Fig. 4A). We also assessed MetaCancer’s performance when using different combinations of omics data. In these cases, when excluding mRNA, we observed the largest decrease in AUC from 91.19 to 78.23, while excluding microRNA produced the smallest decline in the AUC. These results suggest that mRNA data, followed by DNA methylation data, serve better as features to discriminate metastatic status, while microRNA makes the smallest contribution. Nonetheless, integrating all 3-omics types improves the models' ability to distinguish primary tumors and metastatic ones (see Fig. 4B).
Fig. 4.

MetaCancer’s performance. A) Using single-omics vs integrated omics. B) Excluding one omics vs integrated omics.
The finding that the mRNA data layer provides the most significant contribution is consistent with results from a recent cross-cancer (11 cancer types) study performed by Lee and colleagues [43]. They used mRNA and microRNA expression data independently to differentiate primary tumor samples from the metastatic ones. They used the student’s t-test to identify the top 64, 128, and 256 features, which they used to train various models (LASSO, RF, and SVM) evaluated across 100 Monte Carlo cross-validation (MCCV). The RF model with 256 features achieved the highest AUC when predicting metastatic using only mRNA or only microRNA expression data. However, using the mRNA data achieved an AUC of 0.74, while the microRNA achieved a significantly lower AUC of 0.64. They did not report how the ensemble model (using both the mRNA and microRNA expression data as features) would affect the AUC. Nonetheless, as mentioned before, the work reported by Bhalla and colleagues [23] during the same timeframe does report AUC for an ensemble model and the AUC for models based on single omics types. They also showed that using mRNA data only achieved a higher AUC than when using the microRNA and methylation data individually. The surprise was that the ensemble method achieved an AUC slightly lower than when using mRNA data only.
Overall, the approaches that evaluated performance using different omics types are few. Still, they consistently showed that mRNA data-related features make a more significant contribution when distinguishing primary tumors from metastatic ones computationally.
3.3. Comparing our MetaCancer model with an alternative ensemble model
After comparing our model to other models based on mRNA only, we compared our model's performance to a model designed to exploit multiple data layers. In particular, we compared our proposed MetaCancer model’s performance to the ensemble model proposed by Bhalla and colleagues [23]. First, we constructed a model with the same architecture introduced by Bhalla and colleagues [23]. Then we trained and evaluated the model (using the same cross-validation technique used to evaluate our model) with our data.
They trained three independent models in the ensemble model, one for each omics data (mRNA, microRNA, and DNA methylation). They achieved the best performance using SVM with L1 regularization (SVC-L1) [44] as the feature selection method and SVM as the classification method for both mRNA and microRNA. However, for the DNA methylation data, they achieved the best performance using WEKA-FCBF [45] as a feature selection method and logistic regression (LR) as a classification model. After training a model for each omics data type, i.e., mRNA, microRNA, and DNA methylation, the prediction scores were provided as input features to the SVM to give the final prediction. Table 4 presents the results achieved when applying this approach to our data and the results obtained by our model. When comparing the results in Table 3, Table 4, the results suggest that the models using the multiple omics data types outperformed the models that use mRNA only. Moreover, MetaCancer model significantly outperformed the ensemble method based on all five metrics. Here, it is interesting to note that the original model introduced by Bhalla and colleagues [23] reported achieving 87.64% accuracy applied to only one type of cancer. In contrast, our model applied to 11 cancer types achieves a higher accuracy of 88.85%.
Table 4.
Comparing the performance of approaches when integrating omics data (mRNA, microRNA, and DNA methylation).
| Approach (Integrated Omics) | Accuracy | Precision | Sensitivity (Recall) | F1 score | Specificity |
|---|---|---|---|---|---|
| SVM Ensemble | 82.50 (±1.39) | 80.95 (±01.19) | 85.01 (±2.44) | 82.92 (±1.02) | 81.10 (±1.69) |
| CVAE-based (MetaCancer) | 88.85 (±0.74) | 91.65 (±0.86) | 87.69 (±0.22) | 90.44 (±1.23) | 89.61 (±1.17) |
These results show that CVAE possesses distinct advantages over alternative feature extraction methods when exploring the complex nature of the genomic and epigenomic data. CVAE is an entirely data-driven unsupervised approach and does not rely on existing genomic annotations to learn representations; thus, it can identify patterns in the data and extract meaningful knowledge while overcoming data complexities. For most methods, the reliance on existing annotations limits our ability to uncover novel biology as it directs towards the well-known biology with the most molecular data [46]. However, as a data-driven model, CVAE repeatedly adjusts a weighted combination of input features until the model identifies the best possible reconstruction of the input data. This can be thought of as a high dimensional interaction space in which the latent dimensions capture multi-omics features that are similarly associated with the metastatic status. Also, because the CVAE is data-driven and unsupervised, the training process condenses all information about each metastatic sample into a lower-dimensional space from which the inference of novel information is possible.
3.4. Limitations and concluding remarks
Although many studies focused on metastatic status prediction, most of these studies did not consider changes at various genomic layers contributing to the metastasis etiology. In this work, however, we consider the contribution made by mRNA, microRNA, and DNA methylation to the metastasis etiology using the associated omics data to develop a DL model that predicts if a sample has already entered the metastasis phase or not. If developed into a tool, this model's purpose is to help physicians identify metastasis earlier so that treatment regimens can be amended to treat the metastasized cancer as well [47], [48].
While CVAEs can generate useful representations for vast amounts of complex heterogeneous data, in terms of interpretability, the learned representations' biological relevance has to be verified if they are used in clinical decision support systems. Interpretation of these latent vectors representing the features has, however, received little attention. In the near future, we aim to develop a pan-cancer metastasis model using interpretable techniques to unveil the pan-cancer metastasis-related omics signatures. Interpreting these features would reveal more of the core metastasis-related features, providing a better understanding of the biology underpinning metastasis etiology.
Also, the 400 samples used to train and evaluate the model is reasonable, but may not be sufficient to extract all the features that distinguish between samples and enhance the performance. Moreover, we could better show the proposed model’s robustness if we find external test data with all three omics data, which is not currently publicly available to the best of our knowledge.
In the future, we plan to predict the site (or organ) where the cancer would most likely metastasize and collaborate with clinicians to develop case studies that will test and improve the model over time.
Author contributions
S.A., F.N., M.E. and X.G.: Conceptualization; S.A.: Methodology; S.A., F.N., M.T., and M.E.: Formal Analysis; S.A., F.N., M.T., T.G., M.E., and X.G.: Writing - original draft; S.A., T.G., M.E., and X.G.: Writing - review and editing. All authors read and approved the final manuscript.
5. Availability
Code and data can be found at https://github.com/SomayahAlbaradei/MetaCancer.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The research reported in this publication was supported by King Abdullah University of Science and Technology (KAUST) through grant awards Nos. BAS/1/1059-01-01, BAS/1/1624-01-01, FCC/1/1976-20-01, FCC/1/1976-26-01, URF/1/3450-01-01, and URF/1/4098-01-01.
Contributor Information
Magbubah Essack, Email: magbubah.essack@kaust.edu.sa.
Xin Gao, Email: xin.gao@kaust.edu.sa.
References
- 1.Robinson D.R., Wu Y.-M., Lonigro R.J., Vats P., Cobain E., Everett J. Integrative clinical genomics of metastatic cancer. Nature. 2017;548:297–303. doi: 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sinica B. 2015;5:402–418. doi: 10.1016/j.apsb:2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramaswamy S., Ross K.N., Lander E.S., Golub T.R. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 4.Li C., Sun Y.-D., Yu G.-Y., Cui J.-R., Lou Z., Zhang H. Integrated omics of metastatic colorectal cancer. Cancer Cell. 2020;38:734–747.e9. doi: 10.1016/j.ccell.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.v68.610.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 6.He Y., Ma J., Ye X. A support vector machine classifier for the prediction of osteosarcoma metastasis with high accuracy. Int J Mol Med. 2017;40:1357–1364. doi: 10.3892/ijmm.2017.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metri R., Mohan A., Nsengimana J., Pozniak J., Molina-Paris C., Newton-Bishop J. Identification of a gene signature for discriminating metastatic from primary melanoma using a molecular interaction network approach. Sci Rep. 2017;7 doi: 10.1038/s41598-017-17330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei D. A multigene support vector machine predictor for metastasis of cutaneous melanoma. Mol Med Rep. 2018;17:2907–2914. doi: 10.3892/mmr.2017.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuo Y., An N., Zhang M. Feature genes in metastatic breast cancer identified by MetaDE and SVM classifier methods. Mol Med Rep. 2018;17:4281–4290. doi: 10.3892/mmr.2018.8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chereda H., Bleckmann A., Kramer F., Leha A., Beissbarth T. Utilizing molecular network information via graph convolutional neural networks to predict metastatic event in breast cancer. Stud Health Technol Inform. 2019;267:181–186. doi: 10.3233/SHTI190824. [DOI] [PubMed] [Google Scholar]
- 11.Wu J., Xiao Y., Xia C., Yang F., Li H., Shao Z. Identification of biomarkers for predicting lymph node metastasis of stomach cancer using clinical DNA methylation data. Dis Markers. 2017;2017:1–7. doi: 10.1155/2017/5745724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahsen M.E., Boren T.P., Singh N.K., Misganaw B., Mutch D.G., Moore K.N. Sparse feature selection for classification and prediction of metastasis in endometrial cancer. BMC Genomics. 2017;18 doi: 10.1186/s12864-017-3604-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao S., Yu J., Wang L. Machine learning based prediction of brain metastasis of patients with IIIA-N2 lung adenocarcinoma by a three-miRNA signature. Transl Oncol. 2018;11:157–167. doi: 10.1016/j.tranon.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karabulut EM, Ibrikci T. Discriminative deep belief networks for microarray based cancer classification. (2017).
- 15.Albaradei S, Thafar M, Van Neste C, Essack M, Bajic VB, in Proceedings of the 2019 6th International Conference on Bioinformatics Research and Applications 125-130 (Association for Computing Machinery, 2019).
- 16.Hernández-Lemus E., Reyes-Gopar H., Espinal-Enríquez J., Ochoa S. The many faces of gene regulation in cancer: a computational oncogenomics outlook. Genes. 2019;10:865. doi: 10.3390/genes10110865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Anda-Jáuregui G., Hernández-Lemus E. Computational oncology in the multi-omics era: state of the art. Front Oncol. 2020;10:423. doi: 10.3389/fonc.2020.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behring M., Shrestha S., Manne U., Cui X., Gonzalez-Reymundez A., Grueneberg A. Integrated landscape of copy number variation and RNA expression associated with nodal metastasis in invasive ductal breast carcinoma. Oncotarget. 2018;9:36836–36848. doi: 10.18632/oncotarget.v9i9610.18632/oncotarget.26386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernal Rubio YL, et al. Whole-genome multi-omic study of survival in patients with glioblastoma multiforme. G3 2018:8;3627–3636, doi:10.1534/g3.118.200391. [DOI] [PMC free article] [PubMed]
- 20.González-Reymúndez A., Vázquez A.I. Multi-omic signatures identify pan-cancer classes of tumors beyond tissue of origin. Sci Rep. 2020;10:8341. doi: 10.1038/s41598-020-65119-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steeg P.S. Targeting metastasis. Nat Rev Cancer. 2016;16:201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang S., Chaudhary K., Garmire L.X. More is better: recent progress in multi-omics data integration methods. Front Genet. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhalla S., Kaur H., Dhall A., Raghava G.P.S. Prediction and analysis of skin cancer progression using genomics profiles of patients. Sci Rep. 2019;9:15790. doi: 10.1038/s41598-019-52134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gress DM et al. Principles of cancer staging. AJCC Cancer Staging Manual 8, 3-30 (2017).
- 25.Colaprico A et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71, doi:10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed]
- 26.Mounir M. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15 doi: 10.1371/journal.pcbi.1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaudhary K., Poirion O.B., Lu L., Garmire L.X. Deep learning-based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res. 2018;24:1248–1259. doi: 10.1158/1078-0432.CCR-17-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hastie, T., Tibshirani, R., Narasimhan, B. & Chu, G. Impute: impute: Imputation for microarray data. R package version 1.54. 0. (2018).
- 29.Bengio, Y. Learning Deep Architectures for AI. doi:10.1561/9781601982957 (2009).
- 30.Kingma DP, Welling M Auto-encoding variational bayes. arXiv [stat.ML] (2013).
- 31.Blei D.M., Kucukelbir A., McAuliffe J.D. Variational inference: A review for statisticians. J Am Stat Assoc. 2017;112:859–877. [Google Scholar]
- 32.Chollet F, et al., Keras: The Python Deep Learning library. Astrophysics Source Code Library, ascl:1806.1022 (2018).
- 33.Prechelt L. Early stopping - but when? Lect Notes Comput Sci. 1998;55–69 doi: 10.1007/3-540-49430-8_3. [DOI] [Google Scholar]
- 34.Mahmud M., Kaiser M.S., McGinnity T.M., Hussain A. Deep learning in mining biological data. Cogn Comput. 2021;13:1–33. doi: 10.1007/s12559-020-09773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava N., Hinton G., Krizhevsky A., Sutskever I., Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15:1929–1958. [Google Scholar]
- 36.Oughtred R, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res, 2019;47:D529-D541, doi:10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed]
- 37.Prasad TSK, Keshava Prasad TS, Kandasamy K, Pandey A. Human protein reference database and human proteinpedia as discovery tools for systems biology. Methods Mol Biol, 2009;67-79, doi:10.1007/978-1-60761-232-2_6. [DOI] [PubMed]
- 38.Xenarios I. DIP: The Database of Interacting Proteins: 2001 update. Nucleic Acids Res. 2001;29:239–241. doi: 10.1093/nar/29.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohl M., Wiese S., Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 40.Smoot M.E., Ono K., Ruscheinski J., Wang P.-L., Ideker T. Cytoscape 2.8. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedregosa F. Scikit-learn: Machine learning in Python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 42.Ching T., Himmelstein D.S., Beaulieu-Jones B.K., Kalinin A.A., Do B.T., Way G.P. Opportunities and obstacles for deep learning in biology and medicine. J R Soc Interface. 2018;15:20170387. doi: 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S.C., Quinn A., Nguyen T., Venkatesh S., Quinn T.P. A cross-cancer metastasis signature in the microRNA-mRNA axis of paired tissue samples. Mol Biol Rep. 2019;46:5919–5930. doi: 10.1007/s11033-019-05025-w. [DOI] [PubMed] [Google Scholar]
- 44.Jitkrittum W, Hachiya H, Sugiyama M. Feature selection via l1-penalized squared-loss mutual information. IEICE Trans Inf Syst, 2013;E96.D:1513–1524, doi:10.1587/transinf.e96.d.1513.
- 45.Yu L., Liu H. Proceedings of the 20th international conference. 2003. Feature selection for high-dimensional data: A fast correlation-based filter solution. [Google Scholar]
- 46.Haynes W.A., Tomczak A., Khatri P. Gene annotation bias impedes biomedical research. Sci Rep. 2018;8:1362. doi: 10.1038/s41598-018-19333-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cruz JA, Wishart, DS, Applications of machine learning in cancer prediction and prognosis. Cancer Inf, 2006;2, 117693510600200, doi:10.1177/117693510600200030. [PMC free article] [PubMed]
- 48.Kourou K., Exarchos T.P., Exarchos K.P., Karamouzis M.V., Fotiadis D.I. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2015;13:8–17. doi: 10.1016/j.csbj.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]