

Abstract
The ongoing global pandemic caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has prompted worldwide vaccine development. Several vaccines have been authorized by WHO, FDA, or MOH of different countries. However, issues such as need for cold chain, price, and most importantly access problems have limited vaccine usage in some nations especially developing countries. Moreover, the vast global demand justifies further attempts for vaccine development. Multi-epitope polypeptide vaccines enjoy several key features including safety and lower production and transfer costs and could be designed by in silico tools. Spike protein (S), membrane protein (M), and nucleocapsid protein (N), the three major structural proteins of SARS-CoV-2, are ideal candidates for epitope selection. ORF3a (open reading frame3a), a transmembrane protein with pro-apoptotic functions, could be another proper target. Thus, a novel multi-epitope vaccine against SARS-CoV-2 was designed using these four proteins and LL37, a TLR3 agonist adjuvant, through different immunoinformatics and bioinformatics tools. The proposed multi-epitope vaccine is expected to induce robust humoral and cellular immune responses against SARS-CoV-2 with a population coverage of 76.92 % due to containing different immunodominant epitopes and LL37 adjuvant. Selecting epitopes derived from one functional and three structural proteins suggests the protective ability of the vaccine irrespective of probable virus mutations. The computationally observed proper interaction of LL37 with TLR3 implies its ability to induce immune responses effectively. Besides, it showed acceptable structural and physicochemical properties. The in-silico cloning results predicted its high efficiency production in Escherichia coli. Future experimental studies could further confirm its immunological efficacy.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11756-021-00866-y.
Keywords: Adjuvant; Coronavirus; In silico; Multi-epitope; Immunoinformatics, COVID-19
Introduction
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has grown to an ongoing global pandemic, following the first reported case at the end of 2019 in Wuhan, Hubei province, China, and has caused serious healthcare and disease control concerns worldwide (Mosaddeghi et al. 2020). As of 21 May 2021, over 165 million cases of coronavirus disease 2019 (COVID-19) have been confirmed leading to over 3.4 million deaths (WHO, 21 May 2021). SARS-CoV-2 is a β-coronavirus from the family Coronaviridae (Negahdaripour 2020). The genome sequence of SAR-CoV-2 is nearly 80 % similar to that of SARS-CoV, another β-coronavirus responsible for SARS epidemic in 2003 (Zhou et al. 2020).
Many studies have been carried out to identify and investigate the structural and functional proteins of SARS-CoV-2, which could lead to a better understanding of the virus pathology, virulence, and even emergence of new therapeutic and preventive options. SARS-CoV-2 transcriptome encodes four main structural proteins: spike protein (S), membrane protein (M), envelope protein (E), and nucleocapsid protein (N) (Drosten et al. 2003), which are commonly present in all coronaviruses alongside an open reading frame (ORF1a/ORF1ab) (Michel et al. 2020). While M and E proteins of the virus participate in particle assembly and release of the virus (Masters 2006), the S1 subunit of S protein, binds to host cell receptors, and the S2 subunit participates in the process of fusion between viral and host cell membranes thereby facilitating virus entry to host cells (Owji et al. 2020). The key receptor for the receptor binding domain (RBD), which is present in the S protein, is called angiotensin converting enzyme-2 (ACE2). ACE2 is widely distributed in various human organs such as kidneys, liver, small intestine, and lungs (Hamming et al. 2004). Previous publications have discussed the functions of SARS-CoV-2 accessory proteins, including ORF3a, 6, 7a, 7b (Gordon et al. 2020), its pathophysiology, as well as the signaling and immune response pathways involved in this viral infection (Bagheri et al. 2020).
The recent pandemic outbreak prompted worldwide vaccine development, as urgent tactics are needed to prevent further transmission of the virus. Many research groups around the world started development of vaccine candidates, some of which are already approved and some others are in pre-clinical and clinical stages of development (Kaur and Gupta 2020). The US Food and Drug Administration (FDA) issued emergency use authorization (EUA) for Pfizer-BioNTech, Moderna, and Janssen COVID-19 vaccines (FDA US 2020a, b, 2021), while more than 10 vaccines have received an NRA (national regulatory authority) approval to be used in at least one country, and vaccination with these vaccines has been started in some countries (WHO website, accessed on 22 May 2021). However, some challenges such as cold chain maintenance, access issues, and high costs have dampened vaccine usage particularly in developing countries. Moreover, the vast global demand justifies further attempts for development of more vaccines (Negahdaripour, 2021). The efficacy and safety issues are the main challenges for vaccine developers as ever.
Multi-epitope polypeptide vaccines are shown to be safe, needless of complex storage conditions, with lower production and transfer costs (Negahdaripour et al. 2017a, 2018). Still, low immunogenicity is one of the main burdens in the development of polypeptide vaccines. To tackle this issue, employment of adjuvants, as immune response enhancers, could provide beneficiary outcomes by triggering a stronger immune response with fewer doses of the antigen (Coffman et al. 2010).
During recent years, administration of various bioinformatics tools has proved to be a cost-effective and time-saving strategy in modern structural vaccinology, which supports better antigen selection and design (Negahdaripour et al. 2018). In this regard, in silico approaches help to predict the immunogenic epitope regions of various antigenic proteins in order to generate an effective immunological response. Furthermore, the expression level of a gene can be predicted and optimized through in silico cloning before growing the microorganism in vitro (Negahdaripour et al. 2017a, Samad et al. 2020, Yang et al. 2021).
In this study, various bioinformatics servers and computational tools would be employed to design a novel multi-epitope against SARS-CoV-2 using epitope regions from the S, M, and N proteins as well as the ORF3a of SARS-CoV-2. A toll-like receptor (TLR) 3 agonist would also be administered as the vaccine adjuvant to further increase the immunogenic potential of the construct.
Materials and methods
Sequence retrieval
The complete amino acid sequences of S protein (Accession no. YP_009724390.1), M protein (Accession no. YP_009724393.1), N protein (Accession no. YP_009724397.2), and ORF3a (Accession no. YP_009724391.1) were retrieved from the National Centre for Biotechnology Information (NCBI) at www.ncbi.nlm.nih.gov in FASTA format. LL37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) was applied as a TLR3 agonist (Lai et al. 2011b). Moreover, the complete amino acid sequence and structure of TLR3 was retrieved from the PDB database at https://www.rcsb.org/pdb/home/home.do (PDB code: 1ZIW).
Identification of immunodominant epitopes
HLA class I and II binding epitope prediction
For human leukocyte antigen (HLA) class I binding epitope prediction, we selected the 16 most frequent HLA class I alleles in the worldwide population, including HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*11:01, HLA-B*14:02, HLA-A*23:01, HLA-A*24:02, HLA-A*31:01, HLA-A*68:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:02, HLA-B*27:05, HLA-B*35:01, HLA-B*40:01, HLA-B*44:03. The high-score 9-mer epitopes were identified using immune epitope databases (IEDB) (http://tools.iedb.org/mhci). IEDB is an online server that applies different methods for the prediction of HLA class I and II binding epitopes. (Kim et al. 2012). Furthermore, using the IEDB server, the high-ranked 15-mer HLA class II binding epitopes for 22 most frequent HLA class II alleles in the worldwide population were predicted, including H DRB1*01:01, DRB1*01:02, DRB1*03:01, DRB1*04:01, DRB1*04:05, DRB1*07:01, DRB1*07:03, DRB1*08:02, DRB1*08:17, DRB1*09:01, DRB1*12:01, DRB1*11:01, DRB1*11:06, DRB1*13:02, DRB1*13:03, DRB1*13:05, DRB1*14:01, DRB1*15:01, DRB3*01:01, DRB3*02:02, DRB4*01:01, DRB5*01:01. In this study, the IEDB recommended method was utilized to find out the high-ranked epitopes (Wang et al. 2008).
Prediction of CTL epitopes
CTL (cytotoxic T-cell) epitopes were identified using CTLpred (http://crdd.osdd.net/raghava/ctlpred/applying/). CTLpred is an online server that utilizes different methods such as quantitative matrix (QM) and machine learning techniques, namely artificial neural network (ANN) and support vector machine (SVM). The accuracy of QM, ANN, and SVM approaches are similar (70.0 % for QM, 72.2 % for ANN, and 75.2 % for SVM method) (Soria-Guerra et al. 2015). The sensitivity and specificity of QM, ANN, and SVM methods are 0.00, 0.51, and 0.36, respectively.
Population coverage analysis
Population coverage for each individual epitope was predicted by the IEDB population coverage analysis resource (http://tools.iedb.org/population/) against the whole world. This server utilizes LA genotypic frequencies to identify the fraction of individuals that respond to a specific epitope (Bui et al. 2006).
Linear and conformational B-cell epitopes prediction
Linear or continuous B-cell epitopes were predicted using the BCPRED server (http://ailab.ist.psu.edu/bcpred/predict.html). The server accuracy for the prediction of continuous B-cell epitopes is 74.57 %. Predicted regions above the threshold value (2 to 2.5) are considered to be potential B-cell epitopes (EL-Manzalawy et al. 2008).
Conformational B-cell epitopes were identified from the 3D structure of the final construct by the DiscoTope 2.0 server at http://www.cbs.dtu.dk/services/DiscoTope/. This server employs a new epitope propensity amino acid score for predicting epitopes (Kringelum et al. 2012). The default threshold, sensitivity, and specificity were used, which were − 3.7, 0.47, and 0.75, respectively.
Prediction of interferon-gamma inducing epitopes
To predict interferon-gamma inducing epitopes, the IFNepitope server was utilized at http://crdd.osdd.net/raghava/ifnepitope. The server uses various methods for identifying interferon-gamma epitopes, including machine learning methods, motifs-based search, and a hybrid approach. The best predicted method is based on the hybrid approach and achieves maximum accuracy of 81.39 % (Dhanda et al. 2013).
Epitope selection and construction of the construct
The overlapping regions between different predicted epitopes were compared. Several high-ranked and shared parts were selected as the final epitopes from a pool of epitopes. Afterwards, the whole vaccine construct was built by joining the selected epitopes to each other using linkers. A TLR3 agonist was also added to the N-terminal of the construct.
Evaluation of allergenicity and antigenicity
The allergenicity of the designed multi-epitope peptide vaccine was determined using two various servers, namely AlgPred and AllerTOP 2.0 (http://www.ddgpharmfac.net/AllerTOP). The AlgPred server (http://www.imtech.res.in/raghava/algpred) employs six different methods for the prediction of allergens based on the similarity of the known epitopes with any region of proteins. In the present study, the hybrid prediction approach with the highest accuracy was applied as the allergen prediction method (Saha and Raghava 2006). The AllerTOP uses a training set containing 2210 known allergens and 2210 non-allergens of different organisms. The server predicts allergenicity by k nearest neighbors (kNN) algorithm with 85.3 % accuracy (Dimitrov et al. 2014). VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was employed for prediction of the antigenicity. VaxiJen v2.0 predicts antigenicity based on the principal chemical features of proteins using an alignment-independent approach for various target organisms (bacteria, virus, tumor, parasite, and fungi) with 70–89 % accuracy (Doytchinova and Flower 2007). In this study, “virus” was chosen as the target organism (threshold 0.4).
Evaluation of physicochemical parameters
To determine different physicochemical properties of the designed multi-epitope peptide vaccine, the ProtParam server at http://web.expasy.org/protparam was employed. Accordingly, theoretical pI (isoelectric point), amino acid composition, instability index, aliphatic index, molecular weight (MW), in vitro and in vivo half-life, and grand average of hydropathicity (GRAVY) were calculated (Gasteiger et al. 2005).
Homology modeling
The 3D structure of the designed construct was predicted using I-TASSER at http://zhanglab.ccmb.med.umich.edu/I-TASSER. The server works based on iterative threading assembly simulations. The 3D structure modeling of the peptide construct by the I-TASSER consists of several steps: (1) prediction of threading templates, (2) simulation of iterative structure assembly, (3) selection and refinement of the predicted model, and (4) structure-based function annotations (Yang et al. 2015). The quality of the predicted 3D models by I-TASSER is estimated by a C-score (confidence score). A model with a higher C-score poses more confidence (Yang and Zhang 2015).
Refinement of the tertiary modeled structure
The refinement process for the predicted 3D structure was done by GalaxyRefine. The best 3D model obtained from I-TASSER based the C-score was introduced to the GalaxyRefine server at http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE. In this server, mild and aggressive relaxation approaches are employed for refinement of the designed protein (Heo et al. 2013). Both the backbone and side-chain structure qualities are enhanced by repetitive perturbation and relaxation. The server improves the initial models with a high probability (> 50 %) (Shin et al. 2014).
Validation of the refined tertiary structure
To recognize potential errors in the predicted 3D model, the refined structures were evaluated by various servers, including RAMPAGE (Ramachandran Plot Assessment), ProSA-web (Protein Structure Analysis), and verify 3D. The RAMPAGE server at http://mordred.bioc.cam.ac.uk/~rapper/rampage.php estimates phi-psi torsion angles for each residue and classifies them in favored, allowed, and outlier groups (Lovell et al. 2003). The verify 3D at http://services.mbi.ucla.edu/Verify_3D/ compares the predicted model to its residues by assigning a structural class based on its location (Luthy et al. 1992). The ProSA-web at (https://prosa.services.came.sbg.ac.at/prosa.php) estimates an overall quality score for a specific input structure. The server requires only Cα atoms so that low-resolution structures can be evaluated. The interaction energy of each residue with the rest of the structure is analyzed and potential errors in the protein structure are calculated (Wiederstein and Sippl 2007). In this study, discovery studio 3.5 and PyMOL programs were employed for visualization of 3D models.
Molecular docking of the peptide vaccine and TLR3
The Cluspro 2.0 server at http://cluspro.bu.edu/login.php was used for the molecular docking analysis of the designed vaccine as the ligand and the TLR3 as the receptor. This fast rigid-body protein-protein docking server performs docking in three general steps. The first step is rigid-body docking using PIPER, which is based on the fast Fourier transform (FFT) program. The second step is isolating highly populated low energy (clustering) by the pairwise root mean square deviation (RMSD). Finally, the third step is a refinement process via the medium-range optimization method of SDU (Semidefinite Programming-Based Underestimation) (Kozakov et al. 2010).
Codon optimization and in silico cloning
Reverse translation and codon optimization were performed using Codon Usage Wrangler (http://www.mrclmb.cam.ac.uk/ms/methods/codon.html) and the GenScript Rare Codon Analysis tool (https://www.genscript.com/tools/rare-codon-analysis), respectively. Codon adaptation index (CAI), GC content, and codon frequency distribution (CFD) were evaluated using the GenScript Rare Codon Analysis. These key parameters influence the protein expression level in the Eschercia coli host. To prepare the adopted sequences for cloning in E. coli, pET-14b vector was chosen and NdeI and BamHI restriction sites were added to the N and C-terminal of the final construct, respectively.
Results
Identification of immunodominant epitopes and population coverage analysis
The highly-scored epitopes over the 38 most frequent HLA class I and II alleles were identified using IEDB, and the selected segments were compared to each other to find the overlap regions as the high-ranked epitopes (Table S1 and S2). Subsequently, the overlapping regions obtained from the HLA class I and II binding results were compared with the high-ranked results of CTLPred, and their shared regions were exploited (Table S3). Three linear B-cell epitopes were identified by BCPRED, as listed in Table 1. Using DiscoTope server, five conformational B-cell epitopes were predicted in the final 3D model out of a total of 157 residues (Table 1). The population coverage percentages for each epitope and their epitope sets were calculated by the IEDB server (Tables S1, S2, and S3). The epitopes with a higher global population coverage were carefully selected for the final construct to ensure achieving a universal multi-epitope peptide vaccine. Based on the high-ranked and shared regions, six epitopes were selected from the four antigenic proteins (Table 2). The selected epitopes of each protein were fused together by the AAYKK linker. Then, these segments were joined to each other using the GSGSGS linker. LL37 was added as an adjuvant to the N-terminal of the construct using the EAAAK linker. The final designed construct consisted of 157 amino acid residues is illustrated in Fig. 1. Using IFNepitope server, two segments were found as IFN-gamma inducing epitopes on the final vaccine construct (Table 3).
Table 1.
Linear and conformational B-cell epitopes determined by BCPREDS and DiscoTope servers
| Linear B-cell epitopes | ||||
| Position | Epitopes | |||
| 7–17 | RKSKEKIGKEF | |||
| 90–100 | GSGSGSANRNR | |||
| 134–147 | KTFPPTEPKGSGSG | |||
| Conformational B-cell epitopes | ||||
| Residuenumber | Aminoacid | Contactnumber | Propensity score | DiscoTope score |
| 4 | ASP | 2 | -3.408 | -3.246 |
| 7–15 | ARG, LYS,SER, LYS,GLU, LYS, ILE, GLY,LYS | 5,0,0, | -3.163 | -3.029 |
| 18 | LYS | 7 | -2.744 | -3.233 |
| 140 | GLU | 2 | -3.753 | -3.55 |
| 142–146 | LYS, GLY, SER, GLY, SER | 4, 16, 6, 0, 0 | -2.254, -2.061,-2.620, -2.861,-3.228 | -2.455, -3.664, -3.009, -2.532,-2.857 |
Table 2.
The six final epitope segments that were selected based on the results obtained surveying four antigenic viral proteins using several servers
| Protein | Start-End position | Sequence | Length of sequence | Alleles |
|---|---|---|---|---|
| S Protein |
231–259 341–353 |
IGINITRFQTLLALHRSYLTPGDSSSGWT VFNATRFASVYAW |
29 13 |
HLA-B*08:01 HLA-B*14:02 HLA-B*27:02 HLA-B*27:05 HLA-B*40:01 HLA-A*02:01 HLA-A*24:02 HLA-A*31:01 HLA-A*68:01 HLA-A*23:01 HLA-B*08:01 HLA-B*40:01 HLA-B*44:02 HLA-B*44:03 |
| M Protein |
40–49 95–102 |
ANRNRFLYII YFIASFRL |
10 8 |
HLA-A*01:01 HLA-A*02:01 HLA-A*11:01 HLA-A*23:01 HLA-A*24:02 HLA-B*08:01 HLA-B*14:02 HLA-B*27:02 HLA-B*27:05 HLA-B*44:02 HLA-B*35:01 HLA-A*31:01 HLA-A*68:01 HLA-A*23:01 HLA-B*07:01 HLA-B*08:01 |
| N Protein | 352–369 | LLNKHIDAYKTFPPTEPK | 18 |
HLA-A*03:01 HLA-A*11:01 HLA-A*23:01 HLA-B*27:02 HLA-A*31:01 HLA-A*68:01 HLA-DRB1*09:01 HLA-DRB1*12:01 HLA-DRB1*13:02 HLA-DRB1*13:03 HLA-DRB1*14:01 HLA-DRB1*15:01 |
| ORF3a | 103–111 | APFLYLYAL | 9 |
HLA-A*01:01 HLA-A*02:01 HLA-A*03:01 HLA-A*23:01 HLA-B*07:02 HLA-B*08:01 HLA-B*27:02 HLA-B*35:01 HLA-B*44:02 HLA-B*44:03 HLA-DRB1*03:01 HLA-DRB1*07:01 HLA-DRB1*08:02 HLA-DRB1*09:01 HLA-DRB1*11:01 HLA-DRB1*11:06 HLA-DRB1*13:02 HLA-DRB1*13:03 HLA-DRB1*14:01 |
Fig. 1.
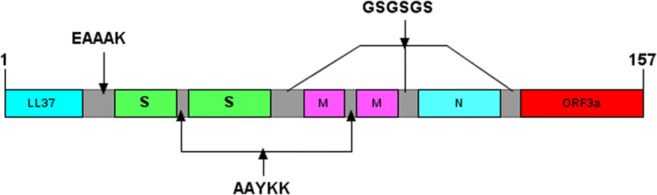
Schematic diagram of the final multi-epitope polypeptide vaccine construct. The sequence consists of 157 residues. The first 37 amino acids are related to LL37 adjuvant, followed by the six immunodominant epitopes from S (spike), M (membrane), N (nucleocapsid), and ORF (open reading frame)3a proteins linked together by AAYKK and GSGSGS linkers
Table 3.
Predicted IFN-gamma inducing epitopes in the final vaccine construct via IFNepitope sever
| Start-End | Sequence | Score |
|---|---|---|
| 352–369 | LLNKHIDAYKTFPPTEPK | 0.064 |
| 103–111 | APFLYLYAL | 0.394 |
Evaluation of allergenicity, antigenicity, and physicochemical parameters
The antigenicity of the whole construct was 0.2197 %. Based on the obtained results, the peptide vaccine could trigger efficient immunity against the pathogen. Furthermore, the designed construct did not induce allergen-specific antibodies. The physicochemical properties of the multi-epitope peptide vaccine indicated that the theoretical pI of the protein was 10.26. Moreover, the MW of the protein was 17.4 kDa. The total numbers of positively and negatively charged residues were 25 and 9, respectively. The estimated half-lives were computed to be 5.5 h (in mammalian reticulocytes, in vitro). The instability index was calculated as 36.43. A protein whose instability index is smaller than 40 is considered as a stable protein. Therefore, the designed peptide vaccine was sTable The aliphatic index was determined as 79.68. The high aliphatic index illustrated the high-protein stability in a wide range of temperatures. Moreover, the GRAVY value of the peptide construct was − 0.234. A negative GRAVY value indicates that the protein is hydrophilic and has an appropriate interaction in an aqueous environment.
Homology modeling, refinement, and molecular docking of the peptide vaccine
Model 1 was selected as the best model with the highest C-score value (-2.06). I-TASSER introduces the top five models based on the C-score. The C-score is in the range of [-5 to 2], and a model with higher confidence has a higher C-score value.
GalaxyRefine introduced five 3D refined models. All obtained models were validated and a high-quality 3D model according to z-score, potential errors, and the overall quality factor (obtained in the next step) was selected. The initial and refined 3D models are compared in Fig. 2. The initial best model of the designed peptide was evaluated for potential errors. Ramachandran analysis was performed before and after refinement processes. In the initial model, 76.1 %, 20.6 %, and 3.2 % of residues were found in the favored, allowed, and outlier regions, respectively. While in the refined model, 92.9 %, 5.8 %, and 1.3 % of residues were located in the favored, allowed, and outlier regions, respectively.
Fig. 2.
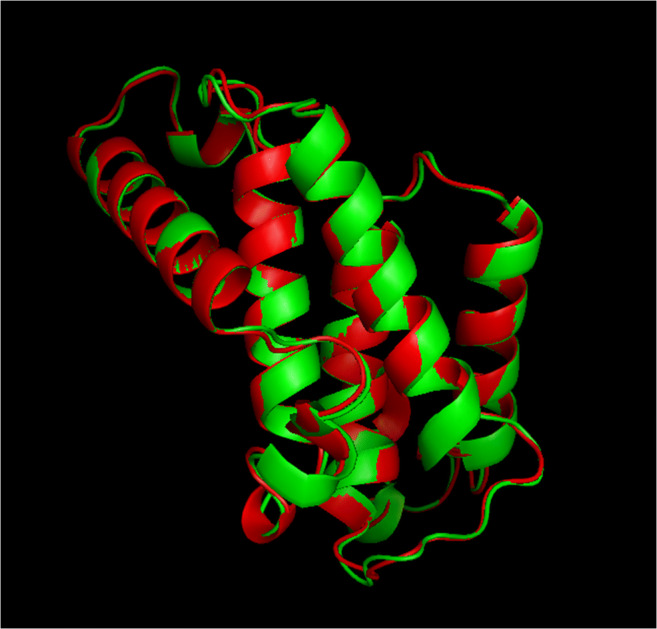
Superimposition of the initial (red) and final (green) 3D multi-epitope peptide vaccine structures, before and after structure refinement
The Z-score of the initial input model and refined model showing the quality of the best model calculated by ProSA-web, were (-6.28) and (-6.25), respectively (Fig. S1a and Fig. S1b). These scores were similar to each other and were within the range of scores typically found in the native proteins of similar sizes. The Verify 3D score indicated that in the initial model, 76.43 % of residues had an average 3D-1D score greater than 0.2 (Fig. 4a). In the refined model, the Verify 3D score was improved to 80.89 %. It means that more amino acid residues were located in an acceptable side chain environment (Fig. S2a). In molecular docking, ten models were generated based on the biophysical properties of the designed vaccine as the ligand and TLR3 as the receptor, using the ClusPro server. Finally, the most plausible docked model structure was selected based on the interaction regions (Fig. 3). PDB formats of the docked structures were visualized using discovery studio 3.5 and the PyMOL software.
Fig. 4.
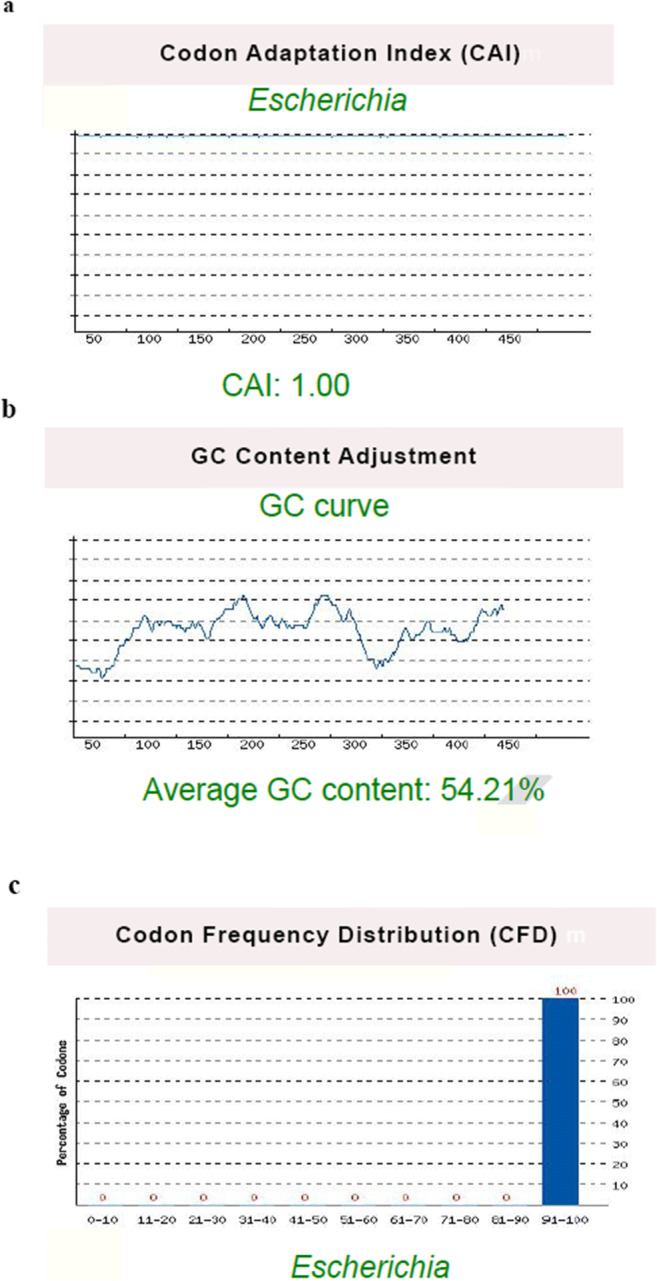
Codon adaptation index (CAI), GC content, and codon frequency distribution (CFD) of the reverse translated gene for cloning of the construct, evaluated by the GenScript Rare Codon Analysis tool. (a) The CAI of the optimized sequence was 1.0, which is ideal for expression in E. coli. (b) The average GC content of the DNA sequence was 54.21 %, which indicates proper protein expression. (c) The CFD value was 100 % for the optimized sequence, which suggests an optimum translational efficiency. These results proposed that the optimized sequence could achieve an acceptable expression in E. coli
Fig. 3.
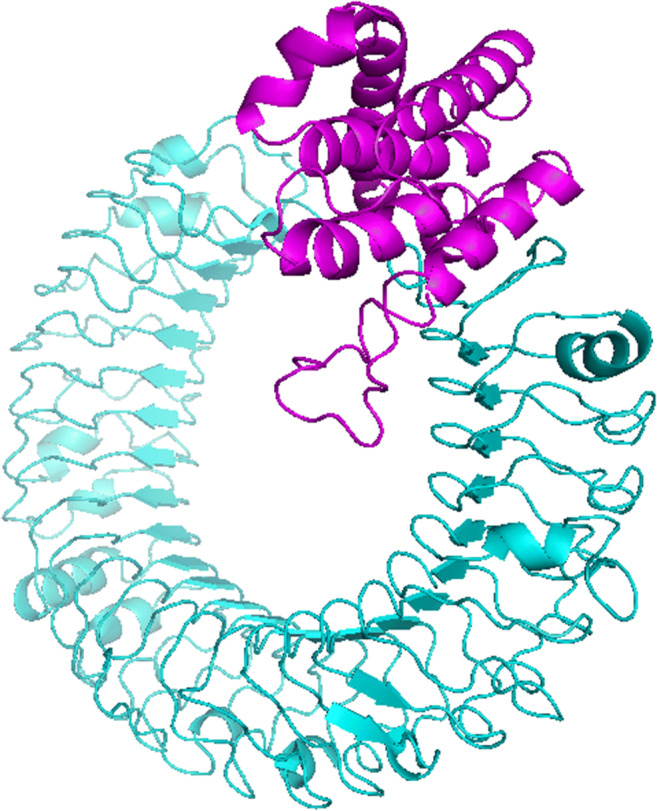
The docked model (cartoon representation) of the multi-epitope peptide vaccine and TLR3 obtained by the Cluspro 2.0 server. TLR3 is shown in cyan and the designed vaccine is in magenta colors. It indicated a significant affinity between the designed vaccine and TLR3
Codon optimization and in silico cloning
CAI of the optimized sequence was 1.0. It is considered ideal for expression in E. coli (ideal value for CAI is between 0.8 and 1.0 (Fig. 4a). The average GC content of the DNA sequence was 54.21 % (Fig. 4b). The ideal range of GC content is between 30 and 70 %. Any region outside of this range will decrease protein expression. The CFD value was 100 % for the optimized sequence. A CFD lower than 30 will negatively affect translational efficiency (Fig. 4c). These results proposed that the optimized sequence could achieve an acceptable expression in E. coli.
Discussion
The advances made in bioinformatics and novel computational and predictive tools have greatly contributed to the development of novel vaccines. Moreover, further understanding of the immune system’s regulatory and functional pathways has proven to improve the development of effective and safe vaccine candidates (Negahdaripour et al. 2018). The current pandemic outbreak of SARS-CoV-2 led to worldwide collaborations in the urgent searches for preventive and therapeutic solutions. Although development of numerous vaccine candidates worldwide is proceeding with unprecedented speed, some vaccines are still in evaluation phases (Krammer 2020). While the health authority approval in different countries has been announced for some vaccines including those developed by Moderna (Jackson et al. 2020; Mahase 2020), Pfizer and BioNTech (Walsh et al. 2020), Janssen, and AstraZeneca and the University of Oxford (Ramasamy et al. 2021), as well as Sputnik V, Sinopharm vaccine, and some others. However, there are several concerns regarding the storage, cold chain transfer conditions, and costs of some of these pioneer vaccines. Moreover, availability is still an issue, which might limit their administration, especially in the developing countries (Ashok et al. 2017). Multi-epitope polypeptide vaccines could be attractive alternatives considering their safety, more convenient development and storage, and less production costs. Their platform also allows inclusion of several different epitopes that can induce both cellular and humoral immune responses (Negahdaripour et al. 2017b). Furthermore, in silico approaches for designing such vaccines could save considerable amounts of time, energy, and costs, which facilitates vaccine production process (O’Hagan and De Gregorio 2009).
In the worldwide struggle for finding solutions to help prevent further loss due to COVID-19, several bioinformatics studies have been carried out to design vaccine candidates for potential clinical studies. For instance, in two computational vaccine designing studies, only the S protein was employed for epitope detection (Samad et al. 2020, Yang et al. 2021). In another in silico study, designing a vaccine construct out of multiple S protein epitopes were performed to induce both CD4 and CD8 T-cell response against SARS-CoV-2 (Abraham Peele et al. 2020). Dong et al. developed a multi-epitope protein vaccine consisting of epitopes from nine different proteins of SARS-CoV-2 (ORF7a protein, ORF8 protein, nsp (nonstructural protein) 9, nsp6, nsp3, endoRNAse, ORF3a protein, membrane glycoprotein, and nucleocapsid phosphoprotein) utilizing bioinformatics and computational tools (Dong et al. 2020). Besides, a reverse vaccinology in silico study employed the viral N protein, ORF3a, and M protein to achieve a multi-epitope vaccine construct (Enayatkhani et al. 2020). In this study, various epitopes found on S, N, and M proteins along with ORF3a were employed simultaneously as a novel approach for designing a multi-epitope vaccine against SARS-CoV-2 to potentially enhance its antigenicity and efficacy. N-glycans present in the structure of viral S protein, play a crucial role in modulation of RBD conformational dynamics thereby facilitating ACE2 receptor recognition by the S protein (Casalino et al. 2020). The SARS-CoV-2 N protein, which takes part in RNA packaging processes, also possesses immunological value, since immunological assays provided data that showed the presence of antibodies against the N protein (Zeng et al. 2020). The viral M protein also participates in virus assembly (Masters 2006). As indicated in previous studies, ORF3a of SARS-CoV is a transmembrane protein consisting of various motifs that provide pro-apoptotic functions (Chan et al. 2009). Moreover, a recent study carried out in China pointed out the structural similarities between ORF3a proteins of SARS-CoV and SARS-CoV-2 and indicated that the ORF3a of SARS-CoV-2 also induced cellular apoptosis (Ren et al. 2020). It was shown that ORF3a accessory protein stimulates signals for NLRP3 (NOD- [nucleotide-binding domain], LRR- [leucine-rich repeat], and pyrin domain-containing protein 3) inflammasome activation and induces the production of IL-1β through induction of the pro-IL-1β gene (Siu et al. 2019). IL-1β, a critical pro-inflammatory cytokine, is secreted in the early stages of the viral infection (De Lang et al. 2007). Furthermore, ORF3a induces in vitro neutralizing antibodies against SARS-CoV-2 (Qiu et al. 2005). Hence, we assumed that using the epitopes on four different viral proteins that are important in pathogenesis could not only grant the vaccine more antigenicity, but also help the vaccine to maintain its efficiency even in the case of viral mutations. Obviously, SARS-CoV-2 as an RNA virus bears a high chance of mutations. Given that mutations may not occur in all the viral proteins at the same time, this is a very useful logical strategy for achieving an efficient vaccine.
In this study, overlapping epitopes were selected from different immunological classes of B-cell and T-cell stimulating antigens through using various servers. It was found that the combination of several promising epitopes in a single peptide construct helps address the low immunogenicity problem associated with peptide vaccines (Abraham et al. 2020, Livingston et al. 2002). Hence, in addition to employment of several T- and B-cell inducing epitopes, the presence of IFN-gamma inducing epitopes was also affirmed. Some cytokines, especially IFN-gamma, play an important role in mediating protection (Samuel 2001).
Moreover, addition of LL37 (residues 17–29), as a TLR3 agonist adjuvant, could reinforce its ability in stimulation of innate immune responses leading to more potent adaptive immune responses. An increasing trend is found in using TLR agonists as potential built-in adjuvants for the development of novel vaccines. TLRs, as a family of pattern recognition receptors (PRRs), recognize conserved pathogenic structures, such as pathogen-associated molecular patterns (PAMPs) (Suresh and Mosser, 2013), and expedite antigen presentation on the cell surface, thereby further promoting the immune response to the vaccine construct (Moyle and Toth, 2013). A number of TLR ligands, such as TLR3, TLR4, TLR5, and TLR9 (Reed et al. 2016), have been identified as adjuvants, which could enhance CTL response via CD4 + T-helper cells (van der Burg et al. 2006). TLR signaling could be carried out by five different adapter proteins (MyD88, MAL, TRIF, TRAM, and SARM). In this regard, TLR3 only signals through TRIF and TRAM protein adapters (O’Neill and Bowie, 2007).
In regards to adjuvant, a TLR-4 agonist, 50 S ribosomal protein L2 was used in a vaccine designed against SARS-CoV-2 (Yang et al. 2021), while 50 S ribosomal protein L7/L12, was employed for the same purpose in another study (Samad et al. 2020). Selection of LL37 as a TLR3 agonist in our study was based on the following rational findings about role of TLR3 in coronaviruses and the nature of LL37. It was confirmed that TLR3 is able to induce strong immune responses against viruses, especially MERS-CoV (Middle East respiratory syndrome coronavirus) and SARS-CoV (Lester and Li 2014). LL37 is an antimicrobial protein with the ability to modulate dendritic cell (DC) activity and also chemotactic properties. LL37 could amplify antigen-specific immune responses (An et al. 2005; Kim et al. 2015) by facilitating viral responses through the activation of TLR3 signaling (Lai et al. 2011). Accordingly, LL37 was incorporated into the designed vaccine sequence to improve the immunogenicity of the construct.
For joining different segments of the vaccine construct, several linkers, namely GSGSGS, AYYKK, and EAAAK, were used. Linkers facilitate antigen processing and avoid formation of the “junctional epitopes”, which could alter the structure of the peptide vaccine (Livingston et al. 2002). The GSGSGS linker allows relatively free rotation of the segments in the vaccine construct. Small amino acids such as glycine and serine provide flexibility for the peptide vaccine (Negahdaripour et al. 2017a). The KKAAY linker increases the probability of epitope presentation in both MHC I and II pathway (Vakili et al. 2018), and EAAAK is a rigid linker that causes a more effective separation between the functional domains (Arai et al. 2004). Thus, these different linkers were employed in various parts of the designed structure. Such linkers are usually employed in different computationally designed vaccines (Hajighahramani et al. 2019, Samad et al. 2020, Yang et al. 2021).
Our designed candidate vaccine comprised of 157 amino acids, which possessed acceptable immunological and structural properties according to the physicochemical results. The length of multi-epitope peptide vaccines could be very different mostly based on the choice of the designers. For example, multi-epitope SARS-CoV-2 vaccines designed by Dong et al. and Yang et al. had a length of 864 and 649 amino acids, respectively (Dong et al. 2020, Yang et al. 2021). Of note, some considerations in respect to convenient cloning, less possibility of conformational instability and huge variations, and formulation concerns should be taken into account in this regard.
The instability index, half-life, and theoretical PI revealed that the peptide vaccine was sTable Furthermore, the construct was soluble, immunogen, and nonallergic (as mentioned in Section 3.2), which means that the designed peptide could be proposed as an appropriate vaccine candidate.
The model refinement and validation of the initial tertiary structure of the vaccine helped to improve the quality of the refined model remarkably. Additionally, results of the docking study, as seen in Fig. 3, indicated that the designed vaccine had a significant affinity towards the TLR3.
Finally, the CAI and GC content of the codon optimized gene sequence suggested ideal expression of the recombinant protein in E. coli host.
All in all, the proposed multi-epitope peptide vaccine is expected to induce robust humoral and cellular immune responses against the virus due to all the mentioned acceptable immunological, structural, and physicochemical properties. Selecting epitopes from three viral structural proteins and another functional protein suggests the vaccine’s protective ability irrespective of probable virus mutations. However, this candidate vaccine construct should be further investigated in a course of various studies including in vitro, in vivo, and clinical trials, respectively, to confirm its efficacy and safety. Application of carriers could also further improve its antigenicity in future evaluations.
Supplementary Information
(PDF 124 kb)
{kind=link}
ProSA validation of the initial (a) and final (b) 3D structure model of peptide vaccine; Z-Score plot contains the z-scores of all experimental protein chains in the PDB database, determined by NMR spectroscopy (dark blue) and X-ray crystallography (light blue). The z-score of the designed peptide vaccine is illustrated in the large black dot. The plot shows results with a z-score ≤ 10. The Z-score of the final models after refinement was −6.25, which is in the range of native protein conformations (PNG 53376 kb)
{kind=link}
Verify3D results for validation of the initial (a) and final (b) 3D structure models of the peptide vaccine. The Verify 3D score of the refined model was 80.89%. Residues with an average 3D-1D score greater than zero are considered reliable (PNG 67312 kb)
Abbreviations
- ACE2
Angiotensin converting enzyme-2
- ANN
Artificial neural network
- CAI
Codon adaptation index
- CFD
Codon frequency distribution
- COVID-19
Coronavirus disease 2019
- C-score
Confidence score
- CTL
Cytotoxic T-cell
- DC
Dendritic cell
- E
Envelope protein
- EUA
Emergency use authorization
- FDA
Food and Drug Administration
- FFT
Fast fourier transform
- GRAVY
Grand average of hydropathicity
- HLA
Human leukocyte antigen
- IEDB
Immune epitope databases
- IFN
Interferon
- IL
Interleukin
- kNN
K nearest neighbors
- LRR
Leucine-rich repeat
- M
Membrane protein
- MAL
MyD88-adaptor-like protein
- MERS-CoV
Middle East respiratory syndrome coronavirus
- MHC
Major histocompatibility complex
- MyD88
Myeloid differentiation primary response 88
- MW
Molecular weight
- N
Nucleocapsid protein
- NCBI
National Centre for Biotechnology Information
- NLRP3
NOD-, LRR- and pyrin domain-containing protein 3
- NOD
Nucleotide-binding domain
- nsp
Nonstructural protein
- ORF
Open reading frame
- PAMPs
Pathogen-associated molecular patterns
- PRRs
Pattern recognition receptors
- pI
isoelectric point
- ProSA-web
Protein Structure Analysis
- QM
Quantitative matrix
- RAMPAGE
Ramachandran Plot Assessment
- RBD
Receptor binding domain
- RMSD
Root mean square deviation
- S
Spike protein
- SARM
Sterile a- and armadillo-motif-containing protein
- SARS-CoV-2
Severe acute respiratory syndrome coronavirus-2
- SDU
Semidefinite Programming-Based Underestimation
- TLR
Support vector machine (SVM); Toll-like receptor
- TRIF
TIR-domain-containing adaptor protein inducing IFN-β
- TRAM
TRIF-related adaptor molecule
Funding
This study was supported by Grant No. 98-01-106-22099 from the Research Council of Shiraz University of Medical Sciences, Shiraz, Iran.
Data availability
More data are available as supplementary materials.
Declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
The approval code of IR.SUMS.REC.1399.056 is dedicated to this project by Shiraz University of Medical Sciences.
Informed consent
Not applicable, since no patient was investigated in this study.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Bahareh Vakili and Ashkan Bagheri contributed equally to this work.
References
- Abraham Peele K, Srihansa T, Krupanidhi S, Ayyagari VS, Venkateswarulu TC (2020) Design of multi-epitope vaccine candidate against SARS-CoV-2: a in-silico study. J Biomol Struct Dyn:1–9. 10.1080/07391102.2020.1770127 [DOI] [PMC free article] [PubMed]
- An LL, Yang YH, Ma XT, Lin YM, Li G, Song YH, Wu KF. LL-37 enhances adaptive antitumor immune response in a murine model when genetically fused with M-CSFRJ6-1 DNA vaccine. Leuk Res. 2005;29:535–543. doi: 10.1016/j.leukres.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Arai R, Wriggers W, Nishikawa Y, Nagamune T, Fujisawa T. Conformations of variably linked chimeric proteins evaluated by synchrotron X-ray small‐angle scattering. Proteins. 2004;57:829–838. doi: 10.1002/prot.20244. [DOI] [PubMed] [Google Scholar]
- Ashok A, Brison M, LeTallec Y. Improving cold chain systems: Challenges and solutions. Vaccine. 2017;35(17):2217–2223. doi: 10.1016/j.vaccine.2016.08.045. [DOI] [PubMed] [Google Scholar]
- Bagheri A, Moezzi SMI, Mosaddeghi P, Parashkouhi SN, Hoseini SMF, Badakhshan F, Negahdaripour M (2020) Interferon-inducer antivirals: potential candidates to combat COVID-19. Int Immunopharmacol:107245. 10.1016/j.intimp.2020.107245 [DOI] [PMC free article] [PubMed]
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7:153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L, Gaieb Z, Goldsmith JA, Hjorth CK, Dommer AC, Harbison AM, Fogarty CA, Barros EP, Taylor BC, McLellan JS. Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein. ACS Cent Sci. 2020;6:1722–1734. doi: 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CM, Tsoi H, Chan WM, Zhai S, Wong CO, Yao X, Chan WY, Tsui SKW, Chan HYE. The ion channel activity of the SARS-coronavirus 3a protein is linked to its pro-apoptotic function. Int J Bioch Cell Biol. 2009;41:2232–2239. doi: 10.1016/j.biocel.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman RL, Sher A, Seder RA. Vaccine adjuvants: Putting innate immunity to work. Immunity. 2010;33(4):492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lang A, Baas T, Teal T, Leijten LM, Rain B, Osterhaus AD, Haagmans BL, Katze MG. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS-CoV–infected macaques. PLOS Pathog. 2007;3:e112. doi: 10.1371/journal.ppat.0030112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v.2–a server for in silico prediction of allergens. J Mol Model. 2014;20:2278. doi: 10.1007/s00894-014-2278-5. [DOI] [PubMed] [Google Scholar]
- Dong R, Chu Z, Yu F, Zha Y. Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front Immunol. 2020;11:1784. doi: 10.3389/fimmu.2020.01784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8(1):4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C, Günther S, Preiser W, van der Werf S, Brodt H-R, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RAM, Berger A, Burguière A-M, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra J-C, Müller S, Rickerts V, Stürmer M, Vieth S, Klenk H-D, Osterhaus ADME, Schmitz H, Doerr HW. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/nejmoa030747. [DOI] [PubMed] [Google Scholar]
- EL-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B‐cell epitopes using string kernels. J Mol Recognit. 2008;21:243–255. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayatkhani M, Hasaniazad M, Faezi S, Guklani H, Davoodian P, Ahmadi N, Einakian MA, Karmostaji A, Ahmadi K (2020) Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: an in silico study. J Biomol Struct Dyn:1–16. 10.1080/07391102.2020.1756411 [DOI] [PMC free article] [PubMed]
- FDA US (2020a) Pfizer-BioNTech COVID-19 vaccine. https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/pfizer-biontech-covid-19-vaccine#additional. Accessed 22 May 2021
- FDA US (2020b) Moderna COVID-19 vaccine. https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/moderna-covid-19-vaccine. Accessed 22 May 2021
- FDA US (2021) Janssen COVID-19 vaccine. https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/janssen-covid-19-vaccine. Accessed 22 May 2021
- Gasteiger E, Hoogland C, Gattiker A, Duvaud Se, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. Methods Mol Biol. 2005;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang XP, Liu YF, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu-Ozturk D, Wang HY, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, García-Sastre A, Shokat KM, Shoichet BK, Krogan NJ. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajighahramani N, Eslami M, Negahdaripour M, Ghoshoon MB, Dehshahri A, Erfani N, Heidari R, Gholami A, Nezafat N, Ghasemi Y. Computational design of a chimeric epitope-based vaccine to protect against Staphylococcus aureus infections. Mol Cell Probes. 2019;46:101414. doi: 10.1016/j.mcp.2019.06.004. [DOI] [PubMed] [Google Scholar]
- Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41(W1):W384–W388. doi: 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, McCullough MP, Chappell JD, Denison MR, Stevens LJ, Pruijssers AJ, McDermott A, Flach B, Doria-Rose NA, Corbett KS, Morabito KM, O’Dell S, Schmidt SD, Swanson PA, Padilla M, Mascola JR, Neuzil KM, Bennett H, Sun W, Peters E, Makowski M, Albert J, Cross K, Buchanan W, Pikaart-Tautges R, Ledgerwood JE, Graham BS, Beigel JH. An mRNA Vaccine against SARS-CoV-2 — Preliminary Report. N Engl J Med. 2020;383:1920–1931. doi: 10.1056/nejmoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur SP, Gupta V. COVID-19 Vaccine: A comprehensive status report. Virus Res. 2020;288:198114. doi: 10.1016/j.virusres.2020.198114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE, Nielsen M, Peters B. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40:W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Yang IY, Kim J, Lee KY, Jang YS. Antimicrobial peptide LL-37 promotes antigen‐specific immune responses in mice by enhancing Th17‐skewed mucosal and systemic immunities. Eur J Immunol. 2015;45:1402–1413. doi: 10.1002/eji.201444988. [DOI] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Beglov D, Brenke R, Comeau SR, Shen Y, Li K, Zheng J, Vakili P, Paschalidis IC, Vajda S. Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins. 2010;78:3124–3130. doi: 10.1002/prot.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516–527. doi: 10.1038/s41586-020-2798-3. [DOI] [PubMed] [Google Scholar]
- Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 2012;8:e1002829. doi: 10.1371/journal.pcbi.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Adhikarakunnathu S, Bhardwaj K, Ranjith-Kumar CT, Wen Y, Jordan JL, Wu LH, Dragnea B, San Mateo L, Kao CC. LL37 and cationic peptides enhance TLR3 signaling by viral double-stranded RNAs. PLoS One. 2011;6:e26632. doi: 10.1371/journal.pone.0026632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester SN, Li K. Toll-like receptors in antiviral innate immunity. J Mol Biol. 2014;426:1246–1264. doi: 10.1016/j.jmb.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston B, Crimi C, Newman M, Higashimoto Y, Appella E, Sidney J, Sette A. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol. 2002;168(11):5499–5506. doi: 10.4049/jimmunol.168.11.5499. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins. 2003;50(3):437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356(6364):83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- Mahase E. Covid-19: Moderna vaccine is nearly 95 % effective, trial involving high risk and elderly people shows. BMJ. 2020;371:m4471. doi: 10.1136/bmj.m4471. [DOI] [Google Scholar]
- Masters PS. The Molecular biology of coronaviruses. Adv Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel CJ, Mayer C, Poch O, Thompson JD. Characterization of accessory genes in coronavirus genomes. Virol J. 2020;17:131. doi: 10.1186/s12985-020-01402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosaddeghi P, Shahabinezhad F, Dorvash M, Goodarzi M, Negahdaripour M (2020) Harnessing the non-specific immunogenic effects of available vaccines to combat COVID-19. Hum Vaccines Immunother:1–12. 10.1080/21645515.2020.1833577 [DOI] [PMC free article] [PubMed]
- Moyle PM, Toth I. Modern subunit vaccines: development, components, and research opportunities. ChemMedChem. 2013;8(3):360–376. doi: 10.1002/cmdc.201200487. [DOI] [PubMed] [Google Scholar]
- Negahdaripour M (2020) The battle against COVID-19: Where do we stand now? IJMS 45(2):81. 10.30476/ijms.2020.46357 [DOI] [PMC free article] [PubMed]
- Negahdaripour M. COVID-19 vaccine global access is an urgency. IJMS. 2021;46(2):79–80. doi: 10.30476/ijms.2021.47336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negahdaripour M, Eslami M, Nezafat N, Hajighahramani N, Ghoshoon MB, Shoolian E, Dehshahri A, Erfani N, Morowvat MH, Ghasemi Y. A novel HPV prophylactic peptide vaccine, designed by immunoinformatics and structural vaccinology approaches. Infect Genet Evol. 2017;54:402–416. doi: 10.1016/j.meegid.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Negahdaripour M, Golkar N, Hajighahramani N, Kianpour S, Nezafat N, Ghasemi Y. Harnessing self-assembled peptide nanoparticles in epitope vaccine design. Biotechnol Adv. 2017;35(5):575–596. doi: 10.1016/j.biotechadv.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negahdaripour M, Nezafat N, Eslami M, Ghoshoon MB, Shoolian E, Najafipour S, Morowvat MH, Dehshahri A, Erfani N, Ghasemi Y. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect Genet Evol. 2018;58:96–109. doi: 10.1016/j.meegid.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Negahdaripour M, Nezafat N, Heidari R, Erfani N, Hajighahramani N, Ghoshoon MB, Shoolian E, Rahbar MR, Najafipour S, Dehshahri A. Production and preliminary in vivo evaluations of a novel in silico-designed L2-based potential HPV vaccine. Curr Pharm Biotechnol. 2020;21:316–324. doi: 10.2174/1389201020666191114104850. [DOI] [PubMed] [Google Scholar]
- O’Hagan DT, De Gregorio E. The path to a successful vaccine adjuvant - ‘The long and winding road’. Drug Discov Today. 2009;14:541–551. doi: 10.1016/j.drudis.2009.02.009. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Owji H, Negahdaripour M, Hajighahramani N. Immunotherapeutic approaches to curtail COVID-19. Int Immunopharmacol. 2020;88:106924. doi: 10.1016/j.intimp.2020.106924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu M, Shi Y, Guo Z, Chen Z, He R, Chen R, Zhou D, Dai E, Wang X, Si B. Antibody responses to individual proteins of SARS coronavirus and their neutralization activities. Microb Infect. 2005;7:882–889. doi: 10.1016/j.micinf.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy MN, Minassian AM, Ewer KJ, Flaxman AL, Folegatti PM, Owens DR, Voysey M, Aley PK, Angus B, Babbage G. Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): a single-blind, randomised, controlled, phase 2/3 trial. Lancet. 2021;396:1979–1993. doi: 10.1016/S0140-6736(20)32466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SG, Hsu F-C, Carter D, Orr MT. The science of vaccine adjuvants: advances in TLR4 ligand adjuvants. Curr Opin Immunol. 2016;41:85–90. doi: 10.1016/j.coi.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, Han Y, Zhang XY, Zhou W, Qiu Y, Zhou X. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol. 2020;17:881–883. doi: 10.1038/s41423-020-0485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Raghava GPS. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34:W202–W209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samad A, Ahammad F, Nain Z, Alam R, Imon RR, Hasan M, Rahman MS (2020) Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J Biomol Struct Dyn 1–17. 10.1080/07391102.2020.1792347 [DOI] [PMC free article] [PubMed]
- Samuel CE (2001) Antiviral actions of interferons. Clin Microbiol Rev 14:778–809 [DOI] [PMC free article] [PubMed]
- Shin WH, Lee GR, Heo L, Lee H, Seok C. Prediction of protein structure and interaction by GALAXY protein modeling programs. Bio Design. 2014;2:1–11. [Google Scholar]
- Siu KL, Yuen KS, Castaño-Rodriguez C, Ye ZW, Yeung ML, Fung SY, Yuan S, Chan C-P, Yuen KY, Enjuanes L. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33:8865–8877. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inform. 2015;53:405–414. doi: 10.1016/j.jbi.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Suresh R, Mosser DM. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv Physiol Educ. 2013;37:284–291. doi: 10.1152/advan.00058.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakili B, Eslami M, Hatam GR, Zare B, Erfani N, Nezafat N, Ghasemi Y. Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int J Biol Macromol. 2018;120:1127–1139. doi: 10.1016/j.ijbiomac. [DOI] [PubMed] [Google Scholar]
- van der Burg SH, Bijker MS, Welters MJ, Offringa R, Melief CJ. Improved peptide vaccine strategies, creating synthetic artificial infections to maximize immune efficacy. Adv Drug Deliv Rev. 2006;58:916–930. doi: 10.1016/j.addr.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Walsh EE, Frenck RW, Falsey AR, Kitchin N, Absalon J, Gurtman A, Lockhart S, Neuzil K, Mulligan MJ, Bailey R, Swanson KA, Li P, Koury K, Kalina W, Cooper D, Fontes-Garfias C, Shi P-Y, Türeci Ö, Tompkins KR, Lyke KE, Raabe V, Dormitzer PR, Jansen KU, Şahin U, Gruber WC. Safety and immunogenicity of two RNA-based Covid-19 vaccine candidates. N Engl J Med. 2020 doi: 10.1056/NEJMoa2027906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):e1000048–e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO website. https://covid19.who.int/. Accessed 22 May 2021
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43:W174–W181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Bogdan P, Nazarian S (2021) An in-silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci Rep 11. 10.21203/rs.3.rs-36528/v1 [DOI] [PMC free article] [PubMed]
- Zeng W, Liu G, Ma H, Zhao D, Yang Y, Liu M, Mohammed A, Zhao C, Yang Y, Xie J. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem Biophys Res Commun. 2020;527:618–623. doi: 10.1016/j.bbrc.2020.04.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 124 kb)
ProSA validation of the initial (a) and final (b) 3D structure model of peptide vaccine; Z-Score plot contains the z-scores of all experimental protein chains in the PDB database, determined by NMR spectroscopy (dark blue) and X-ray crystallography (light blue). The z-score of the designed peptide vaccine is illustrated in the large black dot. The plot shows results with a z-score ≤ 10. The Z-score of the final models after refinement was −6.25, which is in the range of native protein conformations (PNG 53376 kb)
Verify3D results for validation of the initial (a) and final (b) 3D structure models of the peptide vaccine. The Verify 3D score of the refined model was 80.89%. Residues with an average 3D-1D score greater than zero are considered reliable (PNG 67312 kb)
Data Availability Statement
More data are available as supplementary materials.