

Summary
Using existing knowledge to carry out drug-disease associations prediction is a vital method for drug repositioning. However, effectively fusing the biomedical text and biological network information is one of the great challenges for most current drug repositioning methods. In this study, we propose a drug repositioning method based on heterogeneous networks and text mining (HeTDR). This model can combine drug features from multiple drug-related networks, disease features from biomedical corpora with the known drug-disease associations network to predict the correlation scores between drug and disease. Experiments demonstrate that HeTDR has excellent performance that is superior to that of state-of-the-art models. We present the top 10 novel HeTDR-predicted approved drugs for five diseases and prove our model is capable of discovering potential candidate drugs for disease indications.
Keywords: drug repositioning, heterogeneous networks, text mining, drug-disease associations, feature representation
Graphical abstract
Highlights
-
•
We developed a novel DL-based method for drug repositioning (HeTDR)
-
•
HeTDR succeeds in fusing networks topology information and text mining information
-
•
HeTDR obtains high accuracy, excessing most state-of-the-art models
-
•
HeTDR could represent an algorithm integrating multiple sources of information
The bigger picture
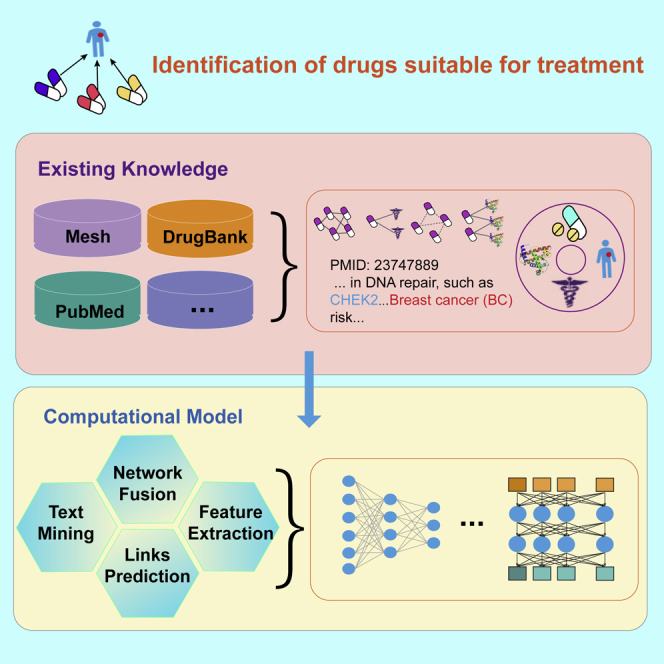
Traditional drug discovery and development are often time consuming and high risk. Drug repositioning aims to expand existing indications or discover new targets by studying the approved drug compounds, thereby reducing the time, costs, and risk of drug development. We propose a novel method in drug repositioning based on heterogeneous networks and text mining (HeTDR), which combines drugs features from multiple networks and diseases features from biomedical corpora to predict the correlation scores between drugs and diseases. This prediction model has provided a potential solution for multiple information fusion and to exhibit accurate performance leading to the discovery of new drugs for indications. This algorithm could contribute a new idea to the acceleration and development of future drug repositioning by using computational methods and provide computer-aided guidance for biologists in clinical settings.
Drug repositioning is a useful way to discover new drug candidates for curing diseases. However, integrating multiple networks and text mining information for drug repositioning is still a major challenge. We propose a drug repositioning method based on heterogeneous networks and text mining (HeTDR), which can combine drug features from multiple networks and disease features from biomedical corpora for drug repositioning. HeTDR obtains high accuracy in predicting drug-disease interactions and is capable of finding novel indications of approved drugs.
Introduction
The time for new drug development has been gradually increasing, and the cost it takes to bring a new drug to market is becoming more expensive up to 2.6 billion dollars.1,2 In recent years, an increasing number of studies have shown that some approved drugs can be used to treat new indications, this process is referred to as drug repositioning.3 Drug repositioning aims to expand the existing indications or discover new targets by studying the approved drug compounds, thereby reducing the time, costs, and risks in drug development.4 In the beginning, most of the repositioned drugs were accidentally discovered in clinical settings. With the development of high-throughput technology, the increase in large-scale genomics and pharmacological and chemical datasets has made it possible to predict the relationship between drug with disease by using systematic and reasonable calculation methods.
Drug repositioning is a long-standing problem and many calculation methods have been proposed to predict drug-disease associations for drug repositioning. These studies could be roughly classified into three groups: machine learning, network-based methods, and literature mining. Most of the machine learning methods formulated drug repositioning as a classification task, and some machine learning classification algorithms are widely used, such as support vector machines (SVMs),5, 6, 7 logistic regression,7,8 and random forest,7 to identify the potential indications for approved drugs. However, those supervised classification methods will randomly generate negative samples, which could lead to biased decisions. The network-based methods are the most widely used methods for drug repositioning. Its mainstream algorithms include: network inference,9, 10, 11, 12 random walk,13,14 and matrix factorization.15,16 These methods usually rely heavily on the richness of interaction network data. In addition, many studies that used such methods proposed the need to construct more complete and large-scale data information to improve prediction performance. The performance of literature mining-based methods relies on biomedical entities' co-occurrence and semantic inference of some keywords of interest. These methods will be limited by the ambiguity of natural language and the limitations of text-mining technology.17, 18, 19, 20 Recently, deep learning has achieved tremendous development, and the models of network representation21,22 and text mining23 have been continuously improved. However, comprehensively considering topological properties and statistical correlation to discover potential drug-disease relationships remains a challenge.
In this study, we develop a novel method, called HeTDR, that allows the incorporation of topological structure information from the heterogeneous networks and the features information from biomedical text mining. In our model, the drug features extraction module is based on similarity network fusion (SNF)24 and sparse autoencode (SAE),25,26 and the disease features extraction module is based on Bidirectional Encoder Representations from Transformers for Biomedical Text Mining (BioBERT).27 Some previous studies proved that combining the drug-disease associations network with feature information could make the prediction model obtain more robust results than using the feature information alone.10,28 Accordingly, we adopt a model that can combine the attribute information with the topological structure information for final prediction. The contributions of HeTDR can be enumerated as follows: (1) HeTDR integrates nine drug-related networks into a low-dimensional and compact feature representation common to all networks, which can better capture the overall information of the drug. (2) HeTDR obtains the information of disease from biomedical corpora, thereby improving the accuracy of drug-disease associations prediction. (3) HeTDR makes full use of both topology and attribute information to overcome the influence of network data often being partially observed. (4) HeTDR combines a network-based method and a text mining-based method to provide a new solution for drug repositioning. In the computational experiments, HeTDR obtains high accuracy in predicting drug-disease interactions, and significantly outperforms existing state-of-the-art methods. Moreover, case studies show that HeTDR can help discover novel associations not included in known drug-disease pairs and find novel indications of approved drugs.
Results
The HeTDR pipeline
The workflow of HeTDR is shown in Figure 1, and consists of three parts: (1) HeTDR uses SNF to integrate nine drug-related networks into one network with global information and then utilizes the SAE to obtain high-quality features representation of the drug. (2) HeTDR uses the BioBERT model to obtain disease features information from biomedical corpora. Specifically, we use the pre-trained parameters of the BioBERT, and select the relation extraction task for fine-tuning training. After the fine-tuning process, we extract the representation of sub-words and obtain the representations of all diseases by frequent sub-words. (3) HeTDR combines the drug-disease associations network with the features information of the drug and disease to infer the potential associations between drug and disease.
Figure 1.

Flowchart of HeTDR
The model consists of three parts: (A) HeTDR integrates nine drug-related networks to obtain global information of drugs. In the heterogeneous interaction networks, we first use the Jaccard similarity coefficient to calculate the similarity network. Then, we fuse these drug-related networks into one network by SNF and apply SAE to obtain low-dimensional features of the drugs. (B) HeTDR obtains vector representation of the disease features by text mining biomedical corpora. In the pre-training stage, we directly use the model parameters pre-trained by BioBERT. Then, we select the relation extraction task for fine-tuning training. After the fine-tuning process has taken place, we extract the representations of sub-words and use the representations of these sub-words to obtain the representations of all diseases. (C) HeTDR predicts potential drug-disease associations by an embedding learning method, which can capture both the drug-disease associations network topological structural proximity and node attributes proximity.
Evaluation of prediction performance by ablation analysis
We partition 6,677 reported drug-disease pairs into three subsets, 80% of the known drug-disease pairs for training, 10% for validation, and 10% for testing. For the validation set and test set, we randomly generate negative samples with a 1:1 ratio matched with the positive samples. To evaluate the performance of HeTDR, we use the following performance metrics: area under the receiver operating characteristic curve (AUROC), the area under the precision-recall curve (AUPR), and F1-measure (F1).
HeTDR showed a high performance (AUROC = 0.959, AUPR = 0.955, and F1 = 0.901) (Figure 2). We conduct ablation studies on HeTDR to explore the effect of drug features and disease features. We run experiments with the same data splits, model parameters, and evaluation protocol to keep the comparison as fair as possible. The result is presented in Figure 2. We implement three simplified variants of HeTDR.
-
(1)
HeTDR_Di: by removing the drug features.
-
(2)
HeTDR_Dr: by removing the disease features.
-
(3)
HeTDR_SNF: by removing the SAE.
Figure 2.

Performance of HeTDR comparing the different features
(A) ROC curves of prediction results by using different features.
(B) PR curves of prediction results by using different attributes.
(C) F1 scores of prediction results by using different features.
Minimal difference is found in the results obtained by using drug features (AUROC = 0.916, AUPR = 0.915, and F1 = 0.830) or disease features (AUROC = 0.921, AUPR = 0.919, and F1 = 0.838) alone, but combining these features information on disease and drug has the highest accuracy of drug-disease associations prediction (AUROC = 0.959, AUPR = 0.955, and F1 = 0.901). The result of using SAE when obtaining drug features is better than the output when SNF is used to obtain the drug features (AUROC = 0.934, AUPR = 0.928, and F1 = 0.862), which illustrates the importance of filtering information when fusing multiple types of networks data. HeTDR makes full use of multiple drug-related information and disease-related information, which accounts for its superior performance over other methods.
Evaluation of prediction performance on cross-validation
To evaluate HeTDR more comprehensively, we conduct 5-fold cross-validation and compare HeTDR with classic methods. We compare our results with seven methods: SVM,29 Katz,30 MBiRW,14 DTINet,31 DRRS,32 DeepDR,10 and HNet-DNN.12 SVM is the traditional machine learning algorithm; Katz is the path-based classic algorithm calculating similarities between nodes in a network for associations prediction; MBiRW utilizes some comprehensive similarity measures and the BiRandom Walk (BiRW) algorithm for drug repositioning; DTINet is a matrix factorization-based model and it can integrate diverse information from heterogeneous networks; DRRS is a computational drug repositioning method using low-rank matrix appropriation and randomized algorithm; DeepDR is a network-based deep learning method to infer potential novel drug-disease associations; HNet-DNN uses the deep neural network (DNN) to predict new drug-disease interactions. We use AUROC and AUPR to evaluate the performance of these methods. In particular, each method is configured to its default setting or best parameter values reported in its paper. In the 5-fold cross-validation results, HeTDR obtains high performance (AUROC = 0.979 and AUPR = 0.976), outperforming other state-of-the-art methods: SVM (AUROC = 0.602 and AUPR = 0.580), Katz (AUROC = 0.701 and AUPR = 0.722), MBiRW (AUROC = 0.812 and AUPR = 0.810), DTINet (AUROC = 0.870 and AUPR = 0.884), DRRS (AUROC = 0.912 and AUPR = 0.877), DeepDR (AUROC = 0.915 and AUPR = 0.927), and HNet-DNN (AUROC = 0.951 and AUPR = 0.908) (Figure 3).
Figure 3.

HeTDR outperforms other state-of-the-art methods for drug-disease associations prediction
(A) ROC curves of prediction results obtained by applying HeTDR and five previously reported methods in 5-fold cross-validation.
(B) PR curves of prediction results obtained by HeTDR and five previously reported methods in 5-fold cross-validation.
HeTDR identifies novel drug-disease associations
We save the results of HeTDR predictions of drug-disease associations in “Evaluate prediction performance by ablation analysis.” We delete 6,677 known drug-disease associations used in the prediction model, and select the novel top 150 pairs with highest similarity drug-disease pairs predicted by HeTDR, and visualize the interactions network (Figure 4). Among the list of top 150 predictions, HeTDR can capture the experimental or clinical reported drug-disease associations. For example, novel predictions show that DB08827 (lomitapide) and DB05528 (mipomersen) can act on C0342882 (familial hypercholesterolemia-heterozygous). The novel prediction was confirmed by previous studies. Lomitapide is an inhibitor of a microsomal triglyceride transfer protein that lowers hepatic low-density lipoprotein cholesterol production, as new therapeutic modalities treating homozygous familial hypercholesterolemia.33,34 Mipomersen decreases the levels of apolipoprotein B, low-density lipoprotein non-high-density lipoprotein cholesterol, and total cholesterol, which are used in patients with homozygous familial hypercholesterolemia as an adjunct to diet and other lipid-lowering medications.34,35 For C0026705 (mucopolysaccharidosis II), the significant association with DB00090 (laronidase) is successfully identified by HeTDR.
Figure 4.

Network visualization of the drug-disease associations predicted by HeTDR
In this network, the predicted novel top 150 drug-disease pairs network is visualized. The label of the node represents the ID of the drugs (Drugbank_ID) or diseases (UMLS_ID). The node size denotes the degree. The weight of edges (drug-disease pairs) denotes the predicted score by HeTDR. The novel top 150 pairs of the highest similarity drug-disease associations can be found in Table S1. This image was generated by Gephi (https://gephi.org).
To interpret the results of the HeTDR model, we analyze the novel associations predicted in the example. We use the features learned by the HeTDR model to calculate the drug-drug and disease-disease similarity relationships in the association prediction module at the model. We select the top 20 diseases predicted to be most related to C0342882 and C0026705 (see Table S1), and use known drug-disease associations to find approved drugs that can be used to treat these diseases (Figure 5). In the model, C0342881 (familial hypercholesterolemia-homozygous) is most related to C0342882, and DB05528 and DB08827 are drugs that have been approved to act on C0342881. The disease most related to C0026705 captured by our model is C0023786 (mucopolysaccharidosis I), and DB00090 is an approved drug that can be used to treat C0023786. Accordingly, DB00090 has a high potential to cure C0026705. These results demonstrate that our model can make full use of features information to capture approximate relationships of drug-drug and disease-disease, and reveal potential drug-disease associations based on these approximate relationships and the known associations. The novel top 150 pairs of the highest similarity drug-disease associations can be found in Table S2.
Figure 5.

The interpretability of HeTDR Identifies novel associations
(A) The upper nodes are the top 20 most relevant diseases of C0342882.
(B) The upper nodes are the top 20 most relevant diseases of C0026705. The edges are the known drug-disease associations, and the heavier color of the edge represents edge linking disease rank higher in the top 20. This image was generated by Gephi (https://gephi.org).
Case studies
To further demonstrate the ability of HeTDR for discovering novel drug-disease associations, we choose five diseases for case studies, namely, Alzheimer's disease, obesity, asthma, epilepsy, and Parkinson’s disease. Similarly, we delete known drug-disease associations used in the prediction model, and select the top 10 drugs according to the predicted highest association scores, which are considered drug candidates for the disease, as shown in Table 1. To provide a more reliable reference for follow-up researchers, we also provide the associated prediction scores for the top 20 drugs of all diseases, and the number of side effects associated with these drugs (see Table S3). All the side effects and the drug-side effect interactions can be found in the data and code availability.
Table 1.
The top 10 related candidate drugs for Alzheimer's disease, obesity, asthma, epilepsy, and Parkinson's disease
| Disease name | Rank | Drug name | Description | Rank | Drug name | Description |
|---|---|---|---|---|---|---|
| Alzheimer's disease | 1 | corticotropin | CTD_I | 6 | ergoloid | 36, 37, 38, 39, 40 |
| 2 | natalizumab | N/A | 7 | fludrocortisone | CTD_I | |
| 3 | dexamethasone | CTD_I | 8 | prednisone | CTD_I | |
| 4 | teriflunomide | N/A | 9 | budesonide | CTD_I | |
| 5 | canakinumab | 41, 42, 43, 44, 45 | 10 | dalfampridine | CTD_I | |
| Obesity | 1 | lisdexamfetamine | CTD_I | 6 | nicotine | CTD_M |
| 2 | methylphenidate | CTD_I | 7 | guanfacine | CTD_I | |
| 3 | dextroamphetamine | CTD_I | 8 | naloxegol | N/A | |
| 4 | atomoxetine | 46, 47, 48, 49, 50, 51 | 9 | methylnaltrexone | 52,53 | |
| 5 | dexmethylphenidate | N/A | 10 | disulfiram | CTD_I | |
| Asthma | 1 | zileuton | CTD_T | 6 | ipratropium bromide | CTD_T |
| 2 | salbutamol | CTD_T | 7 | arformoterol | CTD_T | |
| 3 | montelukast | CTD_T | 8 | fluticasone propionate | CTD_T | |
| 4 | formoterol | CTD_T | 9 | dexpanthenol | CTD_I | |
| 5 | salmeterol | CTD_T | 10 | ephedrine | CTD_T | |
| Epilepsy | 1 | lorazepam | DrugBank_T | 6 | oxcarbazepine | CTD_T |
| 2 | clonazepam | CTD_T | 7 | levetiracetam | CTD_T | |
| 3 | ethosuximide | CTD_T | 8 | tiagabine | CTD_I | |
| 4 | stiripentol | DrugBank_T | 9 | nitrazepam | CTD_T | |
| 5 | topiramate | CTD_T | 10 | ethotoin | 54,55 | |
| Parkinson's disease | 1 | pergolide | CTD_T | 6 | octreotide | CTD_I |
| 2 | metixene | DrugBank_T | 7 | cabergoline | CTD_T | |
| 3 | orphenadrine | CTD_T | 8 | gabapentin | CTD_T | |
| 4 | rivastigmine | CTD_T | 9 | lanreotide | 56 | |
| 5 | gabapentin enacarbil | 57,58 | 10 | pegvisomant | N/A |
CTD_I, inferred; CTD_M, curated (marker/mechanism); CTD_T, curated (therapeutic); DrugBank_T, therapeutic; N/A, could not be confirmed.
We first use the Comparative Toxicogenomics Database (CTD) (http://ctdbase.org/) and the DrugBank database (https://go.drugbank.com) to validate the top 10 drugs predicted by HeTDR. The CTD (http://ctdbase.org/) is a publicly available database resource providing manually curated key information about the chemical-disease, chemical-gene, and gene-disease interactions from the literature. The DrugBank database is comprehensive molecular information that integrates bioinformatics and chemoinformatics resources and provides detailed drugs data, drugs target information, drugs mechanism, and drugs-related clinical trials information. In addition, we verify whether novel associations that could not be identified in CTD and DrugBank can be supported by previously published studies (Table 1). The results demonstrate that the HeTDR is an effective method to find the potential drug-disease associations and can help develop candidate drugs for the disease.
Then, we compute the associated prediction scores by HeTDR_Di and HeTDR_Dr, and screen out the top 20 predicted drugs that may act on the five diseases. Through analysis, it is found that some of the potential drugs with higher rankings predicted by HeTDR, such as zileuton and metixene, are also captured in the top 20 drugs predicted by HeTDR_Di and HeTDR_Dr. Some drugs, such as Prednisone for the treatment of Alzheimer's disease, can only be found in the top 20 results of HeTDR_Dr, and corticotropin also used for the treatment of Alzheimer's disease, can only be found in the results of HeTDR_Di. This also shows that HeTDR combined with network-based and text-based information has complementary effects, and can better reveal potential novel associations.
Discussion
In this article, a novel HeTDR model for drug repositioning was built with both network topology attributes and text mining information. HeTDR first fused diverse information from a multitude of different drug-related networks by using SNF and obtained low-dimensional and compact drug features representation by using SAE. Then, HeTDR used the BioBERT to obtain disease features representation from biomedical corpora. Finally, HeTDR combined clinically reported drug-disease pairs with disease and drug features representations to predict potential drug-disease associations. HeTDR performed better than existing drug repositioning methods because we fused diverse information from a multitude of different drug-related network types and integrated disease-related information from biomedical text mining. The analysis and verification also showed that combining the information from the text and the network can make better predictions compared with only using the data from a single source. In addition, HeTDR could preserve the known drug-disease associations network's topological structure and node attribute proximity to predict novel drug-disease associations. Experiments have shown that the HeTDR model achieved state-of-the-art performance in drug-disease associations prediction. Case studies of five diseases further proved the effectiveness of our model in finding novel drug-disease associations, as validated by database records or literature.
We acknowledge that HeTDR still has some room for improvement. First, we directly used BioBERT's pre-training parameters, which may have certain limitations, and we expect to obtain more effective disease features information from medical records to further improve the predictive ability of our model in the future. Second, our model obtains features through different modules, and developing an end-to-end model that uses downstream tasks to obtain better features for associations prediction is possible. Despite the shortcomings, we tried to avoid the impact of these problems by verifying the effectiveness of each module in obtaining features. The constructed HeTDR model is still the most powerful model that integrates multiple types of information.
In summary, our model could be used as an effective method to predict drug-disease associations, develop a new idea for drug repositioning calculation, and provide computer-aided guidance for biologists in clinical trials.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Xiangrong Liu (xrliu@xmu.edu.cn).
Materials availability
There are no physical materials associated with this study.
Data and code availability
The code and data are available at https://github.com/stjin-XMU/HeTDR, https://doi.org/10.5281/zenodo.4915882.
Data and preparation
We use the drug-related networks constructed by previous research,10 to obtain the drug features and predict the drug-disease associations. Specifically, we use the drug-drug associations network, drug-protein associations network, drug-side effect associations network, and six drug similarity networks obtained from different omics data to obtain drug features. The known drug-disease interactions are used for the final association prediction. The type of all drug similarity networks and the numbers of all drugs, proteins, side effects, diseases, and associations are listed in Table 2. Specific construction details can be found in the supplemental experimental procedures.
Table 2.
Materials of networks
| Node types | Numbers | Edge types | Numbers/Types |
|---|---|---|---|
| Drugs | 1,519 | drug-drug | 290,836 |
| Proteins | 1,025 | drug-protein | 6,744 |
| Side effects | 12,904 | drug-side effect | 382,041 |
| Diseases | 1,229 | drug-disease | 6,677 |
| drug-drug similarities | chemical similarities, therapeutic similarities, drugs' target sequence similarities, gene ontology (GO) biological process, GO cellular component, and GO molecular function |
For the embedding features of disease obtained through text mining, we directly use the pre-trained parameters of the BioBERT model.27 The text corpora used in the training process of the model includes: (1) English Wikipedia, (2) BooksCorpus, (3) PubMed Abstracts, and (4) PMC Full-text articles.
Methods
Obtain drug features based on a heterogeneous network
To better utilize nine multiple drug-related associations networks to capture the features information of the drug, we first obtain the positive pointwise mutual information (PPMI) matrix of the drugs in each association network. Then, we use the SNF to fuse these PPMI matrices obtained from multiple networks. Finally, we use SAE to obtain a high-quality representation of the drug features (Figure 1A).
We randomly sort the vertices in a given drug-related associations network. As to the ith vertex, assume that there is a transition matrix that obtains the transition probabilities between vertices. is denoted as a row of vectors, and the j-th entry represents the probability of reaching the jth vertex after transferring steps. The is the initial one-hot vector with the ith vertex value is 1, and all other values are 0. We can learn the probability of transitions between vertices iteratively. In each iteration process, the random surfing process will continue with probability of , and there is a probability to return to the original vertex and restart this process. The recurrence relation could be described as follows:
| (Equation 1) |
By performing the above process on each node, a probabilistic co-occurrence (PCO) matrix can be obtained. After yielding the PCO matrix, we calculate a shifted PPMI matrix.59 The PPMI matrix-specific calculation method is as follows:
| (Equation 2) |
where represents the original PCO matrix, and and represent the serial numbers of rows and columns, respectively. The process occurs as a pre-processing and the random surfing-based representation is mitigating the sparsity of some individual network types.
For the PPMI matrix obtained by each network, a computational method is needed to better integrate these matrix data to establish a comprehensive view of a given drug. We denote the correlation score between drug and drug in the PPMI matrix as and use the SNF24 to obtain a comprehensive biological view of a given drug. For the fused matrix from multiple of matrices, a full and sparse kernel is defined on the vertex set V. The following formula is used to normalize the full kernel:
| (Equation 3) |
The K nearest neighbors is utilized to measure local affinity in a given network, as follows:
| (Equation 4) |
where is denoted as a set of ’s neighbors, including in network. Where the contains the complete information about the similarity of each drug to all other drugs, and only encodes the similarity to the most similar drugs for each drug. In the SNF process, is always used as the initial state and S is used as the kernel matrix.
The and of each PPMI matrix are obtained by the above two formulas. Let us first take the fusion of the previous two PPMI matrices as a case, we get the status and from two PPMI matrices by Equation 3, and obtain the kernel matrices and by Equation 4. When , we denote the initial two status matrices and . Iteratively updating the similarity matrix corresponding to each network data by using the following formulas is a key step of SNF:
| (Equation 5) |
| (Equation 6) |
After iterations, is the status matrix of , is the similarity matrix of . The status matrices are updated in this procedure with generating two parallel interchanging diffusion processes each time. After t steps, the two matrices fused into a matrix is computed as follows:
| (Equation 7) |
As shown in the framework Figure 1A, we sequentially merge our multiple PPMI matrices pairwise. Based on message passing theory, a non-linear method is used in the process of network fusion to iteratively update each network to make it more similar to other networks in each iteration. Through several iterations, SNF converges multiple networks into a single network. This method of merging multiple network data can reduce the noise of the network and preserve the strongly associated edges in the network.
After fusing multiple drug-related networks, we use the SAE26 method to obtain high-quality and low-dimensional drug features. The encoder function from the input layer to the hidden layer of the SAE is:
| (Equation 8) |
The decoder function from the hidden layer to the output layer is:
| (Equation 9) |
where is the activation function, is the connection parameter, and is the bias vector. The sparse penalty is added to the target function of the autoencoder to capture the effective features of the drug. The activation of the th hidden unit is denoted as , we use the following formula to get the average activation amount of the th hidden unit:
| (Equation 10) |
The loss function of SAE with sparse penalty is as follow:
| (Equation 11) |
| (Equation 12) |
where is the loss function of the neural network, is the hyperparameter to control the weight of sparsity in loss function, is the number of hidden layer units and is a very small value closed to 0 as a sparsity parameter. The is called KL-divergence, which possesses the property that if . Otherwise, it increases monotonically as diverges from , which acts as the sparsity constraint. We adopt the gradient descent algorithm to minimize the to optimize the parameters and .
Obtaining disease features based on text mining
For the disease features acquisition module, we considered, at the early stage of model design, that drugs that could act on a certain disease and may have an effect on related or similar diseases. Therefore, when we obtain disease features, we hope to capture their possible associations through text mining. The proposed BERT model60 has enabled a qualitative leap for the text mining algorithm and brought a milestone change in the natural language-processing field, which uses transformer as the main framework and is pre-trained on BooksCorpus and English Wikipedia. However, the word distribution between general corpus and biomedical corpus is different, which is why the pure BERT model cannot achieve good results in biomedical text mining tasks.
In our work, to obtain effective disease features representation through text mining, we used the BioBERT model,27 which is a biomedical domain-based pre-trained language-representation model. For tokenization, the BioBERT utilizes the WordPiece tokenization.61 With word tokenization, any disease name words can be represented by frequently occurring sub-words. Given the limitation of training costs, we directly use the pre-trained parameters of the BioBERT model and select the relation extraction task in the three representative biomedical text mining tasks for fine-tuning training. The BioBERT used the original BERT sentence classifier. In the fine-tuning process, diseases and genes are anonymized target entities in the datasets to prevent the supervised information used in the fine-tuning process from overlapping with drug-disease relations in the test set and to avoid the possibility that the test data may be contaminated by the text corpora. For the fine-tuning, we select a batch size of 32 and a learning rate of . After the fine-tuning process done, we extract the representations of sub-words and use the representations of these sub-words to obtain the representations of all diseases (Figure 1B). After obtaining the diseases features, we verified their effectiveness of the diseases features. More details for evaluating the effectiveness of diseases features are available in the supplemental experimental procedures.
Drug-disease associations prediction
The features obtained from the first two steps can be used as the attribute information of drugs and diseases. In the process of embedding learning, we need to preserve node attribute proximity and the drug-disease associations network topological structure. We refer to the GATNE-I model62 to predict the drug-disease interactions Figure 1C. We divide the overall embedding of a given node into three parts: node attributes embedding, neighborhood aggregation embedding, and base embedding.
To better integrate the attribute information of heterogeneous network nodes, the drug and disease features obtained in the above two steps are defined as the attribute of nodes. We define the base embedding , which is a parameterized function of ’s attribute , is a transformation function and is node ’s corresponding node type. For neighborhood aggregation embedding, the th level embedding , () of node is aggregated from neighbor's embedding, and is the dimension of neighborhood aggregation embedding. We compute the neighborhood aggregation embedding by the mean aggregator function63 as:
| (Equation 13) |
where is an activation function, is the weight matrices to propagate information between different layers, and are the neighbors of node . The initial neighborhood aggregation embeddings is also a parameterized function of ’s attribute as . The subscript is ’s corresponding node type and is a transformation function.
Finally, the overall embedding of node ’s function is as follows:
| (Equation 14) |
| (Equation 15) |
where is a hyperparameter, is a trainable transformation matrix, is the dimension of overall embedding, is trainable parameter with size , is trainable parameter with size , is a coefficient, the symbol T represents the transposition of the matrix or the vector, and is a feature transformation matrix on ’s corresponding node type .
Next, optimize the model. Because the drug-disease associations network is a heterogeneous network, to ensure the semantic relationship between different types of nodes can be correctly merged into the skip-gram model, meta-path-based random walks are used to obtain node sequence and skip-gram are performed over the node sequence to learn embeddings.64 Specifically, for a given network and a meta-path scheme , the flow of the walker is conditioned on the pre-defined mate-path and the transition probability at step is define as follows:
| (Equation 16) |
where . Supposing the random walk with length follows a path such that ), ’s context is denoted as , where w is the radius of the window size.
Therefore, given a node with its context C of a path, we aim to minimize the following negative log likelihood:
| (Equation 17) |
where denotes all the parameters, and use the heterogeneous softmax function normalized with respect to the node type of node , the probability of given is defined as:
| (Equation 18) |
where , is the context embedding of node and is the overall embedding of node .
Finally, heterogeneous negative sampling is used to approximate the objective function for each node pair as:
| (Equation 19) |
where is the sigmoid function, L is the number of negative samples corresponding to a positive training sample, and is randomly drawn from a noise distribution defined on node ’s corresponding node set .
Acknowledgments
This work is supported by the National Key R&D Program of China (2017YFE0130600), the National Natural Science Foundation of China (grant nos. 61772441, 61872309, 62072384, and 62072385), and the Basic Research Program of Science and Technology of Shenzhen (JCYJ20180306172637807).
Author contributions
Conceptualization, X.Z., X.L., and S.J.; methodology, X.Z., Z.N., and S.J.; investigation, X.L., X.Z., S.J., Z.N., C.J., and W.H.; writing – original draft, S.J., C.J., and F.X.; writing – review & editing, X.Z., S.J., X.J., and W.H.; funding acquisition, X.L. and X.Z.; resources, S.J., X.L., and X.Z.; supervision, X.L. and X.Z.
Declaration of interest
The authors declare no competing interests.
Published: July 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.patter.2021.100307.
Supplemental information
References
- 1.Dickson M., Gagnon J.P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004;3:417–429. doi: 10.1038/nrd1382. [DOI] [PubMed] [Google Scholar]
- 2.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 3.Li J., Zheng S., Chen B., Butte A.J., Swamidass S.J., Lu Z. A survey of current trends in computational drug repositioning. Brief. Bioinformatics. 2016;17:2–12. doi: 10.1093/bib/bbv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashburn T.T., Thor K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2014;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 5.Napolitano F., Zhao Y., Moreira V.M., Tagliaferri R., Kere J., D’Amato M., Greco D. Drug repositioning: a machine-learning approach through data integration. J. Cheminformatics. 2013;5:30. doi: 10.1186/1758-2946-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y., Chen S., Deng N., Wang Y. Drug repositioning by kernel-based integration of molecular structure, molecular activity, and phenotype data. PLoS One. 2013;8:e78518. doi: 10.1371/journal.pone.0078518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim E., Choi A.-s., Nam H. Drug repositioning of herbal compounds via a machine-learning approach. BMC Bioinformatics. 2019;20:33–43. doi: 10.1186/s12859-019-2811-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottlieb A., Stein G.Y., Ruppin E., Sharan R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011;7:496. doi: 10.1038/msb.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez V., Navarro C., Cano C., Fajardo W., Blanco A. DrugNet: network-based drug–disease prioritization by integrating heterogeneous data. Artif. Intellig. Med. 2015;63:41–49. doi: 10.1016/j.artmed.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Zeng X., Zhu S., Liu X., Zhou Y., Nussinov R., Cheng F. deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics. 2019;35:5191–5198. doi: 10.1093/bioinformatics/btz418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu L., Yu H. DR2DI: a powerful computational tool for predicting novel drug-disease associations. J. Comput. Aided Mol. Des. 2018;32:633–642. doi: 10.1007/s10822-018-0117-y. [DOI] [PubMed] [Google Scholar]
- 12.Liu H., Zhang W., Song Y., Deng L., Zhou S. HNet-DNN: inferring new drug-disease associations with deep neural network based on heterogeneous network features. J. Chem. Inf. Model. 2020;60:2367–2376. doi: 10.1021/acs.jcim.9b01008. [DOI] [PubMed] [Google Scholar]
- 13.Liu H., Song Y., Guan J., Luo L., Zhuang Z. Inferring new indications for approved drugs via random walk on drug-disease heterogenous networks. BMC Bioinformatics. 2016;17:269–277. doi: 10.1186/s12859-016-1336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo H., Wang J., Li M., Luo J., Peng X., Wu F.X., Pan Y. Drug repositioning based on comprehensive similarity measures and bi-random walk algorithm. Bioinformatics. 2016;32:2664–2671. doi: 10.1093/bioinformatics/btw228. [DOI] [PubMed] [Google Scholar]
- 15.Dai W., Liu X., Gao Y., Chen L., Song J., Chen D., Gao K., Jiang Y., Yang Y., Chen J. Matrix factorization-based prediction of novel drug indications by integrating genomic space. Comput. Math. Methods Med. 2015;2015 doi: 10.1155/2015/275045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceddia G., Pinoli P., Ceri S., Masseroli M. IEEE; 2019. Non-negative matrix tri-factorization for data integration and network-based drug repositioning; pp. 1–7. (2019 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB)). [DOI] [Google Scholar]
- 17.Brown A.S., Patel C.J. MeSHDD: literature-based drug-drug similarity for drug repositioning. J. Am. Med. Inform. Assoc. 2017;24:614–618. doi: 10.1093/jamia/ocw142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H.-T., Ju J.H., Wong Y.T., Shmulevich I., Chiang J.H. Literature-based discovery of new candidates for drug repurposing. Brief. Bioinformatics. 2017;18:488–497. doi: 10.1093/bib/bbw030. [DOI] [PubMed] [Google Scholar]
- 19.Li F., Zhang M., Fu G., Ji D. A neural joint model for entity and relation extraction from biomedical text. BMC Bioinformatics. 2017;18:1–11. doi: 10.1186/s12859-017-1609-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang P., Hao T., Yan J., Jin L. Large-scale extraction of drug-disease pairs from the medical literature. J. Assoc. Inf. Sci. Technol. 2017;68:2649–2661. doi: 10.1002/asi.23876. [DOI] [Google Scholar]
- 21.Yang C., Xiao Y., Zhang Y., Sun Y., Han J. Heterogeneous network representation learning: a unified framework with survey and benchmark. IEEE Trans. Knowledge Data Eng. 2020 doi: 10.1109/TKDE.2020.3045924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu Z., Dong Y., Wang K., Sun Y. Proceedings of the Web Conference 2020. 2020. Heterogeneous graph transformer. [DOI] [Google Scholar]
- 23.Antons D., Grünwald E., Cichy P., Salge T.O. The application of text mining methods in innovation research: current state, evolution patterns, and development priorities. R&D Manag. 2020;50:329–351. doi: 10.1111/radm.12408. [DOI] [Google Scholar]
- 24.Wang B., Mezlini A.M., Demir F., Fiume M., Tu Z., Brudno M., Haibe-Kains B., Goldenberg A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods. 2014;11:333. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- 25.Ng A. Sparse autoencoder. CS294A Lecture Notes. 2011;72:1–19. [Google Scholar]
- 26.Jiang H.-J., Huang Y.-A., You Z.-H. SAEROF: an ensemble approach for large-scale drug-disease association prediction by incorporating rotation forest and sparse autoencoder deep neural network. Sci. Rep. 2020;10:1–11. doi: 10.1038/s41598-020-61616-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J., Yoon W., Kim S., Kim D., Kim S., So C.H., Kang J. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics. 2020;36:1234–1240. doi: 10.1093/bioinformatics/btz682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moghadam H., Rahgozar M., Gharaghani S. Scoring multiple features to predict drug disease associations using information fusion and aggregation. SAR QSAR Environ. Res. 2016;27:609–628. doi: 10.1080/1062936X.2016.1209241. [DOI] [PubMed] [Google Scholar]
- 29.Cortes C., Vapnik V. Support-vector networks. Machine Learn. 1995;20:273–297. doi: 10.1007/BF00994018. [DOI] [Google Scholar]
- 30.Chen X., Huang Y.A., You Z.H., Yan G.Y., Wang X.S. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics. 2017;33:733–739. doi: 10.1093/bioinformatics/btw715. [DOI] [PubMed] [Google Scholar]
- 31.Luo Y., Zhao X., Zhou J., Yang J., Zhang Y., Kuang W., Peng J., Chen L., Zeng J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017;8:1–13. doi: 10.1038/s41467-017-00680-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo H., Li M., Wang S., Liu Q., Li Y., Wang J. Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics. 2018;34:1904–1912. doi: 10.1093/bioinformatics/bty013. [DOI] [PubMed] [Google Scholar]
- 33.Aljenedil S., Alothman L., Bélanger A.M., Brown L., Lahijanian Z., Bergeron J., Couture P., Baass A., Ruel I., Brisson D. Lomitapide for treatment of homozygous familial hypercholesterolemia: the Québec experience. Atherosclerosis. 2020;310:54–63. doi: 10.1016/j.atherosclerosis.2020.07.028. [DOI] [PubMed] [Google Scholar]
- 34.Bélanger A.M., Akioyamen L., Alothman L., Genest J. Evidence for improved survival with treatment of homozygous familial hypercholesterolemia. Curr. Opin. Lipidol. 2020;31:176–181. doi: 10.1097/MOL.0000000000000686. [DOI] [PubMed] [Google Scholar]
- 35.Reeskamp L.F., Kastelein J.J., Moriarty P.M., Duell P.B., Catapano A.L., Santos R.D., Ballantyne C.M. Safety and efficacy of mipomersen in patients with heterozygous familial hypercholesterolemia. Atherosclerosis. 2019;280:109–117. doi: 10.1016/j.atherosclerosis.2018.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Jenike M.A., Albert M.S., Heller H., LoCastro S., Gunther J. Combination therapy with lecithin and ergoloid mesylates for Alzheimer's disease. J. Clin. Psychiatry. 1986;47:249–251. [PubMed] [Google Scholar]
- 37.Flynn B.L., Ranno A.E. Pharmacologic management of Alzheimer disease part II: antioxidants, antihypertensives, and ergoloid derivatives. Ann. Pharmacother. 1999;33:188–197. doi: 10.1345/aph.17172. [DOI] [PubMed] [Google Scholar]
- 38.Cover C.C., Poulin J.E., Gustafson M.R., Wyant T., Gamble D.N., Kay M.M. Posttranslational changes in band 3 in adult and aging brain following treatment with ergoloid mesylates, comparison to changes observed in Alzheimer's disease. Life Sci. 1996;58:655–664. doi: 10.1016/S0024-3205(96)80004-X. [DOI] [PubMed] [Google Scholar]
- 39.Singer J.M., Hamot H.B., Patin J.R. Aging 2000: Our Health Care Destiny. Springer; 1985. Differential patient response to ergoloid mesylates according to current etiopathic notions of dementia; pp. 405–420. [DOI] [Google Scholar]
- 40.Gu S., Fu W.Y., Fu A.K., Tong E.P.S., Ip F.C., Huang X., Ip N.Y. Identification of new EphA4 inhibitors by virtual screening of FDA-approved drugs. Sci. Rep. 2018;8:1–7. doi: 10.1038/s41598-018-25790-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nizami S., Hall-Roberts H., Warrier S., Cowley S.A., Di Daniel E. Microglial inflammation and phagocytosis in Alzheimer's disease: potential therapeutic targets. Br. J. Pharmacol. 2019;176:3515–3532. doi: 10.1111/bph.14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dansokho C., Heneka M.T. Neuroinflammatory responses in Alzheimer’s disease. J. Neural Transm. 2018;125:771–779. doi: 10.1007/s00702-017-1831-7. [DOI] [PubMed] [Google Scholar]
- 43.Sheridan C. Novartis trial validates inflammasome as chronic disease driver. Nat. Biotechnol. 2017;35:893–894. doi: 10.1038/nbt1017-893. [DOI] [PubMed] [Google Scholar]
- 44.Mitroulis I., Skendros P., Ritis K. Targeting IL-1β in disease; the expanding role of NLRP3 inflammasome. Eur. J. Intern. Med. 2010;21:157–163. doi: 10.1016/j.ejim.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 45.Chauhan D., Vande Walle L., Lamkanfi M. Therapeutic modulation of inflammasome pathways. Immunological Rev. 2020;297:123–138. doi: 10.1111/imr.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McElroy S.L., Guerdjikova A., Kotwal R., Welge J.A., Nelson E.B., Lake K.A., Keck P.E., Jr., Hudson J.I. Atomoxetine in the treatment of binge-eating disorder: a randomized placebo-controlled trial. J. Clin. Psychiatry. 2007;68:390–398. doi: 10.4088/jcp.v68n0306. [DOI] [PubMed] [Google Scholar]
- 47.Gadde K.M., Yonish G.M., Wagner H.R., Foust M.S., Allison D.B. Atomoxetine for weight reduction in obese women: a preliminary randomised controlled trial. Int. J. Obes. 2006;30:1138–1142. doi: 10.1038/sj.ijo.0803223. [DOI] [PubMed] [Google Scholar]
- 48.Pott W., Albayrak Ö., Hinney A., Hebebrand J., Pauli-Pott U. Successful treatment with atomoxetine of an adolescent boy with attention deficit/hyperactivity disorder, extreme obesity, and reduced melanocortin 4 receptor function. Obes. Facts. 2013;6:109–115. doi: 10.1159/000348792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mirbolooki M.R., Constantinescu C.C., Pan M.L., Mukherjee J. Targeting presynaptic norepinephrine transporter in brown adipose tissue: a novel imaging approach and potential treatment for diabetes and obesity. Synapse. 2013;67:79–93. doi: 10.1002/syn.21617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shibao C., Raj S.R., Gamboa A., Diedrich A., Choi L., Black B.K., Robertson D., Biaggioni I. Norepinephrine transporter blockade with atomoxetine induces hypertension in patients with impaired autonomic function. Hypertension. 2007;50:47–53. doi: 10.1161/HYPERTENSIONAHA.107.089961. [DOI] [PubMed] [Google Scholar]
- 51.Spencer T.J., Kratochvil C.J., Sangal R.B., Saylor K.E., Bailey C.E., Dunn D.W., Geller D.A., Casat C.D., Lipetz R.S., Jain R. Effects of atomoxetine on growth in children with attention-deficit/hyperactivity disorder following up to five years of treatment. J. Child Adolesc. Psychopharmacol. 2007;17:689–699. doi: 10.1089/cap.2006.0100. [DOI] [PubMed] [Google Scholar]
- 52.Yuan C.-S., Wang C.Z., Attele A., Zhang L. Methylnaltrexone reduced body weight gain in ob/ob mice. J. Opioid Manag. 2009;5:213–218. doi: 10.5055/jom.2009.0023. [DOI] [PubMed] [Google Scholar]
- 53.Gatti A., Sabato A.F. Management of opioid-induced constipation in cancer patients. Clin. Drug Invest. 2012;32:293–301. doi: 10.2165/11598000-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 54.Biton V., Gates J.R., Ritter F.J., Loewenson R.B. Adjunctive therapy for intractable epilepsy with ethotoin. Epilepsia. 1990;31:433–437. doi: 10.1111/j.1528-1157.1990.tb05499.x. [DOI] [PubMed] [Google Scholar]
- 55.Ashna A., Van Helden D., Laver D. Phenytoin and ethotoin inhibit ryanodine receptor in manner paralleling that of dantrolene. Heart, Lung, Circ. 2018;27:S182. doi: 10.1016/j.hlc.2018.06.313. [DOI] [Google Scholar]
- 56.Iwabuchi M. SAT-473 acromegaly and drug-induced parkinsonism were controlled by lanreotide. J. Endocr. Soc. 2019;3(Suppl. 1) doi: 10.1210/js.2019-SAT-473. SAT-473. [DOI] [Google Scholar]
- 57.Abe K., Chiba Y., Katsuse O., Hirayasu Y. A case of Parkinson disease with both visual hallucination and pain improved by gabapentin. Clin. Neuropharmacol. 2016;39:55–56. doi: 10.1097/WNF.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 58.Fujishiro H. Effects of gabapentin enacarbil on restless legs syndrome and leg pain in dementia with Lewy bodies. Psychogeriatrics. 2014;14:132–134. doi: 10.1111/psyg.12043. [DOI] [PubMed] [Google Scholar]
- 59.Bullinaria J.A., Levy J.P. Extracting semantic representations from word co-occurrence statistics: a computational study. Behav. Res. Methods. 2007;39:510–526. doi: 10.3758/BF03193020. [DOI] [PubMed] [Google Scholar]
- 60.Devlin J., Chang M.W., Lee K., Toutanova K. BERT: pre-training of deep bidirectional transformers for language understanding. arXiv. 2018 1810.04805. [Google Scholar]
- 61.Wu Y., Schuster M., Chen Z., Le Q.V., Norouzi M., Macherey W., Krikun M., Cao Y., Gao Q., Macherey K. Google's neural machine translation system: bridging the gap between human and machine translation. arXiv. 2016 1609.08144. [Google Scholar]
- 62.Cen Y., Zou X., Zhang J., Yang H., Zhou J., Tang J. Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining. 2019. Representation learning for attributed multiplex heterogeneous network; pp. 1358–1368. [DOI] [Google Scholar]
- 63.Hamilton W., Ying Z., Leskovec J. Inductive representation learning on large graphs. arXiv. 2017 1706.02216. [Google Scholar]
- 64.Dong Y., Chawla N.V., Swami A. Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2017. metapath2vec: scalable representation learning for heterogeneous networks; pp. 135–144. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and data are available at https://github.com/stjin-XMU/HeTDR, https://doi.org/10.5281/zenodo.4915882.