

Abstract
Human ornithine aminotransferase (hOAT) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme that was recently found to play an important role in the metabolic reprogramming of hepatocellular carcinoma (HCC) via proline and glutamine metabolic pathways. The selective inhibition of hOAT by compound 10 exhibited potent in vivo antitumor activity. Inspired by the discovery of aminotransferase inactivator (1S,3S)-3-amino-4-(difluoromethylene)cyclopentane-1-carboxylic acid (5), we rationally designed, synthesized, and evaluated a series of six-membered ring analogs. Among them, 14 was identified as a new selective hOAT inactivator, which demonstrated 22 times greater potency than 10. Three different types of protein mass spectrometry and two crystallographic approaches were employed to identify the structure of hOAT-14 and the formation of a remarkable final adduct (28`) in the active site. These spectral studies reveal an enzyme complex, heretofore not observed in a PLP-dependent enzyme, with covalent bonds to two nearby residues. Crystal soaking experiments and molecular dynamics simulations were carried out to identify the structure of active site intermediate 27`, and to elucidate the order of the two covalent bonds formed, leading to 28’. The initial covalent reaction of the activated warhead occurs with *Thr322 from the second subunit, followed by a subsequent nucleophilic attack by catalytic residue Lys292. The turnover mechanism of 14 by hOAT also was supported by mass spectrometric analysis of metabolites and fluoride ion release experiments. This novel mechanism for hOAT with 14 will contribute to the further rational design of selective inactivators and an understanding of potential inactivation mechanisms by aminotransferases.
Keywords: Mechanism-based inactivators, Covalent inhibitors, Inactivation mechanism, Top-down proteomics, Human ornithine aminotransferase, Hepatocellular carcinoma
Graphical Abstract
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common primary liver cancer and the second leading cause of cancer death worldwide.1–4 The disease is highly malignant, radiotherapy-resistant, and refractory to chemotherapy, including the standard-of-care treatment, sorafenib.5–8 Recently, human ornithine aminotransferase (hOAT) was identified as a metabolic regulator of HCC progression via proline and glutamine metabolic pathways.9, 10 OAT (E.C. 2.6.1.13) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme,11 which is found in the mitochondrial matrix of most human and animal tissues.12 Mechanistically, two coupled half-reactions are required to complete one transamination cycle of OAT (Scheme 1).13 In the first half-reaction, ornithine binds to cofactor PLP in the active site of OAT to form Schiff base M1, followed by tautomerization to intermediate M2; the subsequent hydrolysis of M2 generates pyridoxamine 5′-phosphate (PMP) and glutamate γ-semialdehyde (GγS), which spontaneously cyclizes to Δ1-pyrroline-5-carboxylate (P5C). In the second-half reaction, OAT catalyzes the conversion of α-ketoglutarate (α-KG) and PMP to L-glutamate (L-Glu) and PLP. Glutamate and glutamine metabolism have acknowledged roles in supporting the anabolic demands of tumor growth and it is increasingly clear that proline metabolism promotes liver cancer tumorigenesis.14–17 Not only L-Glu and proline have direct effects on metabolic rewiring, but they also induce a hypoxia-inducible factor-1α (HIF1α) transcriptional program in HCC leading to sorafenib resistance and retained proliferative capacity under oxygen starvation.18 hOAT is commonly overexpressed in HCC as a result of dysregulation and activation of the Wnt/β-catenin pathway.19, 20 The selective inhibition of hOAT exhibited potent in vivo anti-tumor activity in the HCC mouse model, along with dramatically reduced alpha-fetoprotein (AFP, a biomarker for HCC) levels.10 Recently, upregulation of hOAT was found in non-small cell lung cancer (NSCLC) cells, which contributes to the promotion of proliferation, invasion, and migration, the inhibition of apoptosis, and the change of the cell cycle.21 The specific knockdown of hOAT in NSCLC inhibited cell proliferation in vitro and suppressed tumor growth in vivo.21 Thus, hOAT can serve as a promising therapeutic target for the treatment of HCC and related cancers with similar underlying metabolic alterations.
Scheme 1.

Catalytic Mechanism of OAT
OAT belongs to the same enzyme subgroup22 as γ-aminobutyric acid aminotransferase (GABA-AT, E.C. 2.6.1.19), which demonstrates a similar catalytic mechanism to that of OAT,23 in which GABA is converted to succinic semialdehyde (SSA) in the first-half reaction.24 In past decades, our laboratory has been focusing on the rational design of mechanism-based inactivators (MBIs) of GABA-AT25 for the potential treatment of epilepsies and addictions. MBIs initially act as substrates for a target enzyme, but after enzyme-catalyzed modification to an active intermediate, they inhibit the target enzymes;25–28 often they demonstrate higher potency and reduced off-target effects when compared with traditional inhibitors. 10, 25, 29, 30 Vigabatrin (1)31, an MBI of GABA-AT, is an FDA-approved drug for the treatment of infantile spasms and refractory complex partial seizures (Scheme 2).32 A mechanistic study33 revealed that 1 initially forms a Schiff base with PLP in the active site of GABA-AT, followed by tautomerization, as with the native substrate, to afford intermediate 3. Lys329, the residue that initially is bound to PLP, attacks the Michael acceptor of 3 to form covalent adduct 4, which accounts for 70% of the inactivation pathway of 1 (Scheme 2).33 On the basis of this mechanism, difluoro-substituted conformationally-rigid compound 5 was synthesized and exhibited 22 times higher efficiency as an inactivator of GABA-AT than 1, determined by their kinact/KI values (Scheme 2).34, 35 Compound 5 produced Michael acceptor intermediate 7 via similar steps as shown for the mechanistic pathway of 1.36 However, highly electrophilic 7 reacted with water molecules in the pocket, instead of Lys329, leading to the formation of tight-binding adduct 8, in which the newly generated carboxylate forms an electrostatic interaction with Arg445 (Scheme 2).36 Compound 5 has been investigated in a clinical trial and is successfully treating a child with infantile spasms.37, 38 Cyclopentene analog 9 (Figure 1) was found to have enhanced improved potency toward GABA-AT (kinact/KI = 342 mM−1min−1) and to undergo the same mechanism as 5, in which the nucleophilic attack of water molecules was potentially assisted by the catalytic lysine.29 Because of the high similarity of the active sites of these two aminotransferases, 9 also exhibited inhibitory activity of hOAT, but with relatively lower efficiency (kinact/KI = 7.6 mM−1min−1).29
Scheme 2.

Inactivation mechanisms of GABA-AT by vigabatrin (1) and CPP-115 (5)
Figure 1.

Structures of analogs 9–15
When comparing the active sites of hOAT and GABA-AT for differences, hOAT has a more flexible and larger active site, resulting from the replacement of Phe351 and Ile72 in GABA-AT with Tyr55 and Tyr85 in hOAT, which can accommodate ornithine that is one carbon longer than GABA.39 In 2015, the relatively bulky bis(trifluoromethyl) analog 10 (Figure 1) was found to show much improved selectivity for hOAT over GABA-AT.10 Furthermore, administration of 10 at low doses (0.1 and 1.0 mg/kg) was shown to effectively reduce AFP levels and suppress HCC tumor growth in vivo.10 Recently, the enlarged-ring strategy has been successful in improving potency and selectivity of hOAT inactivators, leading to the discovery of cyclohexene analogs 11a and 11b (Figure 1).40
Herein, we have designed and synthesized six-membered ring analogs 12-15 based on GABA-AT inactivators 5 and 9 (Figure 1). Among them, analog 14 is 22 times more efficient as an inactivator of hOAT than 10, along with excellent selectivity over other aminotransferases. We also elucidated the inactivation mechanism of 14 with the employment of several crystallography and mass spectrometry methods, leading to the identification of a remarkable, but unexpected, covalent adduct attached to two residues from two different chains in the hOAT active site. In addition, the turnover mechanism was shown to generate an aromatic metabolite. These studies have revealed a potent and selective hOAT inactivator for the treatment of HCC and uncovered its underlying mechanism of inactivation for further improved rational inactivator design.
RESULTS AND DISCUSSION
Syntheses of Analogs 12–15
The synthetic route to 12–15 is shown in Scheme 3. Chirally-pure lactam 1641 was prepared via a reported procedure. Intermediate 16 was treated with silver trifluoroacetate, followed by Dess-Martin oxidation, to afford ketones 17a and 17b. Intermediate 18 was obtained from 17b with the protection of PMB, followed by the treatment34, 35 of CHF2PO(OEt)2 and tBuLi, to provide intermediate 19. Deprotection of PMB with CAN and the ring-opening reaction under acid conditions afforded the desired product 12. Chirally-pure ester 21 was prepared as reported,40 which was treated42 with 2-PySO2CF2H and tBuOK in DMF to give difluoromethylene intermediate 22. Interestingly, the treatment of 22 with PhSeCl and KHMDS afforded bicyclic lactam 23 as the major product. The sequential Boc protection, ring-opening reaction, and oxidative elimination provided two isomers 24a and 24b. The desired products 13–15 were afforded from the corresponding esters under acid conditions, respectively.
Scheme 3.

Syntheses of six-membered ring analogs 12–15
Reagents and conditions: (a) AgOC(O)CF3, CH3NO2, 0 °C – r.t., 16h; then NH3/MeOH, 1 h; (b) DMP, CH3CN, NaHCO3 (b) NaH, DMF; PMBCl, 0 °C – r.t., 16h; (c) CHF2PO(OEt)2, tBuLi (1.7 M in pentane), −100 °C – r.t., 2h; then reflux, 16h; (d) CAN, CH3CN, H2O, 0 °C - r.t., 10 h; (e) HCl (aq. 4 M), 75 °C, 5h. (e) 2-PySO2CF2H, tBuOK, DMF −60 °C, then −40 °C, 2h, NH4Cl (saturated aq.), HCl (3M), overnight; (f) PhSeCl, KHMDS (1M in THF), −78 °C – r.t., 3h; (g) Boc2O, DIPEA, DMAP, DCM, r.t., overnight; (h) K2CO3, EtOH, 0 °C - r.t. 3h; (i) m-CPBA, DCM, r.t., 3h; (j) HCl (aq. 4 M), AcOH, 80 °C, overnight.
Kinetic Studies of Analogs 12–15
As shown in Table 1, six-membered ring analogs 12–15 exhibited lower binding affinity to GABA-AT (greater KI values) than 5 or 9, which is consistent with our initial design strategy. The enlarged ring could potentially obstruct the initial binding with GABA-AT,40 which has a relatively small and rigid active site.39 Although cyclohexane analogs 12 and 13 showed enhanced selectivity for OAT, their potency was greatly decreased. The introduction of the double bond significantly increased their binding affinity to OAT, which may result from the reduced steric hindrance of cyclohexene.40 α, β-unsaturated analog 14 is more potent than cyclohexene analog 15, possibly because of the reduced acidity of the γ-position, which is critical for the tautomerization step and further inactivation.25
Table 1.
Kinetic constants for the inactivation of hOAT and GABA-AT by 1, 5, 9, 10, and 12–15a
| Comp. | hOAT | GABA-AT | ||||
|---|---|---|---|---|---|---|
| KI (mM) | kinact (min−1) | kinact/KI (mM−1min−1) | KI (mM) | kinact (min−1) | kinact/KI (mM−1min−1) | |
|
| ||||||
| 12 | 1.3 ± 0.2 | 0.1 ± 0.007 | 0.08 | 1.3 ± 0.4 | 0.03 ± 0.002 | 0.02 |
| 13 | 1.5 ± 0.2 | 0.1 ± 0.005 | 0.07 | > 13.1 | >0.2 | 0.02 ± 0.001b |
| 14 | 0.01 ± 0.002 | 0.2 ± 0.01 | 20.0 | > 1.2 | > 0.4 | 0.3 ± 0.01b |
| 15 | 0.1 ± 0.01 | 0.05 ± 0.002 | 0.5 | > 2.5 | > 0.2 | 0.09 ± 0.003 b |
| 5 29 | 0.12 | 0.1 | 0.8 | 0.06 | 2.1 | 34.9 |
| 9 29 | 0.003 | 0.03 | 7.6 | 0.01 | 3.3 | 342 |
| 10 40 | 0.07 | 0.06 | 0.9 | - | - | - |
| 1 40 | - | - | - | 0.3 | 0.2 | 0.7 |
kinact and KI values were determined by the equation: kobs = kinact*[I]/(KI +[I]) and are presented as means and standard errors.
The ratio of kinact/KI was determined by the slope of kobs = kinact*[I]/(KI +[I]). kinact is greater than the maximum kobs determined in the time-dependent assay; KI is greater than kobs(max)/the ratio of kinact/KI.
The most potent analog (14) is 22-fold more efficient (kinact/KI = 20.0 min−1mM−1) as an inactivator of hOAT than 10 (kinact/KI = 0.9 min−1mM−1) and has good selectivity over GABA-AT with lower efficiency constants (kinact/KI = 0.3 min−1mM−1). Compound 14 also exhibited little or no inhibitory activity against alanine aminotransferase (Ala-AT) or aspartate aminotransferase (Asp-AT), even at 20 mM concentration (Figure S1). Currently, in vitro and in vivo studies of 14 are ongoing in our lab for the potential treatment of HCC and other related cancers.
Inactivation Mechanism for 14
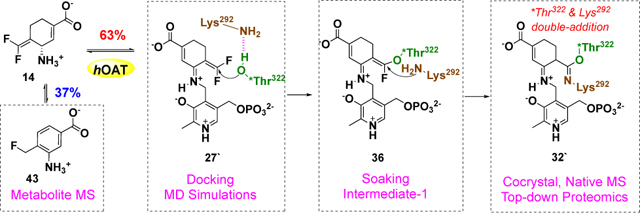
MBIs of aminotransferases inactivate these enzymes through a variety of mechanistic pathways, manifested by the formation of covalent or tight-binding adducts occupying the active site.25 On the basis of previous reports,33,36,43, 44 two reasonable mechanistic pathways (a and b) for the hOAT inactivation by 14 are proposed (Scheme 4). Initially, Schiff base 25 is formed from 14 and the Lys-PLP complex similar to that for the native substrate (Scheme 1), followed by deprotonation at the γ-position to 26. Mechanistic pathway a is similar to the mechanisms for 1 and 5 (Scheme 2), in which reactive Michael acceptor intermediate 27 is generated by protonation at the PLP-C4` position. Nucleophilic attack could then occur by water (Scheme 4, pathway a1) to give potential tight-binding adduct 28 or by Lys292 to give covalent complex 29. The fluorine-substituted olefin in 29 could further react (either directly or via the ketenimine) with a water molecule to afford adduct 30 (Scheme 4, pathway a2). In addition to water, several other nucleophilic residues (Nu) are nearby in the active site of hOAT, such as Tyr55, Tyr85, Glu235, and *Thr322 from the other chain. Although these noncatalytic residues have never been reported to be covalently modified in any inactivation mechanism for aminotransferases, it is possible that 29 could further react with one of them, leading to the formation of double-addition adduct 32 (Scheme 4, pathway a3). Mechanistic pathway b is proposed based on the inactivation mechanism of hOAT by gabaculine (Scheme S1),43, 44 a naturally occurring neurotoxin.45 Intermediate 33 could be formed by protonation at the difluoromethylenyl group of intermediate 26, followed by tautomerization via 34 to 35, a stable aromatic adduct with the PLP, similar to the product of inactivation by gabaculine. To determine the most likely mechanism for the inactivation of hOAT by 14, we carried out dialysis experiments, three types of protein mass spectral experiments, obtained a cocrystal structure and a soaking crystal structure, and conducted molecular docking and molecular dynamic simulation (MD) studies.
Scheme 4.

Possible inactivation mechanisms for 14
Dialysis
Previously, five-membered ring analogs 5 and 9 were identified as partially irreversible inhibitors of GABA-AT, leading to the formation of tight-binding adducts.29, 30 Thus, time-dependent reactivation experiments of hOAT for cyclohexene analog 14 were carried out to determine if similar reversible components were also involved during the inactivation. After hOAT was treated with 0.6–2.0 equivalents of 14, aliquots at different time intervals were taken and assayed for the return of enzyme activity. No matter how many equivalents of 14 were used, no enzyme activity was recovered, even after 91 h of dialysis (Figure S2), supporting a fully irreversible inhibition of hOAT.
Native Mass Spectrometry
To determine the mass of the potential covalent or tight-binding adduct in the active site of hOAT-14, we turned to native mass spectrometry (nMS), which preserves noncovalent substrate binding and protein quaternary structure.46 To evaluate the applicability of nMS for the study of MBIs, native untreated hOAT, hOAT-11b40, and hOAT-gabaculine44 samples were first used as positive controls. In these cases, the active site would be occupied by an unstable covalent adduct, a stable covalent adduct, and a tight-binding adduct, respectively. Theoretical mass shifts for control samples were calculated on the basis of known mechanisms; hOAT-14 mass shifts were calculated from the proposed inactivation pathways a1-a3 and b (Scheme 4, Figure S3). Data for protein dimers were deconvoluted to generate average dimer mass values (Figure S4). The experimental mass difference per monomer was calculated from the observed average mass of the apo-hOAT dimer (92275.0 Da). As shown in Table 2, independent of the binding mechanism for control samples, the experimental adduct masses are consistent with their theoretical masses. Among the proposed adducts for hOAT-14, only adduct 32 from pathway a3 (Scheme 4) shows a mass shift (378.1 Da) that matches the experimental result (378.3 Da), indicating the involvement of a second nucleophilic residue.
Table 2.
The mass differences for the proposed adducts between theoretical and experimental mass values of native/modified hOAT observed in native MS.
| Samples | Theoretical mass difference (per monomer, Da) | Experimental mass (dimer, Da) | Experimental mass difference (per monomer, Da) |
|---|---|---|---|
|
| |||
| Native hOAT | 231.0 | 92736.6 ± 1.8 | 230.8 |
| hOAT- gabaculine | 368.1 | 93014.8 ± 1.5 | 369.9 |
| hOAT-11b | 366.1 | 93009.2 ± 1.0 | 367.1 |
| hOAT-14 | 414.1 (28 in pathway a1) | 93031.6 ± 0.9 | 378.3 |
| 396.1 (30 in pathway a2) | |||
| 378.1 (32 in pathway a3) | |||
| 418.1 (35 in pathway b) | |||
Intact Protein Mass Spectrometry in Denaturing Mode
Protein mass spectrometry has served as an efficient tool for the identification of covalent adducts in studies of inactivation of aminotransferases.40,47, 48 The hydrolysis of unstable groups under acidic conditions, such as imine or enamine groups, has been observed with intact protein mass spectrometry to create masses that correspond to stable covalent adducts alone.42 Accordingly, unstable adducts for 14 may suffer from similar hydrolysis and release of PMP or result in cleavage of newly formed covalent bonds with Lys292/Nu. As shown in Figure S5, adducts and their corresponding theoretical mass differences for pathways a1-a3 and b are depicted with partial adduct loss. For the cases of pathway a1 or b, no mass shift should be observed for tight-binding adduct 28 or 35. A mass shift of +166.0 Da is expected with the hydrolysis of PMP from adduct 30 (pathway a2); a range of mass shifts per monomer (148.0–166.0 Da) is proposed for pathway a3 arising from different degrees of hydrolysis of adduct 32.
By intact protein mass spectrometry, native unmodified, dimeric hOAT presented a deconvoluted mass compatible with the monomeric protein under acidic spray conditions and reversed-phase nanocapillary high performance liquid chromatography (nano HPLC, Figure 2A). 40, 47 However, using the same analytical conditions, analysis of hOAT-14 yielded a deconvoluted mass consistent with dimeric, cross-linked hOAT, accounting for 82% of the total protein abundance (Figure 2B/C), suggesting covalent attachments to two residues, each from a different monomer in complex. Relative to the low abundance, unmodified dimer mass seen in the control (92275.0 ± 1.3 Da), a 305.0 Da mass shift was observed for the hOAT-14 species (92580.0 ± 2.1 Da) (Figure 2D). The observed mass shift is not consistent with any single adduct but may result from a mixture of adducts (296.1–314.1 Da) in pathway a3 (Scheme 4, Figure S5). To determine if modified, monomeric hOAT could be rescued from the observed dimer, the nanoHPLC analytical column temperature was increased from 25 °C to 55 °C. Subsequent analysis of hOAT-14 at 55 °C reduced the dimer to 33% relative abundance and increased the abundance of unmodified hOAT and two additional monomeric forms of the protein (46137.5 ± 1.0 Da; 46304.0 ±2.5 Da; 46470.0 ± 2.2 Da) (Figure 2C/E). The mass shifts of 166.5 Da and 332.5 Da observed for the two hOAT-14 monomer might be single (166.0 Da) and double adducts (2 ×166.0 Da, 332.0 Da), each attached to a single chain (Figure 2E), which would be consistent with the hydrolysis of adducts in pathway a3 (Scheme 4, Figure S5).
Figure 2.

Deconvoluted intact protein mass spectra for hOAT samples. (A) Unmodified hOAT. (B) hOAT-14 analyzed at 25 °C (left); hOAT-14 analyzed at 55 °C (right). (C) The ratio of monomer and dimer in samples as a percent of the total protein signal. (D) Expanded view of unmodified hOAT (red) and hOAT-14 at 25 °C from 92–93 kDa. (E) Expanded view of unmodified hOAT (red) and hOAT-14 at 55 °C from 46–46.6 kDa.
Top-down Mass Spectrometry
We next turned to tandem mass spectrometry (MS/MS) to fragment intact proteins and localize the 14-induced adduct within the hOAT primary sequence. Fragmentation of hOAT-14 was first assessed using electron transfer dissociation (ETD), as this method preserves weaker covalent interactions and cross-linked peptides, and minimizing further adduct loss was desirable in this study.49, 50 ETD fragmentation of unmodified control hOAT gave a set of 54 z-type ions that were readily mapped to the primary sequence as C-terminus containing fragments. In contrast, ETD fragmentation of hOAT-14 created a complex MS/MS (MS2) spectrum with only 6 z-type ions that all mapped to positions after Leu340 (Figure S6). ETD fragmentation of hOAT-14 is not consistent with the expected MS2 spectra for a single amino acid modification and suggests a more complex adduct, possibly because of the cross-linking from two different chains as observed in the intact protein MS (Figure 2). Therefore, as a complementary fragmentation method, higher-energy collisional dissociation (HCD)51 was applied to untreated control hOAT, hOAT-11b40 as a positive control, and hOAT-14 (Figure 3). HCD fragmentation of control hOAT generated y199 and y140 ions corresponding to the cleavage between Val240/Pro241 and Tyr299/Pro300. These ions were used as diagnostic fragments for residue adduction between Lys292 and the C- terminus (Figure 3B, Figure S7/S8). For hOAT-11b, a +366 Da mass shift was observed and could be mapped to y199, consistent with the reported adduct mass (Figure 3C).40 Neither y140 nor any other y-ions corresponding to the cleavage between Lys292 and Val339 were identified for hOAT-11b, possibly because of stabilizing interactions between 11b and active site residues. Nonetheless, fragmentation of hOAT-11b benchmarked these top-down fragmentation methods and their utility to retain and localize adducts. For hOAT-14, an addition of 166 Da in mass was observed on both y199 and y140 fragment ions (Figure 3D, Figure S7/S8). Given the mass shift on y140 and not for any subsequent y-type ions that report on more C-terminal residues (i.e., the y101 and smaller y-type fragment ions), 14-mediated adduction must occur between Pro300 and Glu338 (Figure 3D). Of the nucleophilic residues in the active site of hOAT (Tyr55, Tyr85, Glu235, Lys292, and *Thr322), only *Thr322 occurs in the above range.
Figure 3.

Localization of covalent modifications on hOAT. (A) Proposed structures and masses corresponding to hOAT adducts. HCD fragmentation maps for (B) unmodified hOAT, (C) hOAT-11b, and (D) hOAT-14, with associated scoring metric52 and modified residues colored for each adduct mass. Amino acid residues are numbered according to the full-length protein sequence (Uniprot accession: P04181) used in crystallography studies and not to the recombinantly expressed version of hOAT. For clarity, amino acid cleavage sites generating y199, y140, and y101 are shown in panel (B).
Collectively, the mass spectrometry supports a pathway where hOAT inactivated by 14 produces a covalent, dimeric protein complex, with modification of *Thr322. Native mass spectrometry studies support hOAT-14 adduction between *Thr322 and Lys292, leading to adduct 32 within ±1 Da mass accuracy for the protein dimer. This study also suggests that loss of the adduct occurs as a result of imine hydrolysis47, 48 under the acidic conditions of intact protein MS. Indeed, using a technique we previously developed,42 we could detect PMP as the adduct cofactor following untargeted metabolomics of the flowthrough from fully inactivated, previously desalted hOAT-14, treated with trifluoroacetic acid at 37 °C (Figure S9). A similar loss of adduct cofactor was previously observed when an imine bond was present.42 Together, native mass spectrometry and top-down mass spectrometry serve as tools to directly readout molecular details and thereby help the future investigation of MBI characterization.
Cocrystal structure of hOAT Inactivated by 14
Although the mass spectrometry strongly indicated the formation of adduct 32 (pathway a3, Scheme 4) in the active site of hOAT when inactivated by 14, top-down MS/MS was able to provide direct evidence for the covalent attachment to *Thr322 but not to catalytic residue Lys292. Hence, protein crystallography of hOAT inactivated by 14 was employed to interpret the adduct formed in the active site. The cocrystal structure of hOAT inactivated by 14 was solved by molecular replacement using a monomer from a previously reported structure of hOAT53 (PDB code 1OAT), while water and ligand molecules were deleted. The refined model is shown in Figure 4: two covalent bonds are formed with Lys292 and *Thr322 of the other protein subunit, which is consistent with the mass spectrometry studies. The carboxylate group of the final adduct is located in proximity to Tyr55 with the formation of two hydrogen bonds. A similar arrangement is observed in the cocrystal structure of hOAT-11b. Considering the relative stability of the internal double bonds, adduct 32 was proposed initially, but tautomer 32` (Scheme S2), with an external double bond, was built in the model based on its better fit to the 2Fo-Fc map after refinement (Figure 4). Although the resolution of the cocrystal (1.95Å) does not allow a definitive conclusion regarding the enantiomeric form of 32`, the R-isomer was built into the model on the basis of its lower Rfree factor and better fit to the omit map.
Figure 4.

Omit map (Fo-Fc at 2.5 σ) of the hOAT-14 cocrystal structure. Black dashed lines indicate hydrogen bonds.
Previously, several hOAT inactivators40, 47, 54, 55 were found to form a covalent bond with Lys292, but no inactivator has been found to covalently interact with *Thr322 or any other noncatalytic residues. Given the surprising involvement of *Thr322 during the inactivation process, a multiple sequence alignment was performed for 223 manually annotated and reviewed ornithine aminotransferase enzymes. By this analysis, we find that the active site residue Lys292 and *Thr322 are conserved throughout evolutionary history down to E. coli, suggesting a conserved functional role for these residues (Figure S10A). In contrast, a similar analysis of 18 known human PLP-dependent aminotransferases (EC 2.6.1) demonstrated that Thr322 is poorly conserved across this family of aminotransferases in humans (Figure S10B). Catalytic residue Lys292 plays an important role in the protein native reaction, which is covalently bonded to PLP and located close to *Thr322 (3.8 Å), as shown in the holoenzyme structure of hOAT (Figure S11A). Cocrystal44 structures of hOAT with gabaculine (PDB: 1GBN, Figure S11B) and L-canaline (PDB: 2CAN, Figure S11C) reveal a potential hydrogen bond between the free Lys292 and *Thr322 (2.8 Å), indicating *Thr322 could be “activated” during the native reaction or inactivation process.
Intermediate 27 was proposed to be the active Michael acceptor for the inactivation process, whose difluoromethylene warhead is positioned near the internal H-bond between the protonated imine and oxygen anion (Scheme 4). Surprisingly, the hOAT-14 complex demonstrates that the newly formed covalent linkage is moved to the phosphate side (Figure 4), suggesting an imine isomerization56–59 of the intermediate during the inactivation process. Arg413 is known to form a salt bridge with Glu235 before binding dicarboxylic substrates (L-Glu or α-KG),53 as shown in the crystal structures of native hOAT (Figure S11A) and inactivated hOAT (Figure S11B/C). Disruption of Arg-Glu salt bridges in aminotransferases was observed previously when the final adduct formed a H-bond with another arginine residue29, 36 or when there was full occupation of the active site by a bulky adduct (Figure S11D)47. In the case of hOAT-14 the Arg413-Glu235 salt bridge is disrupted and interactions of Arg413 with both Gln266 and Glu235 can be observed. One of the oxygens in the carboxylate group of Glu235 is 3.6 Å away from the amido group of the Gln266 side chain and potentially could also form a hydrogen bond with this residue. However, neither Arg413 nor Glu235 forms a H-bond with the adduct, and the space proximal to these residues remains unoccupied, indicating that this stable salt bridge might be broken during the inactivation process.
Molecular Docking and Molecular Dynamics (MD) Simulations
Although the structure of the final adduct for hOAT-14 is well supported by mass spectrometry and crystallography studies, the inactivation process is not quite clear. Two covalent bonds are formed with catalytic Lys292 and *Thr322 from the other chain, which potentially resulted from two nucleophilic attacks at the Michael acceptor intermediate (Scheme 4), but it cannot be concluded which covalent bond formed first. Catalytic Lys292 is more nucleophilic than *Thr322, which was found to react with active intermediates previously. However, the highly electrophilic difluoro Michael acceptor was previously found to react with water molecules in the active site of the aminotransferase (Scheme 2),29, 36 supporting its potential to react initially with *Thr322. The cocrystal structure of hOAT inactivated by 14 (Figure 4) provides some clues for further elucidation of the inactivation process. Imine isomerization appears to occur between proposed intermediate 27 and final adduct 32`, which might happen after the nucleophilic attacks by the two residues or result from rotation/tautomerization of adduct 32. It seems highly unlikely that after attachment of two residues a 180° bond rotation would occur readily. However, rotation prior to nucleophilic attack might be possible, transforming 46 to 47, then on to 27’, possibly resulting from steric hindrance between the fluorine of the warhead and the internal H-bond (Scheme S2). The salt-bridge between Arg413 and Glu235 becomes disrupted, as shown in the cocrystal structure (Figure 4), which might play an important role during the rotation and inactivation process. To evaluate the potential and sequence of the nucleophilic addition steps (Lys292 vs. *Thr322), molecular docking and molecular dynamics (MD) simulations of intermediates were conducted in the active site of hOAT, with salt bridge maintained and disrupted, respectively.
Intermediate 25 showed similar docking poses in the active site of hOAT with the salt bridge maintained (Figure S12A) and disrupted (Figure S12B), in which the γ-proton is positioned close to catalytic residue Lys292, facilitating the following deprotonation step. In addition, the salt bridge status demonstrated a significant effect on the docking poses of intermediates 27 and 27`. For the case of the salt bridge maintained, the carboxylate of intermediate 27 establishes hydrogen bonds with Tyr55, and the warhead is located close to Lys292, Arg413, and Glu235 (Figure S12C). No significant change was observed after a 25 ns MD simulation (Figure 5A), in which the CF is slightly closer to Lys292 (5.8 ± 0.4 Å) than to *Thr322 (7.0 ± 0.8 Å), with an average difference of 1.2 Å (Figure 5A). Considering the higher nucleophilicity of the amino group, Lys292 is expected to react with the warhead preferentially if this binding pose were presented (Figure 5A). Furthermore, rotamer intermediate 27` would not fit well if the salt bridge were maintained, in which case the PLP moiety would slip away from its original position.
Figure 5.

Final binding poses and average distances for the nucleophilic additions for 27 and 27` after 25 ns MD simulations. (A) MD simulation of 27 in the active site of hOAT with the intact salt bridge; (B) MD simulation of 27 in the active site of hOAT with the disrupted salt bridge; (C) MD simulation of 27` in the active site of hOAT with the disrupted salt bridge.
For the case where the salt bridge of hOAT is disrupted, there is a strong interaction between Arg413 and the carboxylate of intermediate 27 in the docking studies, which forces the warhead to rotate to the other side near Tyr55 and *Thr322 (Figure S12D). However, the MD simulation showed that it failed to maintain these interactions, and the warhead group stayed away from both Lys292 (7.4 ± 0.3 Å) and *Thr322 (7.3 ± 0.7 Å), indicating that inactivation could not be achieved with this binding pose (Figure 5B). However, molecular docking results of rotamer intermediate 27` showed that its carboxylate formed similar interactions with Arg413 with its internal H-bond maintained in the active site of hOAT when the salt bridge was disrupted (Figure S12E). MD simulations confirmed a stable interaction between 27` and surrounding residues, maintaining its warhead moiety positioned much closer to *Thr322 (3.4 ± 0.3 Å) than to Lys292 (5.9 ± 0.4 Å), which would favor a reaction with the threonine residue (Figure 5C). Notably, Lys292 is positoned near *Thr322 with an average distance of 3.4 ± 0.5 Å (Figure 5C), which is similar to that seen in the cocrystal structure of hOAT with tight-binding adducts (Figures S11B & S11C). Thus, we hypothesize that final adduct 32` could be generated from either intermediate 36 or 49, depending upon the order of nucleophilic additions (Scheme S2).
Soaking structure of hOAT inactivated by 14
As indicated in the above molecular docking and MD simulation studies, the order of nucleophilic addition by the two residues could be determined by different binding poses of 27 (Figure 5A, Figure S12C) and 27` (Figure 5C, Figure S12E), whose carboxylate interacts with either Tyr55 or Arg413 on the basis of the status of the Arg413/Glu235 salt bridge. Previously, we successfully obtained the active intermediate for hOAT inactivated by 10 via soaking experiments.60 To determine the dominant binding orientation, soaking experiments of hOAT with 14 were performed. Holoenzyme hOAT crystals were obtained within four days and then soaked with 5 mM 14 (1 μL added to a 4 μL hanging drop) for different time durations from 17 to 44 minutes. After soaking, the crystals were transferred to a cryo-protectant solution and flash-frozen in liquid nitrogen. The crystal soaked for 44 minutes diffracted to a resolution of 2.1 Å, and its structure was solved using molecular replacement and then compared to the hOAT-14. cocrystal structure.
Satisfyingly, intermediate 36 (Scheme S2) was trapped in the soaking study of hOAT and 14 (Figure 6) based on observed electron density, as found in one out of three copies in an asymmetric unit, in which the connection between the warhead and *Thr322 was confirmed prior to the attachment of Lys292. In the other two protein copies, relatively weak electron density was observed between the warhead and both Lys292 and *Thr322 (Figure S13), suggesting some proportion of Lys292 had already begun to react with the warhead of 36. Importantly, the Arg413-Glu235 salt bridge was disrupted, and the “free” arginine residue established a stable H-bond with the carboxylate of 36, which could explain the broken salt bridge in the cocrystal structure of hOAT inactivated by 14 (Figure 4). Overall, the soaking structure is quite similar to the docking pose of active intermediate 27` (Figure S12E), whose warhead is positioned close to *Thr322 with an internal H-bond maintained. Supported by the above experiments, adduct 36 is proposed to be formed by the initial nucleophilic attack of *Thr322 on the difluoromethylenyl group of intermediate 27`, followed by reaction of 36 with Lys292 to afford final adduct 32` (Scheme S2). This outcome suggests that the inactivation mechanistic pathway follows from rotation of 46 to 47 prior to nucleophilic attack by either active site residue.
Figure 6.

Omit map (Fo-Fc at 2.5 σ) of intermediate 36 within the active site of hOAT. Black dashed lines indicate hydrogen bonds.
Turnover Mechanism
MBIs act as substrates that bind at the active sites of target enzymes initially; therefore, it is common that many MBIs are converted to metabolites during the inactivation process.25 On the basis of the reported turnover mechanisms for 1 and 5 with GABA-AT, 33, 36 two possible turnover pathways for 14 by hOAT are proposed, with the release of PMP or PLP, respectively (Scheme 5). Lys292-assisted deprotonation of Schiff base 25 affords intermediate 26, followed by the protonation at different positions to give intermediate 27` (pathway a) or 33 (pathway b). Intermediate 27` could be further hydrolyzed with the release of PMP and metabolite 37 (pathway a1) or 38 (pathway a2), which is similar to the transamination of ornithine by hOAT (Scheme 1). The reaction of intermediate 33 with Lys292 could afford intermediate 39, followed by the release of PLP and metabolite 40 (pathway b1) or 42 (pathway b2) with hydrolysis to 44. Enamine 40 could be hydrolyzed to give ketone 41; imine 42 could undergo aromatization to generate metabolite 43, and aromatization of 44 would give 45. To identify which of the above-mentioned turnover pathways are involved, partition ratio and fluoride ion release experiments and mass spectrometric analysis of metabolites were carried out.
Scheme 5.

Possible turnover mechanisms for 14 by hOAT
Partition Ratio and Fluoride Ion Release
The partition ratio is used to determine the ratio of degradation to inactivation, calculated by titrating the enzyme with varying equivalents of inactivator. Ideally, a linear relationship (between enzyme activity remaining and equivalents of compound incubated) can be extrapolated to yield the exact equivalents (the intercept with the x-axis, turnover number) required to inactivate the enzyme completely. Since this number includes the one molecule of inactivator required to inactivate one enzyme monomer, the partition ratio is equal to the turnover number minus one. Therefore, the partition ratio of 14 (0.6) was determined by titrating hOAT with varying equivalents of inactivators (Figure S14).
Different equivalents of fluoride ions can be released in PLP and PMP turnover pathways. If PMP is formed in the turnover mechanism, it cannot be converted back to PLP in the absence of α-KG, which results in, at most, two equivalents of fluoride ions released (pathway a, Scheme 5); more equivalents of fluoride ion would be released in the presence of α-KG if PMP is formed after the fluoride ion release step (pathway a1, Scheme 5). If PLP is regenerated during the turnover mechanism, at least two equivalents of fluoride ion will be released in the absence of α-KG (pathway b, Scheme 5 in addition to the fluoride ion released for inactivation), which should be close to the equivalents in the presence of α-KG. On the basis of the partition ratio and the confirmed inactivation mechanism for 14, the theoretical equivalents of fluoride ions that are released per enzyme active site during different turnover pathways can be calculated in the presence and absence of α-KG (Table 3). The concentration of released fluoride ions can be monitored by a fluoride ion-selective electrode. When hOAT was inactivated with an excess of 14, 2.9 equivalents of fluoride ions were released in the presence of α-KG and 2.7 equivalents of fluoride ions were released in the absence of α-KG (Table 3). These results indicate that the regeneration of PLP is involved in the turnover mechanism for 14, in which the experimental fluoride ions released are close to the theoretical calculations for turnover pathway b2 (Scheme 5).
Table 3.
Fluoride ion release during different turnover pathways with and without α-KG
| Conditions | Pathway a1 | Pathway a2 | Pathway b1 | Pathway b2 | Experimental |
|---|---|---|---|---|---|
|
| |||||
| With α-KG | 3.2 equiv | 2.0 equiv | 2.0 equiv | 2.6 equiv | 2.9 equiv |
| Without α-KG | 2.0 equiv | 1.3 equiv | 2.0 equiv | 2.6 equiv | 2.7 equiv |
Mass Spectrometry-Based Analysis of Turnover Products
To identify the potential metabolites generated during the inactivation process of hOAT by 14, the inactivated hOAT sample was filtered through a 10 kDa MWCO filter, followed by the use of untargeted LC-HRMS with alternating +/− mode electrospray to analyze the filtrate obtained. Among the possible metabolites proposed in Scheme 5, only metabolite 43 (observed: 170.0610 m/z, [M+H]+; theoretical: 170.0612 m/z) in pathway b2 was detected, followed by the confirmation of its fragmentation spectrum (Figure 7). Notably, metabolite 45 in pathway b2 was not observed by HRMS, indicating the aromatization step is much faster than the hydrolysis step. Precursor 42 was generated in the active site that is not filled with “free” water molecules. Thus, we believe 42 probably has been converted to amine 43 before being transported out of the pocket, which prevented further hydrolysis. Taken together, both the metabolomic result and the fluoride ion release experiments indicate that 14 undergoes turnover by pathway b2 with the regeneration of PLP and the release of metabolite 43 (Scheme 5).
Figure 7.

Detection of metabolite 43 as the turnover product for hOAT-14. (A) Extracted ion chromatogram for 43 (+ESI, 170.04–170.08 m/z). (B) Confirmation of metabolite 43 using tandem mass spectrometry
Plausible Mechanism for 14 with hOAT
On the basis of the above inactivation and turnover mechanism studies, a modified pathway for 14 with hOAT is proposed in Scheme 6. Initially, Schiff base 25 was generated from 14 and PLP, followed by deprotonation at the γ-position. According to the partition ratio (0.6) of 14 (Figure S14), 63% of 26 is converted to active intermediate 27` (pathway a), which involves rotation about the C-N single bond of 46 and protonation at the PLP-C4` position of 48. Molecular docking and MD simulation studies suggest that the carboxylate of 27` forms a stable interaction with Arg413 (Figure 5C) when the original salt bridge is disrupted. This binding pose allows the warhead to be positioned close to *Thr322 from the other chain (Figure 5C, 3.4 Å vs. 5.9 Å), leading to the formation of 36, which was trapped in the soaking crystal structure (Figure 6). Considering the short distance (3.4 Å) between Lys292 and *Thr322 observed in the MD simulation studies of 27`, Lys292 might play a role in activating *Thr322 for this nucleophilic attack, which is similar to the activation of water molecules by Lys329 in the case of 5 and 9 with GABA-AT (Scheme 2). The covalent attachment to *Thr322 pulls the warhead closer to Lys292 and allows a second nucleophilic attack on 36 to give 50. Finally, adduct 32` is formed to release the potential strain from two covalent attachments (Figure 4), while its carboxylate stays away from Arg413 and forms a H-bond with Tyr55 instead. Interestingly, Arg413 and Glu235 do not form a salt bridge after inactivation but interact with nearby Gln266 (Figure 4). The formation of the double-covalent adduct (32`) in the active site of hOAT-14 is supported by three types of protein MS and X-ray crystallography. With regard to the turnover pathway, 37% of intermediate 26 is converted to intermediate 33 by protonation at the difluoromethylenyl group, followed by Lys292 attack at the PLP iminium group, leading to regeneration of PLP and the release of 43 as the final metabolite. Among the proposed metabolites (Scheme 5), only 43 was detected by HRMS and confirmed by its fragmentation spectrum (Figure 7) and results of fluoride ion release experiments (Table 3).
Scheme 6.

Plausible mechanism for 14 with hOAT
CONCLUSIONS
Recently, selective pharmacological inhibition of human ornithine aminotransferase (hOAT) has been shown as a potential therapeutic approach to treat hepatocellular carcinoma (HCC) and other related cancers. Inspired by the inactivation mechanism for aminotransferase inactivators 5 and 9, a novel series of six-membered ring analogs (12-15) was rationally designed and chirally synthesized on the basis of the difference in the active sites of aminotransferases. Among them, analog 14 was identified as a selective hOAT inactivator and is 22 times more potent than analog 10, which has good in vivo anticancer activity. A plausible mechanism for 14 with hOAT is proposed (Scheme 6) on the basis of four distinct applications of mass spectrometry, two types of protein crystallography, and molecular dynamics (MD) simulations with the aid of a dialysis experiment, total turnover, and measurement of fluoride ion release. Notably, this is the first time in which native mass spectrometry and top-down proteomics have been applied to a mechanistic study of a PLP-dependent enzyme inactivator. Remarkably, double-covalent adduct 32` was formed in the active site of hOAT via sequential nucleophilic attacks, first by the noncatalytic residue *Thr322 and then by the catalytic residue Lys292, at the warhead of active intermediate 27`. To the best of our knowledge, this is the first example of an MBI forming a single adduct with two covalent bonds from two residues in the active site of an enzyme, one of which a noncatalytic residue. Moreover, an unusual turnover mechanism also has been demonstrated, in which aromatic metabolite 43 was generated along with regeneration of cofactor PLP. Interestingly, no water molecule was involved in the inactivation mechanism of hOAT with 14, whereas water molecules play an important role in the mechanisms of 5 and 9 with GABA-AT (Scheme 2). We believe this mechanistic difference derives from the enzymatic machinery of these two aminotransferases, which should contribute to the further rational design of selective inactivators.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (grant R01 DA030604 to R.B.S. and grants P30 DA018310 and S10OD025194 to N.L.K.) for financial support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Comet Bridges Stampede2 through allocation TG-CHE190070, which is supported by National Science Foundation grant number ACI-1548562. This work made use of the IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN). X-ray diffraction data collection used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE- AC02-06CH11357. The use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817). We thank Dr. Joseph Brunzelle at LS-CAT for help on data collection. We thank Dr. Matthew J. Moschitto and Dr. Sida Shen for constructive comments and Dr. Mauricio T. Tavares and Dr. Glaucio M. Ferreira for help on MD simulation studies.
ABBREVIATIONS
- DMP
Dess-Martin periodinane
- PMB
p-methoxybenzyl
- CAN
cerium (IV) ammonium nitrate
- Boc
tert-butyloxycarbonyl
- KHMDS
potassium bis(trimethylsilyl)amide
- DIPEA
N,N-diisopropylethylamine
- Boc2O
di-tert-butyldicarbonate
- m-CPBA
meta-chloroperoxybenzoic acid
- tBuOK
potassium tert-butoxide
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DCM
dichloromethane
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supporting Information is available free of charge on the ACS Publications website. Supplementary figures and tables, experimental methods, syntheses, spectra, and crystallographic data.
Accession Codes
Atomic coordinates and corresponding structure factors for the soaking result and cocrystal complex have been deposited at the Protein Data Bank (PDB) as 7LOM, 7LON and 7LNM, respectively. Authors will release the atomic coordinates upon article publication.
The authors declare no competing financial interest.
REFERENCES
- 1.Sayiner M; Golabi P; Younossi ZM Disease burden of hepatocellular carcinoma: a global perspective. Digest Dis. Sci 2019, 64, 910–917. [DOI] [PubMed] [Google Scholar]
- 2.Personeni N; Rimassa L. Hepatocellular carcinoma: a global disease in need of individualized treatment strategies. J. Oncol. Pract 2017, 13, 368–370. [DOI] [PubMed] [Google Scholar]
- 3.Sherman M; Bruix J; Porayko M; Tran T; Comm APG Screening for hepatocellular carcinoma: The rationale for the American association for the study of liver diseases recommendations. Hepatology 2012, 56, 793–796. [DOI] [PubMed] [Google Scholar]
- 4.Yang JD; Roberts LR Hepatocellular carcinoma: a global view. Nat. Rev. Gastro. Hepat 2010, 7, 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leathers JS; Balderramo D; Prieto J; Diehl F; Gonzalez-Ballerga E; Ferreiro MR; Carrera E; Barreyro F; Diaz-Ferrer J; Singh D; Mattos AZ; Carrilho F; Debes JD Sorafenib for treatment of hepatocellular carcinoma a survival analysis from the aouth american liver research network. J. Clin. Gastroenterol 2019, 53, 464–469. [DOI] [PubMed] [Google Scholar]
- 6.de Rosamel L; Blanc JF Emerging tyrosine kinase inhibitors for the treatment of hepatocellular carcinoma. Expert Opin. Emerg. Dr 2017, 22, 175–190. [DOI] [PubMed] [Google Scholar]
- 7.Milgrom DP; Maluccio MA; Koniaris LG Management of hepatocellular carcinoma (HCC). Curr. Surg. Rep 2016, 4, 20; DOI 10.1007/s40137-016-0143-4. [DOI] [Google Scholar]
- 8.de Lope CR; Tremosini S; Forner A; Reig M; Bruix J. Management of HCC. J. Hepatol 2012, 56, S75–S87. [DOI] [PubMed] [Google Scholar]
- 9.Ginguay A; Cynober L; Curis E; Nicolis I. Ornithine aminotransferase, an important glutamate-metabolizing enzyme at the crossroads of multiple metabolic pathways. Biology (Basel) 2017, 6 (1), 18; doi: 10.3390/biology6010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zigmond E; Ben Ya’acov A; Lee H; Lichtenstein Y; Shalev Z; Smith Y; Zolotarov L; Ziv E; Kalman R; Le HV; Lu HJ; Silverman RB; Ilant Y. Suppression of hepatocellular carcinoma by inhibition of overexpressed ornithine aminotransferase. ACS Med. Chem. Lett 2015, 6, 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.John RA Pyridoxal Phosphate-Dependent Enzymes. Biochim. Biophys. Acta 1995, 1248, 81–96.. [DOI] [PubMed] [Google Scholar]
- 12.Herzfeld A; Knox WE The properties developmental formation and estrogen induction of ornithine aminotransferase in rat tissues. J. Biol. Chem 1968, 243, 3327–3332. [PubMed] [Google Scholar]
- 13.Peraino C; Bunville LG; Tahmisia TN Chemical physical and morphological properties of ornithine aminotransferase from rat liver. J. Biol. Chem 1969, 244, 2241–2249. [PubMed] [Google Scholar]
- 14.Ding Z; Ericksen RE; Escande-Beillard N; Lee QY; Loh A; Denil S; Steckel M; Haegebarth A; Wai Ho TS; Chow P; Toh HC; Reversade B; Gruenewald S; Han W. Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J. Hepatol 2020, 72, 725–735. [DOI] [PubMed] [Google Scholar]
- 15.Altman BJ; Stine ZE; Dang CV From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phang JM; Liu W; Hancock CN; Fischer JW Proline metabolism and cancer: emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phang JM; Liu W; Zabirnyk O. Proline metabolism and microenvironmental stress. Annu. Rev. Nutr 2010, 30, 441–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang L; Zeng J; Geng P; Fang C; Wang Y; Sun M; Wang C; Wang J; Yin P; Hu C; Guo L; Yu J; Gao P; Li E; Zhuang Z; Xu G; Liu Y. Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular varcinoma. Clin. Cancer Res 2018, 24, 474–485. [DOI] [PubMed] [Google Scholar]
- 19.Cadoret A; Ovejero C; Terris B; Souil E; Levy L; Lamers WH; Kitajewski J; Kahn A; Perret C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [DOI] [PubMed] [Google Scholar]
- 20.Colnot S; Decaens T; Niwa-Kawakita M; Godard C; Hamard G; Kahn A; Giovannini M; Perret C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. P. Natl. Acad. Sci. USA 2004, 101, 17216–17221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YF; Wu L; Li K; Liu FR; Wang L; Zhang DL; Zhou J; Ma X; Wang SY; Yang SY Ornithine aminotransferase promoted the proliferation and metastasis of non-small cell lung cancer via upregulation of miR-21. J. Cell Physiol 2019, 234, 12828–12838. [DOI] [PubMed] [Google Scholar]
- 22.Markova M; Peneff C; Hewlins MJE; Schirmer T; John RA Determinants of substrate specificity in omega-aminotransferases. J. Biol. Chem 2005, 280, 36409–36416. [DOI] [PubMed] [Google Scholar]
- 23.Mehta PK; Hale TI; Christen P. Evolutionary relationships among aminotransferases - tyrosine aminotransferase, histidinol-phosphate aminotransferase, and aspartate-aminotransferase are homologous proteins. Eur. J. Biochem 1989, 186, 249–253. [DOI] [PubMed] [Google Scholar]
- 24.Cooper AJL Glutamate-gamma-aminobutyrate transaminase. Methods Enzymol. 1985, 113, 80–82. [DOI] [PubMed] [Google Scholar]
- 25.Silverman RB Design and mechanism of GABA aminotransferase inactivators. treatments for epilepsies and addictions. Chem. Rev 2018, 118, 4037–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silverman RB Mechanism-based enzyme inactivators. Methods Enzymol. 1995, 249, 240–283. [DOI] [PubMed] [Google Scholar]
- 27.Rando RR Mechanism-based enzyme inactivators. Pharmacol. Rev 1984, 36, 111–142. [PubMed] [Google Scholar]
- 28.Walsh CT Suicide Substrates, mechanism based enzyme inactivators - recent developments. Annu. Rev. Biochem 1984, 53, 493–535. [DOI] [PubMed] [Google Scholar]
- 29.Juncosa JI; Takaya K; Le HV; Moschitto MJ; Weerawarna PM; Mascarenhas R; Liu DL; Dewey SL; Silverman RB Design and mechanism of (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid, a highly potent gamma-aminobutyric acid aminotransferase inactivator for the treatment of addiction. J. Am. Chem. Soc 2018, 140, 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Y; Gerasimov MR; Kvist T; Wellendorph P; Madsen KK; Pera E; Lee H; Schousboe A; Chebib M; Brauner-Osborne H; Craft CM; Brodie JD; Schiffer WK; Dewey SL; Miller SR; Silverman RB (1S, 3S)-3-Amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115), a potent gamma-aminobutyric acid aminotransferase inactivator for the treatment of cocaine addiction. J. Med. Chem 2012, 55, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sivenius MR; Ylinen A; Murros K; Matilainen R; Riekkinen P. Double-blind dose reduction study of vigabatrin in complex partial epilepsy. Epilepsia 1987, 28, 688–92. [DOI] [PubMed] [Google Scholar]
- 32.Wheless JW; Ramsay RE; Collins SD Vigabatrin. Neurotherapeutics 2007, 4, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nanavati SM; Silverman RB Mechanisms of inactivation of gamma-aminobutyric-acid aminotransferase by the antiepilepsy drug gamma-vinyl GABA (vigabatrin). J. Am. Chem. Soc 1991, 113, 9341–9349. [Google Scholar]
- 34.Silverman RB The 2011 E. B. Hershberg Award for Important Discoveries in Medicinally Active Substances: (1S,3S)-3-Amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115), a GABA Aminotransferase Inactivator and New Treatment for Drug Addiction and Infantile Spasms. J. Med. Chem 2012, 55, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan Y; Qiu J; Silverman RB Design, synthesis, and biological activity of a difluoro-substituted, conformationally rigid vigabatrin analogue as a potent gamma-aminobutyric acid aminotransferase inhibitor. J. Med. Chem 2003, 46, 5292–5293. [DOI] [PubMed] [Google Scholar]
- 36.Lee H; Doud EH; Wu R; Sanishvili R; Juncosa JI; Liu DL; Kelleher NL; Silverman RB Mechanism of inactivation of gamma-aminobutyric acid aminotransferase by (1S,3S)-3-amino-4-difluoromethylene-1-cyclopentanoic acid (CPP-115). J. Am. Chem. Soc 2015, 137, 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Briggs SW; Mowrey W; Hall CB; Galanopoulou AS CPP-115, a vigabatrin analogue, decreases spasms in the multiple-hit rat model of infantile spasms. Epilepsia 2014, 55, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doumlele K; Conway E; Hedlund J; Tolete P; Devinsky O, A case report on the efficacy of vigabatrin analogue (1S, 3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115) in a patient with infantile spasms. Epilepsy Behav. Case 2016, 6, 67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H; Juncosa JI; Silverman RB Ornithine aminotransferase versus GABA aminotransferase: implications for the design of new anticancer drugs. Med. Res. Rev 2015, 35, 286–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu W; Doubleday PF; Catlin DS; Weerawarna PM; Butrin A; Shen S; Wawrzak Z; Kelleher NL; Liu D; Silverman RB A remarkable difference that one fluorine atom confers on the mechanisms of inactivation of human ornithine aminotransferase by two cyclohexene analogues of gamma-aminobutyric acid. J. Am. Chem. Soc 2020, 142, 4892–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeung YY; Hong S; Corey EJ A short enantioselective pathway for the synthesis of the anti-influenza neuramidase inhibitor oseltamivir from 1,3-butadiene and acrylic acid. J. Am. Chem. Soc 2006, 128, 6310–6311. [DOI] [PubMed] [Google Scholar]
- 42.Moschitto MJ; Silverman RB Synthesis of (S)-3-amino-4-(difluorornethylenyl)-cyclopent-1-ene-1-carboxylic acid (OV329), a potent inactivator of gamma-aminobutyric acid aminotransferase. Org. Lett 2018, 20, 4589–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rando RR Mechanism of irreversible inhibition of gamma-aminobutyric acid-alpha-ketoglutaric acid transaminase by neurotoxin gabaculine. Biochemistry 1977, 16, 4604–4610. [DOI] [PubMed] [Google Scholar]
- 44.Shah SA; Shen BW; Brunger AT Human ornithine aminotransferase complexed with L-canaline and gabaculine: structural basis for substrate recognition. Structure 1997, 5, 1067–1075. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi K; Miyazawa S; Endo A. Isolation and inhibitory activity of gabaculine, a new potent inhibitor of gamma-aminobutyrate aminotransferase produced by a streptomyces. Febs. Lett 1977, 76, 207–210. [DOI] [PubMed] [Google Scholar]
- 46.Skinner OS; Haverland NA; Fornelli L; Melani RD; Do Vale LHF; Seckler HS; Doubleday PF; Schachner LF; Srzentic K; Kelleher NL; Compton PD Top-down characterization of endogenous protein complexes with native proteomics. Nat. Chem. Biol 2018, 14, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moschitto MJ; Doubleday PF; Catlin DS; Kelleher NL; Liu DL; Silverman RB Mechanism of inactivation of ornithine aminotransferase by (1S,3S)-3-amino-4-(hexafluoropropan-2-ylidenyl)cyclopentane-1-carboxylic Acid. J. Am. Chem. Soc 2019, 141, 10711–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen SD; Doubleday PF; Weerawarna PM; Zhu W; Kelleher NL; Silverman RB Mechanism-Based Design of 3-Amino-4-Halocyclopentenecarboxylic Acids as Inactivators of GABA Aminotransferase. ACS Med. Chem. Lett 2020, 11, 1949–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zubarev RA; Horn DM; Fridriksson EK; Kelleher NL; Kruger NA; Lewis MA; Carpenter BK; McLafferty FW Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem 2000, 72, 563–573. [DOI] [PubMed] [Google Scholar]
- 50.Liu F; Lossl P; Scheltema R; Viner R; Heck AJR Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun 2017, 8, 15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olsen JV; Macek B; Lange O; Makarov A; Horning S; Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–12. [DOI] [PubMed] [Google Scholar]
- 52.DeHart CJ; Fellers RT; Fornelli L; Kelleher NL; Thomas PM Bioinformatics analysis of top-down mass spectrometry data with ProSight Lite. Methods Mol. Biol 2017, 1558, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen BW; Hennig M; Hohenester E; Jansonius JN; Schirmer T. Crystal structure of human recombinant ornithine aminotransferase. J .Mol. Biol 1998, 277, 81–102. [DOI] [PubMed] [Google Scholar]
- 54.Mascarenhas R; Le HV; Clevenger KD; Lehrer HJ; Ringe D; Kelleher NL; Silverman RB; Liu D. Selective targeting by a mechanism-based inactivator against pyridoxal 5’-phosphate-dependent enzymes: mechanisms of inactivation and alternative turnover. Biochemistry 2017, 56, 4951–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Storici P; Capitani G; Muller R; Schirmer T; Jansonius JN Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J. Mol. Biol 1999, 285, 297–309. [DOI] [PubMed] [Google Scholar]
- 56.Layer RW The chemistry of imines. Chem. Rev 1963, 63, 489–510. [Google Scholar]
- 57.Johnson JE; Morales NM; Gorczyca AM; Dolliver DD; McAllister MA Mechanisms of acid-catalyzed Z/E isomerization of imines. J. Org. Chem 2001, 66, 7979–7985. [DOI] [PubMed] [Google Scholar]
- 58.Galvez J; Guirado A. A theoretical study of topomerization of imine systems: inversion, rotation or mixed mechanisms? J. Comput. Chem 2010, 31, 520–31. [DOI] [PubMed] [Google Scholar]
- 59.Traven’ VF; Ivanov IV; Panov AV; Safronova OB; Chibisova TA Solvent-induced E/Z(C=N)-isomerization of imines of some hydroxy-substituted formylcoumarins. Russ. Chem.Bull 2008, 57, 1989–1995. [Google Scholar]
- 60.Butrin A; Beaupre BA; Kadamandla N; Zhao P; Shen S; Silverman RB; Moran GR; Liu D. Structural and kinetic analyses reveal the dual inhibition modes of ornithine aminotransferase by (1S,3S)-3-Amino-4-(hexafluoropropan-2-ylidenyl)-cyclopentane-1-carboxylic Acid (BCF3). ACS Chem. Biol 2021, 16, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.